
Mechanisms of Immunotoxicity: Stressors and Evaluators


Abstract
:1. Introduction and Overview of the Immune System
1.1. Immune Cells and Their Development
1.2. Innate and Adaptive Immune Cells and Their Activities
2. Immune Hypersensitivities and Autoimmune Disease
2.1. Autoimmune Diseases
2.2. Hypersensitivity Reactions
3. Mucosal Immunity, the Microbiome and Food Allergies
3.1. Mucosal Immunity
3.2. Microbiota
3.3. Food Allergies
4. Molecular Immunotoxicology of Environmental Stressors
4.1. Physical Stressors
4.2. Chemical Stressors
4.2.1. Metals
4.2.2. Cigarette Smoke
4.2.3. Pesticides and Other Organic Compounds
4.2.4. Endocrine-Disrupting Chemicals
4.2.5. Others
4.3. Psychological Stressors
5. Methods for Assessing Immunotoxicity
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- National Research Council (US) Subcommittee on Immunotoxicology. Biologic Markers in Immunotoxicology; National Academies Press: Washington, DC, USA, 1992. [Google Scholar]
- Picard, C.; Puel, A.; Bustamante, J.; Ku, C.-L.; Casanova, J.-L. Primary immunodeficiencies associated with pneumococcal disease. Curr. Opin. Allergy Clin. Immunol. 2003, 3, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Casale, G.P.; Bavari, S.; Connolly, J.J. Inhibition of human serum complement activity by diisopropylfluorophosphate and selected anticholinesterase insecticides. Fundam. Appl. Toxicol. 1989, 12, 460–468. [Google Scholar] [CrossRef]
- Hepburn, A.; Davies, K. Infection and SLE. Ann. Rheum. Dis. 2002, 61, 668–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falcone, M.; Sarvetnick, N. Cytokines that regulate autoimmune responses. Curr. Opin. Immunol. 1999, 11, 670–676. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Ma, A.; Lipsky, P. Cytokines and autoimmunity. Nat. Rev. Immunol. 2002, 2, 37–45. [Google Scholar] [CrossRef]
- Yadav, D.; Sarvetnick, N. Cytokines and autoimmunity: Redundancy defines their complex nature. Curr. Opin. Immunol. 2003, 15, 697–703. [Google Scholar] [CrossRef]
- Andreakos, E.T.; Foxwell, B.M.; Brennan, F.M.; Maini, R.N.; Feldmann, M. Cytokines and anti-cytokine biologicals in autoimmunity: Present and future. Cytokine Growth Factor Rev. 2002, 13, 299–313. [Google Scholar] [CrossRef]
- Frohman, M.; Francfort, J.; Cowing, C. T-dependent destruction of thyroid isografts exposed to IFN-gamma. J. Immunol. 1991, 146, 2227–2234. [Google Scholar]
- Caturegli, P.; Hejazi, M.; Suzuki, K.; Dohan, O.; Carrasco, N.; Kohn, L.; Rose, N. Hypothyroidism in transgenic mice expressing IFN-γ in the thyroid. Proc. Natl. Acad. Sci. USA 2000, 97, 1719–1724. [Google Scholar] [CrossRef] [Green Version]
- Uetrecht, J.P. Idiosyncratic drug reactions: Possible role of reactive metabolites generated by leukocytes. Pharm. Res. 1989, 6, 265–273. [Google Scholar] [CrossRef]
- Lee, A.Y.; Choi, J.; Chey, W.Y. Patch testing with carbamazepine and its main metabolite carbamazepine epoxide in cutaneous adverse drug reactions to carbamazepine. Contact Dermat. 2003, 48, 137–139. [Google Scholar] [CrossRef]
- Herdeg, C.; Hilt, F.; Büchtemann, A.; Bianchi, L.; Klein, R. Allergic cholestatic hepatitis and exanthema induced by metamizole: Verification by lymphocyte transformation test. Liver 2002, 22, 507–513. [Google Scholar] [CrossRef]
- Choquet-Kastylevsky, G.; Vial, T.; Descotes, J. Allergic adverse reactions to sulfonamides. Curr. Allergy Asthma Rep. 2002, 2, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Christie, P.E.; Henderson, W.R., Jr. Lipid inflammatory mediators: Leukotrienes, prostaglandins, platelet-activating factor. Clin. Allergy Immunol. 2002, 16, 233–254. [Google Scholar]
- Carr, T.F.; Kraft, M. Management of severe asthma before referral to the severe asthma specialist. J. Allergy Clin. Immunol. Pract. 2017, 5, 877–886. [Google Scholar] [CrossRef]
- Salama, A.; Mueller-Eckhardt, C. On the Mechanisms of Sensitization and Attachment of Antibodies to RBC in Drug-Induced Immune Hemolytic Anemia; Elsevier: Amsterdam, The Netherlands, 1987. [Google Scholar]
- Aster, R.H. Drug-induced immune thrombocytopenia: An overview of pathogenesis. Semin. Hematol. 1999, 36, 2–6. [Google Scholar] [PubMed]
- Ropars, A.; Marion, S.; Takorabet, L.; Braun, J.; Charreire, J. Antibodies specific for human thyrotropin receptor induce MHC antigen expression in thyroid cells. J. Immunol. 1994, 153, 3345–3352. [Google Scholar]
- Weber, M.; Andrassy, K.; Pullig, O.; Koderisch, J.; Netzer, K. Antineutrophil-cytoplasmic antibodies and antiglomerular basement membrane antibodies in Goodpasture’s syndrome and in Wegener’s granulomatosis. J. Am. Soc. Nephrol. 1992, 2, 1227–1234. [Google Scholar] [CrossRef]
- Köhl, J.; Gessner, J.E. On the role of complement and Fc γ-receptors in the Arthus reaction. Mol. Immunol. 1999, 36, 893–903. [Google Scholar] [CrossRef]
- LH, C.; GF, D. Drug-induced vasculitis. Curr. Opin. Rheumatol. 1996, 8, 34–40. [Google Scholar]
- Herishanu, Y. Rituximab-Induced Serum Sickness; Wiley Online Library: Hoboken, NJ, USA, 2002. [Google Scholar]
- Nasir, A.; Gaspari, A.A. Contact dermatitis. Clin. Rev. Allergy Immunol. 1996, 14, 151–184. [Google Scholar] [PubMed]
- Cavani, A.; Sebastiani, S.; Nasorri, F.; Albanesi, C.; Girolomoni, G. Allergic contact dermatitis-Effector and regulatory mechanisms. Allergy Clin. Immunol. Int. 2002, 14, 156–160. [Google Scholar] [CrossRef]
- Pichler, W.J. Delayed drug hypersensitivity reactions. Ann. Intern. Med. 2003, 139, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Hari, Y.; Urwyler, A.; Hurni, M.; Yawalkar, N.; Dahinden, C.; Wendland, T.; Braathen, L.R.; Matter, L.; Pichler, W.J. Distinct serum cytokine levels in drug–and measles–induced exanthema. Int. Arch. Allergy Immunol. 1999, 120, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Yawalkar, N.; Egli, F.; Hari, Y.; Nievergelt, H.; Braathen, L.; Pichler, W. Infiltration of cytotoxic T cells in drug-induced cutaneous eruptions. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2000, 30, 847–855. [Google Scholar] [CrossRef]
