
PARP Inhibitors Display Differential Efficacy in Models of BRCA Mutant High-Grade Serous Ovarian Cancer




Abstract
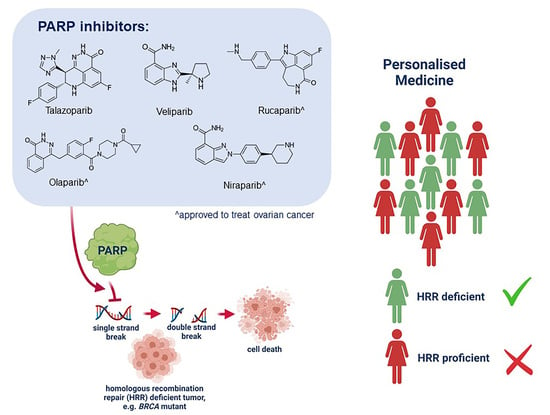
:
1. Introduction
2. Results
2.1. PARPis Display Differential Efficacy on Cell Viability in BRCA Wild-Type and Mutant Ovarian Cancer Cell Line Pairs
2.2. PARPis Display Differential Efficacy on Cell Survival in BRCA Wild-Type and Mutant Ovarian Cancer Cell Line Pairs
2.3. Veliparib Resistant A2780 Cells Retain Resistance to Other PARP Inhibitors
2.4. Down-Regulation of BRCA1 in BRCA1 Wild-Type Ovarian Cancer Cell Lines Sensitises Cells to PARPis
2.5. Down-Regulation of BRCA1 in BRCA1 Wild-Type Ovarian Cancer Cell Lines Decreases Cell Survival
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. BRCA1 Down-Regulation, RNA Extraction and qRT-PCR
4.3. Cell Viability Assays and Calculation of LC50 Doses for PARP Inhibitors
4.4. Clonogenic Cell Survival Analyses
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schettini, F.; Giudici, F.; Bernocchi, O.; Sirico, M.; Corona, S.P.; Giuliano, M.; Locci, M.; Paris, I.; Scambia, G.; De Placido, S.; et al. Poly (ADP-ribose) polymerase inhibitors in solid tumours: Systematic review and meta-analysis. Eur. J. Cancer 2021, 149, 134–152. [Google Scholar] [CrossRef]
- Cerrato, A.; Morra, F.; Celetti, A. Use of poly ADP-ribose polymerase [PARP] inhibitors in cancer cells bearing DDR defects: The rationale for their inclusion in the clinic. J. Exp. Clin. Cancer Res. 2016, 35, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical relevance, mechanisms of action and tumor resistance. Front Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Foo, T.; George, A.; Banerjee, S. PARP inhibitors in ovarian cancer: An overview of the practice-changing trials. Genes Chromosomes Cancer 2021, 60, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.Y.; Rahman, S.; Sparano, J.A. Perspectives on PARP inhibitors as pharmacotherapeutic strategies for breast cancer. Expert Opin Pharm. 2021, 22, 981–1003. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Guo, Z.; Hu, Z. The role of PARP inhibitors in the treatment of prostate cancer: Recent advances in clinical trials. Biomolecules 2021, 11, 722. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.; Chung, S.Y.; Parakrama, R.; Fayyaz, F.; Jose, J.; Saif, M.W. The role of PARP inhibitors in BRCA mutated pancreatic cancer. Therap. Adv. Gastroenterol. 2021, 14. [Google Scholar] [CrossRef] [PubMed]
- Swisher, E.M.; Kwan, T.T.; Oza, A.M.; Tinker, A.V.; Ray-Coquard, I.; Oaknin, A.; Coleman, R.L.; Aghajanian, C.; Konecny, G.E.; O’Malley, D.M.; et al. Molecular and clinical determinants of response and resistance to rucaparib for recurrent ovarian cancer treatment in ARIEL2 (Parts 1 and 2). Nat. Commun. 2021, 12, 2487. [Google Scholar] [CrossRef]
