
Tackling Dysfunction of Mitochondrial Bioenergetics in the Brain



Abstract
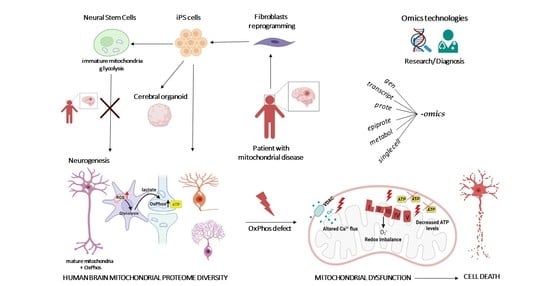
:
1. Introduction
2. Mitoexome, Mitochondrial Proteome, and Mitointeractome
3. Diversity of Bioenergetics Demand in the Brain
4. Structure, Assembly, and Disorders of Bioenergetics Complexes
4.1. NADH–Ubiquinone Oxidoreductase–Complex I
4.2. Succinate–Ubiquinone Oxidoreductase–Complex II
4.3. Ubiquinol: Cytochrome C Oxidoreductase–Complex III
4.4. Cytochrome C Oxidase–Complex IV
4.5. ATP Synthase–Complex V
4.6. Respiratory Supercomplexes
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol. 2014, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Metabolic control of cell death. Science 2014, 345, 1250256. [Google Scholar] [CrossRef] [Green Version]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef]
- Muraresku, C.C.; McCormick, E.M.; Falk, M.J. Mitochondrial disease: Advances in Clinical diagnosis, management, therapeutic development, and preventative strategies. Curr. Genet. Med. Rep. 2018, 6, 62–72. [Google Scholar] [CrossRef]
- Piel, R.B.; Dailey, H.A.; Medlock, A.E. The mitochondrial heme metabolon: Insights into the complex(Ity) of heme synthesis and distribution. Mol. Genet. Metab. 2019, 128, 198–203. [Google Scholar] [CrossRef]
- Miller, W.L. Steroid hormone synthesis in mitochondria. Mol. Cell. Endocrinol. 2013, 379, 62–73. [Google Scholar] [CrossRef]
- Anderson, A.J.; Jackson, T.D.; Stroud, D.A.; Stojanovski, D. Mitochondria—Hubs for regulating cellular biochemistry: Emerging concepts and networks. Open Biol. 2019, 9, 190126. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [Green Version]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef]
- Tiku, V.; Tan, M.-W.; Dikic, I. Mitochondrial functions in infection and immunity. Trends Cell Biol. 2020, 30, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Schapira, A.H. Mitochondrial disease. Lancet 2006, 368, 70–82. [Google Scholar] [CrossRef]
- DiMauro, S.; Schon, E.A. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef]
- Luft, R. The development of mitochondrial medicine. Proc. Natl. Acad. Sci. USA 1994, 91, 8731–8738. [Google Scholar] [CrossRef] [Green Version]
- La Morgia, C.; Maresca, A.; Caporali, L.; Valentino, M.L.; Carelli, V. Mitochondrial diseases in adults. J. Intern. Med. 2020, 287, 592–608. [Google Scholar] [CrossRef]
- Petruzzella, V.; Tiranti, V.; Fernandez, P.; Ianna, P.; Carrozzo, R.; Zeviani, M. Identification and characterization of human CDNAs specific to BCS1, PET112, SCO1, COX15, and COX11—Five genes involved in the formation and function of the mitochondrial respiratory chain. Genomics 1998, 54, 494–504. [Google Scholar] [CrossRef]
- Stenton, S.L.; Prokisch, H. Advancing genomic approaches to the molecular diagnosis of mitochondrial disease. Essays Biochem. 2018, 62, 399–408. [Google Scholar] [CrossRef]
- Stenton, S.L.; Prokisch, H. Genetics of mitochondrial diseases: Identifying mutations to help diagnosis. EBioMedicine 2020, 56, 102784. [Google Scholar] [CrossRef]
- Calvo, S.E.; Compton, A.G.; Hershman, S.G.; Lim, S.C.; Lieber, D.S.; Tucker, E.J.; Laskowski, A.; Garone, C.; Liu, S.; Jaffe, D.B.; et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci. Transl. Med. 2012, 4, 118ra10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plutino, M.; Chaussenot, A.; Rouzier, C.; Ait-El-Mkadem, S.; Fragaki, K.; Paquis-Flucklinger, V.; Bannwarth, S. Targeted next generation sequencing with an extended gene panel does not impact variant detection in mitochondrial diseases. BMC Med. Genet. 2018, 19, 57. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Garone, C.; Donati, M.A.; Sacchini, M.; Garcia-Diaz, B.; Bruno, C.; Calvo, S.; Mootha, V.K.; DiMauro, S. Mitochondrial encephalomyopathy due to a novel mutation in ACAD9. JAMA Neurol. 2013, 70, 1177–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oláhová, M.; Berti, C.C.; Collier, J.J.; Alston, C.L.; Jameson, E.; Jones, S.A.; Edwards, N.; He, L.; Chinnery, P.F.; Horvath, R.; et al. Molecular genetic investigations identify new clinical phenotypes associated with BCS1L-related mitochondrial disease. Hum. Mol. Genet. 2019, 28, 3766–3776. [Google Scholar] [CrossRef] [PubMed]
- Stenton, S.L.; Kremer, L.S.; Kopajtich, R.; Ludwig, C.; Prokisch, H. The diagnosis of inborn errors of metabolism by an integrative “multi-omics’’ approach: A perspective encompassing genomics, transcriptomics, and proteomics. J. Inherit. Metab. Dis. 2020, 43, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Bunkenborg, J.; Olsen, J.V.; Hjerrild, M.; Wisniewski, J.R.; Stahl, E.; Bolouri, M.S.; Ray, H.N.; Sihag, S.; Kamal, M.; et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 2003, 115, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.-E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Gonczarowska-Jorge, H.; Zahedi, R.P.; Sickmann, A. The proteome of baker’s yeast mitochondria. Mitochondrion 2017, 33, 15–21. [Google Scholar] [CrossRef]
- Clamp, M.; Fry, B.; Kamal, M.; Xie, X.; Cuff, J.; Lin, M.F.; Kellis, M.; Lindblad-Toh, K.; Lander, E.S. Distinguishing protein-coding and noncoding genes in the human genome. Proc. Natl. Acad. Sci. USA 2007, 104, 19428–19433. [Google Scholar] [CrossRef] [Green Version]
- Ponomarenko, E.A.; Poverennaya, E.V.; Ilgisonis, E.V.; Pyatnitskiy, M.A.; Kopylov, A.T.; Zgoda, V.G.; Lisitsa, A.V.; Archakov, A.I. The size of the human proteome: The width and depth. Int. J. Anal. Chem. 2016, 2016, 7436849. [Google Scholar] [CrossRef] [Green Version]
- Lopez, M.F.; Kristal, B.S.; Chernokalskaya, E.; Lazarev, A.; Shestopalov, A.I.; Bogdanova, A.; Robinson, M. High-throughput profiling of the mitochondrial proteome using affinity fractionation and automation. Electrophoresis 2000, 21, 3427–3440. [Google Scholar] [CrossRef]
- Karlberg, O.; Canbäck, B.; Kurland, C.G.; Andersson, S.G. The dual origin of the yeast mitochondrial proteome. Yeast 2000, 17, 170–187. [Google Scholar] [CrossRef]
- Cotter, D. MitoProteome: Mitochondrial protein sequence database and annotation system. Nucleic Acids Res. 2004, 32, D463–D467. [Google Scholar] [CrossRef] [Green Version]
- Guda, P.; Subramaniam, S.; Guda, C. Mitoproteome: Human heart mitochondrial protein sequence database. Methods Mol Biol. 2007, 357, 375–383. [Google Scholar] [CrossRef]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.C.; Robinson, A.J. MitoMiner v3.1, an update on the mitochondrial proteomics database. Nucleic Acids Res. 2016, 44, D1258–D1261. [Google Scholar] [CrossRef] [Green Version]
- Doccini, S.; Morani, F.; Nesti, C.; Pezzini, F.; Calza, G.; Soliymani, R.; Signore, G.; Rocchiccioli, S.; Kanninen, K.M.; Huuskonen, M.T.; et al. Proteomic and functional analyses in disease models reveal CLN5 protein involvement in mitochondrial dysfunction. Cell Death Discov. 2020, 6, 18. [Google Scholar] [CrossRef] [Green Version]
- Hung, V.; Lam, S.S.; Udeshi, N.D.; Svinkina, T.; Guzman, G.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Correction: Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. eLife 2019, 8, e50707. [Google Scholar] [CrossRef]
- Geladaki, A.; Kočevar Britovšek, N.; Breckels, L.M.; Smith, T.S.; Vennard, O.L.; Mulvey, C.M.; Crook, O.M.; Gatto, L.; Lilley, K.S. Combining LOPIT with differential ultracentrifugation for high-resolution spatial proteomics. Nat. Commun. 2019, 10, 331. [Google Scholar] [CrossRef] [Green Version]
- Sung, A.Y.; Floyd, B.J.; Pagliarini, D.J. Systems biochemistry approaches to defining mitochondrial protein function. Cell Metab. 2020, 31, 669–678. [Google Scholar] [CrossRef]
- Ohue, M.; Matsuzaki, Y.; Uchikoga, N.; Ishida, T.; Akiyama, Y. MEGADOCK: An all-to-all protein-protein interaction prediction system using tertiary structure data. Protein Pept. Lett. 2013, 21, 766–778. [Google Scholar] [CrossRef] [Green Version]
- Ohue, M.; Shimoda, T.; Suzuki, S.; Matsuzaki, Y.; Ishida, T.; Akiyama, Y. MEGADOCK 4.0: An ultra–high-performance protein–protein docking software for heterogeneous supercomputers. Bioinformatics 2014, 30, 3281–3283. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Matsuzaki, Y.; Yanagisawa, K.; Ohue, M.; Akiyama, Y. MEGADOCK-Web: An integrated database of high-throughput structure-based protein-protein interaction predictions. BMC Bioinform. 2018, 19, 62. [Google Scholar] [CrossRef] [Green Version]
- Floyd, B.J.; Wilkerson, E.M.; Veling, M.T.; Minogue, C.E.; Xia, C.; Beebe, E.T.; Wrobel, R.L.; Cho, H.; Kremer, L.S.; Alston, C.L.; et al. Mitochondrial protein interaction mapping identifies regulators of respiratory chain function. Mol. Cell 2016, 63, 621–632. [Google Scholar] [CrossRef] [Green Version]
- Formosa, L.E.; Dibley, M.G.; Stroud, D.A.; Ryan, M.T. Building a complex complex: Assembly of mitochondrial respiratory chain complex I. Semin. Cell Dev. Biol. 2018, 76, 154–162. [Google Scholar] [CrossRef]
- Labory, J.; Fierville, M.; Ait-El-Mkadem, S.; Bannwarth, S.; Paquis-Flucklinger, V.; Bottini, S. Multi-omics approaches to improve mitochondrial disease diagnosis: Challenges, advances, and perspectives. Front. Mol. Biosci. 2020, 7, 590842. [Google Scholar] [CrossRef]
- Khan, S.; Ince-Dunn, G.; Suomalainen, A.; Elo, L.L. Integrative omics approaches provide biological and clinical insights: Examples from mitochondrial diseases. J. Clin. Investig. 2020, 130, 20–28. [Google Scholar] [CrossRef]
- Yu, Y.; Herman, P.; Rothman, D.L.; Agarwal, D.; Hyder, F. Evaluating the gray and white matter energy budgets of human brain function. J. Cereb. Blood Flow Metab. 2018, 38, 1339–1353. [Google Scholar] [CrossRef]
- Sokoloff, L.; Reivich, M.; Kennedy, C.; Rosiers, M.H.D.; Patlak, C.S.; Pettigrew, K.D.; Sakurada, O.; Shinohara, M. The [14C] deoxyglucose method for the measurement of local cerebral glucose utilization: Theory, procedure, and normal values in the conscious and anesthetized albino rat. J. Neurochem. 1977, 28, 897–916. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barros, L.F.; Courjaret, R.; Jakoby, P.; Loaiza, A.; Lohr, C.; Deitmer, J.W. Preferential transport and metabolism of glucose in bergmann glia over purkinje cells: A multiphoton study of cerebellar slices. Glia 2009, 57, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.K.; Morrison, B.M. Glia-neuron energy metabolism in health and diseases: New insights into the role of nervous system metabolic transporters. Exp. Neurol. 2018, 309, 23–31. [Google Scholar] [CrossRef]
- Parpura, V.; Basarsky, T.A.; Liu, F.; Jeftinija, K.; Jeftinija, S.; Haydon, P.G. Glutamate-mediated astrocyte–neuron signalling. Nature 1994, 369, 744–747. [Google Scholar] [CrossRef]
- Xin, W.; Bonci, A. Functional astrocyte heterogeneity and implications for their role in shaping neurotransmission. Front. Cell. Neurosci. 2018, 12, 141. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, J.F.; Sardinha, V.M.; Guerra-Gomes, S.; Araque, A.; Sousa, N. Do stars govern our actions? Astrocyte involvement in rodent behavior. Trends Neurosci. 2015, 38, 535–549. [Google Scholar] [CrossRef] [Green Version]
- Allen, N.J. Star power: Astrocytes regulate behavior. Cell 2019, 177, 1091–1093. [Google Scholar] [CrossRef]
- Herrero-Mendez, A.; Almeida, A.; Fernández, E.; Maestre, C.; Moncada, S.; Bolaños, J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C–Cdh1. Nat. Cell Biol. 2009, 11, 747–752. [Google Scholar] [CrossRef]
- Bittner, C.X.; Valdebenito, R.; Ruminot, I.; Loaiza, A.; Larenas, V.; Sotelo-Hitschfeld, T.; Moldenhauer, H.; San Martin, A.; Gutierrez, R.; Zambrano, M.; et al. Fast and reversible stimulation of astrocytic glycolysis by K+ and a delayed and persistent effect of glutamate. J. Neurosci. 2011, 31, 4709–4713. [Google Scholar] [CrossRef]
- Bouzier-Sore, A.-K.; Voisin, P.; Canioni, P.; Magistretti, P.J.; Pellerin, L. Lactate is a preferential oxidative energy substrate over glucose for neurons in culture. J. Cereb. Blood Flow Metab. 2003, 23, 1298–1306. [Google Scholar] [CrossRef] [Green Version]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Astrocyte–neuron metabolic relationships: For better and for worse. Trends Neurosci. 2011, 34, 76–87. [Google Scholar] [CrossRef]
- Fecher, C.; Trovò, L.; Müller, S.A.; Snaidero, N.; Wettmarshausen, J.; Heink, S.; Ortiz, O.; Wagner, I.; Kühn, R.; Hartmann, J.; et al. Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat. Neurosci. 2019, 22, 1731–1742. [Google Scholar] [CrossRef]
- Petrova, V.Y.; Drescher, D.; Kujumdzieva, A.V.; Schmitt, M.J. Dual Targeting of yeast catalase A to peroxisomes and mitochondria. Biochem. J. 2004, 380, 393–400. [Google Scholar] [CrossRef]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Fabuel, I.; Le Douce, J.; Logan, A.; James, A.M.; Bonvento, G.; Murphy, M.P.; Almeida, A.; Bolaños, J.P. Complex I Assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 13063–13068. [Google Scholar] [CrossRef] [Green Version]
- Graham, L.C.; Eaton, S.L.; Brunton, P.J.; Atrih, A.; Smith, C.; Lamont, D.J.; Gillingwater, T.H.; Pennetta, G.; Skehel, P.; Wishart, T.M. Proteomic profiling of neuronal mitochondria reveals modulators of synaptic architecture. Mol. Neurodegener. 2017, 12, 77. [Google Scholar] [CrossRef] [Green Version]
- Stauch, K.L.; Purnell, P.R.; Fox, H.S. Quantitative proteomics of synaptic and nonsynaptic mitochondria: Insights for synaptic mitochondrial vulnerability. J. Proteome Res. 2014, 13, 2620–2636. [Google Scholar] [CrossRef]
- Yousefi, R.; Fornasiero, E.F.; Cyganek, L.; Montoya, J.; Jakobs, S.; Rizzoli, S.O.; Rehling, P.; Pacheu-Grau, D. Monitoring mitochondrial translation in living cells. EMBO Rep. 2021, 22, e51635. [Google Scholar] [CrossRef]
- Inak, G.; Rybak-Wolf, A.; Lisowski, P.; Pentimalli, T.M.; Jüttner, R.; Glažar, P.; Uppal, K.; Bottani, E.; Brunetti, D.; Secker, C.; et al. Defective metabolic programming impairs early neuronal morphogenesis in neural cultures and an organoid model of leigh syndrome. Nat. Commun. 2021, 12, 1929. [Google Scholar] [CrossRef]
- Quadalti, C.; Brunetti, D.; Lagutina, I.; Duchi, R.; Perota, A.; Lazzari, G.; Cerutti, R.; Di Meo, I.; Johnson, M.; Bottani, E.; et al. SURF1 knockout cloned pigs: Early onset of a severe lethal phenotype. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 2131–2142. [Google Scholar] [CrossRef]
- Bottani, E.; Lamperti, C.; Prigione, A.; Tiranti, V.; Persico, N.; Brunetti, D. Therapeutic approaches to treat mitochondrial diseases: ‘’One-size-fits-all’’ and ‘’precision medicine’’ strategies. Pharmaceutics 2020, 12, 1083. [Google Scholar] [CrossRef]
- Papa, S.; Martino, P.L.; Capitanio, G.; Gaballo, A.; De Rasmo, D.; Signorile, A.; Petruzzella, V. The oxidative phosphorylation system in mammalian mitochondria. In Advances in Mitochondrial Medicine; Scatena, R., Bottoni, P., Giardina, B., Eds.; Springer: Dordrecht, The Netherlands, 2012; Volume 942, pp. 3–37. [Google Scholar]
- Fernandez-Vizarra, E.; Zeviani, M. Mitochondrial disorders of the OXPHOS system. FEBS Lett. 2021, 595, 1062–1106. [Google Scholar] [CrossRef]
- Tang, J.X.; Thompson, K.; Taylor, R.W.; Oláhová, M. Mitochondrial OXPHOS biogenesis: Co-regulation of protein synthesis, Import, and assembly pathways. Int. J. Mol. Sci. 2020, 21, 3820. [Google Scholar] [CrossRef]
- Bergman, O.; Ben-Shachar, D. Mitochondrial Oxidative Phosphorylation System (OXPHOS) deficits in schizophrenia: Possible interactions with cellular processes. Can. J. Psychiatry 2016, 61, 457–469. [Google Scholar] [CrossRef] [Green Version]
- Chacinska, A.; Koehler, C.M.; Milenkovic, D.; Lithgow, T.; Pfanner, N. Importing mitochondrial proteins: Machineries and mechanisms. Cell 2009, 138, 628–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carapito, C.; Kuhn, L.; Karim, L.; Rompais, M.; Rabilloud, T.; Schwenzer, H.; Sissler, M. Two proteomic methodologies for defining N-termini of mature human mitochondrial aminoacyl-TRNA synthetases. Methods 2017, 113, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Russell, O.M.; Gorman, G.S.; Lightowlers, R.N.; Turnbull, D.M. Mitochondrial diseases: Hope for the future. Cell 2020, 181, 168–188. [Google Scholar] [CrossRef]
- Rusecka, J.; Kaliszewska, M.; Bartnik, E.; Tońska, K. Nuclear genes involved in mitochondrial diseases caused by instability of mitochondrial DNA. J. Appl. Genet. 2018, 59, 43–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasukawa, T.; Kang, D. An overview of mammalian mitochondrial DNA replication mechanisms. J. Biochem. 2018, 164, 183–193. [Google Scholar] [CrossRef]
- Barshad, G.; Marom, S.; Cohen, T.; Mishmar, D. Mitochondrial DNA transcription and its regulation: An evolutionary perspective. Trends Genet. 2018, 34, 682–692. [Google Scholar] [CrossRef]
- Boczonadi, V.; Ricci, G.; Horvath, R. Mitochondrial DNA transcription and translation: Clinical syndromes. Essays Biochem. 2018, 62, 321–340. [Google Scholar] [CrossRef]
- Kummer, E.; Ban, N. Mechanisms and regulation of protein synthesis in mitochondria. Nat. Rev. Mol. Cell Biol. 2021, 22, 307–325. [Google Scholar] [CrossRef]
- Wallace, D.C. Bioenergetics in human evolution and disease: Implications for the origins of biological complexity and the missing genetic variation of common diseases. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120267. [Google Scholar] [CrossRef] [Green Version]
- DiMauro, S.; Schon, E.A.; Carelli, V.; Hirano, M. The clinical maze of mitochondrial neurology. Nat. Rev. Neurol. 2013, 9, 429–444. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Mitochondrial Genetic medicine. Nat. Genet. 2018, 50, 1642–1649. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Rahman, S.; Keller, M.; Zschocke, J.; ICIMD Advisory Group; Abdenur, J.; Ali, H.; Artuch, R.; Ballabio, A.; Barshop, B.; et al. An international classification of inherited metabolic disorders (ICIMD). J. Inherit. Metab. Dis. 2021, 44, 164–177. [Google Scholar] [CrossRef]
- Holt, I.J.; Harding, A.E.; Morgan-Hughes, J.A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988, 331, 717–719. [Google Scholar] [CrossRef]
- Wallace, D.; Singh, G.; Lott, M.; Hodge, J.; Schurr, T.; Lezza, A.; Elsas, L.; Nikoskelainen, E. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef]
- Wallace, D.C.; Zheng, X.; Lott, M.T.; Shoffner, J.M.; Hodge, J.A.; Kelley, R.I.; Epstein, C.M.; Hopkins, L.C. Familial mitochondrial encephalomyopathy (MERRF): Genetic, pathophysiological, and biochemical characterization of a mitochondrial DNA disease. Cell 1988, 55, 601–610. [Google Scholar] [CrossRef]
