
Approaches to Enhance Precise CRISPR/Cas9-Mediated Genome Editing

 ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Significance and Relevance of CRISPR/Cas9 Technology
1.2. Potential Medical Applications of CRISPR/Cas9
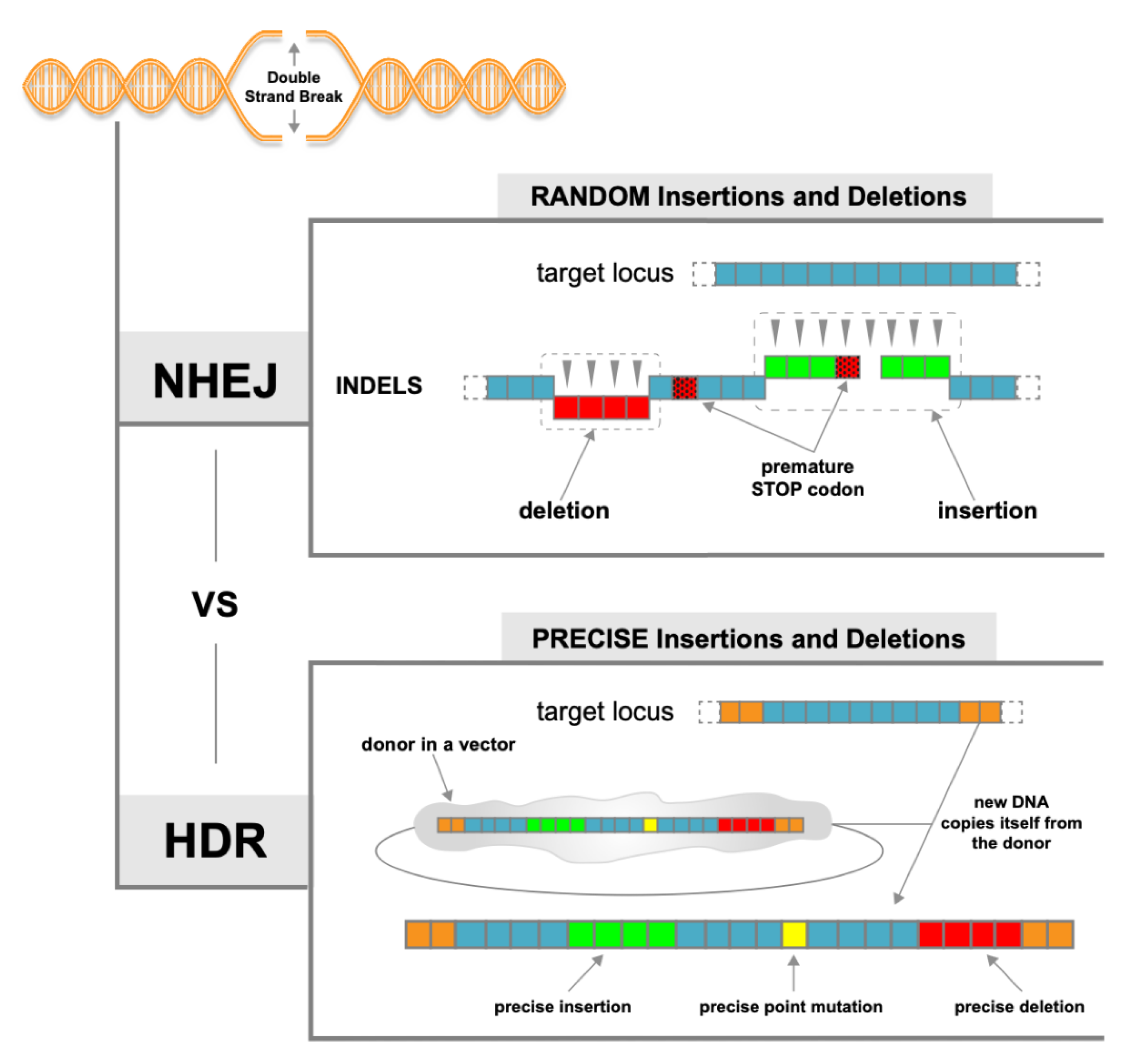
1.3. NHEJ vs. HDR
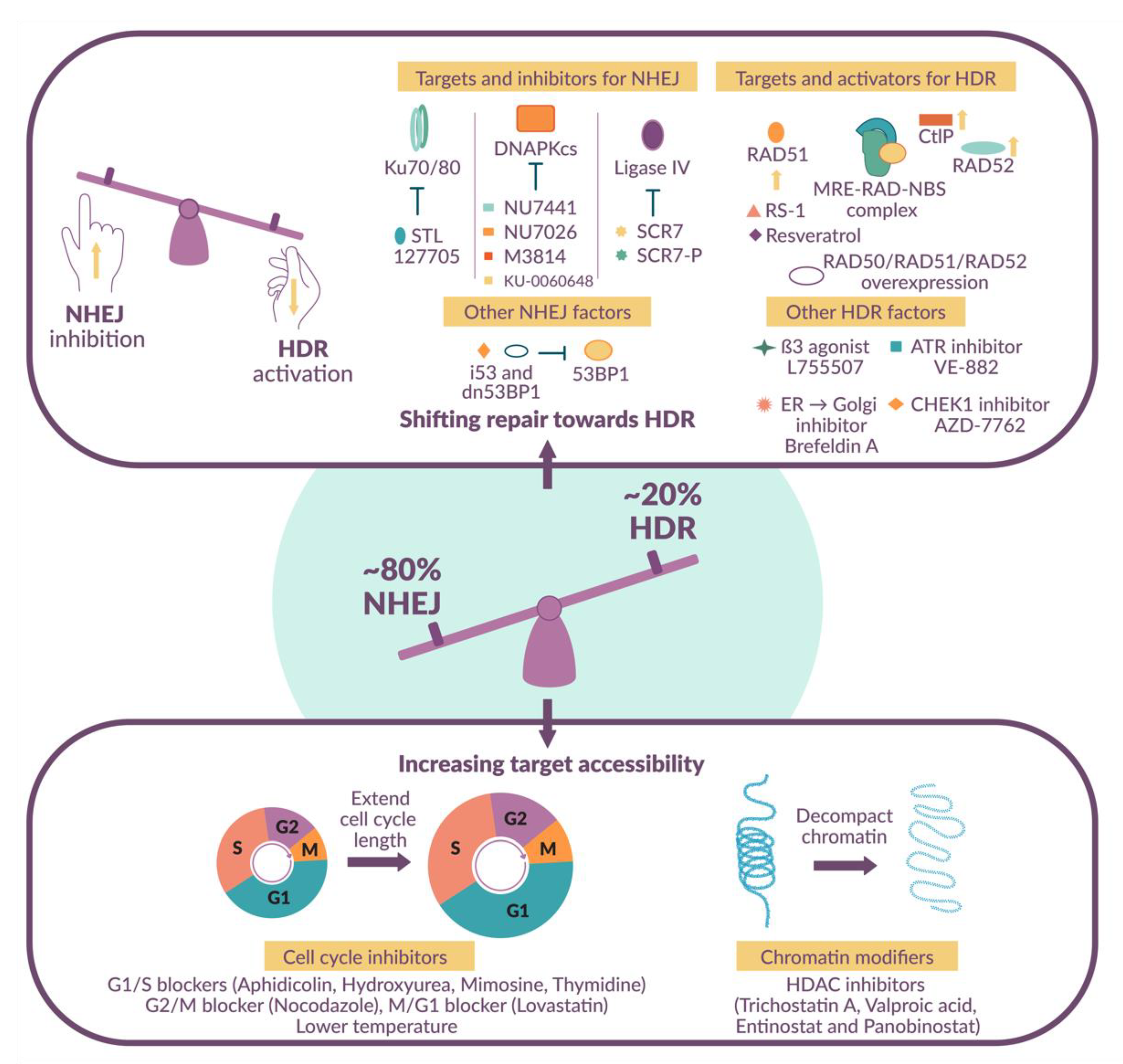
1.4. Shifting the Balance in Favor of HDR-Mediated DNA Repair
2. Small Molecule Modulators
2.1. NHEJ Inhibitors
2.2. HDR Activators
2.3. Cell Cycle Inhibitors
2.4. Histone Deacetylase Inhibitors
2.5. Additional Targets
2.6. Targeting Multiple Pathways Simultaneously
3. Optimized Nucleic Acid Strategies
3.1. dsDNA vs. ssDNA Templates
3.2. Modified Donor Templates
3.2.1. Asymmetric Donor DNA
3.2.2. Phosphorothioate-Modification
3.2.3. Chromatin-Modification
3.2.4. Conjugation of Template and Cas9
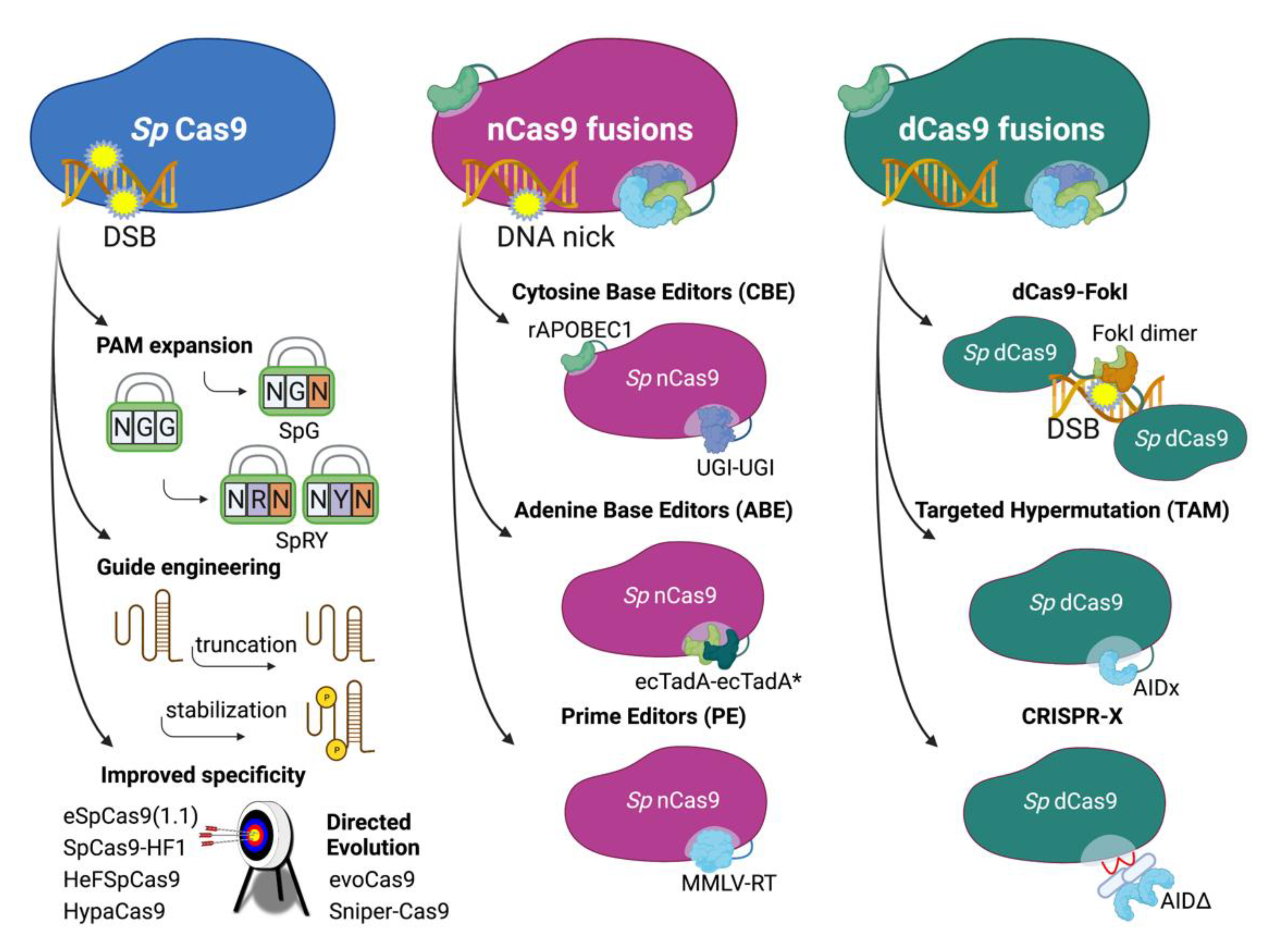
4. Approaches Using Engineered Cas9
4.1. Improved CRISPR/Cas9 Systems
4.2. DNA Base Editors
4.2.1. ABEs and CBEs—Targeted Donor-Free Editing
4.2.2. TAM—Targeted Hypermutation
4.2.3. CRISPR-X—Targeted Hypermutation
4.3. Prime Editing—Comprehensive Targeted Base Editing, Insertions and Deletions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Jiang, F.; Zhou, K.; Ma, L.; Gressel, S.; Doudna, J.A. STRUCTURAL BIOLOGY. A Cas9-guide RNA complex preorganized for target DNA recognition. Science 2015, 348, 1477–1481. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [Green Version]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Gilbert, L.A.; Cimini, B.A.; Schnitzbauer, J.; Zhang, W.; Li, G.W.; Park, J.; Blackburn, E.H.; Weissman, J.S.; Qi, L.S.; et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 2013, 155, 1479–1491. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.S.; Li, Q.C.; Yin, C.Q.; Xue, W.; Song, C.Q. Advances in CRISPR/Cas-based Gene Therapy in Human Genetic Diseases. Theranostics 2020, 10, 4374–4382. [Google Scholar] [CrossRef]
- Xu, L.; Wang, J.; Liu, Y.; Xie, L.; Su, B.; Mou, D.; Wang, L.; Liu, T.; Wang, X.; Zhang, B.; et al. CRISPR-Edited Stem Cells in a Patient with HIV and Acute Lymphocytic Leukemia. N. Engl. J. Med. 2019, 381, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Hutter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Mussig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kucherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’Connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021, 385, 493–502. [Google Scholar] [CrossRef]
- Liang, D.; Gutierrez, N.M.; Chen, T.; Lee, Y.; Park, S.-W.; Ma, H.; Koski, A.; Ahmed, R.; Darby, H.; Li, Y.; et al. Frequent gene conversion in human embryos induced by double strand breaks. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zuccaro, M.V.; Xu, J.; Mitchell, C.; Marin, D.; Zimmerman, R.; Rana, B.; Weinstein, E.; King, R.T.; Smith, M.; Tsang, S.H.; et al. Reading frame restoration at the EYS locus, and allele-specific chromosome removal after Cas9 cleavage in human embryos. bioRxiv 2020. [Google Scholar] [CrossRef]
- Redman, M.; King, A.; Watson, C.; King, D. What is CRISPR/Cas9? Arch. Dis. Child. Educ. Pract. Ed. 2016, 101, 213–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betermier, M.; Bertrand, P.; Lopez, B.S. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet. 2014, 10, e1004086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkman, E.K.; Chen, T.; de Haas, M.; Holland, H.A.; Akhtar, W.; van Steensel, B. Kinetics and Fidelity of the Repair of Cas9-Induced Double-Strand DNA Breaks. Mol. Cell 2018, 70, 801–813. [Google Scholar] [CrossRef] [Green Version]
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2016, 19, 19–23. [Google Scholar] [CrossRef]
- Devkota, S. The road less traveled: Strategies to enhance the frequency of homology-directed repair (HDR) for increased efficiency of CRISPR/Cas-mediated transgenesis. BMB Rep. 2018, 51, 437–443. [Google Scholar] [CrossRef] [Green Version]