- Britschgi, M.; Steiner, U.C.; Schmid, S.; Depta, J.P.; Senti, G.; Bircher, A.; Burkhart, C.; Yawalkar, N.; Pichler, W.J. T-cell involvement in drug-induced acute generalized exanthematous pustulosis. J. Clin. Investig. 2001, 107, 1433–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlumberger, H. Pseudo-allergic reactions to drugs and chemicals. Ann. Allergy 1983, 51, 317–324. [Google Scholar]
- Hochstein, P. Glucose-6-phosphate dehydrogenase deficiency: Mechanisms of drug-induced hemolysis. Exp. Eye Res. 1971, 11, 389–395. [Google Scholar] [CrossRef]
- Shenton, J.M.; Teranishi, M.; Abu-Asab, M.S.; Yager, J.A.; Uetrecht, J.P. Characterization of a potential animal model of an idiosyncratic drug reaction: Nevirapine-induced skin rash in the rat. Chem. Res. Toxicol. 2003, 16, 1078–1089. [Google Scholar] [CrossRef]
- Batchelor, R.; Horne, G.; Rogerson, H. An unusual reaction to procaine penicillin in aqueous suspension. Lancet 1951, 2, 195–198. [Google Scholar] [CrossRef]
- Watkins, J. Markers and mechanisms of anaphylactoid reactions. Monogr. Allergy 1992, 30, 108–129. [Google Scholar] [PubMed]
- McKinnon, R.; Wildsmith, J. Histaminoid reactions in anaesthesia. Br. J. Anaesth. 1995, 74, 217–228. [Google Scholar] [CrossRef]
- Mertes, P.; Laxenaire, M.-C. Allergic reactions occurring during anaesthesia. Eur. J. Anaesthesiol. 2002, 19, 240–262. [Google Scholar] [CrossRef]
- Bowdle, T.A. Adverse effects of opioid agonists and agonist-antagonists in anaesthesia. Drug Saf. 1998, 19, 173–189. [Google Scholar] [CrossRef]
- Renz, C.; Lynch, J.; Thurn, J.; Moss, J. Histamine release during rapid vancomycin administration. Inflamm. Res. 1998, 47, 69–70. [Google Scholar] [CrossRef] [PubMed]
- Dorr, R.T. Pharmacology and toxicology of Cremophor EL diluent. Ann. Pharmacother. 1994, 28, S11–S14. [Google Scholar] [CrossRef] [PubMed]
- Hoffmeister, H.M.; Heller, W. Radiographic contrast media and the coagulation and complement systems. Investig. Radiol. 1996, 31, 591–595. [Google Scholar] [CrossRef]
- Van der Kolk, L.; Grillo-Lopez, A.; Baars, J.; Hack, C.; Van Oers, M. Complement activation plays a key role in the side-effects of rituximab treatment. Br. J. Haematol. 2001, 115, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, I.; Fujihashi, K.; Kiyono, H. Mucosal regulatory cells in the gastrointestinal tract and periodontium. Periodontology 2000 2010, 54, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Kiyono, H.; Azegami, T. The mucosal immune system: From dentistry to vaccine development. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2015, 91, 423–439. [Google Scholar] [CrossRef] [Green Version]
- Erickson, K.L.; Hubbard, N.E. Assessing mucosal immunity with new concepts and innovative, time-honored strategies. Nutr. Rev. 2009, 67, S172–S182. [Google Scholar] [CrossRef] [PubMed]
- Mayer, L. Mucosal immunity. Pediatrics 2003, 111, 1595–1600. [Google Scholar] [CrossRef]
- Feller, L.; Altini, M.; Khammissa, R.A.G.; Chandran, R.; Bouckaert, M.; Lemmer, J. Oral mucosal immunity. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2013, 116, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Stephen-Victor, E.; Chatila, T.A. Regulation of oral immune tolerance by the microbiome in food allergy. Curr. Opin. Immunol. 2019, 60, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Tai, L.; Guo, J.X.; Chen, Y.; Lefever, D.E.; Huang, G.; Lawrence, D.A. Molecular Mechanisms of Immunotoxicity. In Molecular and Biochemical Toxicology, 5th ed.; Robert Smart, E.H., Ed.; John Wiley and Sons: Hoboken, NJ, USA, 2018; pp. 773–822. [Google Scholar]
- Neish, A.S. Mucosal immunity and the microbiome. Ann. Am. Thorac. Soc. 2014, 11 (Suppl. 1), S28–S32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Reddy, D.N. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787. [Google Scholar] [CrossRef]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [Green Version]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Distrutti, E.; Monaldi, L.; Ricci, P.; Fiorucci, S. Gut microbiota role in irritable bowel syndrome: New therapeutic strategies. World J. Gastroenterol. 2016, 22, 2219. [Google Scholar] [CrossRef]
- Macfarlane, G.T.; Macfarlane, S. Human colonic microbiota: Ecology, physiology and metabolic potential of intestinal bacteria. Scand. J. Gastroenterol. 1997, 32, 3–9. [Google Scholar] [CrossRef]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Manco, M.; Putignani, L.; Bottazzo, G.F. Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocr. Rev. 2010, 31, 817–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geuking, M.B.; Köller, Y.; Rupp, S.; McCoy, K.D. The interplay between the gut microbiota and the immune system. Gut Microbes 2014, 5, 411–418. [Google Scholar] [CrossRef]
- Malaguarnera, L. Vitamin D and microbiota: Two sides of the same coin in the immunomodulatory aspects. Int. Immunopharmacol. 2020, 79, 106112. [Google Scholar] [CrossRef]
- Murdaca, G.; Tonacci, A.; Negrini, S.; Greco, M.; Borro, M.; Puppo, F.; Gangemi, S. Emerging role of vitamin D in autoimmune diseases: An update on evidence and therapeutic implications. Autoimmun. Rev. 2019, 18, 102350. [Google Scholar] [CrossRef] [PubMed]
- Murdaca, G.; Pioggia, G.; Negrini, S. Vitamin D and Covid-19: An update on evidence and potential therapeutic implications. Clin. Mol. Allergy 2020, 18, 23. [Google Scholar] [CrossRef]
- Allegra, A.; Musolino, C.; Tonacci, A.; Pioggia, G.; Gangemi, S. Interactions between the MicroRNAs and microbiota in cancer development: Roles and therapeutic opportunities. Cancers 2020, 12, 805. [Google Scholar] [CrossRef] [Green Version]