- Carelli, V.; La Morgia, C. Clinical syndromes associated with MtDNA mutations: Where we stand after 30 years. Essays Biochem. 2018, 62, 235–254. [Google Scholar] [CrossRef] [PubMed]
- Leber, T. Ueber hereditäre und congenital-angelegte sehnervenleiden. Graefe’s Arch. Clin. Exp. Ophthalmol. 1871, 17, 249–291. [Google Scholar] [CrossRef]
- Holt, I.J.; Harding, A.E.; Petty, R.K.; Morgan-Hughes, J.A. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am. J. Hum. Genet. 1990, 46, 428–433. [Google Scholar] [PubMed]
- Tatuch, Y.; Christodoulou, J.; Feigenbaum, A.; Clarke, J.T.; Wherret, J.; Smith, C.; Rudd, N.; Petrova-Benedict, R.; Robinson, B.H. Heteroplasmic MtDNA mutation (T----G) at 8993 can cause leigh disease when the percentage of abnormal MtDNA is high. Am. J. Hum. Genet. 1992, 50, 852–858. [Google Scholar]
- Prezant, T.R.; Agapian, J.V.; Bohlman, M.C.; Bu, X.; Öztas, S.; Qiu, W.-Q.; Arnos, K.S.; Cortopassi, G.A.; Jaber, L.; Rotter, J.I.; et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic–induced and non–syndromic deafness. Nat. Genet. 1993, 4, 289–294. [Google Scholar] [CrossRef]
- Fukuhara, N.; Tokiguchi, S.; Shirakawa, K.; Tsubaki, T. Myoclonus epilepsy associated with ragged-red fibres (mitochondrial abnormalities): Disease entity or a syndrome? J. Neurol. Sci. 1980, 47, 117–133. [Google Scholar] [CrossRef]
- Shoffner, J.M.; Lott, M.T.; Lezza, A.M.S.; Seibel, P.; Ballinger, S.W.; Wallace, D.C. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA TRNALys mutation. Cell 1990, 61, 931–937. [Google Scholar] [CrossRef]
- Pavlakis, S.G.; Phillips, P.C.; DiMauro, S.; De Vivo, D.C.; Rowland, L.P. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: A distinctive clinical syndrome. Ann. Neurol. 1984, 16, 481–488. [Google Scholar] [CrossRef]
- Goto, Y.; Nonaka, I.; Horai, S. A Mutation in the TRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 1990, 348, 651–653. [Google Scholar] [CrossRef]
- Kearns, T.P.; Sayre, G.P. Retinitis pigmentosa, external ophthalmophegia, and complete heart block: Unusual syndrome with histologic study in one of two cases. AMA. Arch. Ophthalmol. 1958, 60, 280–289. [Google Scholar] [CrossRef]
- Zeviani, M.; Moraes, C.T.; DiMauro, S.; Nakase, H.; Bonilla, E.; Schon, E.A.; Rowland, L.P. Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology 1988, 38, 1339. [Google Scholar] [CrossRef]
- Pearson, H.A.; Lobel, J.S.; Kocoshis, S.A.; Naiman, J.L.; Windmiller, J.; Lammi, A.T.; Hoffman, R.; Marsh, J.C. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J. Pediatr. 1979, 95, 976–984. [Google Scholar] [CrossRef]
- Rötig, A.; Cormier, V.; Blanche, S.; Bonnefont, J.P.; Ledeist, F.; Romero, N.; Schmitz, J.; Rustin, P.; Fischer, A.; Saudubray, J.M. Pearson’s marrow-pancreas syndrome. A multisystem mitochondrial disorder in infancy. J. Clin. Investig. 1990, 86, 1601–1608. [Google Scholar] [CrossRef]
- Carelli, V.; Ghelli, A.; Ratta, M.; Bacchilega, E.; Sangiorgi, S.; Mancini, R.; Leuzzi, V.; Cortelli, P.; Montagna, P.; Lugaresi, E.; et al. Leber’s hereditary optic neuropathy: Biochemical effect of 11778/ND4 and 3460/ND1 mutations and correlation with the mitochondrial genotype. Neurology 1997, 48, 1623–1632. [Google Scholar] [CrossRef]
- Tatuch, Y.; Robinson, B.H. The mitochondrial DNA mutation at 8993 associated with NARP slows the rate of ATP synthesis in isolated lymphoblast mitochondria. Biochem. Biophys. Res. Commun. 1993, 192, 124–128. [Google Scholar] [CrossRef]
- Bernes, S.M.; Bacino, C.; Prezant, T.R.; Pearson, M.A.; Wood, T.S.; Fournier, P.; Fischel-Ghodsian, N. Identical mitochondrial DNA deletion in mother with progressive external ophthalmoplegia and son with Pearson marrow-pancreas syndrome. J. Pediatr. 1993, 123, 598–602. [Google Scholar] [CrossRef]
- Shanske, S.; Tang, Y.; Hirano, M.; Nishigaki, Y.; Tanji, K.; Bonilla, E.; Sue, C.; Krishna, S.; Carlo, J.R.; Willner, J.; et al. Identical mitochondrial DNA deletion in a woman with ocular myopathy and in her son with Pearson syndrome. Am. J. Hum. Genet. 2002, 71, 679–683. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Ghosh, A. Molecular mechanism of mitochondrial respiratory chain assembly and its relation to mitochondrial diseases. Mitochondrion 2020, 53, 1–20. [Google Scholar] [CrossRef]
- Dang, Q.-C.L.; Phan, D.H.; Johnson, A.N.; Pasapuleti, M.; Alkhaldi, H.A.; Zhang, F.; Vik, S.B. Analysis of human mutations in the supernumerary subunits of complex I. Life 2020, 10, 296. [Google Scholar] [CrossRef]
- Hirst, J. Mitochondrial complex I. Ann. Rev. Biochem. 2013, 82, 551–575. [Google Scholar] [CrossRef]
- Ripple, M.O.; Kim, N.; Springett, R. Mammalian complex I pumps 4 protons per 2 electrons at high and physiological proton motive force in living cells*. J. Biol. Chem. 2013, 288, 5374–5380. [Google Scholar] [CrossRef] [Green Version]
- Efremov, R.G.; Baradaran, R.; Sazanov, L.A. The architecture of respiratory complex I. Nature 2010, 465, 441–445. [Google Scholar] [CrossRef]
- Zhu, J.; Vinothkumar, K.R.; Hirst, J. Structure of mammalian respiratory complex I. Nature 2016, 536, 354–358. [Google Scholar] [CrossRef] [Green Version]
- Vinothkumar, K.R.; Zhu, J.; Hirst, J. Architecture of mammalian respiratory complex I. Nature 2014, 515, 80–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clason, T.; Ruiz, T.; Schägger, H.; Peng, G.; Zickermann, V.; Brandt, U.; Michel, H.; Radermacher, M. The structure of eukaryotic and prokaryotic complex I. J. Struct. Biol. 2010, 169, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baradaran, R.; Berrisford, J.M.; Minhas, G.S.; Sazanov, L.A. Crystal structure of the entire respiratory complex I. Nature 2013, 494, 443–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agip, A.-N.A.; Blaza, J.N.; Bridges, H.R.; Viscomi, C.; Rawson, S.; Muench, S.P.; Hirst, J. Cryo-EM structures of complex I from mouse heart mitochondria in two biochemically defined states. Nat. Struct. Mol. Biol. 2018, 25, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J.; Roessler, M.M. Energy conversion, redox catalysis and generation of reactive oxygen species by respiratory complex I. Biochim. Biophys. Acta (BBA)-Bioenerg. 2016, 1857, 872–883. [Google Scholar] [CrossRef] [Green Version]
- Stroud, D.A.; Surgenor, E.E.; Formosa, L.E.; Reljic, B.; Frazier, A.E.; Dibley, M.G.; Osellame, L.D.; Stait, T.; Beilharz, T.H.; Thorburn, D.R.; et al. Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature 2016, 538, 123–126. [Google Scholar] [CrossRef] [Green Version]
- Yip, C.; Harbour, M.E.; Jayawardena, K.; Fearnley, I.M.; Sazanov, L.A. Evolution of respiratory complex I. J. Biol. Chem. 2011, 286, 5023–5033. [Google Scholar] [CrossRef] [Green Version]
- Sazanov, L.A. A giant molecular proton pump: Structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 2015, 16, 375–388. [Google Scholar] [CrossRef]
- Zickermann, V.; Wirth, C.; Nasiri, H.; Siegmund, K.; Schwalbe, H.; Hunte, C.; Brandt, U. Mechanistic Insight from the crystal structure of mitochondrial complex I. Science 2015, 347, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Parey, K.; Wirth, C.; Vonck, J.; Zickermann, V. Respiratory complex I—Structure, mechanism and evolution. Curr. Opin. Struct. Biol. 2020, 63, 1–9. [Google Scholar] [CrossRef]
- Kampjut, D.; Sazanov, L.A. The coupling mechanism of mammalian respiratory complex I. Science 2020, 370, eabc4209. [Google Scholar] [CrossRef]
- Grba, D.N.; Hirst, J. Mitochondrial complex I structure reveals ordered water molecules for catalysis and proton translocation. Nat. Struct. Mol. Biol. 2020, 27, 892–900. [Google Scholar] [CrossRef]
- Klusch, N.; Senkler, J.; Yildiz, Ö.; Kühlbrandt, W.; Braun, H.-P. A ferredoxin bridge connects the two arms of plant mitochondrial complex I. Plant Cell 2021, 33, 2072–2091. [Google Scholar] [CrossRef]
- Soufari, H.; Parrot, C.; Kuhn, L.; Waltz, F.; Hashem, Y. Specific features and assembly of the plant mitochondrial complex I revealed by Cryo-EM. Nat. Commun. 2020, 11, 5195. [Google Scholar] [CrossRef]
- Guo, R.; Zong, S.; Wu, M.; Gu, J.; Yang, M. Architecture of human mitochondrial respiratory megacomplex I2III2IV2. Cell 2017, 170, 1247–1257. [Google Scholar] [CrossRef] [Green Version]
- Signes, A.; Fernandez-Vizarra, E. Assembly of mammalian oxidative phosphorylation complexes I–V and supercomplexes. Essays Biochem. 2018, 62, 255–270. [Google Scholar] [CrossRef]
- Vartak, R.S.; Semwal, M.K.; Bai, Y. An update on complex I assembly: The assembly of players. J. Bioenerg. Biomembr. 2014, 46, 323–328. [Google Scholar] [CrossRef] [Green Version]
- Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Post-translational modifications near the quinone binding site of mammalian complex I*. J. Biol. Chem. 2013, 288, 24799–24808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protasoni, M.; Zeviani, M. Mitochondrial structure and bioenergetics in normal and disease conditions. Int. J. Mol. Sci. 2021, 22, 586. [Google Scholar] [CrossRef]
- Nouws, J.; Nijtmans, L.; Houten, S.M.; van den Brand, M.; Huynen, M.; Venselaar, H.; Hoefs, S.; Gloerich, J.; Kronick, J.; Hutchin, T.; et al. Acyl-CoA dehydrogenase 9 is required for the biogenesis of oxidative phosphorylation complex I. Cell Metab. 2010, 12, 283–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haack, T.B.; Danhauser, K.; Haberberger, B.; Hoser, J.; Strecker, V.; Boehm, D.; Uziel, G.; Lamantea, E.; Invernizzi, F.; Poulton, J.; et al. Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat. Genet. 2010, 42, 1131–1134. [Google Scholar] [CrossRef] [PubMed]
- Vogel, R.O.; Janssen, R.J.; van den Brand, M.A.M.; Dieteren, C.E.J.; Verkaart, S.; Koopman, W.J.H.; Willems, P.H.G.M.; Pluk, W.; van den Heuvel, L.P.W.J.; Smeitink, J.A.M.; et al. Cytosolic signaling protein ecsit also localizes to mitochondria where it interacts with chaperone NDUFAF1 and functions in complex I assembly. Genes Dev. 2007, 21, 615–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rendón, O.Z.; Antonicka, H.; Horvath, R.; Shoubridge, E.A. A mutation in the flavin adenine dinucleotide-dependent oxidoreductase FOXRED1 results in cell-type-specific assembly defects in oxidative phosphorylation complexes I and II. Mol. Cell. Biol. 2016, 36, 2132–2140. [Google Scholar] [CrossRef] [Green Version]
- Formosa, L.E.; Mimaki, M.; Frazier, A.E.; McKenzie, M.; Stait, T.L.; Thorburn, D.R.; Stroud, D.A.; Ryan, M.T. Characterization of mitochondrial FOXRED1 in the assembly of respiratory chain complex I. Hum. Mol. Genet. 2015, 24, 2952–2965. [Google Scholar] [CrossRef] [Green Version]
- Calvo, S.E.; Tucker, E.J.; Compton, A.G.; Kirby, D.M.; Crawford, G.; Burtt, N.P.; Rivas, M.; Guiducci, C.; Bruno, D.L.; Goldberger, O.A.; et al. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat. Genet. 2010, 42, 851–858. [Google Scholar] [CrossRef]
- Andrews, B.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Assembly factors for the membrane arm of human complex I. Proc. Natl. Acad. Sci. USA 2013, 110, 18934–18939. [Google Scholar] [CrossRef] [Green Version]
- Čížková, A.; Stránecký, V.; Mayr, J.A.; Tesařová, M.; Havlíčková, V.; Paul, J.; Ivánek, R.; Kuss, A.W.; Hansíková, H.; Kaplanová, V.; et al. TMEM70 mutations cause isolated ATP synthase deficiency and neonatal mitochondrial encephalocardiomyopathy. Nat. Genet. 2008, 40, 1288–1290. [Google Scholar] [CrossRef]
- Sánchez-Caballero, L.; Elurbe, D.M.; Baertling, F.; Guerrero-Castillo, S.; van den Brand, M.; van Strien, J.; van Dam, T.J.P.; Rodenburg, R.; Brandt, U.; Huynen, M.A.; et al. TMEM70 functions in the assembly of complexes I and V. Biochim. Biophys. Acta (BBA)-Bioenerg. 2020, 1861, 148202. [Google Scholar] [CrossRef]
- Catteruccia, M.; Verrigni, D.; Martinelli, D.; Torraco, A.; Agovino, T.; Bonafé, L.; D’Amico, A.; Donati, M.A.; Adorisio, R.; Santorelli, F.M.; et al. Persistent pulmonary arterial hypertension in the newborn (PPHN): A frequent manifestation of TMEM70 defective patients. Mol. Genet. Metab. 2014, 111, 353–359. [Google Scholar] [CrossRef]
- Staretz-Chacham, O.; Wormser, O.; Manor, E.; Birk, O.S.; Ferreira, C.R. TMEM70 deficiency: Novel mutation and hypercitrullinemia during metabolic decompensation. Am. J. Med. Genet. 2019, 179, 1293–1298. [Google Scholar] [CrossRef]
- Hirono, K.; Ichida, F.; Nishio, N.; Ogawa-Tominaga, M.; Fushimi, T.; Feichtinger, R.G.; Mayr, J.A.; Kohda, M.; Kishita, Y.; Okazaki, Y.; et al. Mitochondrial complex deficiency by novel compound heterozygous TMEM70 variants and correlation with developmental delay, undescended testicle, and left ventricular noncompaction in a Japanese patient: A case report. Clin. Case Rep. 2019, 7, 553–557. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, R.; Khayat, M.; Shalev, S.A.; Horovitz, Y.; Mandel, H.; Hershkovitz, E.; Barghuti, F.; Shaag, A.; Saada, A.; Korman, S.H.; et al. TMEM70 mutations are a common cause of nuclear encoded ATP synthase assembly defect: Further delineation of a new syndrome. J. Med. Genet. 2011, 48, 177–182. [Google Scholar] [CrossRef]
- Vogel, R.O.; Janssen, R.J.R.J.; Ugalde, C.; Grovenstein, M.; Huijbens, R.J.; Visch, H.-J.; van den Heuvel, L.P.; Willems, P.H.; Zeviani, M.; Smeitink, J.A.M.; et al. Human mitochondrial complex I assembly is mediated by NDUFAF1. FEBS J. 2005, 272, 5317–5326. [Google Scholar] [CrossRef]
- Dunning, C.J.R.; McKenzie, M.; Sugiana, C.; Lazarou, M.; Silke, J.; Connelly, A.; Fletcher, J.M.; Kirby, D.M.; Thorburn, D.R.; Ryan, M.T. Human CIA30 is involved in the early assembly of mitochondrial complex I and mutations in its gene cause disease. EMBO J. 2007, 26, 3227–3237. [Google Scholar] [CrossRef] [Green Version]
- Ogilvie, I.; Ogilvie, I.; Kennaway, N.G.; Shoubridge, E.A. A molecular chaperone for mitochondrial complex I assembly is mutated in a progressive encephalopathy. J. Clin. Investig. 2005, 115, 2784–2792. [Google Scholar] [CrossRef]
- Saada, A.; Edvardson, S.; Rapoport, M.; Shaag, A.; Amry, K.; Miller, C.; Lorberboum-Galski, H.; Elpeleg, O. C6ORF66 is an assembly factor of mitochondrial complex I. Am. J. Hum. Genet. 2008, 82, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Saada, A.; Vogel, R.O.; Hoefs, S.J.; van den Brand, M.A.; Wessels, H.J.; Willems, P.H.; Venselaar, H.; Shaag, A.; Barghuti, F.; Reish, O.; et al. Mutations in NDUFAF3 (C3ORF60), encoding an NDUFAF4 (C6ORF66)-interacting complex I assembly protein, cause fatal neonatal mitochondrial disease. Am. J. Hum. Genet. 2009, 84, 718–727. [Google Scholar] [CrossRef] [Green Version]
- Sugiana, C.; Pagliarini, D.J.; McKenzie, M.; Kirby, D.M.; Salemi, R.; Abu-Amero, K.K.; Dahl, H.-H.M.; Hutchison, W.M.; Vascotto, K.A.; Smith, S.M.; et al. Mutation of C20orf7 disrupts complex I assembly and causes lethal neonatal mitochondrial disease. Am. J. Hum. Genet. 2008, 83, 468–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhein, V.F.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. NDUFAF5 Hydroxylates NDUFS7 at an early stage in the assembly of human complex I. J. Biol. Chem. 2016, 291, 14851–14860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenzie, M.; Tucker, E.J.; Compton, A.G.; Lazarou, M.; George, C.; Thorburn, D.R.; Ryan, M.T. Mutations in the gene encoding C8orf38 block complex I assembly by inhibiting production of the mitochondria-encoded subunit ND1. J. Mol. Biol. 2011, 414, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Bianciardi, L.; Imperatore, V.; Fernandez-Vizarra, E.; Lopomo, A.; Falabella, M.; Furini, S.; Galluzzi, P.; Grosso, S.; Zeviani, M.; Renieri, A.; et al. Exome sequencing coupled with MRNA analysis identifies NDUFAF6 as a leigh gene. Mol. Genet. Metab. 2016, 119, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Catania, A.; Ardissone, A.; Verrigni, D.; Legati, A.; Reyes, A.; Lamantea, E.; Diodato, D.; Tonduti, D.; Imperatore, V.; Pinto, A.M.; et al. Compound heterozygous missense and deep intronic variants in NDUFAF6 unraveled by exome sequencing and MRNA analysis. J. Hum. Genet. 2018, 63, 563–568. [Google Scholar] [CrossRef]
- Baide-Mairena, H.; Gaudó, P.; Marti-Sánchez, L.; Emperador, S.; Sánchez-Montanez, A.; Alonso-Luengo, O.; Correa, M.; Grau, A.M.; Ortigoza-Escobar, J.D.; Artuch, R.; et al. Mutations in the mitochondrial complex I assembly factor NDUFAF6 cause isolated bilateral striatal necrosis and progressive dystonia in childhood. Mol. Genet. Metab. 2019, 126, 250–258. [Google Scholar] [CrossRef] [Green Version]
- Hartmannová, H.; Piherová, L.; Tauchmannová, K.; Kidd, K.; Acott, P.D.; Crocker, J.F.S.; Oussedik, Y.; Mallet, M.; Hodaňová, K.; Stránecký, V.; et al. Acadian variant of fanconi syndrome is caused by mitochondrial respiratory chain complex I deficiency due to a non-coding mutation in complex I assembly factor NDUFAF6. Hum. Mol. Genet. 2016, 25, 4062–4079. [Google Scholar] [CrossRef]
- Carilla-Latorre, S.; Gallardo, M.E.; Annesley, S.J.; Calvo-Garrido, J.; Graña, O.; Accari, S.L.; Smith, P.K.; Valencia, A.; Garesse, R.; Fisher, P.R.; et al. MidA is a putative methyltransferase that is required for mitochondrial complex I function. J. Cell Sci. 2010, 123, 1674–1683. [Google Scholar] [CrossRef] [Green Version]
- Rhein, V.F.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. NDUFAF7 methylates arginine 85 in the NDUFS2 subunit of human complex I. J. Biol. Chem. 2013, 288, 33016–33026. [Google Scholar] [CrossRef] [Green Version]
- Alston, C.L.; Veling, M.T.; Heidler, J.; Taylor, L.S.; Alaimo, J.T.; Sung, A.Y.; He, L.; Hopton, S.; Broomfield, A.; Pavaine, J.; et al. Pathogenic bi-allelic mutations in NDUFAF8 cause leigh syndrome with an isolated complex I deficiency. Am. J. Hum. Genet. 2020, 106, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Sheftel, A.D.; Stehling, O.; Pierik, A.J.; Netz, D.J.A.; Kerscher, S.; Elsässer, H.-P.; Wittig, I.; Balk, J.; Brandt, U.; Lill, R. Human Ind1, an iron-sulfur cluster assembly factor for respiratory complex I. Mol. Cell. Biol. 2009, 29, 6059–6073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bych, K.; Kerscher, S.; Netz, D.J.A.; Pierik, A.J.; Zwicker, K.; Huynen, M.A.; Lill, R.; Brandt, U.; Balk, J. The iron–sulphur protein Ind1 is required for effective complex I assembly. EMBO J. 2008, 27, 1736–1746. [Google Scholar] [CrossRef] [Green Version]
- Protasoni, M.; Bruno, C.; Donati, M.A.; Mohamoud, K.; Severino, M.; Allegri, A.; Robinson, A.J.; Reyes, A.; Zeviani, M.; Garone, C. Novel compound heterozygous pathogenic variants in nucleotide-binding protein like protein (NUBPL) cause leukoencephalopathy with multi-systemic involvement. Mol. Genet. Metab. 2020, 129, 26–34. [Google Scholar] [CrossRef]