- Carroll, D. Genome editing: Progress and challenges for medical applications. Genome Med. 2016, 8, 120. [Google Scholar] [CrossRef] [Green Version]
- Beumer, K.J.; Trautman, J.K.; Mukherjee, K.; Carroll, D. Donor DNA Utilization During Gene Targeting with Zinc-Finger Nucleases. G3 (Bethesda) 2013, 3, 657–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kuhn, R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shy, B.R.; MacDougall, M.S.; Clarke, R.; Merrill, B.J. Co-incident insertion enables high efficiency genome engineering in mouse embryonic stem cells. Nucleic Acids Res. 2016, 44, 7997–8010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riesenberg, S.; Chintalapati, M.; Macak, D.; Kanis, P.; Maricic, T.; Paabo, S. Simultaneous precise editing of multiple genes in human cells. Nucleic Acids Res. 2019, 47, e116. [Google Scholar] [CrossRef]
- Beumer, K.J.; Trautman, J.K.; Bozas, A.; Liu, J.L.; Rutter, J.; Gall, J.G.; Carroll, D. Efficient gene targeting in Drosophila by direct embryo injection with zinc-finger nucleases. Proc. Natl. Acad. Sci. USA 2008, 105, 19821–19826. [Google Scholar] [CrossRef] [Green Version]
- Canny, M.D.; Moatti, N.; Wan, L.C.K.; Fradet-Turcotte, A.; Krasner, D.; Mateos-Gomez, P.A.; Zimmermann, M.; Orthwein, A.; Juang, Y.C.; Zhang, W.; et al. Inhibition of 53BP1 favors homology-dependent DNA repair and increases CRISPR-Cas9 genome-editing efficiency. Nat. Biotechnol. 2018, 36, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife 2014, 3, e04766. [Google Scholar] [CrossRef]
- Jayathilaka, K.; Sheridan, S.D.; Bold, T.D.; Bochenska, K.; Logan, H.L.; Weichselbaum, R.R.; Bishop, D.K.; Connell, P.P. A chemical compound that stimulates the human homologous recombination protein RAD51. Proc. Natl. Acad. Sci. USA 2008, 105, 15848–15853. [Google Scholar] [CrossRef] [Green Version]
- Pinder, J.; Salsman, J.; Dellaire, G. Nuclear domain ‘knock-in’ screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic Acids Res. 2015, 43, 9379–9392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riesenberg, S.; Maricic, T. Targeting repair pathways with small molecules increases precise genome editing in pluripotent stem cells. Nat. Commun. 2018, 9, 2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohiuddin, I.S.; Kang, M.H. DNA-PK as an Emerging Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 635. [Google Scholar] [CrossRef]
- Robert, F.; Barbeau, M.; Ethier, S.; Dostie, J.; Pelletier, J. Pharmacological inhibition of DNA-PK stimulates Cas9-mediated genome editing. Genome Med. 2015, 7, 93. [Google Scholar] [CrossRef] [Green Version]
- Aksoy, Y.A.; Nguyen, D.T.; Chow, S.; Chung, R.S.; Guillemin, G.J.; Cole, N.J.; Hesselson, D. Chemical reprogramming enhances homology-directed genome editing in zebrafish embryos. Commun. Biol. 2019, 2, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, D.; Nakao, Y. Efficient biallelic knock-in in mouse embryonic stem cells by in vivo-linearization of donor and transient inhibition of DNA Polymerase θ/DNA-PK. bioRxiv 2021. [Google Scholar] [CrossRef]
- Li, G.-L.; Rong, Q.; Wang, H.-Q.; Ruan, X.-F.; Mo, J.-X.; Zhong, C.-L.; Yang, H.-Q.; Li, Z.-C.; Ting, G.; Liu, D.-W. Inhibition of KU70 and KU80 by CRISPR interference, not NgAgo interference, increases the efficiency of homologous recombination in pig fetal fibroblasts. J. Integr. Agric. 2019, 18, 438–448. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Li, L.; Wang, G.; Zhang, W.; Xu, J.; Liang, A. KU70 Inhibition Impairs Both Non-Homologous End Joining and Homologous Recombination DNA Damage Repair Through SHP-1 Induced Dephosphorylation of SIRT1 in T-Cell Acute Lymphoblastic Leukemia (T-ALL) [corrected]. Cell. Physiol. Biochem. 2018, 49, 2111–2123. [Google Scholar] [CrossRef] [PubMed]
- Weterings, E.; Gallegos, A.C.; Dominick, L.N.; Cooke, L.S.; Bartels, T.N.; Vagner, J.; Matsunaga, T.O.; Mahadevan, D. A novel small molecule inhibitor of the DNA repair protein Ku70/80. DNA Repair 2016, 43, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef]