- Heine, R.G. Food allergy prevention and treatment by targeted nutrition. Ann. Nutr. Metab. 2018, 72, 33–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimitrov, V.; White, J.H. Vitamin D signaling in intestinal innate immunity and homeostasis. Mol. Cell. Endocrinol. 2017, 453, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Akimbekov, N.S.; Digel, I.; Sherelkhan, D.K.; Lutfor, A.B.; Razzaque, M.S. Vitamin d and the host-gut microbiome: A brief overview. Acta Histochem. Cytochem. 2020, 53, 33–42. [Google Scholar] [CrossRef]
- Ignacio, A.; Breda, C.N.S.; Camara, N.O.S. Innate lymphoid cells in tissue homeostasis and diseases. World J. Hepatol. 2017, 9, 979. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Waddell, A.; Lin, Y.-D.; Cantorna, M.T. Dysbiosis caused by vitamin D receptor deficiency confers colonization resistance to Citrobacter rodentium through modulation of innate lymphoid cells. Mucosal Immunol. 2015, 8, 618–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantorna, M.T.; Waddell, A. The vitamin D receptor turns off chronically activated T cells. Ann. N. Y. Acad. Sci. 2014, 1317, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Cantorna, M.T.; Lin, Y.-D.; Arora, J.; Bora, S.; Tian, Y.; Nichols, R.G.; Patterson, A.D. Vitamin D regulates the microbiota to control the numbers of RORγt/FoxP3+ regulatory T cells in the colon. Front. Immunol. 2019, 10, 1772. [Google Scholar] [CrossRef] [Green Version]
- Branum, A.M.; Lukacs, S.L. Food Allergy among US Children: Trends in Prevalence and Hospitalizations; NHCS: Atlanta, GA, USA, 2008. [Google Scholar]
- Waserman, S.; Watson, W. Food allergy. Allergy Asthma Clin. Immunol. 2011, 7, S7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, G.; Berti, I.; Burks, A.W.; Krauss, B.; Barbi, E. IgE-mediated food allergy in children. Lancet 2013, 382, 1656–1664. [Google Scholar] [CrossRef]
- Anvari, S.; Miller, J.; Yeh, C.-Y.; Davis, C.M. IgE-mediated food allergy. Clin. Rev. Allergy Immunol. 2019, 57, 244–260. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Chehade, M.; Liu, W.; Xiong, H.; Mayer, L.; Berin, M.C. Allergen-IgE complexes trigger CD23-dependent CCL20 release from human intestinal epithelial cells. Gastroenterology 2007, 133, 1905–1915. [Google Scholar] [CrossRef] [Green Version]
- Lipscomb, M.F.; Wilder, J.A.; Masten, B.J. Dendritic cells and their role in linking innate and adaptive immune responses. In The Biology of Dendritic Cells and HIV Infection; Springer: Berlin/Heidelberg, Germany, 2007; pp. 45–84. [Google Scholar]
- Bellanti, J.A. Cytokines and allergic diseases: Clinical aspects. Allergy Asthma Proc. 1998, 19, 337. [Google Scholar] [CrossRef]
- Herberth, G.; Daegelmann, C.; Röder, S.; Behrendt, H.; Krämer, U.; Borte, M.; Heinrich, J.; Herbarth, O.; Lehmann, I.; LISAplus Study Group. IL-17E but not IL-17A is associated with allergic sensitization: Results from the LISA study. Pediatric Allergy Immunol. 2010, 21, 1086–1090. [Google Scholar] [CrossRef]
- Ngoc, L.P.; Gold, D.R.; Tzianabos, A.O.; Weiss, S.T.; Celedon, J.C. Cytokines, allergy, and asthma. Curr. Opin. Allergy Clin. Immunol. 2005, 5, 161–166. [Google Scholar] [CrossRef]
- Shea-Donohue, T.; Stiltz, J.; Zhao, A.; Notari, L. Mast cells. Curr. Gastroenterol. Rep. 2010, 12, 349–357. [Google Scholar] [CrossRef] [Green Version]
- Tkaczyk, C.; Okayama, Y.; Metcalfe, D.D.; Gilfillan, A.M. Fcγ receptors on mast cells: Activatory and inhibitory regulation of mediator release. Int. Arch. Allergy Immunol. 2004, 133, 305–315. [Google Scholar] [CrossRef]
- Klemann, C.; Ammann, S.; Heizmann, M.; Fuchs, S.; Bode, S.F.; Heeg, M.; Fuchs, H.; Lehmberg, K.; Zur Stadt, U.; Roll, C. Hemophagocytic lymphohistiocytosis as presenting manifestation of profound combined immunodeficiency due to an ORAI1 mutation. J. Allergy Clin. Immunol. 2017, 140, 1721–1724. [Google Scholar] [CrossRef] [Green Version]
- Connors, L.; O’Keefe, A.; Rosenfield, L.; Kim, H. Non-IgE-mediated food hypersensitivity. Allergy Asthma Clin. Immunol. 2018, 14, 56. [Google Scholar] [CrossRef]
- Nowak-Węgrzyn, A.; Katz, Y.; Mehr, S.S.; Koletzko, S. Non-IgE-mediated gastrointestinal food allergy. J. Allergy Clin. Immunol. 2015, 135, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Morita, H.; Nomura, I.; Orihara, K.; Yoshida, K.; Akasawa, A.; Tachimoto, H.; Ohtsuka, Y.; Namai, Y.; Futamura, M.; Shoda, T. Antigen-specific T-cell responses in patients with non–IgE-mediated gastrointestinal food allergy are predominantly skewed to TH2. J. Allergy Clin. Immunol. 2013, 131, 590–592.e596. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.L.; Hwang, J.B.; Park, J.J.; Kim, S.G. Expression of transforming growth factor β1, transforming growth factor type I and II receptors, and TNF-α in the mucosa of the small intestine in infants with food protein–induced enterocolitis syndrome. J. Allergy Clin. Immunol. 2002, 109, 150–154. [Google Scholar] [CrossRef]
- Varga, J.; Frisvad, J.C.; Samson, R. Two new aflatoxin producing species, and an overview of Aspergillus section Flavi. Stud. Mycol. 2011, 69, 57–80. [Google Scholar] [CrossRef] [PubMed]
- Gürbay, A.; Sabuncuoğlu, S.A.; Girgin, G.; Şahin, G.; Yiğit, Ş.; Yurdakök, M.; Tekinalp, G. Exposure of newborns to aflatoxin M1 and B1 from mothers’ breast milk in Ankara, Turkey. Food Chem. Toxicol. 2010, 48, 314–319. [Google Scholar] [CrossRef]
- Pierron, A.; Alassane-Kpembi, I.; Oswald, I.P. Impact of mycotoxin on immune response and consequences for pig health. Anim. Nutr. 2016, 2, 63–68. [Google Scholar] [CrossRef]
- Phillips, T.M. Assessing environmental exposure in children: Immunotoxicology screening. J. Expo. Sci. Environ. Epidemiol. 2000, 10, 769–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duramad, P.; Tager, I.B.; Holland, N.T. Cytokines and other immunological biomarkers in children’s environmental health studies. Toxicol. Lett. 2007, 172, 48–59. [Google Scholar] [CrossRef] [Green Version]