- Guarani, V.; Paulo, J.; Zhai, B.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. TIMMDC1/C3orf1 functions as a membrane-embedded mitochondrial complex I assembly factor through association with the MCIA complex. Mol. Cell. Biol. 2014, 34, 847–861. [Google Scholar] [CrossRef] [Green Version]
- Kremer, L.S.; Bader, D.M.; Mertes, C.; Kopajtich, R.; Pichler, G.; Iuso, A.; Haack, T.B.; Graf, E.; Schwarzmayr, T.; Terrile, C.; et al. Genetic diagnosis of mendelian disorders via RNA sequencing. Nat. Commun. 2017, 8, 15824. [Google Scholar] [CrossRef]
- Désir, J.; Coppieters, F.; Van Regemorter, N.; De Baere, E.; Abramowicz, M.; Cordonnier, M. TMEM126A mutation in a Moroccan family with autosomal recessive optic atrophy. Mol. Vis. 2012, 18, 1849–1857. [Google Scholar]
- Hanein, S.; Perrault, I.; Roche, O.; Gerber, S.; Khadom, N.; Rio, M.; Boddaert, N.; Jean-Pierre, M.; Brahimi, N.; Serre, V.; et al. TMEM126A, encoding a mitochondrial protein, is mutated in autosomal-recessive nonsyndromic optic atrophy. Am. J. Hum. Genet. 2009, 84, 493–498. [Google Scholar] [CrossRef] [Green Version]
- Kloth, K.; Synofzik, M.; Kernstock, C.; Schimpf-Linzenbold, S.; Schuettauf, F.; Neu, A.; Wissinger, B.; Weisschuh, N. Novel Likely Pathogenic Variants in TMEM126A identified in non-syndromic autosomal recessive optic atrophy: Two case reports. BMC Med. Genet. 2019, 20, 62. [Google Scholar] [CrossRef]
- La Morgia, C.; Caporali, L.; Tagliavini, F.; Palombo, F.; Carbonelli, M.; Liguori, R.; Barboni, P.; Carelli, V. First TMEM126A missense mutation in an italian proband with optic atrophy and deafness. Neurol. Genet. 2019, 5, e329. [Google Scholar] [CrossRef] [Green Version]
- Meyer, E.; Michaelides, M.; Tee, L.J.; Robson, A.G.; Rahman, F.; Pasha, S.; Luxon, L.M.; Moore, A.T.; Maher, E.R. Nonsense mutation in TMEM126A causing autosomal recessive optic atrophy and auditory neuropathy. Mol. Vis. 2010, 16, 650–664. [Google Scholar]
- Heide, H.; Bleier, L.; Steger, M.; Ackermann, J.; Dröse, S.; Schwamb, B.; Zörnig, M.; Reichert, A.S.; Koch, I.; Wittig, I.; et al. Complexome profiling identifies TMEM126B as a component of the mitochondrial complex I assembly complex. Cell Metab. 2012, 16, 538–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Caballero, L.; Ruzzenente, B.; Bianchi, L.; Assouline, Z.; Barcia, G.; Metodiev, M.D.; Rio, M.; Funalot, B.; van den Brand, M.A.M.; Guerrero-Castillo, S.; et al. Mutations in complex I assembly factor TMEM126B result in muscle weakness and isolated complex I deficiency. Am. J. Hum. Genet. 2016, 99, 208–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alston, C.L.; Compton, A.G.; Formosa, L.E.; Strecker, V.; Oláhová, M.; Haack, T.B.; Smet, J.; Stouffs, K.; Diakumis, P.; Ciara, E.; et al. Biallelic mutations in TMEM126B cause severe complex I deficiency with a variable clinical phenotype. Am. J. Hum. Genet. 2016, 99, 217–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrero-Castillo, S.; Baertling, F.; Kownatzki, D.; Wessels, H.J.; Arnold, S.; Brandt, U.; Nijtmans, L. The assembly pathway of mitochondrial respiratory chain complex I. Cell Metab. 2017, 25, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Martinez Lyons, A.; Ardissone, A.; Reyes, A.; Robinson, A.J.; Moroni, I.; Ghezzi, D.; Fernandez-Vizarra, E.; Zeviani, M. COA7 (C1orf163/RESA1) mutations associated with mitochondrial leukoencephalopathy and cytochrome C oxidase deficiency. J. Med. Genet. 2016, 53, 846–849. [Google Scholar] [CrossRef] [Green Version]
- Dibley, M.G.; Formosa, L.E.; Lyu, B.; Reljic, B.; McGann, D.; Muellner-Wong, L.; Kraus, F.; Sharpe, A.J.; Stroud, D.A.; Ryan, M.T. The mitochondrial acyl-carrier protein interaction network highlights important roles for LYRM family members in complex I and mitoribosome assembly. Mol. Cell. Proteom. 2020, 19, 65–77. [Google Scholar] [CrossRef]
- Bugiani, M.; Invernizzi, F.; Alberio, S.; Briem, E.; Lamantea, E.; Carrara, F.; Moroni, I.; Farina, L.; Spada, M.; Donati, M.A.; et al. Clinical and molecular findings in children with complex I deficiency. Biochim. Biophys. Acta (BBA)-Bioenerg. 2004, 1659, 136–147. [Google Scholar] [CrossRef] [Green Version]
- Malfatti, E.; Bugiani, M.; Invernizzi, F.; de Souza, C.F.-M.; Farina, L.; Carrara, F.; Lamantea, E.; Antozzi, C.; Confalonieri, P.; Sanseverino, M.T.; et al. Novel mutations of ND genes in complex I deficiency associated with mitochondrial encephalopathy. Brain 2007, 130, 1894–1904. [Google Scholar] [CrossRef]
- Fassone, E.; Rahman, S. Complex I deficiency: Clinical features, biochemistry and molecular genetics. J. Med. Genet. 2012, 49, 578–590. [Google Scholar] [CrossRef] [Green Version]
- Rodenburg, R.J. Mitochondrial complex I-linked disease. Biochim. Biophys. Acta (BBA)-Bioenerg. 2016, 1857, 938–945. [Google Scholar] [CrossRef]
- Man, P.Y.-W.; Griffiths, P.G.; Brown, D.T.; Howell, N.; Turnbull, D.M.; Chinnery, P.F. The epidemiology of Leber hereditary optic neuropathy in the North East of England. Am. J. Hum. Genet. 2003, 72, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Carelli, V.; Rugolo, M.; Sgarbi, G.; Ghelli, A.; Zanna, C.; Baracca, A.; Lenaz, G.; Napoli, E.; Martinuzzi, A.; Solaini, G. Bioenergetics shapes cellular death pathways in Leber’s hereditary optic neuropathy: A model of mitochondrial neurodegeneration. Biochim. Biophys. Acta (BBA)-Bioenerg. 2004, 1658, 172–179. [Google Scholar] [CrossRef] [Green Version]
- Yu-Wai-Man, P.; Griffiths, P.G.; Chinnery, P.F. Mitochondrial optic neuropathies—Disease mechanisms and therapeutic strategies. Prog. Retin. Eye Res. 2011, 30, 81–114. [Google Scholar] [CrossRef] [Green Version]
- Giordano, C.; Iommarini, L.; Giordano, L.; Maresca, A.; Pisano, A.; Valentino, M.L.; Caporali, L.; Liguori, R.; Deceglie, S.; Roberti, M.; et al. Efficient mitochondrial biogenesis drives incomplete penetrance in Leber’s hereditary optic neuropathy. Brain 2014, 137, 335–353. [Google Scholar] [CrossRef] [Green Version]
- Bianco, A.; Martínez-Romero, I.; Bisceglia, L.; D’Agruma, L.; Favia, P.; Ruiz-Pesini, E.; Guerriero, S.; Montoya, J.; Petruzzella, V. Mitochondrial DNA copy number differentiates the Leber’s hereditary optic neuropathy affected individuals from the unaffected mutation carriers. Brain 2016, 139, e1. [Google Scholar] [CrossRef]
- Bianco, A.; Bisceglia, L.; Russo, L.; Palese, L.L.; D’Agruma, L.; Emperador, S.; Montoya, J.; Guerriero, S.; Petruzzella, V. High mitochondrial DNA copy number is a protective factor from vision loss in heteroplasmic Leber’s hereditary optic neuropathy (LHON). Investig. Opthalmol. Vis. Sci. 2017, 58, 2193. [Google Scholar] [CrossRef] [Green Version]
- Bianco, A.; Valletti, A.; Longo, G.; Bisceglia, L.; Montoya, J.; Emperador, S.; Guerriero, S.; Petruzzella, V. Mitochondrial DNA copy number in affected and unaffected LHON mutation carriers. BMC Res. Notes 2018, 11, 911. [Google Scholar] [CrossRef] [Green Version]
- Tun, A.W.; Chaiyarit, S.; Kaewsutthi, S.; Katanyoo, W.; Chuenkongkaew, W.; Kuwano, M.; Tomonaga, T.; Peerapittayamongkol, C.; Thongboonkerd, V.; Lertrit, P. Profiling the mitochondrial proteome of Leber’s hereditary optic neuropathy (LHON) in Thailand: Down-regulation of bioenergetics and mitochondrial protein quality control pathways in fibroblasts with the 11778G>A mutation. PLoS ONE 2014, 9, e106779. [Google Scholar] [CrossRef] [Green Version]
- Lenaz, G.; Baracca, A.; Carelli, V.; D’Aurelio, M.; Sgarbi, G.; Solaini, G. Bioenergetics of mitochondrial diseases associated with MtDNA mutations. Biochim. Biophys. Acta (BBA)-Bioenerg. 2004, 1658, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.D.; Trounce, I.A.; Jun, A.S.; Allen, J.C.; Wallace, D.C. Functional analysis of lymphoblast and cybrid mitochondria containing the 3460, 11778, or 14484 Leber’s hereditary optic neuropathy mitochondrial DNA mutation. J. Biol. Chem. 2000, 275, 39831–39836. [Google Scholar] [CrossRef] [Green Version]
- Floreani, M.; Napoli, E.; Martinuzzi, A.; Pantano, G.; De Riva, V.; Trevisan, R.; Bisetto, E.; Valente, L.; Carelli, V.; Dabbeni-Sala, F. Antioxidant defences in cybrids harboring MtDNA mutations associated with Leber’s hereditary optic neuropathy: Antioxidant defences in LHON cybrids. FEBS J. 2005, 272, 1124–1135. [Google Scholar] [CrossRef]
- Simon, D.K.; Friedman, J.; Breakefield, X.O.; Jankovic, J.; Brin, M.F.; Provias, J.; Bressman, S.B.; Charness, M.E.; Tarsy, D.; Johns, D.R.; et al. A heteroplasmic mitochondrial complex I gene mutation in adult-onset dystonia. Neurogenetics 2003, 4, 199–205. [Google Scholar] [CrossRef]
- Kirby, D.M. Mutations of the mitochondrial ND1 gene as a cause of MELAS. J. Med. Genet. 2004, 41, 784–789. [Google Scholar] [CrossRef] [Green Version]
- Howell, N.; Bindoff, L.A.; McCullough, D.A.; Kubacka, I.; Poulton, J.; Mackey, D.; Taylor, L.; Turnbull, D.M. Leber hereditary optic neuropathy: Identification of the same mitochondrial ND1 mutation in six pedigrees. Am. J. Hum. Genet. 1991, 49, 939–950. [Google Scholar]
- Johns, D.R.; Berman, J. Alternative, simultaneous complex I mitochondrial DNA mutations in Leber’s hereditary optic neuropathy. Biochem. Biophys. Res. Commun. 1991, 174, 1324–1330. [Google Scholar] [CrossRef]
- McFarland, R.; Kirby, D.M.; Fowler, K.J.; Ohtake, A.; Ryan, M.T.; Amor, D.J.; Fletcher, J.M.; Dixon, J.W.; Collins, F.A.; Turnbull, D.M.; et al. De novo mutations in the mitochondrial ND3 gene as a cause of infantile mitochondrial encephalopathy and complex I deficiency. Ann. Neurol. 2004, 55, 58–64. [Google Scholar] [CrossRef]
- Torroni, A.; Petrozzi, M.; D’Urbano, L.; Sellitto, D.; Zeviani, M.; Carrara, F.; Carducci, C.; Leuzzi, V.; Carelli, V.; Barboni, P.; et al. Haplotype and phylogenetic analyses suggest that one european-specific MtDNA background plays a role in the expression of Leber hereditary optic neuropathy by increasing the penetrance of the primary mutations 11778 and 14484. Am. J. Hum. Genet. 1997, 60, 1107–1121. [Google Scholar] [PubMed]
- Lertrit, P.; Noer, A.S.; Jean-Francois, M.J.; Kapsa, R.; Dennett, X.; Thyagarajan, D.; Lethlean, K.; Byrne, E.; Marzuki, S. A new disease-related mutation for mitochondrial encephalopathy lactic acidosis and strokelike episodes (MELAS) syndrome affects the ND4 subunit of the respiratory complex I. Am. J. Hum. Genet. 1992, 51, 457–468. [Google Scholar] [PubMed]
- Brown, M.D.; Starikovskaya, E.; Derbeneva, O.; Hosseini, S.; Allen, J.C.; Mikhailovskaya, I.E.; Sukernik, R.I.; Wallace, D.C. The role of MtDNA background in disease expression: A new primary LHON mutation associated with western eurasian haplogroup. J. Hum. Genet. 2002, 110, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.D.; Voljavec, A.S.; Lott, M.T.; Macdonald, I.; Wallace, D.C. Leber’s hereditary optic neuropathy: A model for mitochondrial neurodegenerative diseases. FASEB J. 1992, 6, 2791–2799. [Google Scholar] [CrossRef]
- Liolitsa, D.; Rahman, S.; Benton, S.; Carr, L.J.; Hanna, M.G. Is the mitochondrial complex I ND5 gene a hot-spot for MELAS causing mutations? Ann. Neurol. 2003, 53, 128–132. [Google Scholar] [CrossRef]
- Ravn, K.; Wibrand, F.; Hansen, F.J.; Horn, N.; Rosenberg, T.; Schwartz, M. An MtDNA mutation, 14453G→A, in the NADH dehydrogenase subunit 6 associated with severe MELAS syndrome. Eur. J. Hum. Genet. 2001, 9, 805–809. [Google Scholar] [CrossRef] [Green Version]
- Schuelke, M.; Smeitink, J.; Mariman, E.; Loeffen, J.; Plecko, B.; Trijbels, F.; Stöckler-Ipsiroglu, S.; van den Heuvel, L. Mutant NDUFV1 subunit of mitochondrial complex I causes leukodystrophy and myoclonic epilepsy. Nat. Genet. 1999, 21, 260–261. [Google Scholar] [CrossRef]
- Bénit, P.; Chretien, D.; Kadhom, N.; de Lonlay-Debeney, P.; Cormier-Daire, V.; Cabral, A.; Peudenier, S.; Rustin, P.; Munnich, A.; Rötig, A. Large-scale deletion and point mutations of the nuclear NDUFV1 and NDUFS1 genes in mitochondrial complex I deficiency. Am. J. Hum. Genet. 2001, 68, 1344–1352. [Google Scholar] [CrossRef] [Green Version]
- Bénit, P.; Beugnot, R.; Chretien, D.; Giurgea, I.; De Lonlay-Debeney, P.; Issartel, J.-P.; Corral-Debrinski, M.; Kerscher, S.; Rustin, P.; Rötig, A.; et al. Mutant NDUFV2 subunit of mitochondrial complex I causes early onset hypertrophic cardiomyopathy and encephalopathy: NDUFV2 and cardiomyopathy/encephalopathy. Hum. Mutat. 2003, 21, 582–586. [Google Scholar] [CrossRef]
- Loeffen, J.; Elpeleg, O.; Smeitink, J.; Smeets, R.; Stöckler-Ipsiroglu, S.; Mandel, H.; Sengers, R.; Trijbels, F.; van den Heuvel, L. Mutations in the complex I NDUFS2 gene of patients with cardiomyopathy and encephalomyopathy. Ann. Neurol. 2001, 49, 195–201. [Google Scholar] [CrossRef]
- Benit, P.; Slama, A.; Cartault, F.; Giurgea, I.; Chretien, D.; Lebon, S.; Marsac, C.; Munnich, A.; Rotig, A.; Rustin, P. Mutant NDUFS3 Subunit of mitochondrial complex I causes leigh syndrome. J. Med. Genet. 2004, 41, 14–17. [Google Scholar] [CrossRef]
- Budde, S.M.S.; van den Heuvel, L.P.W.J.; Janssen, A.J.; Smeets, R.J.P.; Buskens, C.A.F.; DeMeirleir, L.; Van Coster, R.; Baethmann, M.; Voit, T.; Trijbels, J.M.F.; et al. Combined enzymatic complex I and III deficiency associated with mutations in the nuclear encoded NDUFS4 gene. Biochem. Biophys. Res. Commun. 2000, 275, 63–68. [Google Scholar] [CrossRef]
- Spiegel, R.; Shaag, A.; Mandel, H.; Reich, D.; Penyakov, M.; Hujeirat, Y.; Saada, A.; Elpeleg, O.; Shalev, S.A. Mutated NDUFS6 is the cause of fatal neonatal lactic acidemia in caucasus jews. Eur. J. Hum. Genet. 2009, 17, 1200–1203. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.W.; Pyle, A.; Griffin, H.; Blakely, E.L.; Duff, J.; He, L.; Smertenko, T.; Alston, C.L.; Neeve, V.C.; Best, A.; et al. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA 2014, 312, 68. [Google Scholar] [CrossRef] [Green Version]
- Smeitink, J.; van den Heuvel, L. Human mitochondrial complex I in health and disease. Am. J. Hum. Genet. 1999, 64, 1505–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeffen, J.; Smeitink, J.; Triepels, R.; Smeets, R.; Schuelke, M.; Sengers, R.; Trijbels, F.; Hamel, B.; Mullaart, R.; van den Heuvel, L. The first nuclear-encoded complex I mutation in a patient with Leigh syndrome. Am. J. Hum. Genet. 1998, 63, 1598–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, I.; Hershkovitz, E.; Shaag, A.; Edvardson, S.; Saada, A.; Elpeleg, O. Mitochondrial complex I deficiency caused by a deleterious NDUFA11 mutation. Ann. Neurol. 2008, 63, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Moreira, D.; Ugalde, C.; Smeets, R.; Rodenburg, R.J.T.; Lopez-Laso, E.; Ruiz-Falco, M.L.; Briones, P.; Martin, M.A.; Smeitink, J.A.M.; Arenas, J. X-Linked NDUFA1 gene mutations associated with mitochondrial encephalomyopathy. Ann. Neurol. 2007, 61, 73–83. [Google Scholar] [CrossRef]
- Hoefs, S.J.G.; Dieteren, C.E.J.; Distelmaier, F.; Janssen, R.J.R.J.; Epplen, A.; Swarts, H.G.P.; Forkink, M.; Rodenburg, R.J.; Nijtmans, L.G.; Willems, P.H.; et al. NDUFA2 complex I mutation leads to Leigh disease. Am. J. Hum. Genet. 2008, 82, 1306–1315. [Google Scholar] [CrossRef] [Green Version]
- Alston, C.L.; Heidler, J.; Dibley, M.G.; Kremer, L.S.; Taylor, L.S.; Fratter, C.; French, C.E.; Glasgow, R.I.C.; Feichtinger, R.G.; Delon, I.; et al. Bi-Allelic mutations in NDUFA6 establish its role in early-onset isolated mitochondrial complex I deficiency. Am. J. Hum. Genet. 2018, 103, 592–601. [Google Scholar] [CrossRef] [Green Version]
- van den Bosch, B.J.C.; Gerards, M.; Sluiter, W.; Stegmann, A.P.A.; Jongen, E.L.C.; Hellebrekers, D.M.E.I.; Oegema, R.; Lambrichs, E.H.; Prokisch, H.; Danhauser, K.; et al. Defective NDUFA9 as a novel cause of neonatally fatal complex I disease. J. Med. Genet. 2012, 49, 10–15. [Google Scholar] [CrossRef]
- Ostergaard, E.; Rodenburg, R.J.; van den Brand, M.; Thomsen, L.L.; Duno, M.; Batbayli, M.; Wibrand, F.; Nijtmans, L. Respiratory chain complex I deficiency due to NDUFA12 mutations as a new cause of Leigh syndrome. J. Med. Genet. 2011, 48, 737–740. [Google Scholar] [CrossRef]
- Angebault, C.; Charif, M.; Guegen, N.; Piro-Megy, C.; de Camaret, B.M.; Procaccio, V.; Guichet, P.-O.; Hebrard, M.; Manes, G.; Leboucq, N.; et al. Mutation in NDUFA13/GRIM19 leads to early onset hypotonia, dyskinesia and sensorial deficiencies, and mitochondrial complex I instability. Hum. Mol. Genet. 2015, 24, 3948–3955. [Google Scholar] [CrossRef] [Green Version]
- Haack, T.B.; Madignier, F.; Herzer, M.; Lamantea, E.; Danhauser, K.; Invernizzi, F.; Koch, J.; Freitag, M.; Drost, R.; Hillier, I.; et al. Mutation screening of 75 candidate genes in 152 complex I deficiency cases identifies pathogenic variants in 16 genes including NDUFB9. J. Med. Genet. 2012, 49, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Piekutowska-Abramczuk, D.; Assouline, Z.; Mataković, L.; Feichtinger, R.G.; Koňařiková, E.; Jurkiewicz, E.; Stawiński, P.; Gusic, M.; Koller, A.; Pollak, A.; et al. NDUFB8 mutations cause mitochondrial complex I deficiency in individuals with Leigh-like encephalomyopathy. Am. J. Hum. Genet. 2018, 102, 460–467. [Google Scholar] [CrossRef] [Green Version]
- Friederich, M.W.; Erdogan, A.J.; Coughlin, C.R.; Elos, M.T.; Jiang, H.; O’Rourke, C.P.; Lovell, M.A.; Wartchow, E.; Gowan, K.; Chatfield, K.C.; et al. Mutations in the accessory subunit NDUFB10 result in isolated complex I deficiency and illustrate the critical role of intermembrane space import for complex I holoenzyme assembly. Hum. Mol. Genet. 2016, 26, 702–716. [Google Scholar] [CrossRef] [Green Version]
- Van Rahden, V.A.; Fernandez-Vizarra, E.; Alawi, M.; Brand, K.; Fellmann, F.; Horn, D.; Zeviani, M.; Kutsche, K. Mutations in NDUFB11, encoding a complex I component of the mitochondrial respiratory chain, cause microphthalmia with linear skin defects syndrome. Am. J. Hum. Genet. 2015, 96, 640–650. [Google Scholar] [CrossRef] [Green Version]
- Reinson, K.; Kovacs-Nagy, R.; Õiglane-Shlik, E.; Pajusalu, S.; Nõukas, M.; Wintjes, L.T.; van den Brandt, F.C.A.; Brink, M.; Acker, T.; Ahting, U.; et al. Diverse phenotype in patients with complex I deficiency due to mutations in NDUFB11. Eur. J. Med. Genet. 2019, 62, 103572. [Google Scholar] [CrossRef]