- Hu, Z.; Shi, Z.; Guo, X.; Jiang, B.; Wang, G.; Luo, D.; Chen, Y.; Zhu, Y.S. Ligase IV inhibitor SCR7 enhances gene editing directed by CRISPR-Cas9 and ssODN in human cancer cells. Cell Biosci. 2018, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Zhang, X.; Zhong, C.; Mo, J.; Quan, R.; Yang, J.; Liu, D.; Li, Z.; Yang, H.; Wu, Z. Small molecules enhance CRISPR/Cas9-mediated homology-directed genome editing in primary cells. Sci. Rep. 2017, 7, 8943. [Google Scholar] [CrossRef]
- Singh, P.; Schimenti, J.C.; Bolcun-Filas, E. A mouse geneticist’s practical guide to CRISPR applications. Genetics 2015, 199, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Chen, W.; Zhang, X.; Yu, L.; Dong, W.; Pan, S.; Gao, S.; Huang, X.; Zhang, L. Increasing the efficiency of CRISPR/Cas9-mediated precise genome editing in rats by inhibiting NHEJ and using Cas9 protein. RNA Biol. 2016, 13, 605–612. [Google Scholar] [CrossRef] [Green Version]
- Vartak, S.V.; Swarup, H.A.; Gopalakrishnan, V.; Gopinatha, V.K.; Ropars, V.; Nambiar, M.; John, F.; Kothanahally, S.K.S.; Kumari, R.; Kumari, N. Autocyclized and oxidized forms of SCR 7 induce cancer cell death by inhibiting nonhomologous DNA end joining in a Ligase IV dependent manner. FEBS J. 2018, 285, 3959–3976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Killian, T.; Dickopf, S.; Haas, A.K.; Kirstenpfad, C.; Mayer, K.; Brinkmann, U. Disruption of diphthamide synthesis genes and resulting toxin resistance as a robust technology for quantifying and optimizing CRISPR/Cas9-mediated gene editing. Sci. Rep. 2017, 7, 15480. [Google Scholar] [CrossRef] [Green Version]
- Aslan, Y.; Tadjuidje, E.; Zorn, A.M.; Cha, S.W. High-efficiency non-mosaic CRISPR-mediated knock-in and indel mutation in F0 Xenopus. Development 2017, 144, 2852–2858. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Scavuzzo, M.A.; Chmielowiec, J.; Sharp, R.; Bajic, A.; Borowiak, M. Enrichment of G2/M cell cycle phase in human pluripotent stem cells enhances HDR-mediated gene repair with customizable endonucleases. Sci. Rep. 2016, 6, 21264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wienert, B.; Nguyen, D.N.; Guenther, A.; Feng, S.J.; Locke, M.N.; Wyman, S.K.; Shin, J.; Kazane, K.R.; Gregory, G.L.; Carter, M.A.M.; et al. Timed inhibition of CDC7 increases CRISPR-Cas9 Med. iated templated repair. Nat. Commun. 2020, 11, 2109. [Google Scholar] [CrossRef]
- Song, J.; Yang, D.; Xu, J.; Zhu, T.; Chen, Y.E.; Zhang, J. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat. Commun. 2016, 7, 10548. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Chen, X.; Jin, Y.; Ge, W.; Wang, W.; Kong, L.; Ji, J.; Guo, X.; Huang, J.; Feng, X.H.; et al. Small molecules promote CRISPR-Cpf1-mediated genome editing in human pluripotent stem cells. Nat. Commun. 2018, 9, 1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bothmer, A.; Robbiani, D.F.; Feldhahn, N.; Gazumyan, A.; Nussenzweig, A.; Nussenzweig, M.C. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J. Exp. Med. 2010, 207, 855–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunting, S.F.; Callen, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurihara, T.; Kouyama-Suzuki, E.; Satoga, M.; Li, X.; Badawi, M.; Thiha; Baig, D.N.; Yanagawa, T.; Uemura, T.; Mori, T.; et al. DNA repair protein RAD51 enhances the CRISPR/Cas9-mediated knock-in efficiency in brain neurons. Biochem. Biophys. Res. Commun. 2020, 524, 621–628. [Google Scholar] [CrossRef]
- Lamas-Toranzo, I.; Martinez-Moro, A.; Callaghan, E.O.; Millan-Blanca, G.; Sanchez, J.M.; Lonergan, P.; Bermejo-Alvarez, P. RS-1 enhances CRISPR-mediated targeted knock-in in bovine embryos. Mol. Reprod. Dev. 2020, 87, 542–549. [Google Scholar] [CrossRef]