- Norval, M.; Halliday, G.M. The consequences of UV-induced immunosuppression for human health. Photochem. Photobiol. 2011, 87, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.E.; Byrne, S.N. The immunologic revolution: Photoimmunology. J. Investig. Dermatol. 2012, 132, 896–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toda, M.; Wang, L.; Ogura, S.; Torii, M.; Kurachi, M.; Kakimi, K.; Nishikawa, H.; Matsushima, K.; Shiku, H.; Kuribayashi, K. UV irradiation of immunized mice induces type 1 regulatory T cells that suppress tumor antigen specific cytotoxic T lymphocyte responses. Int. J. Cancer 2011, 129, 1126–1136. [Google Scholar] [CrossRef]
- Murphy, G. Ultraviolet radiation and immunosuppression. Br. J. Dermatol. 2009, 161, 90–95. [Google Scholar] [CrossRef] [PubMed]
- De Gruijl, F. Skin cancer and solar UV radiation. Eur. J. Cancer 1999, 35, 2003–2009. [Google Scholar] [CrossRef]
- Setlow, R.; Carrier, W. Pyrimidine dimers in ultraviolet-irradiated DNA’s. J. Mol. Biol. 1966, 17, 237–254. [Google Scholar] [CrossRef]
- Varghese, A.; Patrick, M. Cytosine derived heteroadduct formation in ultraviolet-irradiated DNA. Nature 1969, 223, 299–300. [Google Scholar] [CrossRef]
- Granstein, R.D.; Matsui, M.S. UV radiation-induced immunosuppression and skin cancer. Cutis 2004, 74, 4–9. [Google Scholar]
- Snaidr, V.A.; Damian, D.L.; Halliday, G.M. Nicotinamide for photoprotection and skin cancer chemoprevention: A review of efficacy and safety. Exp. Dermatol. 2019, 28, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Yarovaya, L.; Waranuch, N.; Wisuitiprot, W.; Khunkitti, W. Effect of grape seed extract on skin fibroblasts exposed to UVA light and its photostability in sunscreen formulation. J. Cosmet. Dermatol. 2021, 20, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Hultman, P.; Pollard, K.M. Immunotoxicology of metals. In Handbook on the Toxicology of Metals; Elsevier: Amsterdam, The Netherlands, 2015; pp. 379–398. [Google Scholar]
- Nordstrom, D.K. Worldwide Occurrences of Arsenic in Ground Water; American Association for the Advancement of Science: Washington, DC, USA, 2002. [Google Scholar]
- Dangleben, N.L.; Skibola, C.F.; Smith, M.T. Arsenic immunotoxicity: A review. Environ. Health 2013, 12, 73. [Google Scholar] [CrossRef] [Green Version]
- Centeno, J.A.; Mullick, F.G.; Martinez, L.; Page, N.P.; Gibb, H.; Longfellow, D.; Thompson, C.; Ladich, E.R. Pathology related to chronic arsenic exposure. Environ. Health Perspect. 2002, 110, 883–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, L.S. Immunotoxicology of beryllium lung disease. Environ. Health Prev. Med. 2007, 12, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Liu, H.; Jian, Z.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Immunotoxicity of nickel: Pathological and toxicological effects. Ecotoxicol. Environ. Saf. 2020, 203, 111006. [Google Scholar] [CrossRef] [PubMed]
- Kasten-Jolly, J.; Lawrence, D.A. Lead modulation of macrophages causes multiorgan detrimental health effects. J. Biochem. Mol. Toxicol. 2014, 28, 355–372. [Google Scholar] [CrossRef]
- Daum, J.R.; Shepherd, D.M.; Noelle, R.J. Immunotoxicology of cadmium and mercury on B-lymphocytes—I. Effects on lymphocyte function. Int. J. Immunopharmacol. 1993, 15, 383–394. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
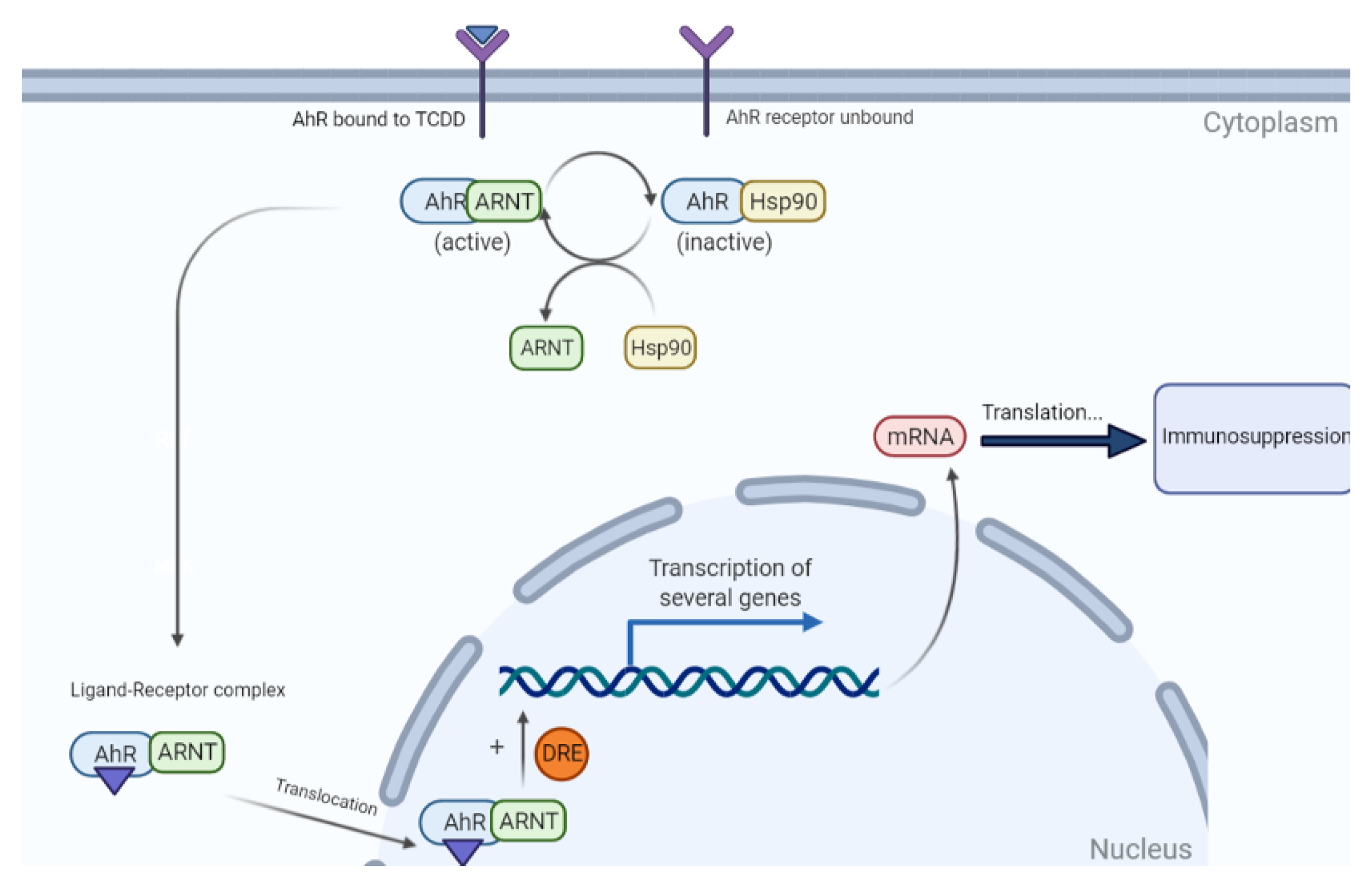
- Moura-Alves, P.; Faé, K.; Houthuys, E.; Dorhoi, A.; Kreuchwig, A.; Furkert, J.; Barison, N.; Diehl, A.; Munder, A.; Constant, P. AhR sensing of bacterial pigments regulates antibacterial defence. Nature 2014, 512, 387–392. [Google Scholar] [CrossRef]
- Smart, R.C.; Hodgson, E. Molecular and Biochemical Toxicology; John Wiley & Sons: Hoboken, NJ, USA, 2018. [Google Scholar]
- Staples, J.E.; Murante, F.G.; Fiore, N.C.; Gasiewicz, T.A.; Silverstone, A.E. Thymic alterations induced by 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin are strictly dependent on aryl hydrocarbon receptor activation in hemopoietic cells. J. Immunol. 1998, 160, 3844–3854. [Google Scholar]
- Vos, J.G.; De Heer, C.; Van Loveren, H. Immunotoxic effects of TCDD and toxic equivalency factors. Teratog. Carcinog. Mutagenesis 1997, 17, 275–284. [Google Scholar] [CrossRef]
- Grassman, J.A.; Masten, S.A.; Walker, N.J.; Lucier, G.W. Animal models of human response to dioxins. Environ. Health Perspect. 1998, 106, 761–775. [Google Scholar] [PubMed]
- Petrulis, J.R.; Perdew, G.H. The role of chaperone proteins in the aryl hydrocarbon receptor core complex. Chem. Biol. Interact. 2002, 141, 25–40. [Google Scholar] [CrossRef]
- Kovalova, N.; Nault, R.; Crawford, R.; Zacharewski, T.R.; Kaminski, N.E. Comparative analysis of TCDD-induced AhR-mediated gene expression in human, mouse and rat primary B cells. Toxicol. Appl. Pharmacol. 2017, 316, 95–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corsini, E.; Sokooti, M.; Galli, C.; Moretto, A.; Colosio, C. Pesticide induced immunotoxicity in humans: A comprehensive review of the existing evidence. Toxicology 2013, 307, 123–135. [Google Scholar] [CrossRef]
- Galloway, T.; Handy, R. Immunotoxicity of organophosphorous pesticides. Ecotoxicology 2003, 12, 345–363. [Google Scholar] [CrossRef] [PubMed]
- Filipov, N.M.; Pinchuk, L.M.; Boyd, B.L.; Crittenden, P.L. Immunotoxic effects of short-term atrazine exposure in young male C57BL/6 mice. Toxicol. Sci. 2005, 86, 324–332. [Google Scholar] [CrossRef]
- Li, L.; Hu, F.; Wang, C.; Wang, X. Enantioselective induction of oxidative stress by acetofenate in rat PC12 cells. J. Environ. Sci. 2010, 22, 1980–1986. [Google Scholar] [CrossRef]