- Hu, Y.; Guo, M. Synthetic lethality strategies: Beyond BRCA1/2 mutations in pancreatic cancer. Cancer Sci. 2020, 111, 3111–3121. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Gentles, L.; Goranov, B.; Matheson, E.; Herriott, A.; Kaufmann, A.; Hall, S.; Mukhopadhyay, A.; Drew, Y.; Curtin, N.J.; O’Donnell, R.L. Exploring the frequency of homologous recombination DNA repair dysfunction in multiple cancer types. Cancers 2019, 11, 354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zheng, K.; Huang, Y.; Xiong, H.; Su, J.; Chen, R.; Zou, Y. PARP inhibitors in gastric cancer: Beacon of hope. J. Exp. Clin. Cancer Res. 2021, 40, 211. [Google Scholar] [CrossRef] [PubMed]
- Fritz, C.; Portwood, S.M.; Przespolewski, A.; Wang, E.S. PARP goes the weasel! Emerging role of PARP inhibitors in acute leukemias. Blood Rev. 2021, 45, 100696. [Google Scholar] [CrossRef]
- Amé, J.C.; Spenlehauer, C.; de Murcia, G. The PARP superfamily. Bioessays 2004, 26, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Jubin, T.; Kadam, A.; Jariwala, M.; Bhatt, S.; Sutariya, S.; Gani, A.R.; Gautam, S.; Begum, R. The PARP family: Insights into functional aspects of poly (ADP-ribose) polymerase-1 in cell growth and survival. Cell Prolif. 2016, 49, 421–437. [Google Scholar] [CrossRef]
- Kuzminov, A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc. Natl. Acad. Sci. USA 2001, 98, 8241–8246. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- del Rivero, J.; Kohn, E.C. PARP Inhibitors: The Cornerstone of DNA Repair-Targeted Therapies. Oncology 2017, 31, 265–273. [Google Scholar]
- Nijman, S.M. Synthetic lethality: General principles, utility and detection using genetic screens in human cells. FEBS Lett. 2011, 585, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Dziadkowiec, K.N.; Gąsiorowska, E.; Nowak-Markwitz, E.; Jankowska, A. PARP inhibitors: Review of mechanisms of action and BRCA1/2 mutation targeting. Prz. Menopauzalny 2016, 15, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Kamaletdinova, T.; Fanaei-Kahrani, Z.; Wang, Z.Q. The enigmatic function of PARP1: From PARylation activity to PAR readers. Cells 2019, 8, 1625. [Google Scholar] [CrossRef] [Green Version]
- Beck, C.; Robert, I.; Reina-San-Martin, B.; Schreiber, V.; Dantzer, F. Poly(ADP-ribose) polymerases in double-strand break repair: Focus on PARP1, PARP2 and PARP3. Exp. Cell Res. 2014, 329, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Pilié, P.G.; Gay, C.M.; Byers, L.A.; O’Connor, M.J.; Yap, T.A. PARP inhibitors: Extending benefit beyond BRCA-mutant cancers. Clin. Cancer Res. 2019, 25, 3759–3771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Antolin, A.A.; Ameratunga, M.; Banerji, U.; Clarke, P.A.; Workman, P.; Al-Lazikani, B. The kinase polypharmacology landscape of clinical PARP inhibitors. Sci. Rep. 2020, 10, 2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Food and Drug Administration. Highlights of Prescribing Information: Lynparza (Revised: January 2018). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/208558s001lbl.pdf (accessed on 15 July 2021).
- Food and Drug Administration. Highlights of prescribing information: Rubraca (Revised: December 2016). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/209115s000lbl.pdf (accessed on 15 July 2021).
- Food and Drug Administration. Highlights of Presribing Information: Zejula (Revised: March 2017). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/208447s015s017lbledt.pdf (accessed on 15 July 2021).
