
NAD(H) Regulates the Permeability Transition Pore in Mitochondria through an External Site



Abstract
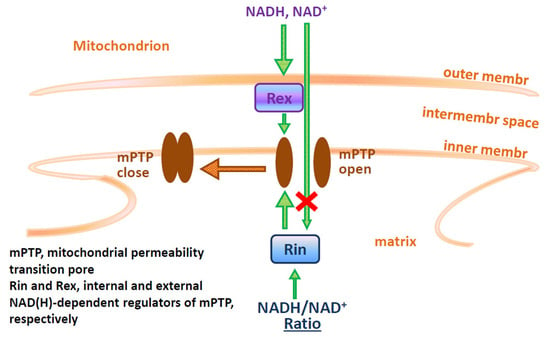
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
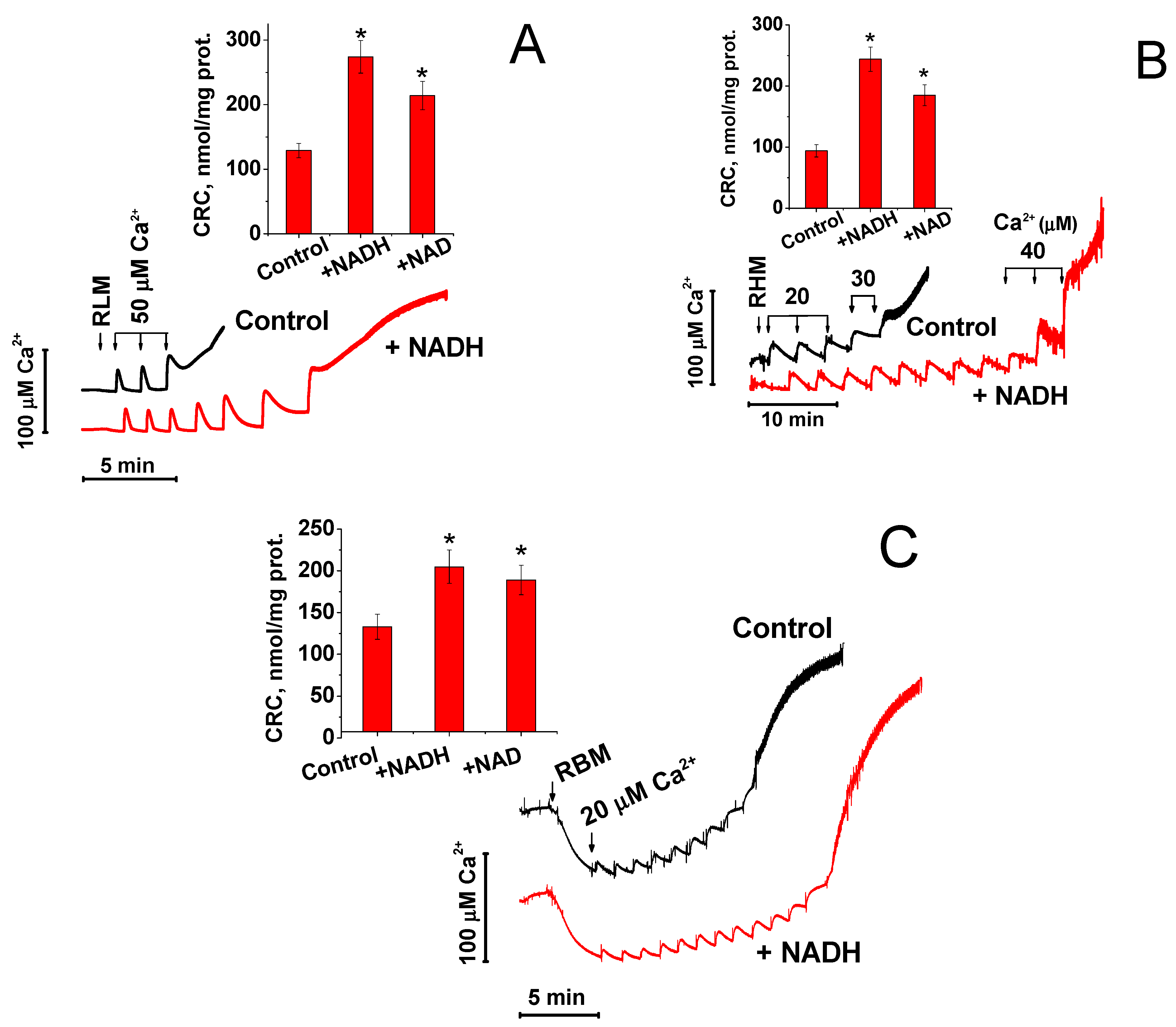
2.1. NAD(H) Increases the Ca2+-Retention Capacity of Liver, Heart, and Brain Mitochondria
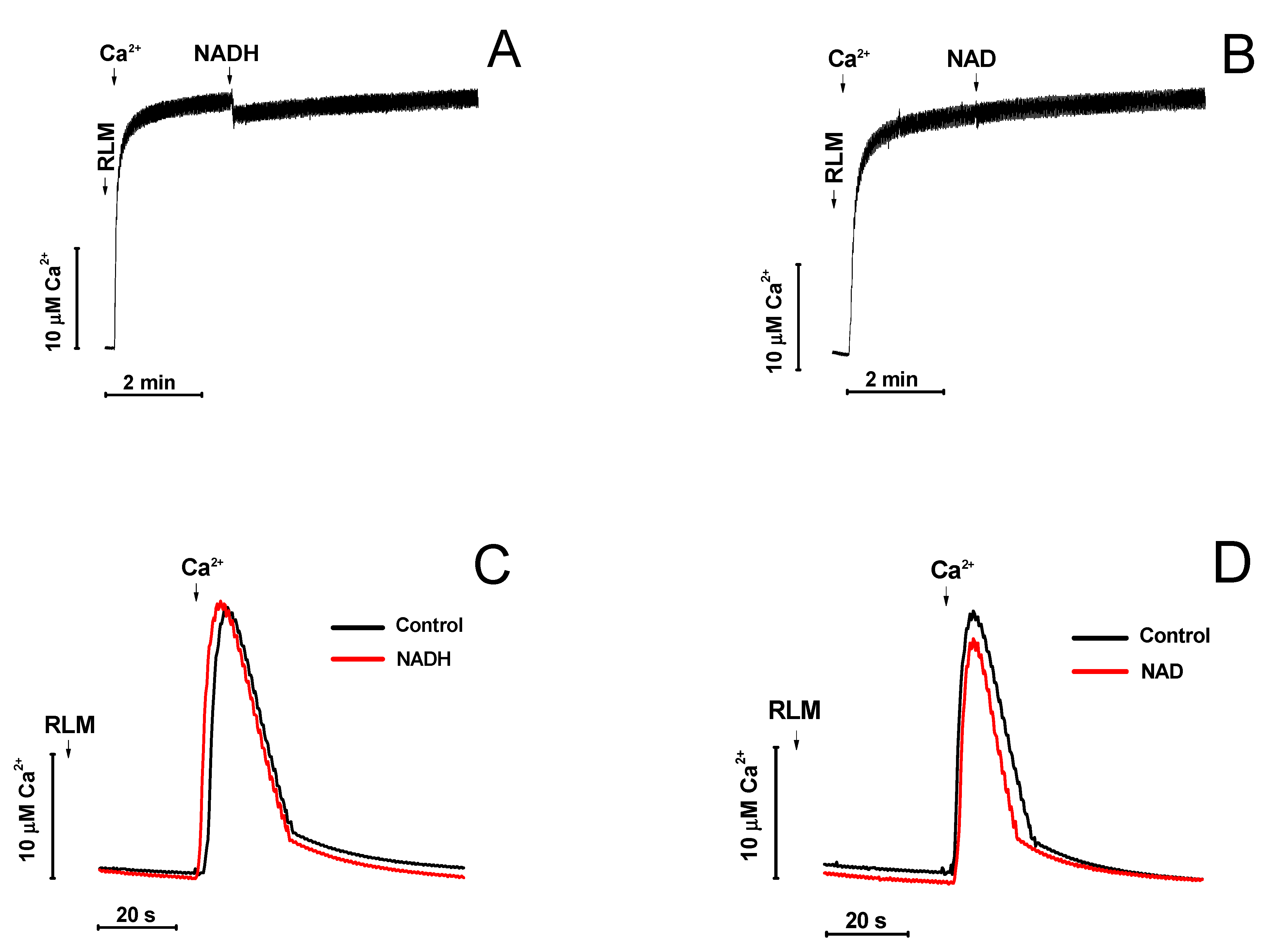
2.2. Effect of NAD(H) on the Concentration of Ca2+ in the Medium and the Rate of Its Uptake by Mitochondria
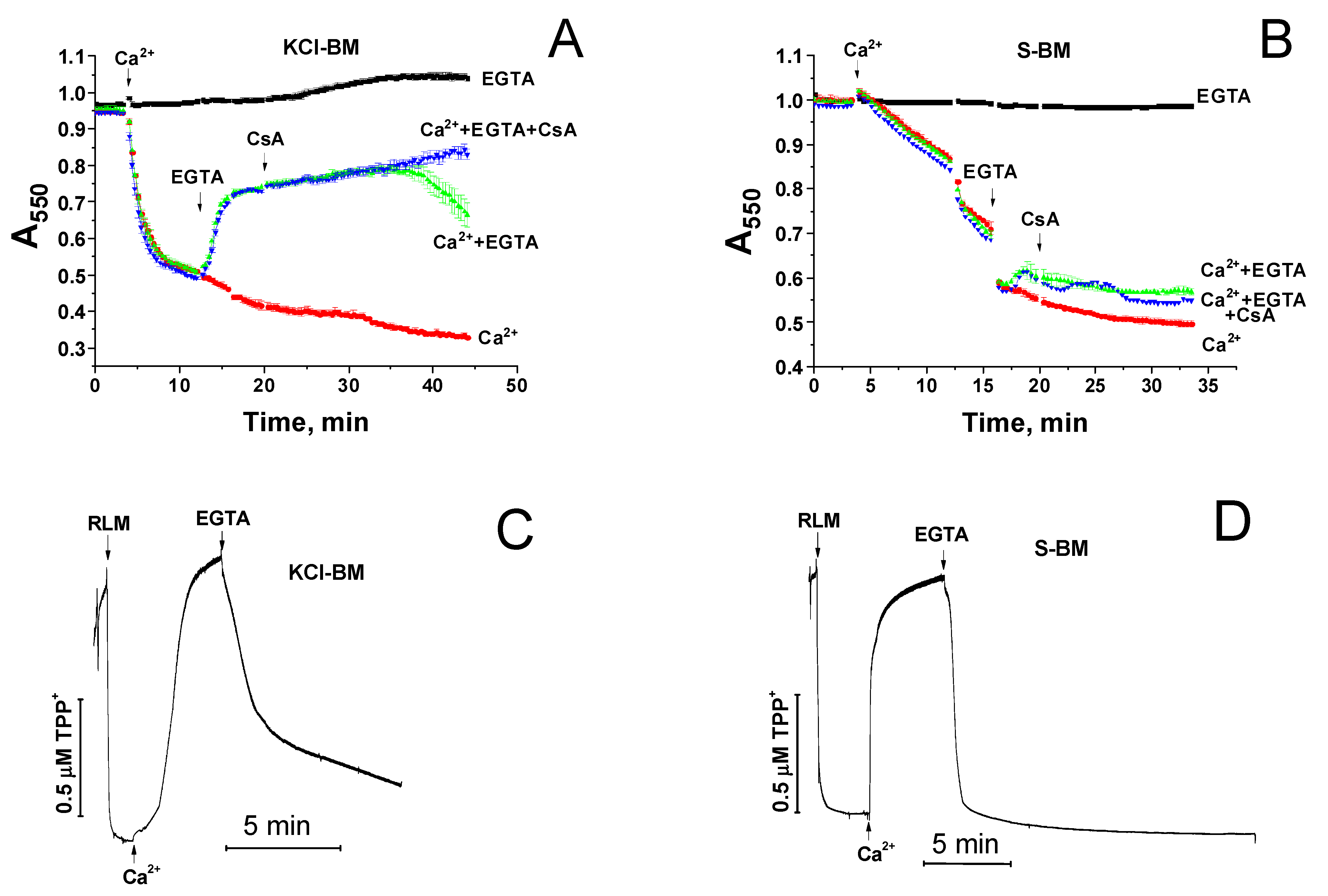
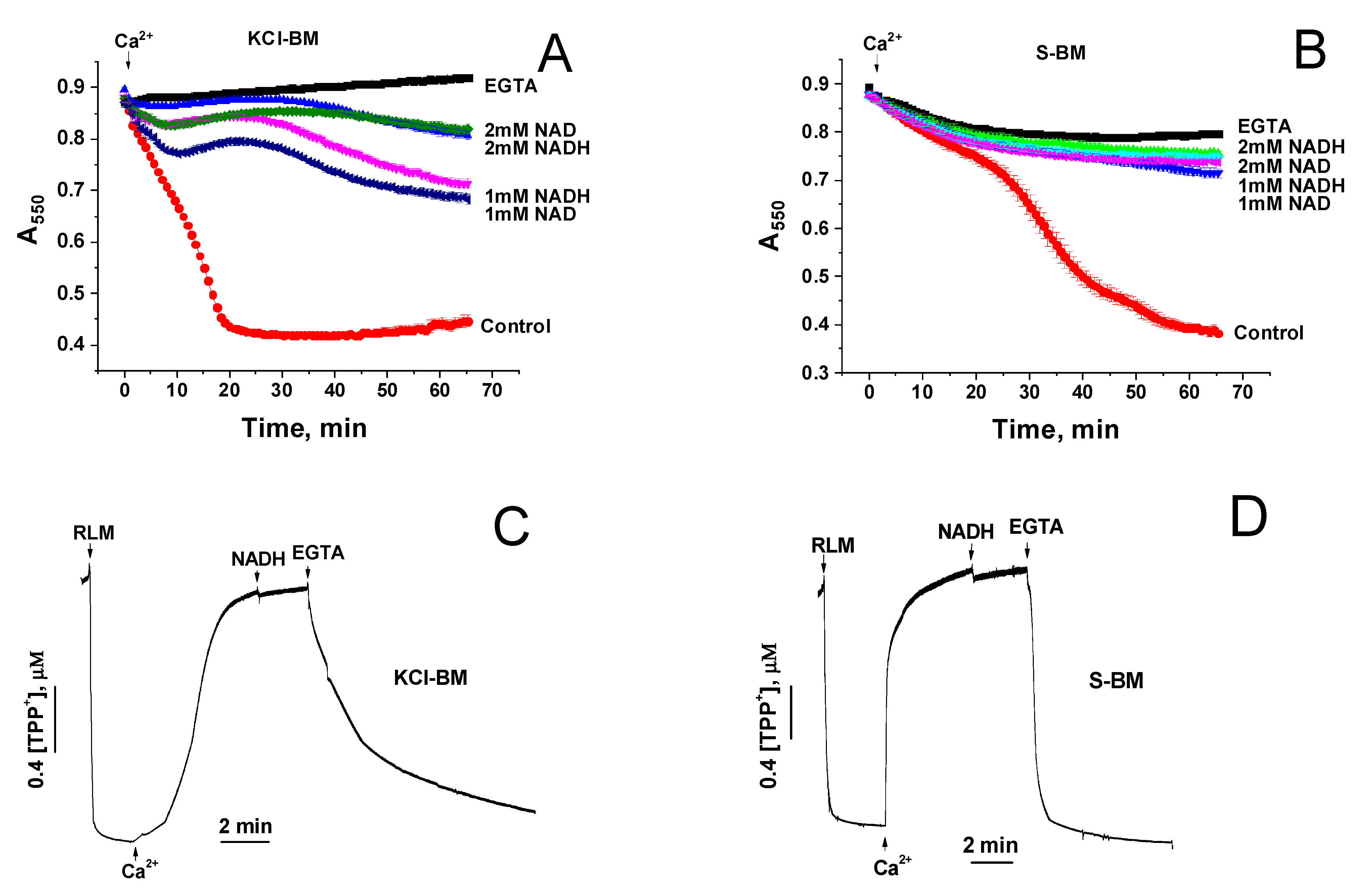
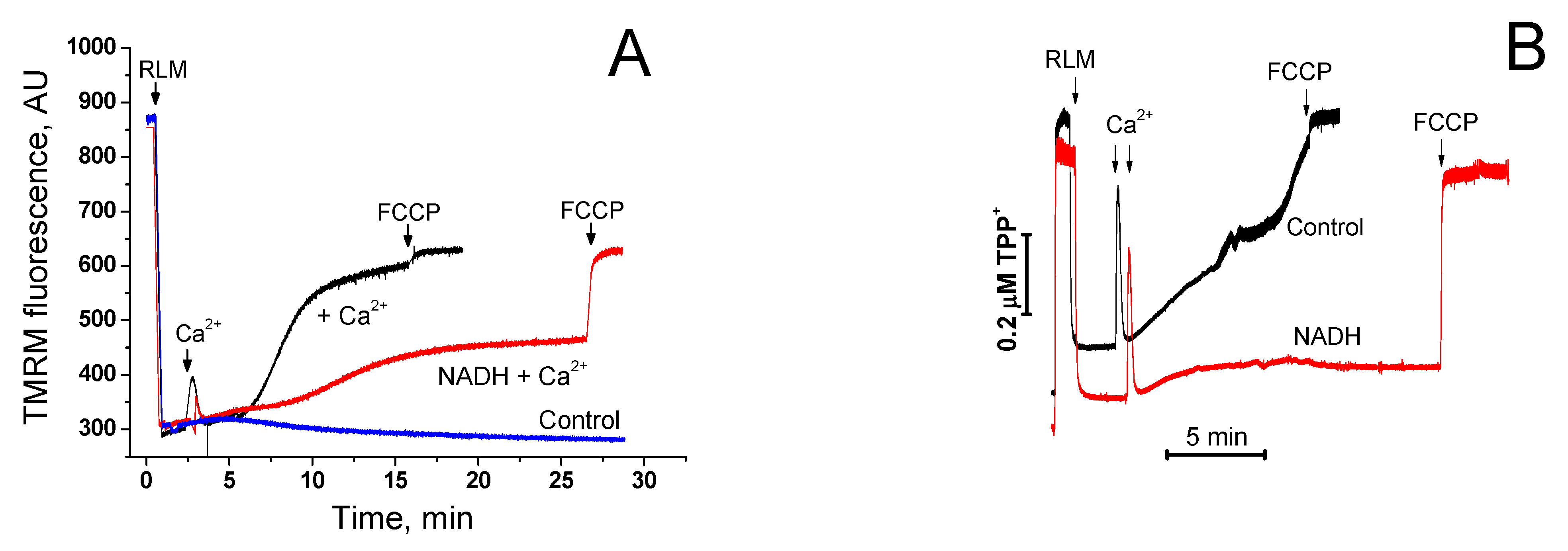
2.3. Divergent Effects of mPTP Closure on the Mitochondrial Shrinkage and ΔΨm Recovery in KCl- and Sucrose-Based Media
2.4. Effect of NAD(H) on the Ca2+-Induced Mitochondrial Swelling in KCl- and Sucrose-Based Media
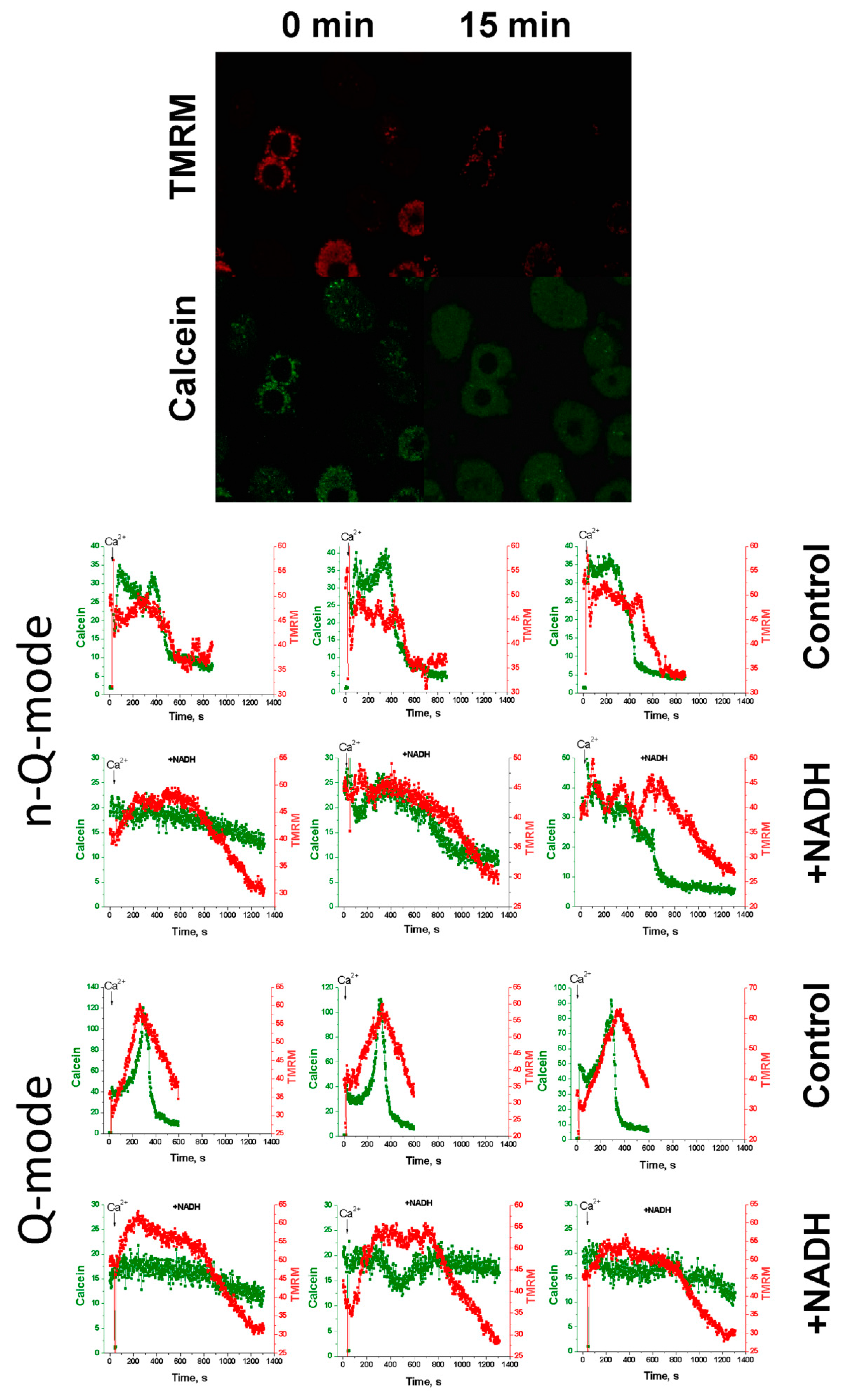
2.5. Effect of NAD(H) on the Ca2+-Induced Loss of ΔΨm and Calcein Release from Individual Mitochondria
2.6. Protective Effect of NAD(H) Is Due to the Suppression of Ca2+-Dependent Permeabilization of the Inner Mitochondrial Membrane
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Isolation of Mitochondria from Rat Liver, Heart, and Brain
4.3. Isolation of Rat Hepatocytes
4.4. Determination of the Ca2+ Uptake Rate and the Ca2+-Retention Capacity of Mitochondria
4.5. Recording of Mitochondrial Swelling
4.6. Recording of the Mitochondrial Membrane Potential
4.7. Registration of mPTP Opening by Confocal Microscopy
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chaudhuri, R.C.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodeling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Jiménez, M.; Hurtado, O.; Cuartero, M.I.; Ballesteros, I.; Moraga, A.; Pradillo, J.M.; McBurney, M.W.; Lizasoain, I.; Moro, M.A. Silent information regulator 1 protects the brain against cerebral ischemic damage. Stroke 2013, 44, 2333–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafner, A.V.; Dai, J.; Gomes, A.P.; Xiao, C.Y.; Palmeira, C.M.; Rosenzweig, A.; Sinclair, D.A. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging 2010, 2, 914–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chini, E.N. CD38 as a regulator of cellular NAD: A novel potential pharmacological target for metabolic conditions. Curr. Pharmm. Des. 2009, 15, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Aksoy, P.; White, T.A.; Thompson, M.; Chini, E.N. Regulation of intracellular levels of NAD: A novel role for CD38. Biochem. Biophys. Res. Commun. 2006, 345, 1386–1392. [Google Scholar] [CrossRef]
- Rasola, A.; Bernardi, P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis 2007, 12, 815–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, D.R.; Haworth, R.A. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch. Biochem. Biophys. 1979, 195, 453–459. [Google Scholar] [CrossRef]
- Haworth, R.A.; Hunter, D.R. Allosteric inhibition of the Ca2+-activated hydrophilic channel of the mitochondrial inner membrane by nucleotides. J. Membr. Biol. 1980, 54, 231–236. [Google Scholar] [CrossRef]
- Bindoli, A.; Callegaro, M.T.; Barzon, E.; Benetti, M.; Rigobello, M.P. Influence of the redox state of pyridine nucleotides on mitochondrial sulfhydryl groups and permeability transition. Arch. Biochem. Biophys. 1997, 342, 22–28. [Google Scholar] [CrossRef]
- Zago, E.B.; Castilho, R.F.; Vercesi, A.E. The redox state of endogenous pyridine nucleotides can determine both the degree of mitochondrial oxidative stress and the solute selectivity of the permeability transition pore. FEBS Lett. 2000, 478, 29–33. [Google Scholar] [CrossRef] [Green Version]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Invest. 2009, 119, 2758–2771. [Google Scholar] [CrossRef] [Green Version]
- Porter, G.A.; Urciuoli, W.R.; Brookes, P.S.; Nadtochiy, S.M. SIRT3 deficiency exacerbates ischemia-reperfusion injury: Implication for aged hearts. Am. J. Physiol. Heart. Circ. Physiol. 2014, 306, H1602–H1609. [Google Scholar] [CrossRef] [Green Version]
- Tao, R.; Kim, S.H.; Honbo, N.; Karliner, J.S.; Alano, C.C. Minocycline protects cardiac myocytes against simulated ischemia–reperfusion injury by inhibiting poly(ADP-ribose) polymerase-1. J. Cardiovasc. Pharmacol. 2010, 56, 659–668. [Google Scholar] [CrossRef]