- Sauer, E.; Gauer, B.; Nascimento, S.; Nardi, J.; Göethel, G.; Costa, B.; Correia, D.; Matte, U.; Charão, M.; Arbo, M. The role of B7 costimulation in benzene immunotoxicity and its potential association with cancer risk. Environ. Res. 2018, 166, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ahn, S.; Zhang, L. Benzene-associated immunosuppression and chronic inflammation in humans: A systematic review. Occup. Environ. Med. 2021, 78, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Verma, Y. Biochemical toxicity of benzene. J. Environ. Biol. 2005, 26, 157–168. [Google Scholar]
- Brouwer, A.; Ahlborg, U.G.; Van den Berg, M.; Birnbaum, L.S.; Boersma, E.R.; Bosveld, B.; Denison, M.S.; Gray, L.E.; Hagmar, L.; Holene, E. Functional aspects of developmental toxicity of polyhalogenated aromatic hydrocarbons in experimental animals and human infants. Eur. J. Pharmacol. Environ. Toxicol. Pharmacol. 1995, 293, 1–40. [Google Scholar] [CrossRef]
- White, K.L., Jr.; Lysy, H.H.; Holsapple, M.P. Immunosuppression by polycyclic aromatic hydrocarbons: A structure-activity relationship in B6C3F1 and DBA/2 mice. Immunopharmacology 1985, 9, 155–164. [Google Scholar] [CrossRef]
- Szczeklik, A.; Szczeklik, J.; Galuszka, Z.; Musial, J.; Kolarzyk, E.; Targosz, D. Humoral immunosuppression in men exposed to polycyclic aromatic hydrocarbons and related carcinogens in polluted environments. Environ. Health Perspect. 1994, 102, 302–304. [Google Scholar] [CrossRef]
- Krieger, J.A.; BoRN, J.L.; Burchiel, S.W. Persistence of calcium elevation in the HPB-ALL human T cell line correlates with immunosuppressive properties of polycyclic aromatic hydrocarbons. Toxicol. Appl. Pharmacol. 1994, 127, 268–274. [Google Scholar] [CrossRef]
- Wuttke, W.; Jarry, H.; Seidlova-Wuttke, D. Definition, classification and mechanism of action of endocrine disrupting chemicals. Hormones 2010, 9, 9–15. [Google Scholar] [CrossRef]
- Vom Saal, F.S.; Nagel, S.C.; Coe, B.L.; Angle, B.M.; Taylor, J.A. The estrogenic endocrine disrupting chemical bisphenol A (BPA) and obesity. Mol. Cell. Endocrinol. 2012, 354, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Rogers, J.A.; Metz, L.; Yong, V.W. Endocrine disrupting chemicals and immune responses: A focus on bisphenol-A and its potential mechanisms. Mol. Immunol. 2013, 53, 421–430. [Google Scholar] [CrossRef]
- Jefferson, W.N.; Padilla-Banks, E.; Newbold, R.R. Disruption of the female reproductive system by the phytoestrogen genistein. Reprod. Toxicol. 2007, 23, 308–316. [Google Scholar] [CrossRef]
- Padilla-Banks, E.; Jefferson, W.N.; Newbold, R.R. Neonatal exposure to the phytoestrogen genistein alters mammary gland growth and developmental programming of hormone receptor levels. Endocrinology 2006, 147, 4871–4882. [Google Scholar] [CrossRef]
- Toporova, L.; Balaguer, P. Nuclear receptors are the major targets of endocrine disrupting chemicals. Mol. Cell. Endocrinol. 2020, 502, 110665. [Google Scholar] [CrossRef]
- Chen, K.-C.; Juo, S.-H.H. MicroRNAs in atherosclerosis. Kaohsiung J. Med Sci. 2012, 28, 631–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, C.-H.; Wu, H.-C.; Kuo, T.-H.; Kuo, C.-H.; Yang, S.-N.; Wang, W.-L.; Chen, H.-N.; Wei, W.-J.; Hung, C.-H. Suppressive effect on MDC and IP-10 expression in monocytes by endocrine disruptor chemicals. Inflammation 2010, 33, 10–17. [Google Scholar] [CrossRef]
- Hong, C.-C.; Shimomura-Shimizu, M.; Muroi, M.; Tanamoto, K.-i. Effect of endocrine disrupting chemicals on lipopolysaccharide-induced tumor necrosis factor-α and nitric oxide production by mouse macrophages. Biol. Pharm. Bull. 2004, 27, 1136–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, T.; Uchikawa, R.; Yamada, M.; Arizono, N.; Oikawa, S.; Kawanishi, S.; Nishio, A.; Nakase, H.; Kuribayashi, K. Environmental pollutant tributyltin promotes Th2 polarization and exacerbates airway inflammation. Eur. J. Immunol. 2004, 34, 1312–1321. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Tada-Oikawa, S.; Takahashi, K.; Saito, K.; Wang, L.; Nishio, A.; Hakamada-Taguchi, R.; Kawanishi, S.; Kuribayashi, K. Endocrine disruptors that deplete glutathione levels in APC promote Th2 polarization in mice leading to the exacerbation of airway inflammation. Eur. J. Immunol. 2006, 36, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Larsson, M.; Hägerhed-Engman, L.; Kolarik, B.; James, P.; Lundin, F.; Janson, S.; Sundell, J.; Bornehag, C.-G. PVC–as flooring material–and its association with incident asthma in a Swedish child cohort study. Indoor Air 2010, 20, 494–501. [Google Scholar] [CrossRef]
- Kolarik, B.; Naydenov, K.; Larsson, M.; Bornehag, C.-G.; Sundell, J. The association between phthalates in dust and allergic diseases among Bulgarian children. Environ. Health Perspect. 2008, 116, 98–103. [Google Scholar] [CrossRef] [Green Version]
- Bornehag, C.-G.; Nanberg, E. Phthalate exposure and asthma in children. Int. J. Androl. 2010, 33, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Chung, S.W.; Kang, B.Y.; Park, J.; Lee, C.H.; Hwang, S.Y.; Kim, T.S. Enhanced interleukin-4 production in CD4+ T cells and elevated immunoglobulin E levels in antigen-primed mice by bisphenol A and nonylphenol, endocrine disruptors: Involvement of nuclear factor-AT and Ca2+. Immunology 2003, 109, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Kim, E.; Kim, T.S. Exposure to 4-tert-octylphenol, an environmentally persistent alkylphenol, enhances interleukin-4 production in T cells via NF-AT activation. Toxicol. Appl. Pharmacol. 2004, 197, 19–28. [Google Scholar] [CrossRef]
- Lee, M.H.; Park, J.; Chung, S.W.; Kang, B.Y.; Kim, S.H.; Kim, T.S. Enhancement of interleukin-4 production in activated CD4+ T cells by diphthalate plasticizers via increased NF-AT binding activity. Int. Arch. Allergy Immunol. 2004, 134, 213–222. [Google Scholar] [CrossRef]