- Liu, B.; Chen, S.; Rose, A.; Chen, D.; Cao, F.; Zwinderman, M.; Kiemel, D.; Aissi, M.; Dekker, F.J.; Haisma, H.J. Inhibition of histone deacetylase 1 (HDAC1) and HDAC2 enhances CRISPR/Cas9 genome editing. Nucleic Acids Res. 2020, 48, 517–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayama, K.; Igai, K.; Hagihara, Y.; Hashimoto, R.; Hanawa, M.; Sakuma, T.; Tachibana, M.; Sakurai, F.; Yamamoto, T.; Mizuguchi, H. Highly efficient biallelic genome editing of human ES/iPS cells using a CRISPR/Cas9 or TALEN system. Nucleic Acids Res. 2017, 45, 5198–5207. [Google Scholar] [CrossRef] [PubMed]
- Defoort, E.N.; Kim, P.M.; Winn, L.M. Valproic acid increases conservative homologous recombination frequency and reactive oxygen species formation: A potential mechanism for valproic acid-induced neural tube defects. Mol. Pharmacol. 2006, 69, 1304–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schimmel, J.; Kool, H.; van Schendel, R.; Tijsterman, M. Mutational signatures of non-homologous and polymerase theta-mediated end-joining in embryonic stem cells. EMBO J. 2017, 36, 3634–3649. [Google Scholar] [CrossRef] [PubMed]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Gelot, C.; Pantelidou, C.; Li, A.; Yucel, H.; Davis, R.E.; Farkkila, A.; Kochupurakkal, B.; Syed, A.; Shapiro, G.I.; et al. A first-in-class Polymerase Theta Inhibitor selectively targets Homologous-Recombination-Deficient Tumors. Nat. Cancer 2021, 2, 598–610. [Google Scholar] [CrossRef]
- Yu, C.; Liu, Y.; Ma, T.; Liu, K.; Xu, S.; Zhang, Y.; Liu, H.; La Russa, M.; Xie, M.; Ding, S.; et al. Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell 2015, 16, 142–147. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Guan, Y.; Wang, L.; Qiu, Z.; Liu, M.; Chen, Y.; Wu, L.; Li, Y.; Ma, X.; Liu, M.; et al. CRISPR/Cas-mediated genome editing in the rat via direct injection of one-cell embryos. Nat. Protoc. 2014, 9, 2493–2512. [Google Scholar] [CrossRef]
- Ishibashi, R.; Abe, K.; Ido, N.; Kitano, S.; Miyachi, H.; Toyoshima, F. Genome editing with the donor plasmid equipped with synthetic crRNA-target sequence. Sci. Rep. 2020, 10, 14120. [Google Scholar] [CrossRef]
- Zhang, J.P.; Li, X.L.; Li, G.H.; Chen, W.; Arakaki, C.; Botimer, G.D.; Baylink, D.; Zhang, L.; Wen, W.; Fu, Y.W.; et al. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol. 2017, 18, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, F.; Stieger, K. Optimizing the DNA Donor Template for Homology-Directed Repair of Double-Strand Breaks. Mol. Ther. Nucleic Acids 2017, 7, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Kanca, O.; Zirin, J.; Garcia-Marques, J.; Knight, S.M.; Yang-Zhou, D.; Amador, G.; Chung, H.; Zuo, Z.; Ma, L.; He, Y.; et al. An efficient CRISPR-based strategy to insert small and large fragments of DNA using short homology arms. eLife 2019, 8, e51539. [Google Scholar] [CrossRef]
- Nakade, S.; Tsubota, T.; Sakane, Y.; Kume, S.; Sakamoto, N.; Obara, M.; Daimon, T.; Sezutsu, H.; Yamamoto, T.; Sakuma, T.; et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat. Commun. 2014, 5, 5560. [Google Scholar] [CrossRef]
- Park, K.E.; Powell, A.; Sandmaier, S.E.; Kim, C.M.; Mileham, A.; Donovan, D.M.; Telugu, B.P. Targeted gene knock-in by CRISPR/Cas ribonucleoproteins in porcine zygotes. Sci. Rep. 2017, 7, 42458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, H.; Liu, L.; An, K.; Lu, X.; Harrison, M.; Zhao, Y.; Yan, R.; Lu, Z.; Li, S.; Lin, S.; et al. CRISPR/Cas9-mediated precise genome modification by a long ssDNA template in zebrafish. BMC Genom. 2020, 21, 67. [Google Scholar] [CrossRef]
- Veneziano, R.; Shepherd, T.R.; Ratanalert, S.; Bellou, L.; Tao, C.; Bathe, M. In vitro synthesis of gene-length single-stranded DNA. Sci. Rep. 2018, 8, 6548. [Google Scholar] [CrossRef] [Green Version]