- Food and Drug Administration. Highlights of Prescribing Information: Talzenna (Revised: October 2018). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/211651s000lbl.pdf (accessed on 15 July 2021).
- Valabrega, G.; Scotto, G.; Tuninetti, V.; Pani, A.; Scaglione, F. Differences in PARP inhibitors for the treatment of ovarian cancer: Mechanisms of action, pharmacology, safety, and efficacy. Int. J. Mol. Sci. 2021, 22, 4203. [Google Scholar] [CrossRef]
- Hopkins, T.A.; Shi, Y.; Rodriguez, L.E.; Solomon, L.R.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Wilsbacher, J.L.; Gao, W.; Olson, A.M.; et al. Mechanistic dissection of PARP1 trapping and the impact on in vivo tolerability and efficacy of PARP inhibitors. Mol. Cancer Res. 2015, 13, 1465–1477. [Google Scholar] [CrossRef] [Green Version]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Goel, N.; Foxall, M.E.; Scalise, C.B.; Wall, J.A.; Arend, R.C. Strategies in overcoming homologous recombination (HR) proficiency and poly (ADP-Ribose) polymerase inhibitor (PARPi) resistance. Mol. Cancer Ther. 2021. [Online ahead of print]. [Google Scholar] [CrossRef]
- Etemadmoghadam, D.; George, J.; Cowin, P.A.; Cullinane, C.; Kansara, M.; Gorringe, K.L.; Smyth, G.K.; Bowtell, D.D. Amplicon-dependent CCNE1 expression is critical for clonogenic survival after cisplatin treatment and is correlated with 20q11 gain in ovarian cancer. PLoS ONE 2010, 5, e15498. [Google Scholar] [CrossRef]
- Alblihy, A.; Alabdullah, M.L.; Toss, M.S.; Algethani, M.; Mongan, N.P.; Rakha, E.A.; Madhusudan, S. RAD50 deficiency is a predictor of platinum sensitivity in sporadic epithelial ovarian cancers. Mol. Biomed. 2020, 1, 19. [Google Scholar] [CrossRef]
- Etemadmoghadam, D.; Weir, B.A.; Au-Yeung, G.; Alsop, K.; Mitchell, G.; George, J.; Davis, S.; D’Andrea, A.D.; Simpson, K.; Hahn, W.C.; et al. Synthetic lethality between CCNE1 amplification and loss of BRCA1. Proc. Natl. Acad. Sci. USA 2013, 110, 19489–19494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledermann, J.A. PARP inhibitors in ovarian cancer. Ann. Oncol. 2016, 27 (Suppl. 1), i40–i44. [Google Scholar] [CrossRef]
- Poveda, A.; Floquet, A.; Ledermann, J.A.; Asher, R.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Pignata, S.; Friedlander, M.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A final analysis of a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2021, 22, 620–631. [Google Scholar] [CrossRef]
- Poveda, A.; Floquet, A.; Ledermann, J.A.; Asher, R.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Pignata, S.; Friedlander, M.; et al. Final overall survival (OS) results from SOLO2/ENGOT-ov21: A phase III trial assessing maintenance olaparib in patients (pts) with platinum-sensitive, relapsed ovarian cancer and a BRCA mutation. J. Clin. Oncol. 2020, 38, 6002. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- Cecere, S.C.; Giannone, G.; Salutari, V.; Arenare, L.; Lorusso, D.; Ronzino, G.; Lauria, R.; Cormio, G.; Carella, C.; Scollo, P.; et al. Olaparib as maintenance therapy in patients with BRCA 1-2 mutated recurrent platinum sensitive ovarian cancer: Real world data and post progression outcome. Gynecol. Oncol. 2020, 156, 38–44. [Google Scholar] [CrossRef]