- Schriewer, J.M.; Peek, C.B.; Bass, J.; Schumacker, P.T. ROS-mediated PARP activity undermines mitochondrial function after permeability transition pore opening during myocardial ischemia-reperfusion. J. Am. Heart Assoc. 2013, 2, 159. [Google Scholar] [CrossRef] [Green Version]
- Kahraman, S.; Siegel, A.; Polster, B.M.; Fiskum, G. Permeability transition pore-dependent and PARP-mediated depletion of neuronal pyridine nucleotides during anoxia and glucose deprivation. J. Bioenerg Biomembr. 2015, 47, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Ying, W. NAD(+) Deficiency is a common central pathological factor of a number of diseases and aging: Mechanisms and therapeutic implications. Antioxid. Redox Signal. 2019, 30, 890–905. [Google Scholar] [CrossRef]
- Lee, C.F.; Chavez, J.D.; Garcia-Menendez, L.; Choi, Y.; Roe, N.D.; Chiao, Y.A.; Edgar, J.S.; Goo, Y.A.; Goodlett, D.R.; Bruce, J.E.; et al. Normalization of NAD+ redox balance as a therapy for heart failure. Circulation 2016, 134, 883–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristian, T.; Balan, I.; Schuh, R.; Onken, M. Mitochondrial dysfunction and nicotinamide dinucleotide catabolism as mechanisms of cell death and promising targets for neuroprotection. J. Neurosci. Res. 2011, 891, 946–1955. [Google Scholar] [CrossRef]
- Kruglov, A.G.; Teplova, V.V.; Saris, N.E. The effect of the lipophilic cation lucigenin on mitochondria depends on the site of its reduction. Biochem. Pharmacol. 2007, 74, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Kruglov, A.G.; Subbotina, K.B.; Saris, N.E. Redox-cycling compounds can cause the permeabilization of mitochondrial membranes by mechanisms other than ROS production. Free Radic. Biol. Med. 2008, 44, 646–656. [Google Scholar] [CrossRef]