- Renaud, J.B.; Boix, C.; Charpentier, M.; De Cian, A.; Cochennec, J.; Duvernois-Berthet, E.; Perrouault, L.; Tesson, L.; Edouard, J.; Thinard, R.; et al. Improved Genome Editing Efficiency and Flexibility Using Modified Oligonucleotides with TALEN and CRISPR-Cas9 Nucleases. Cell Rep. 2016, 14, 2263–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez-Triana, J.A.; Tavhelidse, T.; Thumberger, T.; Thomas, I.; Wittbrodt, B.; Kellner, T.; Anlas, K.; Tsingos, E.; Wittbrodt, J. Efficient single-copy HDR by 5’ modified long dsDNA donors. eLife 2018, 7, e39468. [Google Scholar] [CrossRef]
- Cruz-Becerra, G.; Kadonaga, J.T. Enhancement of homology-directed repair with chromatin donor templates in cells. eLife 2020, 9, e55780. [Google Scholar] [CrossRef]
- Ling, X.; Xie, B.; Gao, X.; Chang, L.; Zheng, W.; Chen, H.; Huang, Y.; Tan, L.; Li, M.; Liu, T. Improving the efficiency of precise genome editing with site-specific Cas9-oligonucleotide conjugates. Sci. Adv. 2020, 6, eaaz0051. [Google Scholar] [CrossRef] [Green Version]
- Lim, D.; Sreekanth, V.; Cox, K.J.; Law, B.K.; Wagner, B.K.; Karp, J.M.; Choudhary, A. Engineering designer beta cells with a CRISPR-Cas9 conjugation platform. Nat. Commun. 2020, 11, 4043. [Google Scholar] [CrossRef] [PubMed]
- Savic, N.; Ringnalda, F.C.; Lindsay, H.; Berk, C.; Bargsten, K.; Li, Y.; Neri, D.; Robinson, M.D.; Ciaudo, C.; Hall, J.; et al. Covalent linkage of the DNA repair template to the CRISPR-Cas9 nuclease enhances homology-directed repair. eLife 2018, 7, e33761. [Google Scholar] [CrossRef]
- Lee, K.; Mackley, V.A.; Rao, A.; Chong, A.T.; Dewitt, M.A.; Corn, J.E.; Murthy, N. Synthetically modified guide RNA and donor DNA are a versatile platform for CRISPR-Cas9 engineering. eLife 2017, 6, e25312. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Topkar, V.V.; Nguyen, N.T.; Zheng, Z.; Gonzales, A.P.; Li, Z.; Peterson, R.T.; Yeh, J.R.; et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 2015, 523, 481–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Walton, R.T.; Christie, K.A.; Whittaker, M.N.; Kleinstiver, B.P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 2020, 368, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Bolukbasi, M.F.; Gupta, A.; Oikemus, S.; Derr, A.G.; Garber, M.; Brodsky, M.H.; Zhu, L.J.; Wolfe, S.A. DNA-binding-domain fusions enhance the targeting range and precision of Cas9. Nat. Methods 2015, 12, 1150–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [Green Version]
- Muller, M.; Lee, C.M.; Gasiunas, G.; Davis, T.H.; Cradick, T.J.; Siksnys, V.; Bao, G.; Cathomen, T.; Mussolino, C. Streptococcus thermophilus CRISPR-Cas9 Systems Enable Specific Editing of the Human Genome. Mol. Ther. 2016, 24, 636–644. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.M.; Cradick, T.J.; Bao, G. The Neisseria meningitidis CRISPR-Cas9 System Enables Specific Genome Editing in Mammalian Cells. Mol. Ther. 2016, 24, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, P.; Jakimo, N.; Lee, J.; Amrani, N.; Rodriguez, T.; Koseki, S.R.T.; Tysinger, E.; Qing, R.; Hao, S.; Sontheimer, E.J.; et al. An engineered ScCas9 with broad PAM range and high specificity and activity. Nat. Biotechnol. 2020, 38, 1154–1158. [Google Scholar] [CrossRef]
- Pausch, P.; Al-Shayeb, B.; Bisom-Rapp, E.; Tsuchida, C.A.; Li, Z.; Cress, B.F.; Knott, G.J.; Jacobsen, S.E.; Banfield, J.F.; Doudna, J.A. CRISPR-CasPhi from huge phages is a hypercompact genome editor. Science 2020, 369, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, S.Q.; Wyvekens, N.; Khayter, C.; Foden, J.A.; Thapar, V.; Reyon, D.; Goodwin, M.J.; Aryee, M.J.; Joung, J.K. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat. Biotechnol. 2014, 32, 569–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilinger, J.P.; Thompson, D.B.; Liu, D.R. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat. Biotechnol. 2014, 32, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Kiani, S.; Chavez, A.; Tuttle, M.; Hall, R.N.; Chari, R.; Ter-Ovanesyan, D.; Qian, J.; Pruitt, B.W.; Beal, J.; Vora, S.; et al. Cas9 gRNA engineering for genome editing, activation and repression. Nat. Methods 2015, 12, 1051–1054. [Google Scholar] [CrossRef] [Green Version]