- Kohda, M.; Tokuzawa, Y.; Kishita, Y.; Nyuzuki, H.; Moriyama, Y.; Mizuno, Y.; Hirata, T.; Yatsuka, Y.; Yamashita-Sugahara, Y.; Nakachi, Y.; et al. A comprehensive genomic analysis reveals the genetic landscape of mitochondrial respiratory chain complex deficiencies. PLoS Genet. 2016, 12, e1005679. [Google Scholar] [CrossRef]
- Alahmad, A.; Nasca, A.; Heidler, J.; Thompson, K.; Oláhová, M.; Legati, A.; Lamantea, E.; Meisterknecht, J.; Spagnolo, M.; He, L.; et al. Bi-allelic pathogenic variants in NDUFC2 cause early-onset Leigh syndrome and stalled biogenesis of complex I. EMBO Mol. Med. 2020, 12, e12619. [Google Scholar] [CrossRef]
- Baertling, F.; Sánchez-Caballero, L.; Timal, S.; van den Brand, M.A.; Ngu, L.H.; Distelmaier, F.; Rodenburg, R.J.; Nijtmans, L.G. Mutations in mitochondrial complex I assembly factor NDUFAF3 cause Leigh syndrome. Mol. Genet. Metab. 2017, 120, 243–246. [Google Scholar] [CrossRef]
- Baertling, F.; Sánchez-Caballero, L.; van den Brand, M.A.M.; Wintjes, L.T.; Brink, M.; van den Brandt, F.A.; Wilson, C.; Rodenburg, R.J.T.; Nijtmans, L.G.J. NDUFAF4 variants are associated with Leigh syndrome and cause a specific mitochondrial complex I assembly defect. Eur. J. Hum. Genet. 2017, 25, 1273–1277. [Google Scholar] [CrossRef]
- Ishiyama, A.; Muramatsu, K.; Uchino, S.; Sakai, C.; Matsushima, Y.; Makioka, N.; Ogata, T.; Suzuki, E.; Komaki, H.; Sasaki, M.; et al. NDUFAF3 variants that disrupt mitochondrial complex I assembly may associate with cavitating leukoencephalopathy. Clin. Genet. 2018, 93, 1103–1106. [Google Scholar] [CrossRef]
- Ugarteburu, O.; Teresa Garcia-Silva, M.; Aldamiz-Echevarria, L.; Gort, L.; Garcia-Villoria, J.; Tort, F.; Ribes, A. Complex I deficiency, due to NDUFAF4 mutations, causes severe mitochondrial dysfunction and is associated to early death and dysmorphia. Mitochondrion 2020, 55, 78–84. [Google Scholar] [CrossRef]
- Petruzzella, V.; Vergari, R.; Puzziferri, I.; Boffoli, D.; Lamantea, E.; Zeviani, M.; Papa, S. A nonsense mutation in the NDUFS4 gene encoding the 18 KDa (AQDQ) subunit of complex I abolishes assembly and activity of the complex in a patient with Leigh-like syndrome. Hum. Mol. Genet. 2001, 10, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Petruzzella, V.; Papa, S. Mutations in human nuclear genes encoding for subunits of mitochondrial respiratory complex I: The NDUFS4 gene. Gene 2002, 286, 149–154. [Google Scholar] [CrossRef]
- Petruzzella, V.; Panelli, D.; Torraco, A.; Stella, A.; Papa, S. Mutations in the NDUFS4 gene of mitochondrial complex I alter stability of the splice variants. FEBS Lett. 2005, 579, 3770–3776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scacco, S.; Petruzzella, V.; Budde, S.; Vergari, R.; Tamborra, R.; Panelli, D.; van den Heuvel, L.P.; Smeitink, J.A.; Papa, S. Pathological mutations of the human NDUFS4 gene of the 18-KDa (AQDQ) subunit of complex I affect the expression of the protein and the assembly and function of the complex. J. Biol. Chem. 2003, 278, 44161–44167. [Google Scholar] [CrossRef] [Green Version]
- Lamont, R.E.; Beaulieu, C.L.; Bernier, F.P.; Sparkes, R.; Innes, A.M.; Jackel-Cram, C.; Ober, C.; Parboosingh, J.S.; Lemire, E.G. A novel NDUFS4 frameshift mutation causes Leigh disease in the Hutterite population. Am. J. Med. Genet. 2017, 173, 596–600. [Google Scholar] [CrossRef]
- Budde, S.M.S.; van den Heuvel, L.P.W.J.; Smeets, R.J.P.; Skladal, D.; Mayr, J.A.; Boelen, C.; Petruzzella, V.; Papa, S.; Smeitink, J.A.M. Clinical heterogeneity in patients with mutations in the NDUFS4 gene of mitochondrial complex I. J. Inherit. Metab. Dis. 2003, 26, 813–815. [Google Scholar] [CrossRef] [PubMed]
- Leshinsky-Silver, E.; Lebre, A.-S.; Minai, L.; Saada, A.; Steffann, J.; Cohen, S.; Rötig, A.; Munnich, A.; Lev, D.; Lerman-Sagie, T. NDUFS4 mutations cause Leigh syndrome with predominant brainstem involvement. Mol. Genet. Metab. 2009, 97, 185–189. [Google Scholar] [CrossRef]
- Ortigoza-Escobar, J.D.; Oyarzabal, A.; Montero, R.; Artuch, R.; Jou, C.; Jiménez, C.; Gort, L.; Briones, P.; Muchart, J.; López-Gallardo, E.; et al. Ndufs4 related Leigh syndrome: A case report and review of the literature. Mitochondrion 2016, 28, 73–78. [Google Scholar] [CrossRef]
- Finsterer, J.; Zarrouk-Mahjoub, S. NDUFS4-related Leigh syndrome in Hutterites. Am. J. Med. Genet. 2017, 173, 1450–1451. [Google Scholar] [CrossRef]
- Rahman, S.; Blok, R.B.; Dahl, H.-H.M.; Danks, D.M.; Kirby, D.M.; Chow, C.W.; Christodoulou, J.; Thorburn, D.R. Leigh Syndrome: Clinical features and biochemical and DNA abnormalities. Ann. Neurol. 1996, 39, 343–351. [Google Scholar] [CrossRef]
- Leigh, D. Subacute necrotizing encephalomyelopathy in an infant. J. Neurol. Neurosurg. Psychiatry 1951, 14, 216–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assouline, Z.; Jambou, M.; Rio, M.; Bole-Feysot, C.; de Lonlay, P.; Barnerias, C.; Desguerre, I.; Bonnemains, C.; Guillermet, C.; Steffann, J.; et al. A constant and similar assembly defect of mitochondrial respiratory chain complex I allows rapid identification of NDUFS4 mutations in patients with Leigh syndrome. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis 2012, 1822, 1062–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarou, M.; McKenzie, M.; Ohtake, A.; Thorburn, D.R.; Ryan, M.T. Analysis of the assembly profiles for mitochondrial—and nuclear-DNA-encoded subunits into complex I. Mol. Cell. Biol. 2007, 27, 4228–4237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breuer, M.E.; Willems, P.H.G.M.; Smeitink, J.A.M.; Koopman, W.J.H.; Nooteboom, M. Cellular and animal models for mitochondrial complex I deficiency: A focus on the NDUFS4 subunit. IUBMB Life 2013, 65, 202–208. [Google Scholar] [CrossRef] [Green Version]
- Ingraham, C.A.; Burwell, L.S.; Skalska, J.; Brookes, P.S.; Howell, R.L.; Sheu, S.-S.; Pinkert, C.A. NDUFS4: Creation of a mouse model mimicking a complex I disorder. Mitochondrion 2009, 9, 204–210. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Folmes, C.D.L.; Wu, J.; Morey, R.; Mora-Castilla, S.; Ocampo, A.; Ma, L.; Poulton, J.; Wang, X.; Ahmed, R.; et al. Metabolic rescue in pluripotent cells from patients with MtDNA disease. Nature 2015, 524, 234–238. [Google Scholar] [CrossRef]
- Galera-Monge, T.; Zurita-Díaz, F.; Canals, I.; Grønning Hansen, M.; Rufián-Vázquez, L.; Ehinger, J.K.; Elmér, E.; Martin, M.A.; Garesse, R.; Ahlenius, H.; et al. Mitochondrial dysfunction and calcium dysregulation in Leigh syndrome induced pluripotent stem cell derived neurons. Int. J. Mol. Sci. 2020, 21, 3191. [Google Scholar] [CrossRef]
- Zheng, X.; Boyer, L.; Jin, M.; Kim, Y.; Fan, W.; Bardy, C.; Berggren, T.; Evans, R.M.; Gage, F.H.; Hunter, T. Alleviation of neuronal energy deficiency by MTOR inhibition as a treatment for mitochondria-related neurodegeneration. eLife 2016, 5, e13378. [Google Scholar] [CrossRef]
- Lorenz, C.; Lesimple, P.; Bukowiecki, R.; Zink, A.; Inak, G.; Mlody, B.; Singh, M.; Semtner, M.; Mah, N.; Auré, K.; et al. Human IPSC-derived neural progenitors are an effective drug discovery model for neurological MtDNA disorders. Cell Stem Cell 2017, 20, 659–674.e9. [Google Scholar] [CrossRef] [Green Version]
- Romero-Morales, A.; Rastogi, A.; Temuri, H.; Rasmussen, M.; McElroy, G.S.; Hsu, L.; Almonacid, P.M.; Milis, B.A.; Chandel, N.; Cartailler, J.-P.; et al. Human iPSC-derived cerebral organoids model features of leigh syndrome and reveal abnormal corticogenesis. Cell Biol. 2020. [Google Scholar] [CrossRef]
- Zhu, Z.; Yao, J.; Johns, T.; Fu, K.; Bie, I.D.; Macmillan, C.; Cuthbert, A.P.; Newbold, R.F.; Wang, J.; Chevrette, M.; et al. SURF1, Encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat. Genet. 1998, 20, 337–343. [Google Scholar] [CrossRef]
- Tiranti, V.; Hoertnagel, K.; Carrozzo, R.; Galimberti, C.; Munaro, M.; Granatiero, M.; Zelante, L.; Gasparini, P.; Marzella, R.; Rocchi, M.; et al. Mutations of SURF-1 in Leigh disease associated with cytochrome c oxidase deficiency. Am. J. Hum. Genet. 1998, 63, 1609–1621. [Google Scholar] [CrossRef] [Green Version]
- Saneto, R.; Ruhoy, I. The genetics of Leigh syndrome and its implications for clinical practice and risk management. Appl. Clin. Genet. 2014, 7, 221. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.; Huo, X.; Zhai, Y.; Wang, A.; Xu, J.; Su, D.; Bartlam, M.; Rao, Z. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell 2005, 121, 1043–1057. [Google Scholar] [CrossRef] [Green Version]
- Van Vranken, J.G.; Na, U.; Winge, D.R.; Rutter, J. Protein-mediated assembly of succinate dehydrogenase and its cofactors. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 168–180. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, D.; Goffrini, P.; Uziel, G.; Horvath, R.; Klopstock, T.; Lochmüller, H.; D’Adamo, P.; Gasparini, P.; Strom, T.M.; Prokisch, H.; et al. SDHAF1, encoding a LYR complex-II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nat. Genet. 2009, 41, 654–656. [Google Scholar] [CrossRef]
- Munnich, A.; Rustin, P. Clinical spectrum and diagnosis of mitochondrial disorders. Am. J. Med. Genet. 2001, 106, 4–17. [Google Scholar] [CrossRef]
- Bourgeron, T.; Rustin, P.; Chretien, D.; Birch-Machin, M.; Bourgeois, M.; Viegas-Péquignot, E.; Munnich, A.; Rötig, A. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat. Genet. 1995, 11, 144–149. [Google Scholar] [CrossRef]
- Parfait, B.; Chretien, D.; Rötig, A.; Marsac, C.; Munnich, A.; Rustin, P. Compound heterozygous mutations in the flavoprotein gene of the respiratory chain complex II in a patient with Leigh syndrome. Hum. Genet. 2000, 106, 236–243. [Google Scholar] [CrossRef]
- Van Coster, R.; Seneca, S.; Smet, J.; Van Hecke, R.; Gerlo, E.; Devreese, B.; Van Beeumen, J.; Leroy, J.G.; De Meirleir, L.; Lissens, W. Homozygous Gly555Glu mutation in the nuclear-encoded 70 KDa flavoprotein gene causes instability of the respiratory chain complex II. Am. J. Med. Genet. 2003, 120A, 13–18. [Google Scholar] [CrossRef]
- Pagnamenta, A.T.; Hargreaves, I.P.; Duncan, A.J.; Taanman, J.-W.; Heales, S.J.; Land, J.M.; Bitner-Glindzicz, M.; Leonard, J.V.; Rahman, S. Phenotypic variability of mitochondrial disease caused by a nuclear mutation in complex II. Mol. Genet. Metab. 2006, 89, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Horvath, R.; Abicht, A.; Holinski-Feder, E.; Laner, A.; Gempel, K.; Prokisch, H.; Lochmüller, H.; Klopstock, T.; Jaksch, M. Leigh syndrome caused by mutations in the flavoprotein (Fp) subunit of succinate dehydrogenase (SDHA). J. Neurol. Neurosurg. Psychiatry 2006, 77, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Jain-Ghai, S.; Cameron, J.M.; Al Maawali, A.; Blaser, S.; MacKay, N.; Robinson, B.; Raiman, J. Complex II deficiency—A case report and review of the literature. Am. J. Med. Genet. 2013, 161, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Nesti, C.; Meschini, M.C.; Meunier, B.; Sacchini, M.; Doccini, S.; Romano, A.; Petrillo, S.; Pezzini, I.; Seddiki, N.; Rubegni, A.; et al. Additive effect of nuclear and mitochondrial mutations in a patient with mitochondrial encephalomyopathy. Hum. Mol. Genet. 2015, 24, 3248–3256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levitas, A.; Muhammad, E.; Harel, G.; Saada, A.; Caspi, V.C.; Manor, E.; Beck, J.C.; Sheffield, V.; Parvari, R. Familial neonatal isolated cardiomyopathy caused by a mutation in the flavoprotein subunit of succinate dehydrogenase. Eur. J. Hum. Genet. 2010, 18, 1160–1165. [Google Scholar] [CrossRef]
- Burnichon, N.; Brière, J.-J.; Libé, R.; Vescovo, L.; Rivière, J.; Tissier, F.; Jouanno, E.; Jeunemaitre, X.; Bénit, P.; Tzagoloff, A.; et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum. Mol. Genet. 2010, 19, 3011–3020. [Google Scholar] [CrossRef] [Green Version]
- Astuti, D.; Latif, F.; Dallol, A.; Dahia, P.L.M.; Douglas, F.; George, E.; Sköldberg, F.; Husebye, E.S.; Eng, C.; Maher, E.R. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am. J. Hum. Genet. 2001, 69, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Janeway, K.A.; Kim, S.Y.; Lodish, M.; Nosé, V.; Rustin, P.; Gaal, J.; Dahia, P.L.M.; Liegl, B.; Ball, E.R.; Raygada, M.; et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc. Natl. Acad. Sci. USA 2011, 108, 314–318. [Google Scholar] [CrossRef] [Green Version]
- Baysal, B.E.; Willett-Brozick, J.; Filho, P.; Lawrence, E.C.; Myers, E.N.; Ferrell, R. An alu-mediated partial SDHC deletion causes familial and sporadic paraganglioma. J. Med. Genet. 2004, 41, 703–709. [Google Scholar] [CrossRef] [Green Version]
- McWhinney, S.R.; Pasini, B.; Stratakis, C.A. Familial gastrointestinal stromal tumors and germ-line mutations. N. Engl. J. Med. 2007, 357, 1054–1056. [Google Scholar] [CrossRef]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; Van Der Mey, A.; Taschner, P.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef]
- Hao, H.-X.; Khalimonchuk, O.; Schraders, M.; Dephoure, N.; Bayley, J.-P.; Kunst, H.; Devilee, P.; Cremers, C.W.R.J.; Schiffman, J.D.; Bentz, B.G.; et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 2009, 325, 1139–1142. [Google Scholar] [CrossRef] [Green Version]
- Sköldberg, F.; Grimelius, L.; Woodward, E.R.; Rorsman, F.; Van Schothorst, E.W.; Winqvist, O.; Karlsson, F.A.; Åkerström, G.; Kämpe, O.; Husebye, E.S. A family with hereditary extra-adrenal paragangliomas without evidence for mutations in the von Hippel-Lindau disease or Ret genes: Hereditary extra-adrenal paraganglioma. Clin. Endocrinol. 1998, 48, 11–16. [Google Scholar] [CrossRef]
- Lussey-Lepoutre, C.; Buffet, A.; Gimenez-Roqueplo, A.-P.; Favier, J. Mitochondrial deficiencies in the predisposition to paraganglioma. Metabolites 2017, 7, 17. [Google Scholar] [CrossRef]
- Ghezzi, D.; Zeviani, M. Human diseases associated with defects in assembly of OXPHOS complexes. Essays Biochem. 2018, 62, 271–286. [Google Scholar] [CrossRef]
- Dwight, T.; Na, U.; Kim, E.; Zhu, Y.; Richardson, A.L.; Robinson, B.G.; Tucker, K.M.; Gill, A.J.; Benn, D.E.; Clifton-Bligh, R.J.; et al. Analysis of SDHAF3 in familial and sporadic pheochromocytoma and paraganglioma. BMC Cancer 2017, 17, 497. [Google Scholar] [CrossRef]
- Kudryavtseva, A.V.; Kalinin, D.V.; Pavlov, V.S.; Savvateeva, M.V.; Fedorova, M.S.; Pudova, E.A.; Kobelyatskaya, A.A.; Golovyuk, A.L.; Guvatova, Z.G.; Razmakhaev, G.S.; et al. Mutation profiling in eight cases of vagal paragangliomas. BMC Med. Genom. 2020, 13, 115. [Google Scholar] [CrossRef]
- Iwata, S.; Lee, J.W.; Okada, K.; Lee, J.K.; Iwata, M.; Rasmussen, B.; Link, T.A.; Ramaswamy, S.; Jap, B.K. Complete structure of the 11-subunit bovine mitochondrial cytochrome Bc1 complex. Science 1998, 281, 64–71. [Google Scholar] [CrossRef]
- Hildenbeutel, M.; Hegg, E.L.; Stephan, K.; Gruschke, S.; Meunier, B.; Ott, M. Assembly factors monitor sequential hemylation of cytochrome b to regulate mitochondrial translation. J. Cell Biol. 2014, 205, 511–524. [Google Scholar] [CrossRef] [Green Version]
- Tucker, E.J.; Wanschers, B.F.J.; Szklarczyk, R.; Mountford, H.S.; Wijeyeratne, X.W.; van den Brand, M.A.M.; Leenders, A.M.; Rodenburg, R.J.; Reljić, B.; Compton, A.G.; et al. Mutations in the UQCC1-interacting protein, UQCC2, cause human complex III deficiency associated with perturbed cytochrome b protein expression. PLoS Genet. 2013, 9, e1004034. [Google Scholar] [CrossRef] [Green Version]
- Wanschers, B.F.J.; Szklarczyk, R.; van den Brand, M.A.M.; Jonckheere, A.; Suijskens, J.; Smeets, R.; Rodenburg, R.J.; Stephan, K.; Helland, I.B.; Elkamil, A.; et al. A mutation in the human CBP4 ortholog UQCC3 impairs complex III assembly, activity and cytochrome b stability. Hum. Mol. Genet. 2014, 23, 6356–6365. [Google Scholar] [CrossRef] [Green Version]
- Bottani, E.; Cerutti, R.; Harbour, M.E.; Ravaglia, S.; Dogan, S.A.; Giordano, C.; Fearnley, I.M.; D’Amati, G.; Viscomi, C.; Fernandez-Vizarra, E.; et al. TTC19 plays a husbandry role on UQCRFS1 turnover in the biogenesis of mitochondrial respiratory complex III. Mol. Cell 2017, 67, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, A.; Smith, P.; Fox, J.L.; Cui, T.-Z.; Khalimonchuk, O.; Winge, D.R. The LYR protein Mzm1 functions in the insertion of the rieske Fe/S protein in yeast mitochondria. Mol. Cell. Biol. 2011, 31, 3988–3996. [Google Scholar] [CrossRef] [Green Version]
- Cui, T.-Z.; Smith, P.M.; Fox, J.L.; Khalimonchuk, O.; Winge, D.R. Late-stage maturation of the Rieske Fe/S protein: Mzm1 stabilizes Rip1 but does not facilitate its translocation by the AAA ATPase Bcs1. Mol. Cell. Biol. 2012, 32, 4400–4409. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, E.; Lobo, T.; Fox, J.L.; Zeviani, M.; Winge, D.R.; Fernández-Vizarra, E. LYRM7/MZM1L is a UQCRFS1 chaperone involved in the last steps of mitochondrial complex III assembly in human cells. Biochim. Biophys. Acta (BBA)-Bioenerg. 2013, 1827, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Cruciat, C.-M.; Hell, K.; Fölsch, H.; Neupert, W.; Stuart, R.A. Bcs1p, an AAA-family member, is a chaperone for the assembly of the cytochrome Bc1 complex. EMBO J. 1999, 18, 5226–5233. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Vizarra, E.; Bugiani, M.; Goffrini, P.; Carrara, F.; Farina, L.; Procopio, E.; Donati, A.; Uziel, G.; Ferrero, I.; Zeviani, M. Impaired complex III assembly associated with BCS1L gene mutations in isolated mitochondrial encephalopathy. Hum. Mol. Genet. 2007, 16, 1241–1252. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.K.; Borgnia, M.J.; Hsu, A.L.; Esser, L.; Fox, T.; de Val, N.; Xia, D. Structures of AAA protein translocase Bcs1 suggest translocation mechanism of a folded protein. Nat. Struct. Mol. Biol. 2020, 27, 202–209. [Google Scholar] [CrossRef]
- Wagener, N.; Ackermann, M.; Funes, S.; Neupert, W. A pathway of protein translocation in mitochondria mediated by the AAA-ATPase Bcs1. Mol. Cell 2011, 44, 191–202. [Google Scholar] [CrossRef]
- Peruzzo, R.; Corrà, S.; Costa, R.; Brischigliaro, M.; Varanita, T.; Biasutto, L.; Rampazzo, C.; Ghezzi, D.; Leanza, L.; Zoratti, M.; et al. Exploiting pyocyanin to treat mitochondrial disease due to respiratory complex III dysfunction. Nat. Commun. 2021, 12, 2103. [Google Scholar] [CrossRef] [PubMed]
- Andreu, A.L.; Bruno, C.; Dunne, T.C.; Tanji, K.; Shanske, S.; Sue, C.M.; Krishna, S.; Hadjigeorgiou, G.M.; Shtilbans, A.; Bonilla, E.; et al. A nonsense mutation (G15059A) in the cytochrome b gene in a patient with exercise intolerance and myoglobinuria. Ann. Neurol. 1999, 45, 127–130. [Google Scholar] [CrossRef]