- Abedi-Valugerdi, M.; Nilsson, C.; Zargari, A.; Gharibdoost, F.; DePierre, J.; Hassan, M. Bacterial lipopolysaccharide both renders resistant mice susceptible to mercury-induced autoimmunity and exacerbates such autoimmunity in susceptible mice. Clin. Exp. Immunol. 2005, 141, 238–247. [Google Scholar] [CrossRef]
- Sobel, E.S.; Gianini, J.; Butfiloski, E.J.; Croker, B.P.; Schiffenbauer, J.; Roberts, S.M. Acceleration of autoimmunity by organochlorine pesticides in (NZB× NZW) F1 mice. Environ. Health Perspect. 2005, 113, 323–328. [Google Scholar] [CrossRef] [Green Version]
- Jugan, M.-L.; Levi, Y.; Blondeau, J.-P. Endocrine disruptors and thyroid hormone physiology. Biochem. Pharmacol. 2010, 79, 939–947. [Google Scholar] [CrossRef] [Green Version]
- Bodin, J.; Bølling, A.K.; Becher, R.; Kuper, F.; Løvik, M.; Nygaard, U.C. Transmaternal bisphenol A exposure accelerates diabetes type 1 development in NOD mice. Toxicol. Sci. 2014, 137, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.L.; Germolec, D.R.; Zheng, J.F.; Kooistra, L.; Auttachoat, W.; Smith, M.J.; White, K.L.; Elmore, S.A. Genistein protects female nonobese diabetic mice from developing type 1 diabetes when fed a soy-and alfalfa-free diet. Toxicol. Pathol. 2015, 43, 435–448. [Google Scholar] [CrossRef] [Green Version]
- Lang, I.A.; Galloway, T.S.; Scarlett, A.; Henley, W.E.; Depledge, M.; Wallace, R.B.; Melzer, D. Association of urinary bisphenol A concentration with medical disorders and laboratory abnormalities in adults. JAMA 2008, 300, 1303–1310. [Google Scholar] [CrossRef]
- Snedeker, S.M.; Hay, A.G. Do interactions between gut ecology and environmental chemicals contribute to obesity and diabetes? Environ. Health Perspect. 2012, 120, 332–339. [Google Scholar] [CrossRef]
- Cox, L.M.; Yamanishi, S.; Sohn, J.; Alekseyenko, A.V.; Leung, J.M.; Cho, I.; Kim, S.G.; Li, H.; Gao, Z.; Mahana, D. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 2014, 158, 705–721. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Santiago, Y.; Nava-Castro, K.E.; Morales-Montor, J. Environmental pollution as a risk factor to develop colorectal cancer: The role of endocrine-disrupting chemicals in the inflammatory process as a risk factor to develop colorectal cancer. In Immunotherapy in Resistant Cancer: From the Lab Bench Work to Its Clinical Perspectives; Elsevier: Amsterdam, The Netherlands, 2021; pp. 131–148. [Google Scholar]
- Faulds, D.; Goa, K.L.; Benfield, P. Cyclosporin. Drugs 1993, 45, 953–1040. [Google Scholar] [CrossRef] [PubMed]
- Randak, C.; Brabletz, T.; Hergenröther, M.; Sobotta, I.; Serfling, E. Cyclosporin A suppresses the expression of the interleukin 2 gene by inhibiting the binding of lymphocyte-specific factors to the IL-2 enhancer. EMBO J. 1990, 9, 2529–2536. [Google Scholar] [CrossRef]
- Zipfel, P.; Irving, S.; Kelly, K.; Siebenlist, U. Complexity of the primary genetic response to mitogenic activation of human T cells. Mol. Cell. Biol. 1989, 9, 1041. [Google Scholar] [CrossRef] [Green Version]
- Fiolka, M.J. Immunosuppressive effect of cyclosporin A on insect humoral immune response. J. Invertebr. Pathol. 2008, 98, 287–292. [Google Scholar] [CrossRef]
- Thomson, A. The effects of cyclosporin A on non-T cell components of the immune system. J. Autoimmun. 1992, 5, 167–176. [Google Scholar] [CrossRef]
- Ameratunga, S.; Hijar, M.; Norton, R. Road-traffic injuries: Confronting disparities to address a global-health problem. Lancet 2006, 367, 1533–1540. [Google Scholar] [CrossRef]
- Marx, J. How the glucocorticoids suppress immunity. Science 1995, 270, 232–233. [Google Scholar] [CrossRef]
- Cain, D.W.; Cidlowski, J.A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247. [Google Scholar] [CrossRef]
- Baschant, U.; Tuckermann, J. The role of the glucocorticoid receptor in inflammation and immunity. J. Steroid Biochem. Mol. Biol. 2010, 120, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Fowles, J.R.; Fairbrother, A.; Fix, M.; Schiller, S.; Kerkvliet, N.I. Glucocorticoid effects on natural and humoral immunity in mallards. Dev. Comp. Immunol. 1993, 17, 165–177. [Google Scholar] [CrossRef]
- Guyre, P.; Girard, M.; Morganelli, P.; Manganiello, P. Glucocorticoid effects on the production and actions of immune cytokines. J. Steroid Biochem. 1988, 30, 89–93. [Google Scholar] [CrossRef]
- MUNCK, A.; GUYRE, P.M. Glucocorticoids and immune function. In Psychoneuroimmunology; Elsevier: Amsterdam, The Netherlands, 1991; pp. 447–474. [Google Scholar]
- Gershwin, M.E.; Goetzl, E.J.; STEINBERG, A.D. Cyclophosphamide: Use in practice. Ann. Intern. Med. 1974, 80, 531–540. [Google Scholar] [CrossRef]
- Fraiser, L.H.; Kanekal, S.; Kehrer, J.P. Cyclophosphamide toxicity. Drugs 1991, 42, 781–795. [Google Scholar] [CrossRef]
- Wanner, A.; Salathé, M.; O’Riordan, T.G. Mucociliary clearance in the airways. Am. J. Respir. Crit. Care Med. 1996, 154, 1868–1902. [Google Scholar] [CrossRef] [PubMed]
- Winkelstein, A. Mechanisms of immunosuppression: Effects of cyclophosphamide on cellular immunity. Blood 1973, 41, 273–284. [Google Scholar] [CrossRef]
- Stockman, G.D.; Heim, L.R.; South, M.A.; Trentin, J.J. Differential effects of cyclophosphamide on the B and T cell compartments of adult mice. J. Immunol. 1973, 110, 277–282. [Google Scholar]
- Croom, E. Metabolism of xenobiotics of human environments. Prog. Mol. Biol. Transl. Sci. 2012, 112, 31–88. [Google Scholar]
- Zabrodskii, P.F. Actual Problems of Immunotoxicology. The Main Mechanisms of Xenobiotics Immunotoxicity. Acta Sci. Microbiol. 2019, 44–47. [Google Scholar]
- Pallardy, M.; Biola, A.; Lebrec, H.; Bréard, J. Assessment of apoptosis in xenobiotic-induced immunotoxicity. Methods 1999, 19, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Bigazzi, P.E. Autoimmunity caused by xenobiotics. Toxicology 1997, 119, 1–21. [Google Scholar] [CrossRef]