- Nikiforova, A.B.; Saris, N.E.; Kruglov, A.G. External mitochondrial NADH-dependent reductase of redox cyclers: VDAC1 or Cyb5R3? Free Radic. Biol. Med. 2014, 74, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Kharechkina, E.S.; Nikiforova, A.B.; Teplova, V.V.; Odinokova, I.V.; Krestinina, O.V.; Baburina, Y.L.; Kruglova, S.A.; Kruglov, A.G. Regulation of permeability transition pore opening in mitochondria by external NAD(H). Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Stein, L.R.; Imai, S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab. 2012, 23, 420–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barile, M.; Passarella, S.; Danese, G.; Quagliariello, E. Rat liver mitochondria can synthesize nicotinamide adenine dinucleotide from nicotinamide mononucleotide and ATP via a putative matrix nicotinamide mononucleotide adenylyltransferase. Biochem. Mol. Biol. Int. 1996, 38, 297–306. [Google Scholar] [PubMed]
- Al-Nasser, I.; Crompton, M. The reversible Ca2+-induced permeabilization of rat liver mitochondria. Biochem. J. 1986, 239, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Broekemeier, K.M.; Schmid, P.C.; Schmid, H.H.; Pfeiffer, D.R. Effects of phospholipase A2 inhibitors on ruthenium red-induced Ca2+ release from mitochondria. J. Biol. Chem. 1985, 260, 105–113. [Google Scholar] [CrossRef]
- Novgorodov, S.A.; Gudz, T.I.; Milgrom, Y.M.; Brierley, G.P. The permeability transition in heart mitochondria is regulated synergistically by ADP and cyclosporin A. J. Biol. Chem. 1992, 267, 16262–16274. [Google Scholar] [CrossRef]
- Broekemeier, K.M.; Klocek, C.K.; Pfeiffer, D.R. Proton selective substate of the mitochondrial permeability transition pore: Regulation by the redox state of the electron transport chain. Biochemistry 1998, 37, 13059–13065. [Google Scholar] [CrossRef]
- Kharechkina, E.S.; Nikiforova, A.B.; Kruglov, A.G. Pyridine nucleotides regulate the superoxide anion flash upon permeabilization of mitochondrial membranes: An MCLA-based study. Free Radic. Biol. Med. 2018, 124, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Komarov, A.; Bezrukov, S.M.; Colombini, M. Dynamics of nucleotides in VDAC channels: Structure-specific noise generation. Biophys. J. 2002, 82, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Szabo, I.; Bernardi, P.; Zoratti, M. Modulation of the mitochondrial megachannel by divalent cations and protons. J. Biol. Chem. 1992, 267, 2940–2946. [Google Scholar] [CrossRef]