- Pothuri, B.; O’Cearbhaill, R.; Eskander, R.; Armstrong, D. Frontline PARP inhibitor maintenance therapy in ovarian cancer: A Society of Gynecologic Oncology practice statement. Gynecol. Oncol. 2020, 159, 8–12. [Google Scholar] [CrossRef]
- Markowski, M.C.; Antonarakis, E.S. BRCA1 versus BRCA2 and PARP inhibitor sensitivity in prostate cancer: More different than alike? J. Clin. Oncol. 2020, 38, 3735–3739. [Google Scholar] [CrossRef] [PubMed]
- Antolín, A.A.; Mestres, J. Linking off-target kinase pharmacology to the differential cellular effects observed among PARP inhibitors. Oncotarget 2014, 5, 3023–3028. [Google Scholar] [CrossRef] [Green Version]
- Chiappa, M.; Guffanti, F.; Bertoni, F.; Colombo, I.; Damia, G. Overcoming PARPi resistance: Preclinical and clinical evidence in ovarian cancer. Drug Resist. Updates 2021, 55, 100744. [Google Scholar] [CrossRef]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef] [PubMed]
- Burgess, B.T.; Anderson, A.M.; McCorkle, J.R.; Wu, J.; Ueland, F.R.; Kolesar, J.M. Olaparib combined with an ATR or Chk1 inhibitor as a treatment strategy for acquired olaparib-resistant BRCA1 mutant ovarian cells. Diagnostics 2020, 10, 121. [Google Scholar] [CrossRef] [Green Version]
- Haynes, B.; Murai, J.; Lee, J.M. Restored replication fork stabilization, a mechanism of PARP inhibitor resistance, can be overcome by cell cycle checkpoint inhibition. Cancer Treat. Rev. 2018, 71, 1–7. [Google Scholar] [CrossRef]
- Hill, S.J.; Decker, B.; Roberts, E.A.; Horowitz, N.S.; Muto, M.G.; Worley, M.J., Jr.; Feltmate, C.M.; Nucci, M.R.; Swisher, E.M.; Nguyen, H.; et al. Prediction of DNA repair inhibitor response in short-term patient-derived ovarian cancer organoids. Cancer Discov. 2018, 8, 1404–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topp, M.D.; Hartley, L.; Cook, M.; Heong, V.; Boehm, E.; McShane, L.; Pyman, J.; McNally, O.; Ananda, S.; Harrell, M.; et al. Molecular correlates of platinum response in human high-grade serous ovarian cancer patient-derived xenografts. Mol. Oncol. 2014, 8, 656–668. [Google Scholar] [CrossRef]
- DelloRusso, C.; Welcsh, P.L.; Wang, W.; Garcia, R.L.; King, M.C.; Swisher, E.M. Functional characterization of a novel BRCA1-null ovarian cancer cell line in response to ionizing radiation. Mol. Cancer Res. 2007, 5, 35–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anglesio, M.S.; Wiegand, K.C.; Melnyk, N.; Chow, C.; Salamanca, C.; Prentice, L.M.; Senz, J.; Yang, W.; Spillman, M.A.; Cochrane, D.R.; et al. Type-specific cell line models for type-specific ovarian cancer research. PLoS ONE 2013, 8, e72162. [Google Scholar]