- Crinnion, W.J. Maternal levels of xenobiotics that affect fetal development and childhood health. Altern. Med. Rev. 2009, 14, 212–222. [Google Scholar] [PubMed]
- Twomey, J.J. The Pathophysiology of Human Immunologic Disorders; Urban & Schwarzenberg: Baltimore, MD, USA, 1982. [Google Scholar]
- Banerjee, B.D.; Chakraborti, A.; Suke, S.G.; Ahmed, R.S.; Tripathi, A. Xenobiotic-Induced Immune Alterations: Implications in Health and Disease; Semantic Scholar: Seattle, DC, USA, 2008. [Google Scholar]
- Murdaca, G.; Colombo, B.M.; Cagnati, P.; Gulli, R.; Spanò, F.; Puppo, F. Endothelial dysfunction in rheumatic autoimmune diseases. Atherosclerosis 2012, 224, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Xu, X.; Belmadani, S.; Park, Y.; Tang, Z.; Feldman, A.M.; Chilian, W.M.; Zhang, C. TNF-alpha contributes to endothelial dysfunction by upregulating arginase in ischemia/reperfusion injury. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1269–1275. [Google Scholar] [CrossRef] [Green Version]
- Ciprandi, G.; Murdaca, G.; Colombo, B.M.; De Amici, M.; Marseglia, G.L. Serum vascular endothelial growth factor in allergic rhinitis and systemic lupus erythematosus. Hum. Immunol. 2008, 69, 510–512. [Google Scholar] [CrossRef]
- Hrvat, F.; Spahić, L.; Pokvić, L.G.; Badnjević, A. Artificial neural networks for prediction of medical device performance based on conformity assessment data: Infusion and perfusor pumps case study. In Proceedings of the 2020 9th Mediterranean Conference on Embedded Computing (MECO), Budva, Montenegro, 8–11 June 2020; pp. 1–4. [Google Scholar]
- Smith, M.; Segal, R.; Segal, J. Stress Symptoms, Signs & Causes: Effects of Stress Overload. HelpGuide.Org. 2013. Available online: https://www.helpguide.org/articles/stress/stress-symptoms-signs-and-causes.htm (accessed on 29 July 2021).
- Dohms, J.E.; Metz, A. Stress—Mechanisms of immunosuppression. Vet. Immunol. Immunopathol. 1991, 30, 89–109. [Google Scholar] [CrossRef]
- Shini, S.; Huff, G.; Shini, A.; Kaiser, P. Understanding stress-induced immunosuppression: Exploration of cytokine and chemokine gene profiles in chicken peripheral leukocytes. Poult. Sci. 2010, 89, 841–851. [Google Scholar] [CrossRef]
- Dhabhar, F.S. Effects of stress on immune function: The good, the bad, and the beautiful. Immunol. Res. 2014, 58, 193–210. [Google Scholar] [CrossRef]
- Marshall, G.D., Jr.; Agarwal, S.K.; Lloyd, C.; Cohen, L.; Henninger, E.M.; Morris, G.J. Cytokine dysregulation associated with exam stress in healthy medical students. Brain Behav. Immun. 1998, 12, 297–307. [Google Scholar] [CrossRef]
- Xiang, L.; Del Ben, K.S.; Rehm, K.E.; Marshall, G.D., Jr. Effects of acute stress-induced immunomodulation on TH1/TH2 cytokine and catecholamine receptor expression in human peripheral blood cells. Neuropsychobiology 2012, 65, 12–19. [Google Scholar] [CrossRef]
- Cheng, C.; Pickler, R. Perinatal stress, fatigue, depressive symptoms, and immune modulation in late pregnancy and one month postpartum. Sci. World J. 2014, 2014, 652630. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.J.; Visness, C.M.; Calatroni, A.; Grayson, M.H.; Gold, D.R.; Sandel, M.T.; Lee-Parritz, A.; Wood, R.A.; Kattan, M.; Bloomberg, G.R. Prenatal maternal stress and cord blood innate and adaptive cytokine responses in an inner-city cohort. Am. J. Respir. Crit. Care Med. 2010, 182, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Dantzer, R.; Kelley, K.W. Stress and immunity: An integrated view of relationships between the brain and the immune system. Life Sci. 1989, 44, 1995–2008. [Google Scholar] [CrossRef]
- Kiecolt-Glaser, J.K.; Glaser, R. Stress and immunity: Age enhances the risks. Curr. Dir. Psychol. Sci. 2001, 10, 18–21. [Google Scholar] [CrossRef]
- Howarth, P. Is allergy increasing?—Early life influences. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 1998, 28, 2–7. [Google Scholar] [CrossRef]
- Eisner, M.D. Environmental tobacco smoke and adult asthma. Clin. Chest Med. 2002, 23, 749–761. [Google Scholar] [CrossRef]
- Herrström, P.; Högstedt, B. Allergic diseases, dental health, and socioeconomic situation of Swedish teenagers: Allergy, dental health, and social situation. Scand. J. Prim. Health Care 1994, 12, 57–61. [Google Scholar] [CrossRef]
- Carmichael, A.J. Skin sensitivity and transdermal drug delivery. Drug Saf. 1994, 10, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Chapter 4 Mechanisms of immunotoxic effects. In Immunotoxicology of Drugs and Chemicals: An Experimental and Clinical Approach; Descotes, J. (Ed.) Elsevier: Amsterdam, The Netherlands, 2004; Volume 1, pp. 127–162. [Google Scholar]
- Katz, M.D.; Lor, E. Acute interstitial nephritis associated with intermittent rifampin use. Drug Intell. Clin. Pharm. 1986, 20, 789–792. [Google Scholar] [CrossRef]
- Evans, E.; Casinghino, S. Clinical Pathology as a Tool to Assess Immunotoxicity; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Reagan, W.; Poitout-Belissent, F.; Rovira, A. Design and Methods Used for Preclinical Hematotoxicity Studies; Weiss, D.J., Wardop, K.J., Eds.; Wiley-Blackwell: Ames, IA, USA, 2010. [Google Scholar]
- Hall, R.L.; Everds, N.E. Factors affecting the interpretation of canine and nonhuman primate clinical pathology. Toxicol. Pathol. 2003, 31, 6–10. [Google Scholar] [CrossRef]
- Latimer, K.S. Duncan and Prasse’s Veterinary Laboratory Medicine: Clinical Pathology; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- O’Connell, K.E.; Mikkola, A.M.; Stepanek, A.M.; Vernet, A.; Hall, C.D.; Sun, C.C.; Yildirim, E.; Staropoli, J.F.; Lee, J.T.; Brown, D.E. Practical murine hematopathology: A comparative review and implications for research. Comp. Med. 2015, 65, 96–113. [Google Scholar]
- Dietert, R.R. Immunotoxicity Testing; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Murata, H.; Shimada, N.; Yoshioka, M. Current research on acute phase proteins in veterinary diagnosis: An overview. Vet. J. 2004, 168, 28–40. [Google Scholar] [CrossRef]