- Hüser, J.; Rechenmacher, C.E.; Blatter, L.A. Imaging the permeability pore transition in single mitochondria. Biophys. J. 1998, 74, 2129–2137. [Google Scholar] [CrossRef] [Green Version]
- Hüser, J.; Blatter, L.A. Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem J. 1999, 343, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Vergun, O.; Votyakova, T.V.; Reynolds, I.J. Spontaneous Changes in Mitochondrial Membrane Potential in Single Isolated Brain Mitochondria. Biophys. J. 2003, 85, 3358–3366. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Kwong, J.Q.; Molkentin, J.D.; Bers, D.M. Individual Cardiac Mitochondria Undergo Rare Transient Permeability Transition Pore Openings. Circ. Res. 2016, 118, 834–841. [Google Scholar] [CrossRef] [Green Version]
- Chernyak, B.V.; Bernardi, P. The mitochondrial permeability transition pore is modulated by oxidative agents through both pyridine nucleotides and glutathione at two separate sites. Eur. J. Biochem. 1996, 238, 623–630. [Google Scholar] [CrossRef]
- Nieminen, A.L.; Byrne, A.M.; Herman, B.; Lemasters, J.J. Mitochondrial permeability transition in hepatocytes induced by t-BuOOH: NAD(P)H and reactive oxygen species. Am. J. Physiol. 1997, 272, 1286–1294. [Google Scholar] [CrossRef]
- Busanello, E.N.B.; Figueira, T.R.; Marques, A.C.; Navarro, C.D.C.; Oliveira, H.C.F.; Vercesi, A.E. Facilitation of Ca2+-induced opening of the mitochondrial permeability transition pore either by nicotinamide nucleotide transhydrogenase deficiency or statins treatment. Cell Biol. Int. 2018, 42, 742–746. [Google Scholar] [CrossRef] [PubMed]
- Nightigale, E.R. Phenomenological theory of ion solvation. Effective radii of hydrated ions. J. Phys. Chem. 1959, 63, 1381–1387. [Google Scholar] [CrossRef]
- Schultz, S.G.; Solomon, A.K. Determination of the Effective Hydrodynamic Radii of Small Molecules by Viscometry. J. Gen. Phys. 1961, 44, 1189–1199. [Google Scholar] [CrossRef] [Green Version]