- Langdon, S.P.; Lawrie, S.S.; Hay, F.G.; Hawkes, M.M.; McDonald, A.; Hayward, I.P.; Schol, D.J.; Hilgers, J.; Leonard, R.C.; Smyth, J.F. Characterization and properties of nine human ovarian adenocarcinoma cell lines. Cancer Res. 1988, 48, 6166–6172. [Google Scholar]
- Stordal, B.; Timms, K.; Farrelly, A.; Gallagher, D.; Busschots, S.; Renaud, M.; Thery, J.; Williams, D.; Potter, J.; Tran, T.; et al. BRCA1/2 mutation analysis in 41 ovarian cell lines reveals only one functionally deleterious BRCA1 mutation. Mol. Oncol. 2013, 7, 567–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, W.; Swisher, E.M.; Jacquemont, C.; Chandramohan, K.V.; Couch, F.J.; Langdon, S.P.; Wurz, K.; Higgins, J.; Villegas, E.; Taniguchi, T. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009, 69, 6381–6386. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.; Zheng, X.; Li, M.; Ye, F.; Song, C.; Xu, C.; Zhang, X.; Li, W.; Wang, Y.; Zeng, S.; et al. C/EBPβ promotes poly(ADP-ribose) polymerase inhibitor resistance by enhancing homologous recombination repair in high-grade serous ovarian cancer. Oncogene 2021, 40, 3845–3858. [Google Scholar] [CrossRef] [PubMed]
- Sebaugh, J.L. Guidelines for accurate EC50/IC50 estimation. Pharm. Stat. 2011, 10, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Crowley, L.C.; Christensen, M.E.; Waterhouse, N.J. Measuring survival of adherent cells with the colony-forming assay. Cold Spring Harb. Protoc. 2016, 721–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unkel, S.; Belka, C.; Lauber, K. On the analysis of clonogenic survival data: Statistical alternatives to the linear-quadratic model. Radiat. Oncol. 2016, 11, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LC50 (µM) | ^ Fold Change | ||
|---|---|---|---|
| PARPi | |||
| PEO1 vs. PEO4 | |||
| PE01 | PE04 | ||
| niraparib | 1.9300 | 5.0640 | 2.62 |
| rucaparib | 2.6370 | 3.2140 | 1.22 |
| olaparib | 2.3560 | 2.5710 | 1.09 |
| talazoparib | 0.0729 | 0.0557 | 0.76 |
| veliparib | 47.5900 | 28.1300 | 0.59 |
| UWB1.289 vs. UWB1.289 + BRCA1 | |||
| UWB1.289 | UWB1.289 + BRCA1 | ||
| niraparib | 0.6936 | 2.4620 | 3.55 |
| rucaparib | 2.2560 | 7.9120 | 3.51 |
| veliparib | 4.8490 | 15.7200 | 3.24 |
| olaparib | 0.1679 | 0.4920 | 2.93 |
| talazoparib | 0.0025 | 0.0062 | 2.52 |
| A2780 vs. A2780VeliR | |||
| A2780 | A2780VeliR | ||
| talazoparib | 0.0024 | 0.0347 | 14.41 |
| rucaparib | 1.1440 | 6.3480 | 5.55 |
| veliparib | 16.62 | 57.49 | 3.46 |
| niraparib | 0.2934 | 0.8718 | 2.97 |
| olaparib | 0.8735 | 2.2230 | 2.54 |
| LC50 (µM) | ^ Fold Change | ||
|---|---|---|---|
| PARPi | |||
| PEO1 vs. PEO4 | |||
| PEO1 | PEO4 | ||
| rucaparib | 0.0417 | 0.5332 | 12.78 |
| niraparib | 0.2168 | 0.9263 | 4.27 |
| olaparib | 0.1405 | 0.4935 | 3.51 |
| talazoparib | 0.00008 | 0.00017 | 2.23 |
| veliparib | 4.445 | 8.2154 | 1.85 |
| UWB1.289 vs. UWB1.289 + BRCA1 | |||
| UWB1.289 | UWB1.289 + BRCA1 | ||
| veliparib | 0.1745 | 8.2640 | 47.36 |