- Rose, J.C.; Popp, N.A.; Richardson, C.D.; Stephany, J.J.; Mathieu, J.; Wei, C.T.; Corn, J.E.; Maly, D.J.; Fowler, D.M. Suppression of unwanted CRISPR-Cas9 editing by co-administration of catalytically inactivating truncated guide RNAs. Nat. Commun. 2020, 11, 2697. [Google Scholar] [CrossRef]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [Green Version]
- Kulcsar, P.I.; Talas, A.; Huszar, K.; Ligeti, Z.; Toth, E.; Weinhardt, N.; Fodor, E.; Welker, E. Crossing enhanced and high fidelity SpCas9 nucleases to optimize specificity and cleavage. Genome Biol. 2017, 18, 190. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.S.; Dagdas, Y.S.; Kleinstiver, B.P.; Welch, M.M.; Sousa, A.A.; Harrington, L.B.; Sternberg, S.H.; Joung, J.K.; Yildiz, A.; Doudna, J.A. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 2017, 550, 407–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casini, A.; Olivieri, M.; Petris, G.; Montagna, C.; Reginato, G.; Maule, G.; Lorenzin, F.; Prandi, D.; Romanel, A.; Demichelis, F.; et al. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat. Biotechnol. 2018, 36, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Jeong, E.; Lee, J.; Jung, M.; Shin, E.; Kim, Y.H.; Lee, K.; Jung, I.; Kim, D.; Kim, S.; et al. Directed evolution of CRISPR-Cas9 to increase its specificity. Nat. Commun. 2018, 9, 3048. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.; Kim, H.K.; Lee, S.; Seo, J.H.; Choi, J.W.; Park, J.; Min, S.; Yoon, S.; Cho, S.R.; Kim, H.H. Prediction of the sequence-specific cleavage activity of Cas9 variants. Nat. Biotechnol. 2020, 38, 1328–1336. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Ren, C.; Liu, Z.; Bai, Y.; Chen, Z.; Wei, Z.; Wang, X.; Zhang, Z.; Xu, K. Enhancing CRISPR/Cas9-mediated homology-directed repair in mammalian cells by expressing Saccharomyces cerevisiae Rad52. Int. J. Biochem. Cell Biol. 2017, 92, 43–52. [Google Scholar] [CrossRef]
- Charpentier, M.; Khedher, A.H.Y.; Menoret, S.; Brion, A.; Lamribet, K.; Dardillac, E.; Boix, C.; Perrouault, L.; Tesson, L.; Geny, S.; et al. CtIP fusion to Cas9 enhances transgene integration by homology-dependent repair. Nat. Commun. 2018, 9, 1133. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Seebeck, T.; Feng, Y.; Jiang, Y.; Davis, G.D.; Chen, F. Improving CRISPR-Cas9 Genome Editing Efficiency by Fusion with Chromatin-Modulating Peptides. CRISPR J. 2019, 2, 51–63. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schellenberger, V.; Wang, C.W.; Geething, N.C.; Spink, B.J.; Campbell, A.; To, W.; Scholle, M.D.; Yin, Y.; Yao, Y.; Bogin, O.; et al. A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner. Nat. Biotechnol. 2009, 27, 1186–1190. [Google Scholar] [CrossRef]
- Mol, C.D.; Arvai, A.S.; Sanderson, R.J.; Slupphaug, G.; Kavli, B.; Krokan, H.E.; Mosbaugh, D.W.; Tainer, J.A. Crystal structure of human uracil-DNA glycosylase in complex with a protein inhibitor: Protein mimicry of DNA. Cell 1995, 82, 701–708. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Leete, T.C.; Born, D.A.; Young, L.; Barrera, L.A.; Lee, S.J.; Rees, H.A.; Ciaramella, G.; Gaudelli, N.M. Cytosine base editors with minimized unguided DNA and RNA off-target events and high on-target activity. Nat. Commun. 2020, 11, 2052. [Google Scholar] [CrossRef]
- Komor, A.C.; Zhao, K.T.; Packer, M.S.; Gaudelli, N.M.; Waterbury, A.L.; Koblan, L.W.; Kim, Y.B.; Badran, A.H.; Liu, D.R. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv. 2017, 3, eaao4774. [Google Scholar] [CrossRef] [Green Version]