- Broekemeier, K.M.; Pfeiffer, D.R. Inhibition of the mitochondrial permeability transition by cyclosporin A during long time frame experiments: Relationship between pore opening and the activity of mitochondrial phospholipases. Biochemistry 1995, 34, 16440–16449. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, J.; Schweizer, M.; Richter, C. “Pore” formation is not required for the hydroperoxide-induced Ca2+ release from rat liver mitochondria. Biochem. J. 1992, 285, 65–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushnareva, Y.E.; Sokolove, P.M. Prooxidants open both the mitochondrial permeability transition pore and a low conductance channel in the inner mitochondrial membrane. Arch. Biochem. Biophys. 2000, 376, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Toninello, A.; Salvi, M.; Schweizer, M.; Richter, C. Menadione induces a low conductance state of the mitochondrial inner membrane sensitive to bongkrekic acid. Free Radic Biol Med. 2004, 37, 1073–1080. [Google Scholar] [CrossRef]
- Brierley, G.P.; Baysal, K.; Jung, D.W. Cation transport systems in mitochondria: Na+ and K+ uniports and exchangers. J. Bioenerg Biomembr. 1994, 26, 519–526. [Google Scholar] [CrossRef]
- Li, X.; Weinman, S.A. Chloride channels and hepatocellular function: Prospects for molecular identification. Annu. Rev. Physiol. 2002, 64, 609–633. [Google Scholar] [CrossRef]
- Drummond-Main, C.D.; Rho, J.M. Electrophysiological characterization of a mitochondrial inner membrane chloride channel in rat brain. FEBS Lett. 2018, 592, 1545–1553. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P. Mitochondrial transport of cations: Channels, exchangers, and permeability transition. Physiol. Rev. 1999, 79, 1127–1155. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.; Zizi, M.; Colombini, M. Beta-NADH decreases the permeability of the mitochondrial outer membrane to ADP by a factor of 6. J. Biol. Chem. 1994, 269, 30974–30980. [Google Scholar] [CrossRef]
- Lee, A.C.; Xu, X.; Colombini, M. The role of pyridine dinucleotides in regulating the permeability of the mitochondrial outer membrane. J. Biol. Chem. 1996, 271, 26724–26731. [Google Scholar] [CrossRef] [Green Version]