- Andreu, A.L.; Hanna, M.G.; Reichmann, H.; Bruno, C.; Penn, A.S.; Tanji, K.; Pallotti, F.; Iwata, S.; Bonilla, E.; Lach, B.; et al. Exercise intolerance due to mutations in the cytochrome b gene of mitochondrial DNA. N. Engl. J. Med. 1999, 341, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- De Coo, I.F.; Renier, W.O.; Ruitenbeek, W.; Ter Laak, H.J.; Bakker, M.; Schägger, H.; Van Oost, B.A.; Smeets, H.J. A 4-base pair deletion in the mitochondrial cytochrome b gene associated with parkinsonism/MELAS overlap syndrome. Ann. Neurol. 1999, 45, 130–133. [Google Scholar] [CrossRef]
- Keightley, J.A.; Anitori, R.; Burton, M.D.; Quan, F.; Buist, N.R.M.; Kennaway, N.G. Mitochondrial encephalomyopathy and complex III deficiency associated with a stop-codon mutation in the cytochrome b gene. Am. J. Hum. Genet. 2000, 67, 1400–1410. [Google Scholar] [CrossRef] [Green Version]
- Andreu, A.L.; Bruno, C.; Shanske, S.; Shtilbans, A.; Hirano, M.; Krishna, S.; Hayward, L.; Systrom, D.S.; Brown, R.H.; DiMauro, S. Missense mutation in the MtDNA cytochrome b gene in a patient with myopathy. Neurology 1998, 51, 1444–1447. [Google Scholar] [CrossRef]
- Lamantea, E.; Carrara, F.; Mariotti, C.; Morandi, L.; Tiranti, V.; Zeviani, M. A novel nonsense mutation (Q352X) in the mitochondrial cytochrome b gene associated with a combined deficiency of complexes I and III. Neuromuscul. Disord. 2002, 12, 49–52. [Google Scholar] [CrossRef]
- Mancuso, M.; Filosto, M.; Stevens, J.C.; Patterson, M.; Shanske, S.; Krishna, S.; DiMauro, S. Mitochondrial myopathy and complex III deficiency in a patient with a new stop-codon mutation (G339X) in the cytochrome b gene. J. Neurol. Sci. 2003, 209, 61–63. [Google Scholar] [CrossRef]
- Andreu, A.L.; Checcarelli, N.; Iwata, S.; Shanske, S.; Dimauro, S. A missense mutation in the mitochondrial cytochrome b gene in a revisited case with histiocytoid cardiomyopathy. Pediatr. Res. 2000, 48, 311–314. [Google Scholar] [CrossRef] [Green Version]
- Wibrand, F.; Ravn, K.; Schwartz, M.; Rosenberg, T.; Horn, N.; Vissing, J. Multisystem disorder associated with a missense mutation in the mitochondrial cytochromeb gene. Ann. Neurol. 2001, 50, 540–543. [Google Scholar] [CrossRef]
- Schuelke, M.; Krude, H.; Finckh, B.; Mayatepek, E.; Janssen, A.; Schmelz, M.; Trefz, F.; Trijbels, F.; Smeitink, J. Septo-optic dysplasia associated with a new mitochondrialcytochrome b mutation. Ann. Neurol. 2002, 51, 388–392. [Google Scholar] [CrossRef]
- Ghelli, A.; Tropeano, C.V.; Calvaruso, M.A.; Marchesini, A.; Iommarini, L.; Porcelli, A.M.; Zanna, C.; De Nardo, V.; Martinuzzi, A.; Wibrand, F.; et al. The cytochrome b p.278Y>C mutation causative of a multisystem disorder enhances superoxide production and alters supramolecular interactions of respiratory chain complexes. Hum. Mol. Genet. 2013, 22, 2141–2151. [Google Scholar] [CrossRef] [Green Version]
- Carossa, V.; Ghelli, A.; Tropeano, C.V.; Valentino, M.L.; Iommarini, L.; Maresca, A.; Caporali, L.; La Morgia, C.; Liguori, R.; Barboni, P.; et al. A novel in-frame 18-Bp microdeletion in MT-CYB causes a multisystem disorder with prominent exercise intolerance. Hum. Mutat. 2014, 35, 954–958. [Google Scholar] [CrossRef]
- Fernández-Vizarra, E.; Zeviani, M. Nuclear gene mutations as the cause of mitochondrial complex III deficiency. Front. Genet. 2015, 6, 134. [Google Scholar] [CrossRef]
- Ghezzi, D.; Arzuffi, P.; Zordan, M.; Da Re, C.; Lamperti, C.; Benna, C.; D’Adamo, P.; Diodato, D.; Costa, R.; Mariotti, C.; et al. Mutations in TTC19 cause mitochondrial complex III deficiency and neurological impairment in humans and flies. Nat. Genet. 2011, 43, 259–263. [Google Scholar] [CrossRef]
- Invernizzi, F.; Tigano, M.; Dallabona, C.; Donnini, C.; Ferrero, I.; Cremonte, M.; Ghezzi, D.; Lamperti, C.; Zeviani, M. A homozygous mutation in LYRM 7/ MZM 1 L associated with early onset encephalopathy, lactic acidosis, and severe reduction of mitochondrial complex III activity. Hum. Mutat. 2013, 34, 1619–1622. [Google Scholar] [CrossRef] [Green Version]
- de Lonlay, P.; Valnot, I.; Barrientos, A.; Gorbatyuk, M.; Tzagoloff, A.; Taanman, J.-W.; Benayoun, E.; Chrétien, D.; Kadhom, N.; Lombès, A.; et al. A mutant mitochondrial respiratory chain assembly protein causes complex III deficiency in patients with tubulopathy, encephalopathy and liver failure. Nat. Genet. 2001, 29, 57–60. [Google Scholar] [CrossRef]
- Lin, C.-H.; Tsai, P.-I.; Lin, H.-Y.; Hattori, N.; Funayama, M.; Jeon, B.; Sato, K.; Abe, K.; Mukai, Y.; Takahashi, Y.; et al. Mitochondrial UQCRC1 mutations cause autosomal dominant parkinsonism with polyneuropathy. Brain 2020, 143, 3352–3373. [Google Scholar] [CrossRef]
- Miyake, N.; Yano, S.; Sakai, C.; Hatakeyama, H.; Matsushima, Y.; Shiina, M.; Watanabe, Y.; Bartley, J.; Abdenur, J.E.; Wang, R.Y.; et al. Mitochondrial complex III deficiency caused by a homozygous UQCRC2 mutation presenting with neonatal-onset recurrent metabolic decompensation. Hum. Mutat. 2013, 34, 446–452. [Google Scholar] [CrossRef]
- Brown, M.D.; Voljavec, A.S.; Lott, M.T.; Torroni, A.; Yang, C.C.; Wallace, D.C. Mitochondrial DNA complex I and III mutations associated with Leber’s hereditary optic neuropathy. Genetics 1992, 130, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Bouzidi, M.F.; Schägger, H.; Collombet, J.-M.; Carrier, H.; Flocard, F.; Quard, S.; Mousson, B.; Godinot, C. Decreased expression o ubiquinol-cytochrome c reductase subunits in patients exhibiting mitochondrial myopathy with progressive exercise intolerance. Neuromuscul. Disord. 1993, 3, 599–604. [Google Scholar] [CrossRef]
- Gaignard, P.; Menezes, M.; Schiff, M.; Bayot, A.; Rak, M.; de Baulny, H.O.; Su, C.-H.; Gilleron, M.; Lombes, A.; Abida, H.; et al. Mutations in CYC1, encoding cytochrome C1 subunit of respiratory chain complex III, cause insulin-responsive hyperglycemia. Am. J. Hum. Genet. 2013, 93, 384–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gusic, M.; Schottmann, G.; Feichtinger, R.G.; Du, C.; Scholz, C.; Wagner, M.; Mayr, J.A.; Lee, C.-Y.; Yépez, V.A.; Lorenz, N.; et al. Bi-allelic UQCRFS1 variants are associated with mitochondrial complex iii deficiency, cardiomyopathy, and alopecia totalis. Am. J. Hum. Genet. 2020, 106, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Haut, S.; Brivet, M.; Touati, G.; Rustin, P.; Lebon, S.; Garcia-Cazorla, A.; Saudubray, J.M.; Boutron, A.; Legrand, A.; Slama, A. A deletion in the human QP-C gene causes a complex III deficiency resulting in hypoglycaemia and lactic acidosis. Hum. Genet. 2003, 113, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Barel, O.; Shorer, Z.; Flusser, H.; Ofir, R.; Narkis, G.; Finer, G.; Shalev, H.; Nasasra, A.; Saada, A.; Birk, O.S. Mitochondrial complex III deficiency associated with a homozygous mutation in UQCRQ. Am. J. Hum. Genet. 2008, 82, 1211–1216. [Google Scholar] [CrossRef] [Green Version]
- Feichtinger, R.G.; Brunner-Krainz, M.; Alhaddad, B.; Wortmann, S.B.; Kovacs-Nagy, R.; Stojakovic, T.; Erwa, W.; Resch, B.; Windischhofer, W.; Verheyen, S.; et al. Combined respiratory chain deficiency and UQCC2 mutations in neonatal encephalomyopathy: Defective supercomplex assembly in complex III deficiencies. Oxidative Med. Cell. Longev. 2017, 2017, 7202589. [Google Scholar] [CrossRef] [Green Version]
- Hausman-Kedem, M.; Ben-Shachar, S.; Menascu, S.; Geva, K.; Sagie, L.; Fattal-Valevski, A. VPS53 gene is associated with a new phenotype of complicated hereditary spastic paraparesis. Neurogenetics 2019, 20, 187–195. [Google Scholar] [CrossRef]
- Baker, R.A.; Priestley, J.R.C.; Wilstermann, A.M.; Reese, K.J.; Mark, P.R. Clinical spectrum of BCS1L mitopathies and their underlying structural relationships. Am. J. Med. Genet. 2019, 179, 373–380. [Google Scholar] [CrossRef]
- Visapää, I.; Fellman, V.; Varilo, T.; Palotie, A.; Raivio, K.O.; Peltonen, L. Assignment of the locus for a new lethal neonatal metabolic syndrome to 2q33-37. Am. J. Hum. Genet. 1998, 63, 1396–1403. [Google Scholar] [CrossRef] [Green Version]
- Siddiqi, S.; Siddiq, S.; Mansoor, A.; Oostrik, J.; Ahmad, N.; Kazmi, S.A.R.; Kremer, H.; Qamar, R.; Schraders, M. Novel mutation in AAA domain of BCS1L causing Bjornstad syndrome. J. Hum. Genet. 2013, 58, 819–821. [Google Scholar] [CrossRef]
- Hinson, J.T.; Fantin, V.R.; Schönberger, J.; Breivik, N.; Siem, G.; McDonough, B.; Sharma, P.; Keogh, I.; Godinho, R.; Santos, F.; et al. Missense mutations in the BCS1L gene as a cause of the Björnstad syndrome. N. Engl. J. Med. 2007, 356, 809–819. [Google Scholar] [CrossRef]
- Gil-Borlado, M.C.; González-Hoyuela, M.; Blázquez, A.; García-Silva, M.T.; Gabaldón, T.; Manzanares, J.; Vara, J.; Martín, M.A.; Seneca, S.; Arenas, J.; et al. Pathogenic Mutations in the 5′ untranslated region of BCS1L MRNA in mitochondrial complex III deficiency. Mitochondrion 2009, 9, 299–305. [Google Scholar] [CrossRef]
- Blázquez, A.; Gil-Borlado, M.C.; Morán, M.; Verdú, A.; Cazorla-Calleja, M.R.; Martín, M.A.; Arenas, J.; Ugalde, C. Infantile mitochondrial encephalomyopathy with unusual phenotype caused by a novel BCS1L mutation in an isolated complex III-deficient patient. Neuromuscul. Disord. 2009, 19, 143–146. [Google Scholar] [CrossRef]
- Dallabona, C.; Abbink, T.E.M.; Carrozzo, R.; Torraco, A.; Legati, A.; van Berkel, C.G.M.; Niceta, M.; Langella, T.; Verrigni, D.; Rizza, T.; et al. LYRM7 mutations cause a multifocal cavitating leukoencephalopathy with distinct MRI appearance. Brain 2016, 139, 782–794. [Google Scholar] [CrossRef] [Green Version]
- Hempel, M.; Kremer, L.S.; Tsiakas, K.; Alhaddad, B.; Haack, T.B.; Löbel, U.; Feichtinger, R.G.; Sperl, W.; Prokisch, H.; Mayr, J.A.; et al. LYRM7—Associated complex III deficiency: A clinical, molecular genetic, MR tomographic, and biochemical study. Mitochondrion 2017, 37, 55–61. [Google Scholar] [CrossRef]
- Kremer, L.S.; L’hermitte-Stead, C.; Lesimple, P.; Gilleron, M.; Filaut, S.; Jardel, C.; Haack, T.B.; Strom, T.M.; Meitinger, T.; Azzouz, H.; et al. Severe respiratory complex III defect prevents liver adaptation to prolonged Fasting. J. Hepatol. 2016, 65, 377–385. [Google Scholar] [CrossRef]
- Morino, H.; Miyamoto, R.; Ohnishi, S.; Maruyama, H.; Kawakami, H. Exome sequencing reveals a novel TTC19 mutation in an autosomal recessive spinocerebellar ataxia patient. BMC Neurol. 2014, 14, 5. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, C.; Barros, J.; Sá, M.J.; Azevedo, L.; Taipa, R.; Torraco, A.; Meschini, M.C.; Verrigni, D.; Nesti, C.; Rizza, T.; et al. Novel TTC19 mutation in a family with severe psychiatric manifestations and complex III deficiency. Neurogenetics 2013, 14, 153–160. [Google Scholar] [CrossRef]
- Habibzadeh, P.; Inaloo, S.; Silawi, M.; Dastsooz, H.; Farazi Fard, M.A.; Sadeghipour, F.; Faghihi, Z.; Rezaeian, M.; Yavarian, M.; Böhm, J.; et al. A novel TTC19 mutation in a patient with neurological, psychological, and gastrointestinal impairment. Front. Neurol. 2019, 10, 944. [Google Scholar] [CrossRef]
- Mordaunt, D.A.; Jolley, A.; Balasubramaniam, S.; Thorburn, D.R.; Mountford, H.S.; Compton, A.G.; Nicholl, J.; Manton, N.; Clark, D.; Bratkovic, D.; et al. Phenotypic variation of TTC19—deficient mitochondrial complex III deficiency: A case report and literature review. Am. J. Med. Genet. 2015, 167, 1330–1336. [Google Scholar] [CrossRef]
- Balsa, E.; Marco, R.; Perales-Clemente, E.; Szklarczyk, R.; Calvo, E.; Landázuri, M.O.; Enríquez, J.A. NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell Metab. 2012, 16, 378–386. [Google Scholar] [CrossRef] [Green Version]
- Pitceathly, R.D.S.; Rahman, S.; Wedatilake, Y.; Polke, J.M.; Cirak, S.; Foley, A.R.; Sailer, A.; Hurles, M.E.; Stalker, J.; Hargreaves, I.; et al. NDUFA4 mutations underlie dysfunction of a cytochrome c oxidase subunit linked to human neurological disease. Cell Rep. 2013, 3, 1795–1805. [Google Scholar] [CrossRef] [Green Version]
- Zong, S.; Wu, M.; Gu, J.; Liu, T.; Guo, R.; Yang, M. Structure of the intact 14-subunit human cytochrome c oxidase. Cell Res. 2018, 28, 1026–1034. [Google Scholar] [CrossRef] [Green Version]
- Hill, B.C. The sequence of electron carriers in the reaction of cytochromec oxidase with oxygen. J. Bioenerg. Biomembr. 1993, 25, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Ala-Vannesluoma, P.; Vattulainen, I.; Wikström, M.; Róg, T. Role of subunit III and its lipids in the molecular mechanism of cytochrome c oxidase. Biochim. Biophys. Acta (BBA)-Bioenerg. 2015, 1847, 690–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinkler, C.A.; Kalpage, H.; Shay, J.; Lee, I.; Malek, M.H.; Grossman, L.I.; Hüttemann, M. Tissue- and condition-specific isoforms of mammalian cytochrome c oxidase subunits: From function to human disease. Oxidative Med. Cell. Longev. 2017, 2017, 1534056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadenbach, B.; Hüttemann, M. The subunit composition and function of mammalian cytochrome c oxidase. Mitochondrion 2015, 24, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Nijtmans, L.G.J.; Taanman, J.-W.; Muijsers, A.O.; Speijer, D.; Van den Bogert, C. Assembly of cytochrome-c oxidase in cultured human cells. Eur. J. Biochem. 1998, 254, 389–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidoni, S.; Harbour, M.E.; Guerrero-Castillo, S.; Signes, A.; Ding, S.; Fearnley, I.M.; Taylor, R.W.; Tiranti, V.; Arnold, S.; Fernandez-Vizarra, E.; et al. MR-1S interacts with pet100 and pet117 in module-based assembly of human cytochrome c oxidase. Cell Rep. 2017, 18, 1727–1738. [Google Scholar] [CrossRef] [Green Version]
- Rak, M.; Bénit, P.; Chrétien, D.; Bouchereau, J.; Schiff, M.; El-Khoury, R.; Tzagoloff, A.; Rustin, P. Mitochondrial cytochrome c oxidase deficiency. Clin. Sci. 2016, 130, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Massa, V.; Fernandez-Vizarra, E.; Alshahwan, S.; Bakhsh, E.; Goffrini, P.; Ferrero, I.; Mereghetti, P.; D’Adamo, P.; Gasparini, P.; Zeviani, M. Severe infantile encephalomyopathy caused by a mutation in COX6B1, a nucleus-encoded subunit of cytochrome c oxidase. Am. J. Hum. Genet. 2008, 82, 1281–1289. [Google Scholar] [CrossRef] [Green Version]
- Abdulhag, U.N.; Soiferman, D.; Schueler-Furman, O.; Miller, C.; Shaag, A.; Elpeleg, O.; Edvardson, S.; Saada, A. Mitochondrial complex IV deficiency, caused by mutated COX6B1, is associated with encephalomyopathy, hydrocephalus and cardiomyopathy. Eur. J. Hum. Genet. 2015, 23, 159–164. [Google Scholar] [CrossRef]
- Lamperti, C.; Diodato, D.; Lamantea, E.; Carrara, F.; Ghezzi, D.; Mereghetti, P.; Rizzi, R.; Zeviani, M. MELAS-like encephalomyopathy caused by a new pathogenic mutation in the mitochondrial DNA encoded cytochrome c oxidase subunit I. Neuromuscul. Disord. 2012, 22, 990–994. [Google Scholar] [CrossRef]
- Valente, L.; Piga, D.; Lamantea, E.; Carrara, F.; Uziel, G.; Cudia, P.; Zani, A.; Farina, L.; Morandi, L.; Mora, M.; et al. Identification of novel mutations in five patients with mitochondrial encephalomyopathy. Biochim. Biophys. Acta (BBA)-Bioenerg. 2009, 1787, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Comi, G.P.; Bordoni, A.; Salani, S.; Franceschina, L.; Sciacco, M.; Prelle, A.; Fortunato, F.; Zeviani, M.; Napoli, L.; Bresolin, N.; et al. Cytochromec oxidase subunit I microdeletion in a patient with motor neuron disease. Ann. Neurol. 1998, 43, 110–116. [Google Scholar] [CrossRef]
- D’Aurelio, M.; Pallotti, F.; Barrientos, A.; Gajewski, C.D.; Kwong, J.Q.; Bruno, C.; Beal, M.F.; Manfredi, G. In vivo regulation of oxidative phosphorylation in cells harboring a stop-codon mutation in mitochondrial DNA-encoded cytochrome c oxidase subunit I. J. Biol. Chem. 2001, 276, 46925–46932. [Google Scholar] [CrossRef] [Green Version]
- Nishigaki, Y.; Ueno, H.; Coku, J.; Koga, Y.; Fujii, T.; Sahashi, K.; Nakano, K.; Yoneda, M.; Nonaka, M.; Tang, L.; et al. Extensive screening system using suspension array technology to detect mitochondrial DNA point mutations. Mitochondrion 2010, 10, 300–308. [Google Scholar] [CrossRef]
- Clark, K.M.; Taylor, R.W.; Johnson, M.A.; Chinnery, P.F.; Chrzanowska-Lightowlers, Z.M.A.; Andrews, R.M.; Nelson, I.P.; Wood, N.W.; Lamont, P.J.; Hanna, M.G.; et al. An MtDNA mutation in the initiation codon of the cytochrome c oxidase subunit II gene results in lower levels of the protein and a mitochondrial encephalomyopathy. Am. J. Hum. Genet. 1999, 64, 1330–1339. [Google Scholar] [CrossRef] [Green Version]
- Abu-Amero, K.K.; Bosley, T.M. Mitochondrial abnormalities in patients with LHON-like optic neuropathies. Investig. Opthalmol. Vis. Sci. 2006, 47, 4211–4220. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S.; Taanman, J.-W.; Cooper, J.M.; Nelson, I.; Hargreaves, I.; Meunier, B.; Hanna, M.G.; García, J.J.; Capaldi, R.A.; Lake, B.D.; et al. A missense mutation of cytochrome oxidase subunit II causes defective assembly and myopathy. Am. J. Hum. Genet. 1999, 65, 1030–1039. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.-L.; Yu, C.-A.; Yang, P.; Li, A.-L.; Wen, J.-Y.; Zhao, S.-M.; Liu, H.-X.; Ke, Y.-N.; Campbell, W.; Zhang, Y.-G.; et al. Novel mitochondrial DNA mutations associated with chinese familial hypertrophic cardiomyopathy. Clin. Exp. Pharmacol. Physiol. 2009, 36, 933–939. [Google Scholar] [CrossRef]
- Tabebi, M.; Mkaouar-Rebai, E.; Mnif, M.; Kallabi, F.; Ben Mahmoud, A.; Ben Saad, W.; Charfi, N.; Keskes-Ammar, L.; Kamoun, H.; Abid, M.; et al. A novel mutation MT-COIII m.9267G>C and MT-COI m.5913G>A mutation in mitochondrial genes in a Tunisian family with maternally inherited diabetes and deafness (MIDD) associated with sever nephropathy. Biochem. Biophys. Res. Commun. 2015, 459, 353–360. [Google Scholar] [CrossRef]
- Horvath, R.; Scharfe, C.; Hoeltzenbein, M.; Do, B.H.; Schröder, C.; Warzok, R.; Vogelgesang, S.; Lochmüller, H.; Müller-Höcker, J.; Gerbitz, K.D.; et al. Childhood onset mitochondrial myopathy and lactic acidosis caused by a stop mutation in the mitochondrial cytochrome c oxidase III gene. J. Med. Genet. 2002, 39, 812–816. [Google Scholar] [CrossRef] [Green Version]
- Mkaouar-Rebai, E.; Ellouze, E.; Chamkha, I.; Kammoun, F.; Triki, C.; Fakhfakh, F. Molecular-clinical correlation in a family with a novel heteroplasmic Leigh syndrome missense mutation in the mitochondrial cytochrome c oxidase III gene. J. Child Neurol. 2011, 26, 12–20. [Google Scholar] [CrossRef]
- Bosley, T.M.; Brodsky, M.C.; Glasier, C.M.; Abu-Amero, K.K. Sporadic bilateral optic neuropathy in children: The role of mitochondrial abnormalities. Investig. Opthalmol. Vis. Sci. 2008, 49, 5250. [Google Scholar] [CrossRef]