- Doman, J.L.; Raguram, A.; Newby, G.A.; Liu, D.R. Evaluation and minimization of Cas9-independent off-target DNA editing by cytosine base editors. Nat. Biotechnol. 2020, 38, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Kantor, A.; McClements, M.E.; MacLaren, R.E. CRISPR-Cas9 DNA Base-Editing and Prime-Editing. Int. J. Mol. Sci. 2020, 21, 6240. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Richter, M.F.; Zhao, K.T.; Eton, E.; Lapinaite, A.; Newby, G.A.; Thuronyi, B.W.; Wilson, C.; Koblan, L.W.; Zeng, J.; Bauer, D.E.; et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020, 38, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Lam, D.K.; Rees, H.A.; Sola-Esteves, N.M.; Barrera, L.A.; Born, D.A.; Edwards, A.; Gehrke, J.M.; Lee, S.J.; Liquori, A.J.; et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 2020, 38, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Nguyen Tran, M.T.; Mohd Khalid, M.K.N.; Wang, Q.; Walker, J.K.R.; Lidgerwood, G.E.; Dilworth, K.L.; Lisowski, L.; Pebay, A.; Hewitt, A.W. Engineering domain-inlaid SaCas9 adenine base editors with reduced RNA off-targets and increased on-target DNA editing. Nat. Commun. 2020, 11, 4871. [Google Scholar] [CrossRef]
- Rees, H.A.; Komor, A.C.; Yeh, W.H.; Caetano-Lopes, J.; Warman, M.; Edge, A.S.B.; Liu, D.R. Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat. Commun. 2017, 8, 15790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.B.; Komor, A.C.; Levy, J.M.; Packer, M.S.; Zhao, K.T.; Liu, D.R. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017, 35, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Koblan, L.W.; Doman, J.L.; Wilson, C.; Levy, J.M.; Tay, T.; Newby, G.A.; Maianti, J.P.; Raguram, A.; Liu, D.R. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018, 36, 843–846. [Google Scholar] [CrossRef]
- Lee, H.K.; Willi, M.; Miller, S.M.; Kim, S.; Liu, C.; Liu, D.R.; Hennighausen, L. Targeting fidelity of adenine and cytosine base editors in mouse embryos. Nat. Commun. 2018, 9, 4804. [Google Scholar] [CrossRef]
- Zafra, M.P.; Schatoff, E.M.; Katti, A.; Foronda, M.; Breinig, M.; Schweitzer, A.Y.; Simon, A.; Han, T.; Goswami, S.; Montgomery, E.; et al. Optimized base editors enable efficient editing in cells, organoids and mice. Nat. Biotechnol. 2018, 36, 888–893. [Google Scholar] [CrossRef]
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z.; et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 2018, 556, 57–63. [Google Scholar] [CrossRef]
- Li, X.; Wang, Y.; Liu, Y.; Yang, B.; Wang, X.; Wei, J.; Lu, Z.; Zhang, Y.; Wu, J.; Huang, X.; et al. Base editing with a Cpf1-cytidine deaminase fusion. Nat. Biotechnol. 2018, 36, 324–327. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, X.; Wang, L.; Yin, S.; Zhu, B.; Xie, L.; Duan, Q.; Hu, H.; Zheng, R.; Wei, Y.; et al. Increasing targeting scope of adenosine base editors in mouse and rat embryos through fusion of TadA deaminase with Cas9 variants. Protein Cell 2018, 9, 814–819. [Google Scholar] [CrossRef] [Green Version]
- Kurt, I.C.; Zhou, R.; Iyer, S.; Garcia, S.P.; Miller, B.R.; Langner, L.M.; Grunewald, J.; Joung, J.K. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat. Biotechnol. 2021, 39, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, L.; Tao, R.; Liu, N.; Long, J.; Qin, F.; Tang, W.; Yang, Y.; Chen, Q.; Yao, S. sgBE: A structure-guided design of sgRNA architecture specifies base editing window and enables simultaneous conversion of cytosine and adenosine. Genome Biol. 2020, 21, 222. [Google Scholar] [CrossRef]
- Sakata, R.C.; Ishiguro, S.; Mori, H.; Tanaka, M.; Tatsuno, K.; Ueda, H.; Yamamoto, S.; Seki, M.; Masuyama, N.; Nishida, K.; et al. Base editors for simultaneous introduction of C-to-T and A-to-G mutations. Nat. Biotechnol. 2020, 38, 865–869. [Google Scholar] [CrossRef]
- Xie, J.; Huang, X.; Wang, X.; Gou, S.; Liang, Y.; Chen, F.; Li, N.; Ouyang, Z.; Zhang, Q.; Ge, W.; et al. ACBE, a new base editor for simultaneous C-to-T and A-to-G substitutions in mammalian systems. BMC Biol. 2020, 18, 131. [Google Scholar] [CrossRef]
- Grunewald, J.; Zhou, R.; Garcia, S.P.; Iyer, S.; Lareau, C.A.; Aryee, M.J.; Joung, J.K. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 2019, 569, 433–437. [Google Scholar] [CrossRef] [PubMed]