- Han, D.; Antunes, F.; Canali, R.; Rettori, D.; Cadenas, E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol. Chem. 2003, 278, 5557–5563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lustgarten, M.S.; Bhattacharya, A.; Muller, F.L.; Jang, Y.C.; Shimizu, T.; Shirasawa, T.; Richardson, A.; Van Remmen, H. Complex I generated, mitochondrial matrix-directed superoxide is released from the mitochondria through voltage dependent anion channels. Biochem. Biophys. Res. Commun. 2012, 422, 515–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villinger, S.; Giller, K.; Bayrhuber, M.; Lange, A.; Griesinger, C.; Becker, S.; Zweckstetter, M. Nucleotide interactions of the human voltage-dependent anion channel. J. Biol. Chem. 2014, 289, 13397–13406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinopoulos, C. Mitochondrial permeability transition pore: Back to the drawing board. Neurochem. Int. 2018, 117, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Tarze, A.; Deniaud, A.; Le Bras, M.; Maillier, E.; Molle, D.; Larochette, N.; Zamzami, N.; Jan, G.; Kroemer, G.; Brenner, C. GAPDH, a novel regulator of the pro-apoptotic mitochondrial membrane permeabilization. Oncogene 2007, 26, 2606–2620. [Google Scholar] [CrossRef] [Green Version]
- Allouche, M.; Pertuiset, C.; Robert, J.L.; Martel, C.; Veneziano, R.; Henry, C.; dein, O.S.; NSaint Brenner, C.; Chopineau, J. ANT-VDAC1 interaction is direct and depends on ANT isoform conformation in vitro. Biochem. Biophys. Res. Commun. 2012, 429, 12–17. [Google Scholar] [CrossRef]
- Vyssokikh, M.; Brdiczka, D. VDAC and peripheral channelling complexes in health and disease. Mol. Cell. Biochem. 2004, 256–257, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Anflous-Pharayra, K.; Cai, Z.J.; Craigen, W.J. VDAC1 serves as a mitochondrial binding site for hexokinase in oxidative muscles. Biochim. Biophys. Acta. 2007, 1767, 136–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Hamad, S.; Zaid, H.; Israelson, A.; Nahon, E.; Shoshan-Barmatz, V. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: Mapping the site of binding. J. Biol. Chem. 2008, 283, 13482–13490. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.; Lardy, H. Isolation of liver or kidney mitochondria. Methods Enzymol. 1967, 10, 94–96. [Google Scholar]