| rucaparib | 0.0066 | 0.2592 | 39.30 |
| olaparib | 0.0091 | 0.2446 | 26.77 |
| niraparib | 0.0178 | 0.1889 | 10.63 |
| talazoparib | 0.00005 | 0.00026 | 4.86 |
| A2780 vs. A2780VeliR | |||
| A2780 | A2780VeliR | ||
| olaparib | 0.0052 | 0.1206 | 23.03 |
| rucaparib | 0.0071 | 0.0915 | 12.97 |
| veliparib | 0.5395 | 4.7707 | 8.84 |
| talazoparib | 0.00003 | 0.0001 | 2.99 |
| niraparib | 0.0194 | 0.0484 | 2.49 |
| LC50 (µM) | |||
|---|---|---|---|
| AS | BRCA1 si#13 (fold change) | BRCA1 si#14 (fold change ^) | |
| PARPi | |||
| A2780 BRCA1 KD (cell viability) | |||
| olaparib | 1.2330 | 0.1994 (6.18) | 0.3539 (3.48) |
| rucaparib | 0.8272 | 0.1886 (4.39) | 0.3320 (2.49) |
| talazoparib | 0.0009 | 0.0003 (3.32) | 0.0004 (2.32) |
| veliparib | 16.9900 | 5.3730 (3.16) | 8.0910 (2.10) |
| niraparib | 0.4010 | 0.1910 (2.10) | 0.1927 (2.08) |
| OVCAR-3 BRCA1 KD (cell viability) | |||
| talazoparib | 0.0058 | 0.0006 (10.24) | 0.0005 (11.81) |
| rucaparib | 2.4500 | 0.1345 (18.22) | 0.2293 (10.68) |
| niraparib | 3.6650 | 0.4730 (7.75) | 0.3468 (10.57) |
| olaparib | 2.9110 | 0.4229 (6.88) | 0.4687 (6.21) |
| veliparib | 20.5600 | 3.2850 (6.26) | 3.6540 (5.63) |
| A2780 BRCA1 KD (cell survival) | |||
| olaparib | 0.0275 | 0.0075 (3.67) | 0.0110 (2.50) |
| niraparib | 0.0471 | 0.0204 (2.30) | 0.0311 (1.51) |
| veliparib | 1.1726 | 0.5215 (2.25) | 0.8487 (1.38) |
| talazoparib | 0.0006 | 0.0003 (2.10) | 0.0004 (1.26) |
| rucaparib | 0.0283 | 0.0119 (2.38) | 0.0229 (1.24) |
| OVCAR-3 BRCA1 KD (cell survival) | |||
| rucaparib | 0.0173 | 0.0085 (2.04) | 0.0074 (2.33) |
| veliparib | 1.5195 | 1.3455 (1.13) | 0.7451 (2.04) |
| olaparib | 0.0089 | 0.0062 (1.43) | 0.0056 (1.59) |
| niraparib | 0.0319 | 0.0263 (1.22) | 0.0206 (1.55) |
| talazoparib | 0.0003 | 0.0003 (0.94) | 0.0003 (0.93) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dickson, K.-A.; Xie, T.; Evenhuis, C.; Ma, Y.; Marsh, D.J. PARP Inhibitors Display Differential Efficacy in Models of BRCA Mutant High-Grade Serous Ovarian Cancer. Int. J. Mol. Sci. 2021, 22, 8506. https://doi.org/10.3390/ijms22168506
Dickson K-A, Xie T, Evenhuis C, Ma Y, Marsh DJ. PARP Inhibitors Display Differential Efficacy in Models of BRCA Mutant High-Grade Serous Ovarian Cancer. International Journal of Molecular Sciences. 2021; 22(16):8506. https://doi.org/10.3390/ijms22168506
Chicago/Turabian StyleDickson, Kristie-Ann, Tao Xie, Christian Evenhuis, Yue Ma, and Deborah J. Marsh. 2021. "PARP Inhibitors Display Differential Efficacy in Models of BRCA Mutant High-Grade Serous Ovarian Cancer" International Journal of Molecular Sciences 22, no. 16: 8506. https://doi.org/10.3390/ijms22168506
APA StyleDickson, K. -A., Xie, T., Evenhuis, C., Ma, Y., & Marsh, D. J. (2021). PARP Inhibitors Display Differential Efficacy in Models of BRCA Mutant High-Grade Serous Ovarian Cancer. International Journal of Molecular Sciences, 22(16), 8506. https://doi.org/10.3390/ijms22168506