- Marotta, R.; Chin, J.; Kirby, D.M.; Chiotis, M.; Cook, M.; Collins, S.J. Novel single base pair COX III subunit deletion of mitochondrial DNA associated with rhabdomyolysis. J. Clin. Neurosci. 2011, 18, 290–292. [Google Scholar] [CrossRef]
- Hanna, M.G.; Nelson, I.P.; Rahman, S.; Lane, R.J.M.; Land, J.; Heales, S.; Cooper, M.J.; Schapira, A.H.V.; Morgan-Hughes, J.A.; Wood, N.W. Cytochrome c oxidase deficiency associated with the first stop-codon point mutation in human MtDNA. Am. J. Hum. Genet. 1998, 63, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Abu-Libdeh, B.; Douiev, L.; Amro, S.; Shahrour, M.; Ta-Shma, A.; Miller, C.; Elpeleg, O.; Saada, A. Mutation in the COX4I1 gene is associated with short stature, poor weight gain and increased chromosomal breaks, simulating fanconi anemia. Eur. J. Hum. Genet. 2017, 25, 1142–1146. [Google Scholar] [CrossRef]
- Pillai, N.R.; AlDhaheri, N.S.; Ghosh, R.; Lim, J.; Streff, H.; Nayak, A.; Graham, B.H.; Hanchard, N.A.; Elsea, S.H.; Scaglia, F. Biallelic variants in COX4I1 associated with a novel phenotype resembling Leigh syndrome with developmental regression, intellectual disability, and seizures. Am. J. Med. Genet. 2019, 179, 2138–2143. [Google Scholar] [CrossRef]
- Shteyer, E.; Saada, A.; Shaag, A.; Al-Hijawi, F.A.; Kidess, R.; Revel-Vilk, S.; Elpeleg, O. Exocrine pancreatic insufficiency, dyserythropoeitic anemia, and calvarial hyperostosis are caused by a mutation in the COX4I2 gene. Am. J. Hum. Genet. 2009, 84, 412–417. [Google Scholar] [CrossRef] [Green Version]
- Baertling, F.; Al-Murshedi, F.; Sánchez-Caballero, L.; Al-Senaidi, K.; Joshi, N.P.; Venselaar, H.; van den Brand, M.A.; Nijtmans, L.G.; Rodenburg, R.J. Mutation in mitochondrial complex IV subunit COX5A causes pulmonary arterial hypertension, lactic acidemia, and failure to thrive. Hum. Mutat. 2017, 38, 692–703. [Google Scholar] [CrossRef]
- Tamiya, G.; Makino, S.; Hayashi, M.; Abe, A.; Numakura, C.; Ueki, M.; Tanaka, A.; Ito, C.; Toshimori, K.; Ogawa, N.; et al. A mutation of COX6A1 causes a recessive axonal or mixed form of charcot-marie-tooth disease. Am. J. Hum. Genet. 2014, 95, 294–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, M.; Uchino, S.; Iida, A.; Noguchi, S.; Hayashi, S.; Takahashi, T.; Fujii, K.; Komaki, H.; Takeshita, E.; Nonaka, I.; et al. COX6A2 variants cause a muscle-specific cytochrome c oxidase deficiency. Ann. Neurol. 2019, 86, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Vondrackova, A.; Vesela, K.; Hansikova, H.; Docekalova, D.Z.; Rozsypalova, E.; Zeman, J.; Tesarova, M. High-resolution melting analysis of 15 genes in 60 patients with cytochrome-c oxidase deficiency. J. Hum. Genet. 2012, 57, 442–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indrieri, A.; van Rahden, V.A.; Tiranti, V.; Morleo, M.; Iaconis, D.; Tammaro, R.; D’Amato, I.; Conte, I.; Maystadt, I.; Demuth, S.; et al. Mutations in COX7B cause microphthalmia with linear skin lesions, an unconventional mitochondrial disease. Am. J. Hum. Genet. 2012, 91, 942–949. [Google Scholar] [CrossRef] [Green Version]
- Hallmann, K.; Kudin, A.P.; Zsurka, G.; Kornblum, C.; Reimann, J.; Stüve, B.; Waltz, S.; Hattingen, E.; Thiele, H.; Nürnberg, P.; et al. Loss of the smallest subunit of cytochrome c oxidase, COX8A, causes Leigh-like syndrome and epilepsy. Brain 2016, 139, 338–345. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Ghezzi, D.; Chassagne, M.; Mayencon, M.; Padet, S.; Melchionda, L.; Rouvet, I.; Lannes, B.; Bozon, D.; Latour, P.; et al. SURF1 deficiency causes demyelinating charcot-marie-tooth disease. Neurology 2013, 81, 1523–1530. [Google Scholar] [CrossRef] [Green Version]
- Valnot, I.; Von Kleist-Retzow, J.-C.; Barrientos, A.; Gorbatyuk, M.; Taanman, J.-W.; Mehaye, B.; Rustin, P.; Tzagoloff, A.; Munnich, A.; Rotig, A. A mutation in the human heme A: Farnesyltransferase gene (COX10) causes cytochrome c oxidase deficiency. Hum. Mol. Genet. 2000, 9, 1245–1249. [Google Scholar] [CrossRef] [Green Version]
- Antonicka, H.; Pankratz, N.; Nichols, W.C.; Uniacke, S.K.; Halter, C.; Murrell, J.; Rudolph, A.; Shults, C.W.; Conneally, P.M.; Foroud, T. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum. Mol. Genet. 2003, 12, 2693–2702. [Google Scholar] [CrossRef] [Green Version]
- Coenen, M.J.H.; van den Heuvel, L.P.; Ugalde, C.; ten Brinke, M.; Nijtmans, L.G.J.; Trijbels, F.J.M.; Beblo, S.; Maier, E.M.; Muntau, A.C.; Smeitink, J.A.M. Cytochromec oxidase biogenesis in a patient with a mutation in COX10 gene. Ann. Neurol. 2004, 56, 560–564. [Google Scholar] [CrossRef]
- Antonicka, H.; Mattman, A.; Carlson, C.G.; Glerum, D.M.; Hoffbuhr, K.C.; Leary, S.C.; Kennaway, N.G.; Shoubridge, E.A. Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am. J. Hum. Genet. 2003, 72, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Alfadhel, M.; Lillquist, Y.P.; Waters, P.J.; Sinclair, G.; Struys, E.; McFadden, D.; Hendson, G.; Hyams, L.; Shoffner, J.; Vallance, H.D. Infantile cardioencephalopathy due to a COX15 gene defect: Report and review. Am. J. Med. Genet. 2011, 155, 840–844. [Google Scholar] [CrossRef]
- Bugiani, M.; Tiranti, V.; Farina, L.; Uziel, G.; Zeviani, M. Novel mutations in COX15 in a long surviving Leigh syndrome patient with cytochrome c oxidase deficiency. J. Med. Genet. 2005, 42, e28. [Google Scholar] [CrossRef]
- Fernández-Vizarra, E.; Tiranti, V.; Zeviani, M. Assembly of the oxidative phosphorylation system in humans: What we have learned by studying its defects. Biochim. Biophys. Acta (BBA)-Bioenerg. 2009, 1793, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Leary, S.C.; Antonicka, H.; Sasarman, F.; Weraarpachai, W.; Cobine, P.A.; Pan, M.; Brown, G.K.; Brown, R.; Majewski, J.; Ha, K.C.H.; et al. Novel mutations in SCO1 as a cause of fatal infantile encephalopathy and lactic acidosis. Hum. Mutat. 2013, 34, 1366–1370. [Google Scholar] [CrossRef]
- Brix, N.; Jensen, J.M.; Pedersen, I.S.; Ernst, A.; Frost, S.; Bogaard, P.; Petersen, M.B.; Bender, L. Mitochondrial disease caused by a novel homozygous mutation (Gly106del) in the SCO1 gene. Neonatology 2019, 116, 290–294. [Google Scholar] [CrossRef]
- Papadopoulou, L.C.; Sue, C.M.; Davidson, M.M.; Tanji, K.; Nishino, I.; Sadlock, J.E.; Krishna, S.; Walker, W.; Selby, J.; Glerum, D.M.; et al. Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat. Genet. 1999, 23, 333–337. [Google Scholar] [CrossRef]
- Jaksch, M. Mutations in SCO2 are associated with a distinct form of hypertrophic cardiomyopathy and cytochrome c oxidase deficiency. Hum. Mol. Genet. 2000, 9, 795–801. [Google Scholar] [CrossRef] [Green Version]
- Sue, C.M.; Karadimas, C.; Checcarelli, N.; Tanji, K.; Papadopoulou, L.C.; Pallotti, F.; Guo, F.L.; Shanske, S.; Hirano, M.; De Vivo, D.C.; et al. Differential features of patients with mutations in two COX assembly genes, SURF-1 and SCO2. Ann. Neurol. 2000, 47, 589–595. [Google Scholar] [CrossRef]
- Jaksch, M.; Horvath, R.; Horn, N.; Auer, D.P.; Macmillan, C.; Peters, J.; Gerbitz, K.-D.; Kraegeloh-Mann, I.; Muntau, A.; Karcagi, V.; et al. Homozygosity (E140K) in SCO2 causes delayed infantile onset of cardiomyopathy and neuropathy. Neurology 2001, 57, 1440–1446. [Google Scholar] [CrossRef]
- Pronicki, M.; Kowalski, P.; Piekutowska-Abramczuk, D.; Taybert, J.; Karkucinska-Wieckowska, A.; Szymanska-Debinska, T.; Karczmarewicz, E.; Pajdowska, M.; Migdal, M.; Milewska-Bobula, B.; et al. A homozygous mutation in the SCO2 gene causes a spinal muscular atrophy like presentation with stridor and respiratory insufficiency. Eur. J. Paediatr. Neurol. 2010, 14, 253–260. [Google Scholar] [CrossRef]
- Pronicka, E.; Piekutowska-Abramczuk, D.; Szymańska-Dębińska, T.; Bielecka, L.; Kowalski, P.; Łuczak, S.; Karkucińska-Więckowska, A.; Migdał, M.; Kubalska, J.; Zimowski, J.; et al. The natural history of SCO2 deficiency in 36 Polish children confirmed the genotype-phenotype correlation. Mitochondrion 2013, 13, 810–816. [Google Scholar] [CrossRef]
- Rebelo, A.P.; Saade, D.; Pereira, C.V.; Farooq, A.; Huff, T.C.; Abreu, L.; Moraes, C.T.; Mnatsakanova, D.; Mathews, K.; Yang, H.; et al. SCO2 mutations cause early-onset axonal charcot-marie-tooth disease associated with cellular copper deficiency. Brain 2018, 141, 662–672. [Google Scholar] [CrossRef] [Green Version]
- Barcia, G.; Assouline, Z.; Pennisi, A.; Gitiaux, C.; Schiff, M.; Boddaert, N.; Munnich, A.; Bonnefont, J.-P.; Rötig, A. Cytochrome c oxidase deficiency caused by biallelic SCO2 mutations in two sibs with cerebellar ataxia and progressive peripheral axonal neuropathy. Mol. Genet. Metab. Rep. 2019, 21, 100528. [Google Scholar] [CrossRef]
- Ghosh, A.; Trivedi, P.P.; Timbalia, S.A.; Griffin, A.T.; Rahn, J.J.; Chan, S.S.L.; Gohil, V.M. Copper supplementation restores cytochrome c oxidase assembly defect in a mitochondrial disease model of coa6 deficiency. Hum. Mol. Genet. 2014, 23, 3596–3606. [Google Scholar] [CrossRef]
- Baertling, F.; Brand, M.A.V.D.; Hertecant, J.L.; Al-Shamsi, A.; Heuvel, L.P.V.D.; Distelmaier, F.; Mayatepek, E.; Smeitink, J.A.; Nijtmans, L.G.; Rodenburg, R. Mutations in COA6 cause cytochrome c oxidase deficiency and neonatal hypertrophic cardiomyopathy. Hum. Mutat. 2015, 36, 34–38. [Google Scholar] [CrossRef]
- Salvador-Severo, K.; Gómez-Caudillo, L.; Quezada, H.; de García-Trejo, J.J.; Cárdenas-Conejo, A.; Vázquez-Memije, M.E.; Minauro-Sanmiguel, F. Mitochondrial proteomic profile of complex IV deficiency fibroblasts: Rearrangement of oxidative phosphorylation complex/supercomplex and other metabolic pathways. Bol. Méd. Del Hosp. Infant. México 2017, 74, 175–180. [Google Scholar] [CrossRef]
- Weraarpachai, W.; Antonicka, H.; Sasarman, F.; Seeger, J.; Schrank, B.; Kolesar, J.E.; Lochmüller, H.; Chevrette, M.; Kaufman, B.A.; Horvath, R.; et al. Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset leigh syndrome. Nat. Genet. 2009, 41, 833–837. [Google Scholar] [CrossRef]
- Oktay, Y.; Güngör, S.; Zeltner, L.; Wiethoff, S.; Schöls, L.; Sonmezler, E.; Yilmaz, E.; Munro, B.; Bender, B.; Kernstock, C.; et al. Confirmation of TACO1 as a Leigh syndrome disease gene in two additional families. J. Neuromuscul. Dis. 2020, 7, 301–308. [Google Scholar] [CrossRef]
- Xu, F.; Morin, C.; Mitchell, G.; Ackerley, C.; Robinson, B.H. The role of the LRPPRC (Leucine-Rich pentatricopeptide repeat cassette) gene in cytochrome oxidase assembly: Mutation causes lowered levels of COX (cytochrome c oxidase) I and COX III MRNA. Biochem. J. 2004, 382, 331–336. [Google Scholar] [CrossRef] [Green Version]
- Antonicka, H.; Shoubridge, E.A. Mitochondrial RNA granules are centers for posttranscriptional RNA processing and ribosome biogenesis. Cell Rep. 2015, 10, 920–932. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, D.; Saada, A.; D’Adamo, P.; Fernandez-Vizarra, E.; Gasparini, P.; Tiranti, V.; Elpeleg, O.; Zeviani, M. FASTKD2 nonsense mutation in an infantile mitochondrial encephalomyopathy associated with cytochrome c oxidase deficiency. Am. J. Hum. Genet. 2008, 83, 415–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Du, M.; Li, D.; Wen, S.; Xie, J.; Li, Y.; Chen, A.; Zhang, K.; Xu, P.; Jia, M.; et al. Mutations in FASTKD2 are associated with mitochondrial disease with multi-OXPHOS deficiency. Hum. Mutat. 2020, 41, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Nobrega, M.P.; Nobrega, F.G.; Tzagoloff, A. COX10 codes for a protein homologous to the ORF1 product of paracoccus denitrificans and is required for the synthesis of yeast cytochrome oxidase. J. Biol. Chem. 1990, 265, 14220–14226. [Google Scholar] [CrossRef]
- Valnot, I.; Osmond, S.; Gigarel, N.; Mehaye, B.; Amiel, J.; Cormier-Daire, V.; Munnich, A.; Bonnefont, J.-P.; Rustin, P.; Rötig, A. Mutations of the SCO1 gene in mitochondrial cytochrome c oxidase deficiency with neonatal-onset hepatic failure and encephalopathy. Am. J. Hum. Genet. 2000, 67, 1104–1109. [Google Scholar] [CrossRef] [Green Version]
- Glerum, D.M.; Muroff, I.; Jin, C.; Tzagoloff, A. COX15 codes for a mitochondrial protein essential for the assembly of yeast cytochrome oxidase. J. Biol. Chem. 1997, 272, 19088–19094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oquendo, C.E.; Antonicka, H.; Shoubridge, E.; Reardon, W.; Brown, G.K. Functional and genetic studies demonstrate that mutation in the COX15 gene can cause Leigh syndrome. J. Med. Genet. 2004, 41, 540–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.; Gray, J.; Mitchell, L.; Antholine, W.E.; Hosler, J.P. Assembly of cytochrome-c oxidase in the absence of assembly protein Surf1p leads to loss of the active site heme. J. Biol. Chem. 2005, 280, 17652–17656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huigsloot, M.; Nijtmans, L.G.; Szklarczyk, R.; Baars, M.J.H.; van den Brand, M.A.M.; HendriksFranssen, M.G.M.; van den Heuvel, L.P.; Smeitink, J.A.M.; Huynen, M.A.; Rodenburg, R.J.T. A mutation in C2orf64 causes impaired cytochrome c oxidase assembly and mitochondrial cardiomyopathy. Am. J. Hum. Genet. 2011, 88, 488–493. [Google Scholar] [CrossRef] [Green Version]
- Stroud, D.A.; Maher, M.J.; Lindau, C.; Vögtle, F.-N.; Frazier, A.E.; Surgenor, E.; Mountford, H.; Singh, A.P.; Bonas, M.; Oeljeklaus, S.; et al. COA6 is a mitochondrial complex IV assembly factor critical for biogenesis of MtDNA-encoded COX2. Hum. Mol. Genet. 2015, 24, 5404–5415. [Google Scholar] [CrossRef] [Green Version]
- Leary, S.C.; Kaufman, B.A.; Pellecchia, G.; Guercin, G.-H.; Mattman, A.; Jaksch, M.; Shoubridge, E.A. Human SCO1 and SCO2 have independent, cooperative functions in copper delivery to cytochrome c oxidase. Hum. Mol. Genet. 2004, 13, 1839–1848. [Google Scholar] [CrossRef] [Green Version]
- Stiburek, L.; Vesela, K.; Hansikova, H.; Hulkova, H.; Zeman, J. Loss of function of Sco1 and its interaction with cytochrome c oxidase. Am. J. Physiol. Cell Physiol. 2009, 296, C1218–C1226. [Google Scholar] [CrossRef]
- Hiser, L.; Di Valentin, M.; Hamer, A.G.; Hosler, J.P. Cox11p is required for stable formation of the CuBand magnesium centers of cytochrome c oxidase. J. Biol. Chem. 2000, 275, 619–623. [Google Scholar] [CrossRef] [Green Version]
- Cerqua, C.; Morbidoni, V.; Desbats, M.A.; Doimo, M.; Frasson, C.; Sacconi, S.; Baldoin, M.C.; Sartori, G.; Basso, G.; Salviati, L.; et al. COX16 is required for assembly of cytochrome c oxidase in human cells and is involved in copper delivery to COX2. Biochim. Biophys. Acta (BBA)-Bioenerg. 2018, 1859, 244–252. [Google Scholar] [CrossRef]
- Carlson, C.G.; Barrientos, A.; Tzagoloff, A.; Glerum, D.M. Cox16 encodes a novel protein required for the assembly of cytochrome oxidase in saccharomyces cerevisiae. J. Biol. Chem. 2003, 278, 3770–3775. [Google Scholar] [CrossRef] [Green Version]
- Glerum, D.M.; Shtanko, A.; Tzagoloff, A. Characterization of, a yeast gene involved in copper metabolism and assembly of cytochrome oxidase. J. Biol. Chem. 1996, 271, 14504–14509. [Google Scholar] [CrossRef] [Green Version]
- Bode, M.; Woellhaf, M.W.; Bohnert, M.; van der Laan, M.; Sommer, F.; Jung, M.; Zimmermann, R.; Schroda, M.; Herrmann, J.M. Redox-regulated dynamic interplay between Cox19 and the copper-binding protein Cox11 in the intermembrane space of mitochondria facilitates biogenesis of cytochrome c oxidase. Mol. Biol Cell 2015, 26, 2385–2401. [Google Scholar] [CrossRef]
- Nobrega, M.P.; Bandeira, S.C.B.; Beers, J.; Tzagoloff, A. Characterization of COX19, a widely distributed gene required for expression of mitochondrial cytochrome oxidase. J. Biol. Chem. 2002, 277, 40206–40211. [Google Scholar] [CrossRef] [Green Version]
- Naess, K.; Bruhn, H.; Stranneheim, H.; Freyer, C.; Wibom, R.; Mourier, A.; Engvall, M.; Nennesmo, I.; Lesko, N.; Wredenberg, A.; et al. Clinical presentation, genetic etiology, and coenzyme Q10 levels in 55 children with combined enzyme deficiencies of the mitochondrial respiratory chain. J. Pediatr. 2021, 228, 240–251.e2. [Google Scholar] [CrossRef]
- Szklarczyk, R.; Wanschers, B.F.J.; Nijtmans, L.G.; Rodenburg, R.J.; Zschocke, J.; Dikow, N.; van den Brand, M.A.M.; Hendriks-Franssen, M.G.M.; Gilissen, C.; Veltman, J.A.; et al. A mutation in the FAM36A gene, the human ortholog of COX20, impairs cytochrome c oxidase assembly and is associated with ataxia and muscle hypotonia. Hum. Mol. Genet. 2013, 22, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Doss, S.; Lohmann, K.; Seibler, P.; Arns, B.; Klopstock, T.; Zühlke, C.; Freimann, K.; Winkler, S.; Lohnau, T.; Drungowski, M.; et al. Recessive dystonia-ataxia syndrome in a Turkish family caused by a COX20 (FAM36A) mutation. J. Neurol. 2014, 261, 207–212. [Google Scholar] [CrossRef]
- Clemente, P.; Peralta, S.; Cruz-Bermudez, A.; Echevarría, L.; Fontanesi, F.; Barrientos, A.; Fernandez-Moreno, M.A.; Garesse, R. HCOA3 stabilizes cytochrome c oxidase 1 (COX1) and promotes cytochrome c oxidase assembly in human mitochondria*. J. Biol. Chem. 2013, 288, 8321–8331. [Google Scholar] [CrossRef] [Green Version]
- Mick, D.U.; Vukotic, M.; Piechura, H.; Meyer, H.E.; Warscheid, B.; Deckers, M.; Rehling, P. Coa3 and Cox14 are essential for negative feedback regulation of COX1 translation in mitochondria. J. Cell Biol. 2010, 191, 141–154. [Google Scholar] [CrossRef] [Green Version]
- Mick, D.U.; Dennerlein, S.; Wiese, H.; Reinhold, R.; Pacheu-Grau, D.; Lorenzi, I.; Sasarman, F.; Weraarpachai, W.; Shoubridge, E.A.; Warscheid, B.; et al. MITRAC links mitochondrial protein translocation to respiratory-chain assembly and translational regulation. Cell 2012, 151, 1528–1541. [Google Scholar] [CrossRef] [Green Version]
- Ostergaard, E.; Weraarpachai, W.; Ravn, K.; Born, A.P.; Jønson, L.; Duno, M.; Wibrand, F.; Shoubridge, E.A.; Vissing, J. Mutations in COA3 cause isolated complex IV deficiency associated with neuropathy, exercise intolerance, obesity, and short stature. J. Med. Genet. 2015, 52, 203–207. [Google Scholar] [CrossRef]
- Higuchi, Y.; Okunushi, R.; Hara, T.; Hashiguchi, A.; Yuan, J.; Yoshimura, A.; Murayama, K.; Ohtake, A.; Ando, M.; Hiramatsu, Y.; et al. Mutations in COA7 cause spinocerebellar ataxia with axonal neuropathy. Brain 2018, 141, 1622–1636. [Google Scholar] [CrossRef]
- Weraarpachai, W.; Sasarman, F.; Nishimura, T.; Antonicka, H.; Auré, K.; Rötig, A.; Lombès, A.; Shoubridge, E.A. Mutations in C12orf62, a factor that couples COX I synthesis with cytochrome c oxidase assembly, cause fatal neonatal lactic acidosis. Am. J. Hum. Genet. 2012, 90, 142–151. [Google Scholar] [CrossRef] [Green Version]