- Pepys, M.B.; Hirschfield, G.M. C-reactive protein: A critical update. J. Clin. Investig. 2003, 111, 1805–1812. [Google Scholar] [CrossRef]
- Finn, O. Immuno-oncology: Understanding the function and dysfunction of the immune system in cancer. Ann. Oncol. 2012, 23, viii6–viii9. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Nilsson, J. Introduction: Atherosclerosis as inflammation: A controversial concept becomes accepted. J. Intern. Med. 2008, 263, 462–463. [Google Scholar] [CrossRef]
- Chou, M.Y.; Hartvigsen, K.; Hansen, L.F.; Fogelstrand, L.; Shaw, P.X.; Boullier, A.; Binder, C.J.; Witztum, J.L. Oxidation-specific epitopes are important targets of innate immunity. J. Intern. Med. 2008, 263, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Stiegel, M.A.; Pleil, J.D.; Sobus, J.R.; Morgan, M.K.; Madden, M.C. Analysis of inflammatory cytokines in human blood, breath condensate, and urine using a multiplex immunoassay platform. Biomarkers 2015, 20, 35–46. [Google Scholar] [CrossRef]
- Tomar, N.; De, R.K. Immunoinformatics: An integrated scenario. Immunology 2010, 131, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Baken, K.A.; Vandebriel, R.J.; Pennings, J.L.; Kleinjans, J.C.; van Loveren, H. Toxicogenomics in the assessment of immunotoxicity. Methods 2007, 41, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Salam, M.T.; Gauderman, W.J.; McConnell, R.; Lin, P.-C.; Gilliland, F.D. Transforming growth factor-β1 C-509T polymorphism, oxidant stress, and early-onset childhood asthma. Am. J. Respir. Crit. Care Med. 2007, 176, 1192–1199. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Ramos, M.C.; Engel, P.; Bastos, R. Towards a comprehensive human cell-surface immunome database. Immunol. Lett. 2011, 134, 183–187. [Google Scholar] [CrossRef]
- Blythe, M.J.; Doytchinova, I.A.; Flower, D.R. JenPep: A database of quantitative functional peptide data for immunology. Bioinformatics 2002, 18, 434–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rammensee, H.-G.; Bachmann, J.; Emmerich, N.P.N.; Bachor, O.A.; Stevanović, S. SYFPEITHI: Database for MHC ligands and peptide motifs. Immunogenetics 1999, 50, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Feldhahn, M.; Dönnes, P.; Thiel, P.; Kohlbacher, O. FRED—A framework for T-cell epitope detection. Bioinformatics 2009, 25, 2758–2759. [Google Scholar] [CrossRef] [Green Version]
- Lefranc, M.-P.; Giudicelli, V.; Ginestoux, C.; Jabado-Michaloud, J.; Folch, G.; Bellahcene, F.; Wu, Y.; Gemrot, E.; Brochet, X.; Lane, J. IMGT®, the international ImMunoGeneTics information system®. Nucleic Acids Res. 2009, 37, D1006–D1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, B.; Sidney, J.; Bourne, P.; Bui, H.-H.; Buus, S.; Doh, G.; Fleri, W.; Kronenberg, M.; Kubo, R.; Lund, O. The immune epitope database and analysis resource: From vision to blueprint. PLoS Biol. 2005, 3, e91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Bhasin, M.; Raghava, G.P. Bcipep: A database of B-cell epitopes. BMC Genom. 2005, 6, 79. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Honda, W. CED: A conformational epitope database. BMC Immunol. 2006, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Schlessinger, A.; Ofran, Y.; Yachdav, G.; Rost, B. Epitome: Database of structure-inferred antigenic epitopes. Nucleic Acids Res. 2006, 34, D777–D780. [Google Scholar] [CrossRef]


{kind=link}
| T-Helper 1 Cell | T-Helper 2 Cell | T-Helper 17 Cell | T-Regulatory | |
|---|---|---|---|---|
| Secretes | IFN-γ, IL-2 | IL-4, IL-5, IL-6, IL-10, IL-13 | IL-17, IL-21, IL-22 | TGF-β, IL-10, IL-35 |
| Function | Activates macrophages and cytotoxic T cells to kill phagocytosed microbes | Activates eosinophils and promotes production of IgE for parasite defense | Immunity against extracellular microbes, through induction of neutrophilic inflammation | Prevents autoimmunity by maintaining tolerance to self-antigens |
| Induced by | IFN-γ, IL-12 | IL-2, IL-4 | TGF-β,IL-1, IL-6 | TGF-β, IL-2 |
| Inhibited by | IL-4, IL-10 (from T-helper 2 cell) | IFN-γ (from T-helper 1 cell) | IFN-γ, IL-4 | IL-6 |
| Peripheral Blood | Serum Chemistry | Supportive Data | |
|---|---|---|---|
| Stress leukogram: corticosteroid mediated | ↓Lymphocytes, eosinophils ↑Neutrophils, monocytes Neutrophils are mature, may be hypersegmented | Hyperglycemia Lymphoid depletion in thymus Adrenal cortical hypertrophy | Overt organ toxicity Weight loss, anorexia Deaths in other animals in dose group. Findings only seen at doses at or higher than the maximum tolerated dose |
| Immunotoxicity: inflammation and infection | ↑Neutrophils, monocytes ↓Lymphocytes (nonrodents) Immature neutrophils, toxic change | Mild nonregenerative anemia Inflammation in tissues Organisms Hypercellular bone marrow, increased M/E ratio | Clinical signs of infection, fever, anorexia, weight loss |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bou Zerdan, M.; Moussa, S.; Atoui, A.; Assi, H.I. Mechanisms of Immunotoxicity: Stressors and Evaluators. Int. J. Mol. Sci. 2021, 22, 8242. https://doi.org/10.3390/ijms22158242
Bou Zerdan M, Moussa S, Atoui A, Assi HI. Mechanisms of Immunotoxicity: Stressors and Evaluators. International Journal of Molecular Sciences. 2021; 22(15):8242. https://doi.org/10.3390/ijms22158242
Chicago/Turabian StyleBou Zerdan, Maroun, Sara Moussa, Ali Atoui, and Hazem I. Assi. 2021. "Mechanisms of Immunotoxicity: Stressors and Evaluators" International Journal of Molecular Sciences 22, no. 15: 8242. https://doi.org/10.3390/ijms22158242
APA StyleBou Zerdan, M., Moussa, S., Atoui, A., & Assi, H. I. (2021). Mechanisms of Immunotoxicity: Stressors and Evaluators. International Journal of Molecular Sciences, 22(15), 8242. https://doi.org/10.3390/ijms22158242