- McCann, J.L.; Salamango, D.J.; Law, E.K.; Brown, W.L.; Harris, R.S. MagnEdit-interacting factors that recruit DNA-editing enzymes to single base targets. Life Sci. Alliance 2020, 3, e201900606. [Google Scholar] [CrossRef]
- Kuscu, C.; Parlak, M.; Tufan, T.; Yang, J.; Szlachta, K.; Wei, X.; Mammadov, R.; Adli, M. CRISPR-STOP: Gene silencing through base-editing-induced nonsense mutations. Nat. Methods 2017, 14, 710–712. [Google Scholar] [CrossRef]
- Billon, P.; Bryant, E.E.; Joseph, S.A.; Nambiar, T.S.; Hayward, S.B.; Rothstein, R.; Ciccia, A. CRISPR-Mediated Base Editing Enables Efficient Disruption of Eukaryotic Genes through Induction of STOP Codons. Mol. Cell 2017, 67, 1068–1079.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.P.; Zhao, K.T.; Miller, S.M.; Gaudelli, N.M.; Oakes, B.L.; Fellmann, C.; Savage, D.F.; Liu, D.R. Circularly permuted and PAM-modified Cas9 variants broaden the targeting scope of base editors. Nat. Biotechnol. 2019, 37, 626–631. [Google Scholar] [CrossRef]
- Porto, E.M.; Komor, A.C.; Slaymaker, I.M.; Yeo, G.W. Base editing: Advances and therapeutic opportunities. Nat. Rev. Drug Discov. 2020, 19, 839–859. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, J.; Yin, W.; Zhang, Z.; Song, Y.; Chang, X. Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nat. Methods 2016, 13, 1029–1035. [Google Scholar] [CrossRef]
- Nachman, M.W.; Crowell, S.L. Estimate of the mutation rate per nucleotide in humans. Genetics 2000, 156, 297–304. [Google Scholar] [CrossRef]
- Di Noia, J.M.; Neuberger, M.S. Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem. 2007, 76, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Peled, J.U.; Kuang, F.L.; Iglesias-Ussel, M.D.; Roa, S.; Kalis, S.L.; Goodman, M.F.; Scharff, M.D. The biochemistry of somatic hypermutation. Annu. Rev. Immunol. 2008, 26, 481–511. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Nagaoka, H.; Shinkura, R.; Begum, N.; Muramatsu, M.; Nakata, M.; Honjo, T. Activation-induced cytidine deaminase shuttles between nucleus and cytoplasm like apolipoprotein B mRNA editing catalytic polypeptide 1. Proc. Natl. Acad. Sci. USA 2004, 101, 1975–1980. [Google Scholar] [CrossRef] [Green Version]
- Hess, G.T.; Fresard, L.; Han, K.; Lee, C.H.; Li, A.; Cimprich, K.A.; Montgomery, S.B.; Bassik, M.C. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat. Methods 2016, 13, 1036–1042. [Google Scholar] [CrossRef] [Green Version]
- Devilder, M.C.; Moyon, M.; Gautreau-Rolland, L.; Navet, B.; Perroteau, J.; Delbos, F.; Gesnel, M.C.; Breathnach, R.; Saulquin, X. Ex vivo evolution of human antibodies by CRISPR-X: From a naive B cell repertoire to affinity matured antibodies. BMC Biotechnol. 2019, 19, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.Y.; Moon, S.B.; Ko, J.H.; Kim, Y.S.; Kim, D. Unbiased investigation of specificities of prime editing systems in human cells. Nucleic Acids Res. 2020, 48, 10576–10589. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Denes, C.E.; Cole, A.J.; Aksoy, Y.A.; Li, G.; Neely, G.G.; Hesselson, D. Approaches to Enhance Precise CRISPR/Cas9-Mediated Genome Editing. Int. J. Mol. Sci. 2021, 22, 8571. https://doi.org/10.3390/ijms22168571
Denes CE, Cole AJ, Aksoy YA, Li G, Neely GG, Hesselson D. Approaches to Enhance Precise CRISPR/Cas9-Mediated Genome Editing. International Journal of Molecular Sciences. 2021; 22(16):8571. https://doi.org/10.3390/ijms22168571
Chicago/Turabian StyleDenes, Christopher E., Alexander J. Cole, Yagiz Alp Aksoy, Geng Li, Graham Gregory Neely, and Daniel Hesselson. 2021. "Approaches to Enhance Precise CRISPR/Cas9-Mediated Genome Editing" International Journal of Molecular Sciences 22, no. 16: 8571. https://doi.org/10.3390/ijms22168571
APA StyleDenes, C. E., Cole, A. J., Aksoy, Y. A., Li, G., Neely, G. G., & Hesselson, D. (2021). Approaches to Enhance Precise CRISPR/Cas9-Mediated Genome Editing. International Journal of Molecular Sciences, 22(16), 8571. https://doi.org/10.3390/ijms22168571