- Bourens, M.; Barrientos, A. A CMC 1 knockout reveals translation-independent control of human mitochondrial complex IV biogenesis. EMBO Rep. 2017, 18, 477–494. [Google Scholar] [CrossRef] [Green Version]
- Hell, K.; Tzagoloff, A.; Neupert, W.; Stuart, R.A. Identification of Cox20p, a novel protein involved in the maturation and assembly of cytochrome oxidase subunit 2. J. Biol. Chem. 2000, 275, 4571–4578. [Google Scholar] [CrossRef] [Green Version]
- Church, C.; Chapon, C.; Poyton, R.O. Cloning and characterization of PET100, a gene required for the assembly of yeast cytochrome c oxidase. J. Biol. Chem. 1996, 271, 18499–18507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.C.; Smith, K.R.; Stroud, D.A.; Compton, A.G.; Tucker, E.J.; Dasvarma, A.; Gandolfo, L.C.; Marum, J.E.; McKenzie, M.; Peters, H.L.; et al. A founder mutation in PET100 causes isolated complex IV deficiency in Lebanese individuals with leigh syndrome. Am. J. Hum. Genet. 2014, 94, 209–222. [Google Scholar] [CrossRef] [Green Version]
- Oláhová, M.; Haack, T.B.; Alston, C.L.; Houghton, J.A.; He, L.; Morris, A.A.; Brown, G.K.; McFarland, R.; Chrzanowska-Lightowlers, Z.M.; Lightowlers, R.N.; et al. A truncating PET100 variant causing fatal infantile lactic acidosis and isolated cytochrome c oxidase deficiency. Eur. J. Hum. Genet. 2015, 23, 935–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, N.G.; Swenson, S.; Harris, N.J.; Germany, E.M.; Fox, J.L.; Khalimonchuk, O. The assembly factor Pet117 couples heme a synthase activity to cytochrome oxidase assembly. J. Biol. Chem. 2017, 292, 1815–1825. [Google Scholar] [CrossRef] [Green Version]
- McEwen, J.E.; Hong, K.H.; Park, S.; Preciado, G.T. Sequence and chromosomal localization of two PET genes required for cytochrome c oxidase assembly in saccharomyces cerevisiae. Curr. Genet. 1993, 23, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Renkema, G.H.; Visser, G.; Baertling, F.; Wintjes, L.T.; Wolters, V.M.; van Montfrans, J.; de Kort, G.A.P.; Nikkels, P.G.J.; van Hasselt, P.M.; van der Crabben, S.N.; et al. Mutated PET117 causes complex IV deficiency and is associated with neurodevelopmental regression and medulla oblongata lesions. Hum. Genet. 2017, 136, 759–769. [Google Scholar] [CrossRef] [Green Version]
- Signes, A.; Cerutti, R.; Dickson, A.S.; Benincá, C.; Hinchy, E.C.; Ghezzi, D.; Carrozzo, R.; Bertini, E.; Murphy, M.P.; Nathan, J.A.; et al. APOPT 1/ COA 8 assists COX assembly and is oppositely regulated by UPS and ROS. EMBO Mol. Med. 2019, 11, e9582. [Google Scholar] [CrossRef]
- Melchionda, L.; Damseh, N.S.; Abu Libdeh, B.Y.; Nasca, A.; Elpeleg, O.; Zanolini, A.; Ghezzi, D. A novel mutation in TTC19 associated with isolated Complex III deficiency, cerebellar hypoplasia, and bilateral basal ganglia lesions. Front. Genet. 2014, 5, 397. [Google Scholar] [CrossRef] [Green Version]
- Hedberg-Oldfors, C.; Darin, N.; Thomsen, C.; Lindberg, C.; Oldfors, A. COX deficiency and leukoencephalopathy due to a novel homozygous APOPT1/COA8 mutation. Neurol. Genet. 2020, 6, e464. [Google Scholar] [CrossRef]
- Souza, R.L.; Green-Willms, N.S.; Fox, T.D.; Tzagoloff, A.; Nobrega, F.G. Cloning and characterization of COX18, ASaccharomyces cerevisiae PET gene required for the assembly of cytochrome oxidase. J. Biol. Chem. 2000, 275, 14898–14902. [Google Scholar] [CrossRef] [Green Version]
- Bourens, M.; Barrientos, A. Human mitochondrial cytochrome c oxidase assembly factor COX18 acts transiently as a membrane insertase within the subunit 2 maturation module. J. Biol. Chem. 2017, 292, 7774–7783. [Google Scholar] [CrossRef] [Green Version]
- Sacconi, S.; Trevisson, E.; Pistollato, F.; Baldoin, M.C.; Rezzonico, R.; Bourget, I.; Desnuelle, C.; Tenconi, R.; Basso, G.; DiMauro, S.; et al. HCOX18 and HCOX19: Two human genes involved in cytochrome c oxidase assembly. Biochem. Biophys. Res. Commun. 2005, 337, 832–839. [Google Scholar] [CrossRef]
- Legati, A.; Reyes, A.; Nasca, A.; Invernizzi, F.; Lamantea, E.; Tiranti, V.; Garavaglia, B.; Lamperti, C.; Ardissone, A.; Moroni, I.; et al. New genes and pathomechanisms in mitochondrial disorders unraveled by NGS technologies. Biochim. Biophys. Acta (BBA)-Bioenerg. 2016, 1857, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, A.I.; Smeitink, J.A.M.; Rodenburg, R.J.T. Mitochondrial ATP synthase: Architecture, function and pathology. J. Inherit. Metab. Dis. 2012, 35, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Abrahams, J.P.; Leslie, A.G.W.; Lutter, R.; Walker, J.E. Structure at 2.8 Â resolution of F1-ATPase from bovine heart mitochondria. Nature 1994, 370, 621–628. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Ford, H.C.; Carroll, J.; Douglas, C.; Gonzales, E.; Ding, S.; Fearnley, I.M.; Walker, J.E. Assembly of the membrane domain of ATP synthase in human mitochondria. Proc. Natl. Acad. Sci. USA 2018, 115, 2988–2993. [Google Scholar] [CrossRef] [Green Version]
- Spikes, T.E.; Montgomery, M.G.; Walker, J.E. Structure of the dimeric ATP synthase from bovine mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 23519–23526. [Google Scholar] [CrossRef]
- Pinke, G.; Zhou, L.; Sazanov, L.A. Cryo-EM structure of the entire mammalian F-type ATP synthase. Nat. Struct. Mol. Biol. 2020, 27, 1077–1085. [Google Scholar] [CrossRef]
- Nijtmans, L.G.J.; Klement, P.; Houštěk, J.; van den Bogert, C. Assembly of mitochondrial ATP synthase in cultured human cells: Implications for mitochondrial diseases. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 1995, 1272, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Wittig, I.; Meyer, B.; Heide, H.; Steger, M.; Bleier, L.; Wumaier, Z.; Karas, M.; Schägger, H. Assembly and oligomerization of human ATP synthase lacking mitochondrial subunits a and A6L. Biochim. Biophys. Acta (BBA)-Bioenerg. 2010, 1797, 1004–1011. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.G.; Ackerman, S.H. The assembly factor Atp11p binds to the beta-subunit of the mitochondrial F(1)-ATPase. J. Biol. Chem. 2000, 275, 5767–5772. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.-G.; Sheluho, D.; Gatti, D.L.; Ackerman, S.H. The alpha -subunit of the mitochondrial F1 ATPase interacts directly with the assembly factor Atp12p. EMBO J. 2000, 19, 1486–1493. [Google Scholar] [CrossRef] [Green Version]
- Shoffner, J.M.; Fernhoff, P.M.; Krawiecki, N.S.; Caplan, D.B.; Holt, P.J.; Koontz, D.A.; Takei, Y.; Newman, N.J.; Ortiz, R.G.; Polak, M.; et al. Subacute necrotizing encephalopathy: Oxidative phosphorylation defects and the ATPase 6 point mutation. Neurology 1992, 42, 2168. [Google Scholar] [CrossRef] [PubMed]
- White, S.L.; Shanske, S.; Biros, I.; Warwick, L.; Dahl, H.M.; Thorburn, D.R.; Di Mauro, S. Two cases of prenatal analysis for the pathogenic T to G substitution at nucleotide 8993 in mitochondrial DNA. Prenat. Diagn. 1999, 19, 1165–1168. [Google Scholar] [CrossRef]
- Burrage, L.C.; Tang, S.; Wang, J.; Donti, T.R.; Walkiewicz, M.; Luchak, J.M.; Chen, L.-C.; Schmitt, E.S.; Niu, Z.; Erana, R.; et al. Mitochondrial myopathy, lactic acidosis, and sideroblastic anemia (MLASA) plus associated with a novel de novo mutation (m.8969G>A) in the mitochondrial encoded ATP6 gene. Mol. Genet. Metab. 2014, 113, 207–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rantamäki, M.T.; Soini, H.K.; Finnilä, S.M.; Majamaa, K.; Udd, B. Adult-onset ataxia and polyneuropathy caused by mitochondrial 8993T-C mutation. Ann. Neurol. 2005, 58, 337–340. [Google Scholar] [CrossRef]
- Craig, K.; Elliott, H.R.; Keers, S.M.; Lambert, C.; Pyle, A.; Graves, T.D.; Woodward, C.; Sweeney, M.G.; Davis, M.B.; Hanna, M.G.; et al. Episodic ataxia and hemiplegia caused by the 8993T->C mitochondrial DNA mutation. J. Med. Genet. 2007, 44, 797–799. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, G.; Blakely, E.L.; Alston, C.L.; Hassani, A.; Boggild, M.; Horvath, R.; Samuels, D.C.; Taylor, R.W.; Chinnery, P.F. Adult-onset spinocerebellar ataxia syndromes due to MTATP6 mutations. J. Neurol. Neurosurg. Psychiatry 2012, 83, 883–886. [Google Scholar] [CrossRef] [Green Version]
- De Meirleir, L.; Seneca, S.; Lissens, W.; Schoentjes, E.; Desprechins, B. Bilateral striatal necrosis with a novel point mutation in the mitochondrial ATPase 6 gene. Pediatr. Neurol. 1995, 13, 242–246. [Google Scholar] [CrossRef]
- Thyagarajan, D.; Shanske, S.; Vazquez -Memije, M.; Devivo, D.; Dimauro, S. A novel mitochondrial ATPase 6 point mutation in familial bilateral striatal necrosis. Ann. Neurol. 1995, 38, 468–472. [Google Scholar] [CrossRef]
- Brum, M.; Semedo, C.; Guerreiro, R.; Pinto Marques, J. Motor neuron syndrome as a new phenotypic manifestation of mutation 9185T>C in gene MTATP6. Case Rep. Neurol. Med. 2014, 2014, 701761. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Makita, Y.; Oki, J.; Miyamoto, A.; Yanagawa, J.; Naito, E.; Goto, Y.; Okuno, A. De novo MtDNA Nt 8993 (T—G) mutation resulting in leigh syndrome. Am. J. Hum. Genet. 1998, 62, 717–719. [Google Scholar] [CrossRef] [Green Version]
- Vilarinho, L.; Barbot, C.; Carrozzo, R.; Calado, E.; Tessa, A.; Dionisi-Vici, C.; Guimarães, A.; Santorelli, F.M. Clinical and molecular findings in four new patients harbouring the MtDNA 8993T’C mutation. J. Inherit. Metab. Dis. 2001, 24, 883–884. [Google Scholar] [CrossRef]
- Carrozzo, R.; Tessa, A.; Vazquez-Memije, M.E.; Piemonte, F.; Patrono, C.; Malandrini, A.; Dionisi-Vici, C.; Vilarinho, L.; Villanova, M.; Schagger, H.; et al. The T9176G MtDNA mutation severely affects ATP production and results in Leigh syndrome. Neurology 2001, 56, 687–690. [Google Scholar] [CrossRef]
- Lopez-Gallardo, E.; Solano, A.; Herrero-Martin, M.D.; Martinez-Romero, I.; Castano-Perez, M.D.; Andreu, A.L.; Herrera, A.; Lopez-Perez, M.J.; Ruiz-Pesini, E.; Montoya, J. NARP syndrome in a patient harbouring an insertion in the MT-ATP6 gene that results in a truncated protein. J. Med. Genet. 2008, 46, 64–67. [Google Scholar] [CrossRef]
- D’Aurelio, M.; Vives-Bauza, C.; Davidson, M.M.; Manfredi, G. Mitochondrial DNA background modifies the bioenergetics of NARP/MILS ATP6 mutant cells. Hum. Mol. Genet. 2010, 19, 374–386. [Google Scholar] [CrossRef] [Green Version]
- de Coo, I.F.; Smeets, H.J.; Gabreëls, F.J.; Arts, N.; van Oost, B.A. Isolated case of mental retardation and ataxia due to a de novo mitochondrial T8993G mutation. Am. J. Hum. Genet. 1996, 58, 636–638. [Google Scholar]
- Ware, S.M.; El-Hassan, N.; Kahler, S.G.; Zhang, Q.; Ma, Y.-W.; Miller, E.; Wong, B.; Spicer, R.L.; Craigen, W.J.; Kozel, B.A.; et al. Infantile cardiomyopathy caused by a mutation in the overlapping region of mitochondrial ATPase 6 and 8 genes. J. Med. Genet. 2009, 46, 308–314. [Google Scholar] [CrossRef]
- Galimberti, C.A.; Diegoli, M.; Sartori, I.; Uggetti, C.; Brega, A.; Tartara, A.; Arbustini, E. Brain pseudoatrophy and mental regression on valproate and a mitochondrial DNA mutation. Neurology 2006, 67, 1715–1717. [Google Scholar] [CrossRef]
- Jonckheere, A.I.; Hogeveen, M.; Nijtmans, L.; van den Brand, M.; Janssen, A.; Diepstra, H.; van den Brandt, F.; van den Heuvel, B.; Hol, F.; Hofste, T.; et al. A novel mitochondrial ATP8 gene mutation in a patient with apical hypertrophic cardiomyopathy and neuropathy. J Med Genet. 2008, 45, 129–133. [Google Scholar] [CrossRef]
- Jonckheere, A.I.; Renkema, G.H.; Bras, M.; van den Heuvel, L.P.; Hoischen, A.; Gilissen, C.; Nabuurs, S.B.; Huynen, M.A.; de Vries, M.C.; Smeitink, J.A.M.; et al. A complex V ATP5A1 defect causes fatal neonatal mitochondrial encephalopathy. Brain 2013, 136, 1544–1554. [Google Scholar] [CrossRef] [Green Version]
- Lieber, D.S.; Calvo, S.E.; Shanahan, K.; Slate, N.G.; Liu, S.; Hershman, S.G.; Gold, N.B.; Chapman, B.A.; Thorburn, D.R.; Berry, G.T.; et al. Targeted exome sequencing of suspected mitochondrial disorders. Neurology 2013, 80, 1762–1770. [Google Scholar] [CrossRef] [Green Version]
- Oláhová, M.; Yoon, W.H.; Thompson, K.; Jangam, S.; Fernandez, L.; Davidson, J.M.; Kyle, J.E.; Grove, M.E.; Fisk, D.G.; Kohler, J.N.; et al. Biallelic mutations in ATP5F1D, which encodes a subunit of ATP synthase, cause a metabolic disorder. Am. J. Hum. Genet. 2018, 102, 494–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayr, J.A.; Havlickova, V.; Zimmermann, F.; Magler, I.; Kaplanova, V.; Jesina, P.; Pecinova, A.; Nuskova, H.; Koch, J.; Sperl, W.; et al. Mitochondrial ATP synthase deficiency due to a mutation in the ATP5E gene for the F1 subunit. Hum. Mol. Genet. 2010, 19, 3430–3439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schauberger, E.M.; Ewart, S.L.; Arshad, S.H.; Huebner, M.; Karmaus, W.; Holloway, J.W.; Friderici, K.H.; Ziegler, J.T.; Zhang, H.; Rose-Zerilli, M.J.; et al. Identification of ATPAF1 as a novel candidate gene for asthma in children. J. Allergy Clin. Immunol. 2011, 128, 753–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Meirleir, L. Respiratory Chain Complex V Deficiency due to a mutation in the assembly gene ATP12. J. Med. Genet. 2004, 41, 120–124. [Google Scholar] [CrossRef] [Green Version]
- de Vries, D.D.; van Engelen, B.G.M.; Gabreëls, F.J.M.; Ruitenbeek, W.; van Oost, B.A. A second missense mutation in the mitochondrial ATPase 6 gene in Leigh’s syndrome: MtDNA mutation in Leigh’s syndrome. Ann. Neurol. 1993, 34, 410–412. [Google Scholar] [CrossRef]
- White, S.L.; Shanske, S.; McGill, J.J.; Mountain, H.; Geraghty, M.T.; DiMauro, S.; Dahl, H.-H.M.; Thorburn, D.R. Mitochondrial DNA mutations at nucleotide 8993 show a lack of tissue- or age-related variation. J. Inherit. Metab. Dis. 1999, 22, 899–914. [Google Scholar] [CrossRef]
- houštěk, j.; pícková, a.; vojtíšková, a.; mráček, t.; pecina, p.; ješina, p. mitochondrial diseases and genetic defects of ATP synthase. Biochim. Biophys. Acta (BBA)-Bioenerg. 2006, 1757, 1400–1405. [Google Scholar] [CrossRef] [Green Version]
- Schägger, H. Respiratory chain supercomplexes of mitochondria and bacteria. Biochim. Biophys. Acta (BBA)-Bioenerg. 2002, 1555, 154–159. [Google Scholar] [CrossRef] [Green Version]
- Wittig, I.; Schägger, H. Structural organization of mitochondrial ATP synthase. Biochim. Biophys. Acta (BBA)-Bioenerg. 2008, 1777, 592–598. [Google Scholar] [CrossRef] [Green Version]
- Mourier, A.; Matic, S.; Ruzzenente, B.; Larsson, N.-G.; Milenkovic, D. The respiratory chain supercomplex organization is independent of COX7a2l isoforms. Cell Metab. 2014, 20, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Acín-Pérez, R.; Fernández-Silva, P.; Peleato, M.L.; Pérez-Martos, A.; Enriquez, J.A. Respiratory active mitochondrial supercomplexes. Mol. Cell 2008, 32, 529–539. [Google Scholar] [CrossRef]
- Gu, J.; Wu, M.; Guo, R.; Yan, K.; Lei, J.; Gao, N.; Yang, M. The architecture of the mammalian respirasome. Nature 2016, 537, 639–643. [Google Scholar] [CrossRef]
- Wu, M.; Gu, J.; Guo, R.; Huang, Y.; Yang, M. Structure of mammalian respiratory supercomplex I 1 III 2 IV 1. Cell 2016, 167, 1598–1609. [Google Scholar] [CrossRef] [Green Version]
- Letts, J.A.; Fiedorczuk, K.; Sazanov, L.A. The architecture of respiratory supercomplexes. Nature 2016, 537, 644–648. [Google Scholar] [CrossRef]
- Sousa, J.S.; Mills, D.J.; Vonck, J.; Kühlbrandt, W. Functional asymmetry and electron flow in the bovine respirasome. eLife 2016, 5, e21290. [Google Scholar] [CrossRef]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acín-Pérez, R.; Latorre-Pellicer, A.; Colás, C.; Balsa, E.; Perales-Clemente, E.; Quirós, P.M.; Calvo, E.; Rodríguez-Hernández, M.A.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef]
- Genova, M.L.; Lenaz, G. Functional role of mitochondrial respiratory supercomplexes. Biochim. Biophys. Acta (BBA)-Bioenerg. 2014, 1837, 427–443. [Google Scholar] [CrossRef] [Green Version]
- Lenaz, G.; Tioli, G.; Falasca, A.I.; Genova, M.L. Complex I function in mitochondrial supercomplexes. Biochim. Biophys. Acta (BBA)-Bioenerg. 2016, 1857, 991–1000. [Google Scholar] [CrossRef]
- Calvo, E.; Cogliati, S.; Hernansanz-Agustín, P.; Loureiro-López, M.; Guarás, A.; Casuso, R.A.; García-Marqués, F.; Acín-Pérez, R.; Martí-Mateos, Y.; Silla-Castro, J.; et al. Functional role of respiratory supercomplexes in mice: SCAF1 relevance and segmentation of the Q pool. Sci. Adv. 2020, 6, eaba7509. [Google Scholar] [CrossRef]
- Capitanio, G.; Papa, F.; Papa, S. The allosteric protein interactions in the proton-motive function of mammalian redox enzymes of the respiratory chain. Biochimie 2021, 189, 1–12. [Google Scholar] [CrossRef]
- Loeffen, J.L.C.M.; Smeitink, J.A.M.; Trijbels, J.M.F.; Janssen, A.J.M.; Triepels, R.H.; Sengers, R.C.A.; van den Heuvel, L.P. Isolated complex I deficiency in children: Clinical, biochemical and genetic aspects. Hum. Mutat. 2000, 15, 123–134. [Google Scholar] [CrossRef]
- Protasoni, M.; Pérez-Pérez, R.; Lobo-Jarne, T.; Harbour, M.E.; Ding, S.; Peñas, A.; Diaz, F.; Moraes, C.T.; Fearnley, I.M.; Zeviani, M.; et al. Respiratory supercomplexes act as a platform for complex III -mediated maturation of human mitochondrial complexes I and IV. EMBO J. 2020, 39, e102817. [Google Scholar] [CrossRef] [PubMed]
- Pernas, L.; Scorrano, L. Mito-morphosis: Mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial cristae: Where beauty meets functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Silva Ramos, E.; Larsson, N.-G.; Mourier, A. Bioenergetic roles of mitochondrial fusion. Biochim. Biophys. Acta (BBA)-Bioenerg. 2016, 1857, 1277–1283. [Google Scholar] [CrossRef]
- Letts, J.A.; Sazanov, L.A. Clarifying the supercomplex: The higher-order organization of the mitochondrial electron transport chain. Nat. Struct. Mol. Biol. 2017, 24, 800–808. [Google Scholar] [CrossRef]




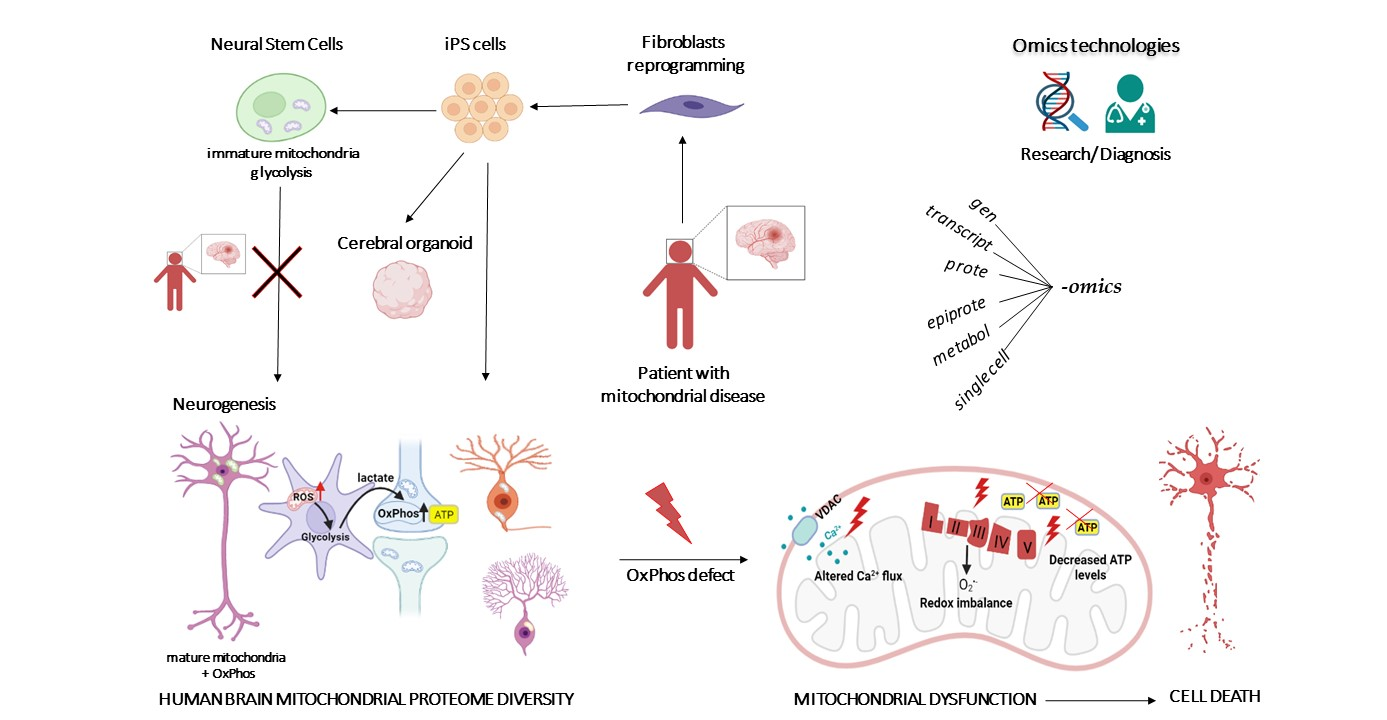{kind=link}
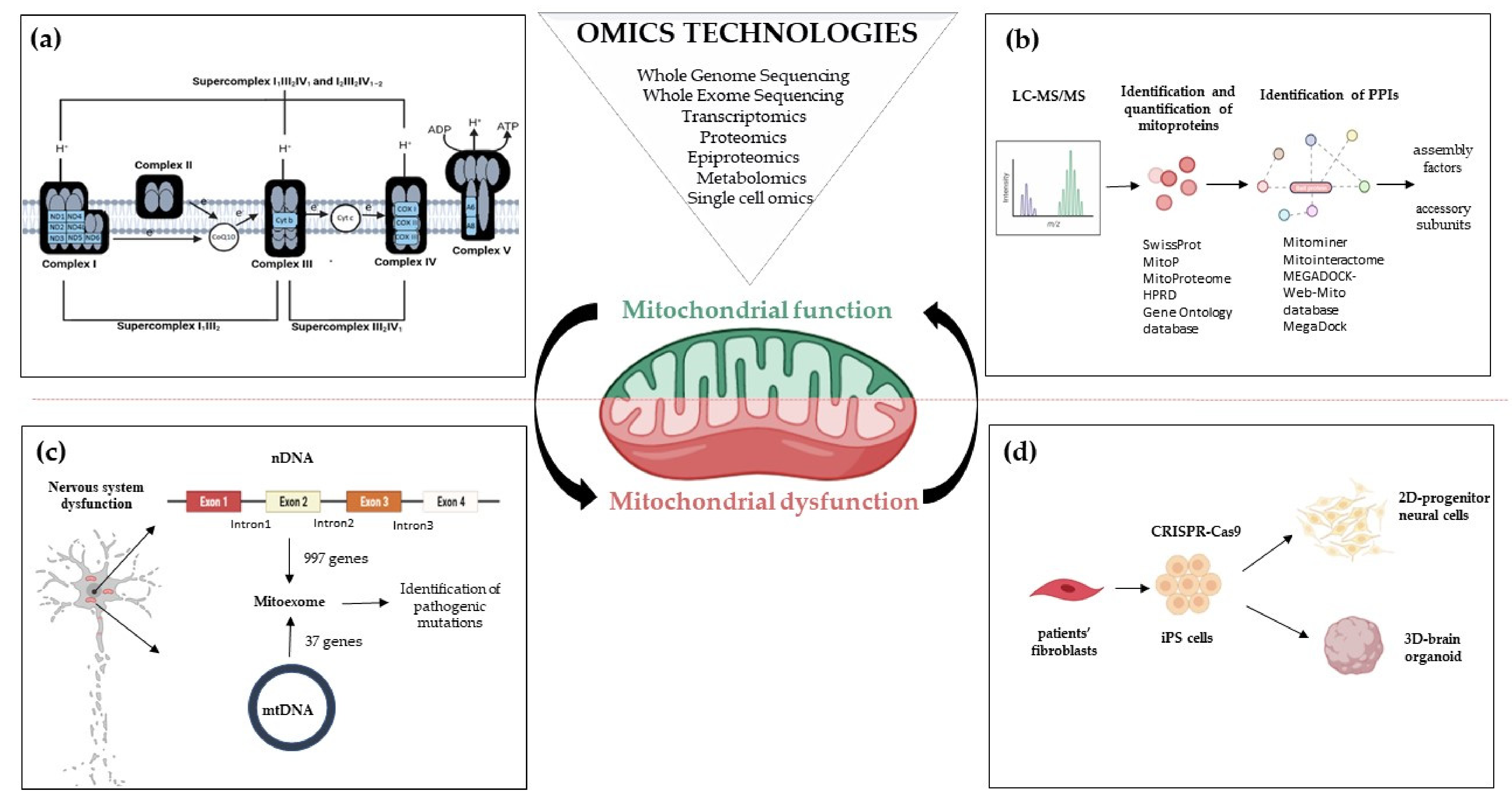{kind=link}
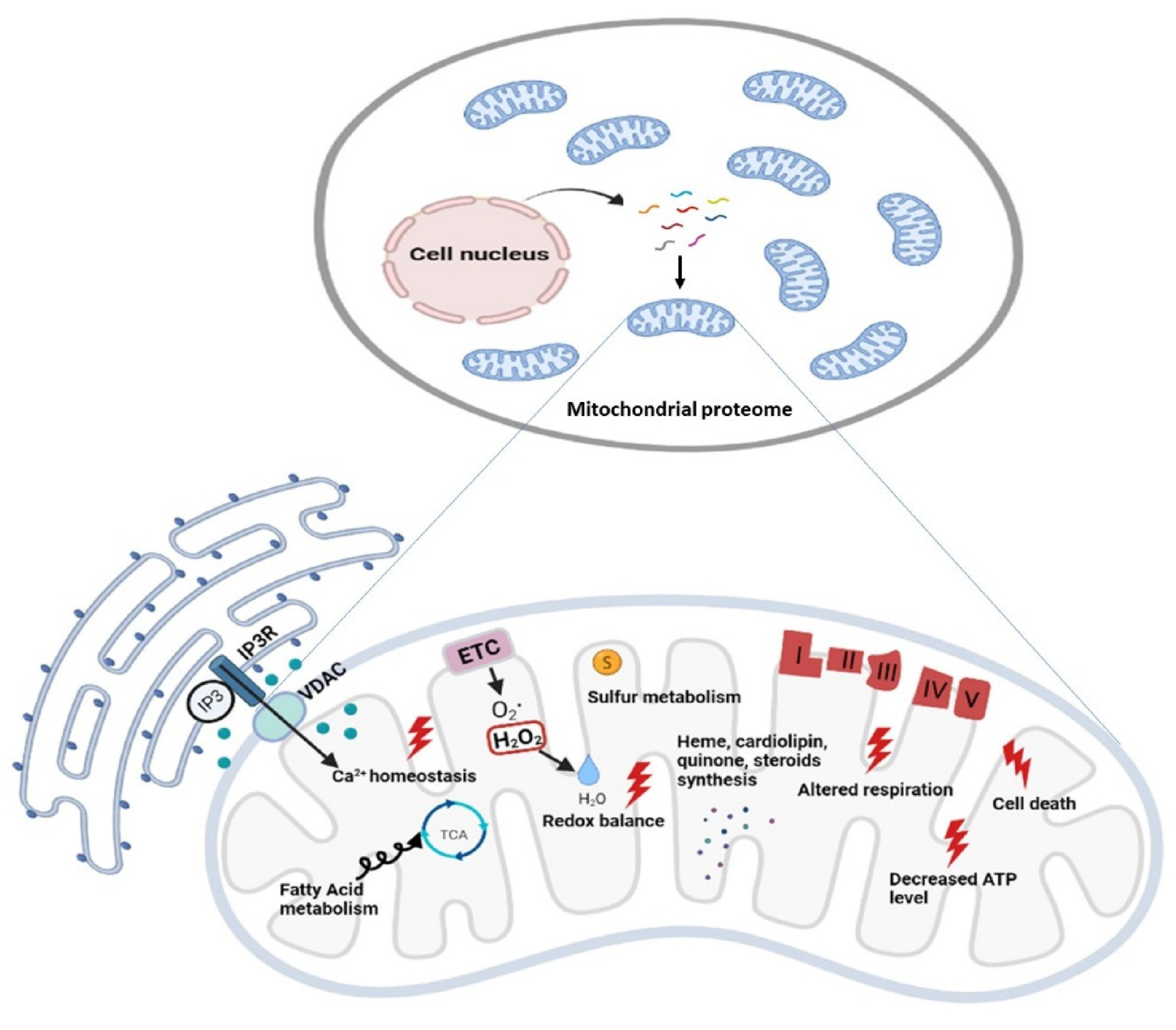{kind=link}
{kind=link}
| Assembly Factors | CI Interacting Module/Function | Associated Clinical Phenotypes | References |
|---|---|---|---|
| ACAD9 | ND2/PP-b module Component of MCIA complex, necessary for insertion of ND2 | Cardiorespiratory depression, hypertrophic cardiomyopathy, encephalopathy, and severe lactic acidosis | [134,135] |
| ECSIT | ND2/PP-b module Component of MCIA complex, necessary for insertion of ND2 | - | [136] |
| FOXRED1 | ND4/PD module | Leigh syndrome, congenital lactic acidosis, athetoid movements of the limbs in early childhood, hypotonia and cerebellar atrophy, mitochondrial respiratory CI deficiency associated with Leigh syndrome, encephalocardiomyopathy, or ataxia | [137,138,139] |