- Krestinina, O.; Azarashvili, T.; Baburina, Y.; Galvita, A.; Grachev, D.; Stricker, R.; Reiser, G. In aging, the vulnerability of rat brain mitochondria is enhanced due to reduced level of 2′,3′-cyclic nucleotide-3′-phosphodiesterase (CNP) and subsequently increased permeability transition in brain mitochondria in old animals. Neurochem. Int. 2015, 80, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Gornall, A.G.; Bardawill, C.J.; David, M.M. Determination of serum proteins by means of the biuret reaction. J. Biol. Chem. 1949, 177, 751–766. [Google Scholar] [CrossRef]
- Herman, B.; Nieminen, A.L.; Gores, G.J.; Lemasters, J.J. Irreversible injury in anoxic hepatocytes precipitated by an abrupt increase in plasma membrane permeability. FASEB J. 1988, 2, 146–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kharechkina, E.; Nikiforova, A.; Kruglov, A. NAD(H) Regulates the Permeability Transition Pore in Mitochondria through an External Site. Int. J. Mol. Sci. 2021, 22, 8560. https://doi.org/10.3390/ijms22168560
Kharechkina E, Nikiforova A, Kruglov A. NAD(H) Regulates the Permeability Transition Pore in Mitochondria through an External Site. International Journal of Molecular Sciences. 2021; 22(16):8560. https://doi.org/10.3390/ijms22168560
Chicago/Turabian StyleKharechkina, Ekaterina, Anna Nikiforova, and Alexey Kruglov. 2021. "NAD(H) Regulates the Permeability Transition Pore in Mitochondria through an External Site" International Journal of Molecular Sciences 22, no. 16: 8560. https://doi.org/10.3390/ijms22168560
APA StyleKharechkina, E., Nikiforova, A., & Kruglov, A. (2021). NAD(H) Regulates the Permeability Transition Pore in Mitochondria through an External Site. International Journal of Molecular Sciences, 22(16), 8560. https://doi.org/10.3390/ijms22168560