| ATP5SL/DMAC2 | ND4/PD module | - | [140] |
| TMEM70 | ND4/PD module | Neonatal mitochondrial encephalocardiomyopathy, mitochondrial CV deficiency, nuclear type 2, occasionally facial dysmorphisms and CI deficiency | [141,142,143,144,145,146] |
| NDUFAF1 | N module, ND1 Component of MCIA complex, necessary for insertion of ND2 | Hypertrophic cardiomyopathy, developmental delay, lactic acidosis, hypotonia, and Wolff–Parkinson–White syndrome | [147,148] |
| NDUFAF2 | N module. Stabilization of pre-CI or 830 kDa subcomplex | Ataxia, lethargy, nystagmus, hypotonia, optic atrophy, and episodic respiratory, insufficiency, generic encephalopathic syndromes, or Leigh syndrome | [149] |
| NDUFAF3/C3ORF60 | Q module | Macrocephaly, weak cry, no eye contact, wide anterior fontanel and axial hypotonia | [150] |
| NDUFAF4/C6ORF66 | Q module | Severe encephalopathy and antenatal Cardiomyopathy | [151] |
| NDUFAF5/C20ORF7 | Not known. Catalyze hydroxylation of NDUFS7 and dimethylation of NDUFS2 of the Q module | Facial dysmorphism, progressive lactic acidosis and neurological defects, severe early-onset encephalopathy | [152,153] |
| NDUFAF6 | Not known. Maintain a normal level of mt-ND1 subunit | Focal seizures, decreased movement and strength, ataxia, lactic acidosis, and Leigh syndrome | [29,154,155,156,157,158] |
| NDUFAF7 | Not known. Catalyze dimethylation of NDUFS2 of the Q module | - | [159,160] |
| NDUFAF8/C17ORF89 | Not known. Stabilization of NDUFAF5 | Leigh syndrome | [161] |
| NUBPL | Supposed to interact with the developing N module and possibly Q module. Insertion of iron-sulfur clusters in N and Q module subunits | Infantile onset hepatopathy, renal tubular acidosis, developmental delay, short stature, leukoencephalopathy, myopathy, nystagmus, and ataxia | [162,163,164] |
| TIMMDC1/C3ORF1 | ND1/PP-a Insertion of ND1 | Infantile onset hypotonia, failure to thrive, delayed or minimal psychomotor development, sensorineural deafness, dysmetria, dyskinetic movements, peripheral neuropathy, nystagmus, and Leigh syndrome | [140,165,166] |
| TMEM126A | ND4 module Component of MCIA complex, necessary for building the intermediate ND2 module | Autosomal recessive optic atrophy | [167,168,169,170,171] |
| TMEM126B | ND2/PP-b module Component of MCIA complex, necessary for building the intermediate ND2 module | Exercise intolerance, muscle weakness, myalgia, early-onset renal tubular acidosis, and hypertrophic cardiomyopathy | [172,173,174] |
| TMEM186 | ND2/PP-b module- Interact strongly with newly synthesized ND3 | - | [175] |
| DMAC1/TMEM261 | ND5/PD-b | - | [120] |
| COA1/MITRAC15 | ND2/PP-b module | - | [175] |
| COA7 | - | Autosomal recessive spinocerebellar ataxia with axonal neuropathy type 3 | [176] |
| LYRM-2 | NADH-Dehydrogenase module Maturation of N-module | - | [177] |
| Subunits | Location | Associated Clinical Phenotypes | References |
|---|---|---|---|
| MTND1 | ND1-module | Leber optic atrophy, MELAS syndrome, dystonia, spasticity, and myopathy | [193,194,195] |
| MTND2 | ND2-module | Leber optic atrophy | [196] |
| MTND3 | ND2-module | Infantile encephalopathy and Leigh syndrome | [197] |
| MTND4 | ND4-module | Leber optic atrophy and MELAS syndrome | [198,199] |
| MTND4L | ND2-module | Leber optic atrophy | [200] |
| MTND5 | ND5-module | Leber optic atrophy and MELAS syndrome | [201,202] |
| MTND6 | ND2-module | Leber optic atrophy and MELAS syndrome | [201,203] |
| NDUFV1 | N-module | Severe encephalopathy and neurologic abnormalities | [204,205] |
| NDUFV2 | N-module | Hypertrophic cardiomyopathy, truncal hypotonia, and encephalopathy | [206] |
| NDUFV3 | N-module | Complex I deficiency | - |
| NDUFS1 | N-module | Growth retardation, axial hypotonia, hepatomegaly, dystonia, and persistent hyperlactatemia | [205] |
| NDUFS2 | Q-module | Neonatal lactic acidosis and hypertrophic cardiomyopathy | [207] |
| NDUFS3 | Q-module | Leigh syndrome, severe axial dystonia with oral and pharyngeal motor dysfunction, dysphagia and a tetraparetic syndrome | [208] |
| NDUFS4 | Q-module | Muscular hypotonia, absence of visual and auditive attention, and cardiac defects | [209] |
| NDUFS6 | Q-module | Fatal infantile lactic acidosis, neonatal myopathy, encephalopathy, and lactic acidosis | [210,211] |
| NDUFS7 | Q-module | Leigh syndrome, feeding problems, dysarthria, and ataxia | [212] |
| NDUFS8 | Q-module | Leigh syndrome, poor feeding, and episodes of apnea and cyanosis | [213] |
| NDUFA11 | ND2-module | Fatal infantile metabolic acidosis, brain atrophy, no motor development and hypertrophic cardiomyopathy | [214] |
| NDUFA1 | ND1-module | Leigh syndrome, hypotonia, nystagmus, generalized choreoathetosis, and decreased reflexes | [215] |
| NDUFA2 | N-module | Leigh syndrome, hypertrophic cardiomyopathy, and developmental delay | [216] |
| NDUFA3 | ND1-module | - | - |
| NDUFA5 | Q-module | - | - |
| NDUFA6/LYRM-6 | LYR protein | Auditory and optic neuropathy, mitochondrial-related infantile death, brain disorder, leukoencephalopathy | [217] |
| NDUFA7 | N-module | - | - |
| NDUFA8 | IMS protein (ND1-module) | Intrauterine growth retardation, respiratory insufficiency, lactic acidosis and hypoglycemia | [178] |
| NDUFA9 | Q-module | Severe neonatal hypotonia, dysmorphic features, epilepsy, and signs of brainstem involvement | [218] |
| NDUFA10 | ND2-module | Leigh syndrome | - |
| NDUFA11 | ND2-module | Encephalocardiomyopathy and fatal infantile lactic acidemia, neuromuscular disorder | - |
| NDUFA12 | N-module | Respiratory and metabolic acidosis, hearing loss, apneas, and retinitis pigmentosa | [219] |
| NDUFA13 | ND1-module | Leigh syndrome, progressive loss of motor abilities, scoliosis, and dystonia | [220] |
| NDUFB1 | ND4-module | - | - |
| NDUFB2 | ND5-module | - | - |
| NDUFB3 | ND5-module | Delayed development, hypotonia, poor eye contact, abnormal eye movements, poor feeding, encephalopathy, and hearing loss | [221] |
| NDUFB4 | ND4-module | - | - |
| NDUFB5 | ND4-module | - | - |
| NDUFB6 | ND5-module | - | - |
| NDUFB7 | ND5-module | - | - |
| NDUFB8 | ND5-module | Encephalopathy, myopathy, hypotonia, developmental delay, and lactic acidosis, mitochondrial Complex I Deficiency in Individuals with Leigh-like Encephalomyopathy | [222] |
| NDUFB9/LYRM-3 | LYR protein | Leigh syndrome, respiratory failure, seizures, hypotonia, cardiac hypertrophy, failureto thrive and severely delayed psychomotor development | [221] |
| NDUFB10 | IMS protein(ND4 module) | Progressive hypotonia associated with increased serum lactate | [223] |
| NDUFB11 | ND4-module | Lethal complex I deficiency, X-linked microphthalmia with linear skin defects (MLS) syndrome | [224,225,226] |
| NDUFC1 | ND2-module | - | - |
| NDUFC2 | ND2-module | X-linked microphthalmia with linear skin defects (MLS) syndrome, cardiomyopathy and other congenital anomalies | [227] |
| NDUFS5 | IMS protein (ND2 module) | - | - |
| Subunits | Function | Associated Clinical Phenotypes | References |
|---|---|---|---|
| SDHA | CII subunit | Leigh syndrome, neonatal dilated cardiomyopathy, catecholamine-secreting extra-adrenal paraganglioma | [259,260,261,262,263,264,265,266,267] |
| SDHB | CII subunit | Paraganglioma, pheochromocytoma, gastrointestinal stromal tumors | [268,269] |
| SDHC | CII subunit | Paraganglioma, gastric stromal sarcoma | [270,271] |
| SDHD | CII subunit | Paraganglioma, pheochromocytoma, gastric stromal sarcoma | [271,272] |
| Assembly Factors | |||
| SDHAF1/LYRM-8 | Insert Fe/S clusters into mature SDHB | Leukoencephalopathy, spastic quadriplegia, psychomotor regression | [257] |
| SDHAF2 | Insert FAD cofactor into apo-protein SDHA | Paraganglioma and pheochromocytomas | [270,272,273,274,275,276] |
| SDHAF3/NDUFV1/LYRM-10 | Maintain SHDB stability | Familial and sporadic pheochromocytomas and paraganglioma | [277] |
| SDHAF4 | Protect the subunit from auto-oxidation and facilitates the assembly with SDHB | Vagal paragangliomas | [278] |
| Subunits | Function | Associated Clinical Phenotypes | References |
|---|---|---|---|
| UQCRC1 | CIII subunit | Parkinsonism with polyneuropathy | [308] |
| UQCRC2 | CIII subunit | Hypoglycemia, lactic acidosis, ketosis, and hyperammonemia | [309] |
| MT-CYB | CIII subunit | Leber optic atrophy, exercise intolerance, encephalomyopathy, cardiomyopathy, and multisystemic disorder, histiocytosis cardiomyopathy, parkinsonism, and MELAS overlap syndrome | [293,294,299,300,310,311] |
| CYC1 | CIII subunit | Neurologic deterioration, insulin-responsive hyperglycemia, ketoacidosis with increased serum lactate, liver failure, and hyperammonemia | [312] |
| UQCRFS1 | CIII subunit | Cardiomyopathy and alopecia totalis | [313] |
| UQCRH | CIII subunit | - | - |
| UQCRB | CIII subunit | Gastroenteritis, liver enlargement, hypoglycemia, and metabolic acidosis but normal psychomotor development at age 4, hepatopathy | [314] |
| UQCRQ | CIII subunit | Severe neurologic phenotype, early-onset severe encephalopathy | [315] |
| UQCR10 | CIII subunit | - | - |
| UQCR11 | CIII subunit | - | - |
| Assembly Factors | |||
| UQCC1 | Cytochrome b assembly factor | - | - |
| UQCC2 | Cytochrome b assembly factor | Intrauterine growth retardation, neonatal lactic acidosis and renal tubular dysfunction | [281,316] |
| UQCC3 | Cytochrome b assembly factor | Lactic acidosis, hypoglycemia, hypotonia, and delayed development | [282] |
| VPS53 | Heme lyase (Cytochrome c1) | Complicated hereditary spastic paraparesis | [317] |
| BCS1L | AAA-ATPase involved in Rieske protein incorporation. Stabilization, incorporation, and metabolism of UQCRFS1 | GRACILE Syndrome, Bjornstad Syndrome, myopathy, encephalopathy, proximal tubulopathy, and liver failure | [26,288,304,318,319,320,321,322,323] |
| MZM1L/LYRM-7 | Matrix protein involved in Rieske protein incorporation. Stabilization, incorporation, and metabolism of UQCRFS1 | Neurological decompensation and regression, leukoencephalopathy and liver failure, infantile CIII deficiency associated with cavitating leukoencephalopathy metabolic decompensation | [306,324,325,326] |
| TTC19 | Rieske protein metabolism Stabilization, incorporation, and metabolism of UQCRFS1 | Progressive encephalopathy, ataxia, spastic paraparesis, and psychiatric phenotype | [305,327,328,329,330] |
| Subunits | Associated Clinical Phenotypes | References |
|---|---|---|
| MTCO1 | MELAS syndrome, myopathy, myoglobinuria, motor neuron disease, exercise intolerance, epilepsy, multisystem disorders, deafness, LHON, or mitochondrial sensorineural hearing loss | [343,344,345,346,347] |
| MTCO2 | Encephalomyopathy, LHON, myopathy, hypertrophic cardiomyopathy | [348,349,350,351] |
| MTCO3 | MIDD, LHON, myopathy, Leigh disease, myoglobinuria, sporadic bilateral optic neuropathy, rhabdomyolysis, encephalopathy | [352,353,354,355,356,357] |
| COX4I1 | Short stature, poor weight gain, mild dysmorphic features, Fanconi anemia, Leigh-like syndrome | [358,359] |
| COX4I2 | Exocrine pancreatic insufficiency, dyserythropoietic anemia, calvarial hyperostosis | [360] |
| COX5A | Early-onset pulmonary arterial hypertension, lactic acidemia, failure to thrive | [361] |
| COX6A1 | Charcot–Marie–Tooth disease | [362] |
| COX6A2 | Muscle weakness and hypotonia, cardiomyopathy | [363] |
| COX6B1 | Severe infantile encephalomyopathy | [341,342] |
| COX7A1 | Failure to thrive, encephalopathy, hypotonia | [364] |
| COX7B | Microphthalmia with linear skin lesions | [365] |
| COX8A | Leigh-like syndrome presenting with leukodystrophy and severe epilepsy | [366] |
| NDUFA4 | Leigh syndrome | [331] |
| Assembly Factors | Function | Associated Clinical Phenotypes | References |
|---|---|---|---|
| RNA Stability and Translation | |||
| TACO1 | Translational activator of mitochondria encoded MTCO1 | Leigh syndrome | [388,389] |
| LRPPRC | Mitochondrial mRNA stability | French Canadian type of Leigh syndrome | [390] |
| FASTKD2 | Involved in post-transcriptional RNA maturation, ribosome biogenesis and translation | Brain atrophy, epilepsy, delayed psychomotor development, bilateral optic atrophy, spastic hemiparesis, cardiomyopathy | [391,392,393] |
| Heme a Biosynthesis and Insertion | |||
| COX10 | Heme a synthesis (conversion of heme b into heme o) | Leigh syndrome, encephalopathy, cardiomyopathy, sensorineural deafness, and metabolic acidosis | [369,370,394,395] |
| COX15 | Heme a synthesis (conversion of heme o into heme a) | Leigh syndrome, encephalopathy, cardiomyopathy, sensorineural deafness, and metabolic acidosis | [369,371,373,396,397] |
| SURF1 | Involved in the insertion or stabilization of heme a3 | Leigh syndrome, Charcot–Marie–Tooth disease | [252,253,276,367,398] |
| Copper Metabolism and Insertion | |||
| COA5/C2ORF64 | Involved in the unknown step of CIV biogenesis | Fatal infantile cardioencephalomyopathy | [399] |
| COA6/C1ORF31 | Copper homeostasis and transport to CIV | Fatal infantile cardioencephalopathy | [385,386,400] |
| SCO1 | Incorporation of copper atoms (biogenesis of CuA center) | Cardioencephalomyopathy, Leigh syndrome-like symptoms, spinal muscular atrophy-like presentations, Charcot–Marie–Tooth disease type 4, CIV deficiency, neonatal hepatopathy, encephalopathy with hepatopathy and cardiomyopathy, pure encephalopathy, metabolic syndrome with exclusively fatal lactic acidosis | [375,381,383,395,401,402] |
| SCO2 | Incorporation of copper atoms (biogenesis of CuA center) | Cardioencephalomyopathy, Leigh syndrome-like symptoms, spinal muscular atrophy-like presentations, Charcot–Marie–Tooth disease type 4, CIV deficiency, cardiac hypertrophy | [377,378,379,380,381] |
| COX11 | Copper chaperone | Coloboma, Ocular, With or Without Hearing Impairment, Cleft Lip/Palate, And/Or Mental Retardation and Spinal Muscular Atrophy, Distal, X-Linked 3 | [403] |
| COX16 | MTCO2 maturation | - | [404,405] |
| COX17 | Copper transfer | - | [406] |
| COX19 | Stabilization of COX11 | - | [407,408] |
| COX20 | Stabilization of MT-CO2 | Cerebellar ataxia | [409,410,411] |
| Assembly | |||
| COA3/MITRAC12 | Required for MTCO1 stability and assembly, involved in translational regulation of MTCO1 and prevention of MTCO1 aggregation before assembly | Mild phenotype, exercise intolerance, peripheral neuropathy, obesity, and short stature | [412,413,414,415] |
| COA7 | Unknown | Ataxia and peripheral neuropathy, cognitive impairments, leukodystrophy | [176,416] |
| COX14/C12ORF62 | MTCO1 stability and assembly; avoids MTCO1 aggregation | Severe lactic acidosis and dysmorphic features | [417] |
| CMC1 | Stabilizes the interaction between MTCO1, COX14, and COA3 | [418] | |
| COX20/FAM36A | MTCO2 chaperone for copper metalation | Growth delay, hypotonia, cerebellar ataxia | [410,411,419] |
| PET100 | Stabilizes MT-CO2 module | Early-onset psychomotor delay, seizures, hypotonia, Leigh syndrome, CIV deficiency, and fatal infantile lactic acidosis | [420,421,422] |
| PET117 | Assembly factor: possible role in Cox15 oligomerization and function, stabilizes MT-CO2 module | Neurodevelopmental regression and bulbar lesions | [423,424,425] |
| MR-1S | Interacts with PET117 and PET100, | - | [339] |
| APOPT1/COA8 | intermediates assembly steps Putative role in CIV protection from ROS damage, enhances CIV biogenesis | Leukodystrophy, neurological signs | [426,427,428] |
| COX18 | Promotes the translocation of MTCO2 globular domain through the IMM | Isolated COX deficiency in infancy | [429,430,431] |
| COX19 | Stabilization of COX11 | Isolated COX deficiency in infancy | [407,408,431] |
| COA-X | Putative assembly factor | - | [432] |
| HIGD2A | Promotes incorporation of MT-CO3 module | - | - |
| Subunits | Location | Associated Clincial Phenotypes | References |
|---|---|---|---|
| MT-ATP6 | Fo domain | Mitochondrial CV deficiency Neuropathy, Ataxia and Retinitis Pigmentosa (NARP) syndrome Leigh syndrome Adult-onset ataxia and polyneuropathy Bilateral striatal necrosis Motor neuron syndrome Mitochondrial myopathy, lactic acidosis, and sideroblastic anemia | [94,95,442,443,444,445,446,447,448,449,450,451,452,453,454,455,456,457] |
| MT-ATP8 | Fo domain | Mitochondrial CV deficiency Valproate-induced reversible brain atrophy Hypertrophic cardiomyopathy | [458,459] |
| MT-ATP6/8 overlap region | Fo domain | Mitochondrial CV deficiency Infantile hypertrophic cardiomyopathy | [457] |
| ATP5F1A | F1 domain | Mitochondrial CV deficiency Combined OXPHOS deficiency Fatal infantile encephalopathy | [460,461] |
| ATP5F1D | F1 domain | Mitochondrial CV deficiency Metabolic decompensation with lactic acidosis, hypoglycemia, hyperammonemia, and 3-methylglutaconic aciduria, encephalopathy | [462] |
| ATP5F1E | F1 domain | Mitochondrial CV deficiency Neonatal-onset lactic acidosis, 3-methylglutaconic aciduria, mild mental retardation, hypertrophic cardiomyopathy, and peripheral neuropathy | [463] |
| Assembly Factors | |||
| ATPAF1 | Binds and stabilIzes subunit beta of F1 Domain | Asthma in children | [464] |
| ATPAF2 | Binds and stabilizes subunit alpha of F1 domain | Degenerative encephalopathy, elevated lactate levels, developmental delay | [465] |
| TMEM70 | Unknown | Neonatal mitochondrial encephalocardiomyopathy Mitochondrial CV deficiency, nuclear type 2 Occasionally facial dysmorphisms CI deficiency | [141,142,143,144,145,146] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanfardino, P.; Doccini, S.; Santorelli, F.M.; Petruzzella, V. Tackling Dysfunction of Mitochondrial Bioenergetics in the Brain. Int. J. Mol. Sci. 2021, 22, 8325. https://doi.org/10.3390/ijms22158325
Zanfardino P, Doccini S, Santorelli FM, Petruzzella V. Tackling Dysfunction of Mitochondrial Bioenergetics in the Brain. International Journal of Molecular Sciences. 2021; 22(15):8325. https://doi.org/10.3390/ijms22158325
Chicago/Turabian StyleZanfardino, Paola, Stefano Doccini, Filippo M. Santorelli, and Vittoria Petruzzella. 2021. "Tackling Dysfunction of Mitochondrial Bioenergetics in the Brain" International Journal of Molecular Sciences 22, no. 15: 8325. https://doi.org/10.3390/ijms22158325
APA StyleZanfardino, P., Doccini, S., Santorelli, F. M., & Petruzzella, V. (2021). Tackling Dysfunction of Mitochondrial Bioenergetics in the Brain. International Journal of Molecular Sciences, 22(15), 8325. https://doi.org/10.3390/ijms22158325