
Glycosphingolipids with Very Long-Chain Fatty Acids Accumulate in Fibroblasts from Adrenoleukodystrophy Patients


Abstract
:1. Introduction
2. Results
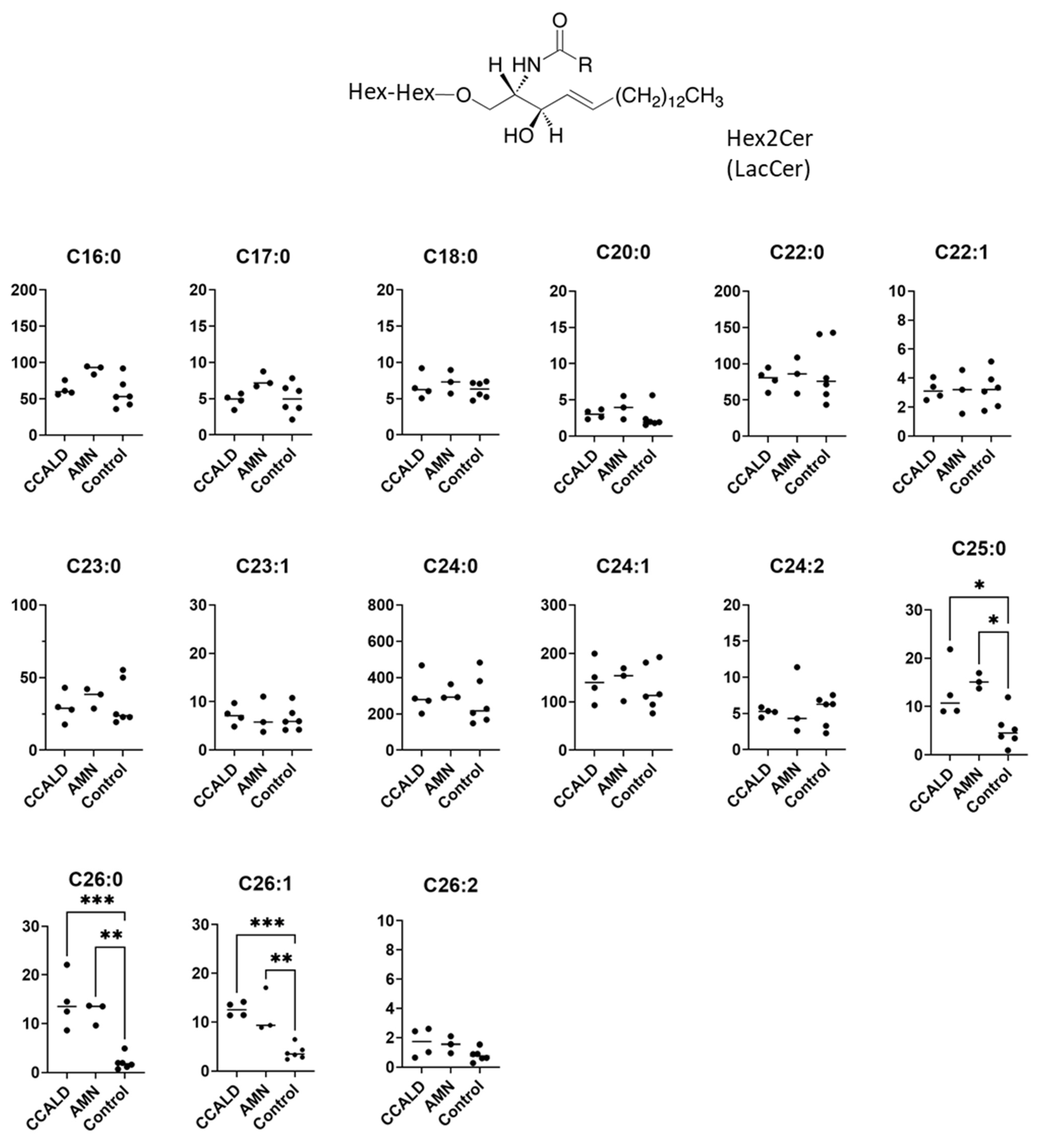
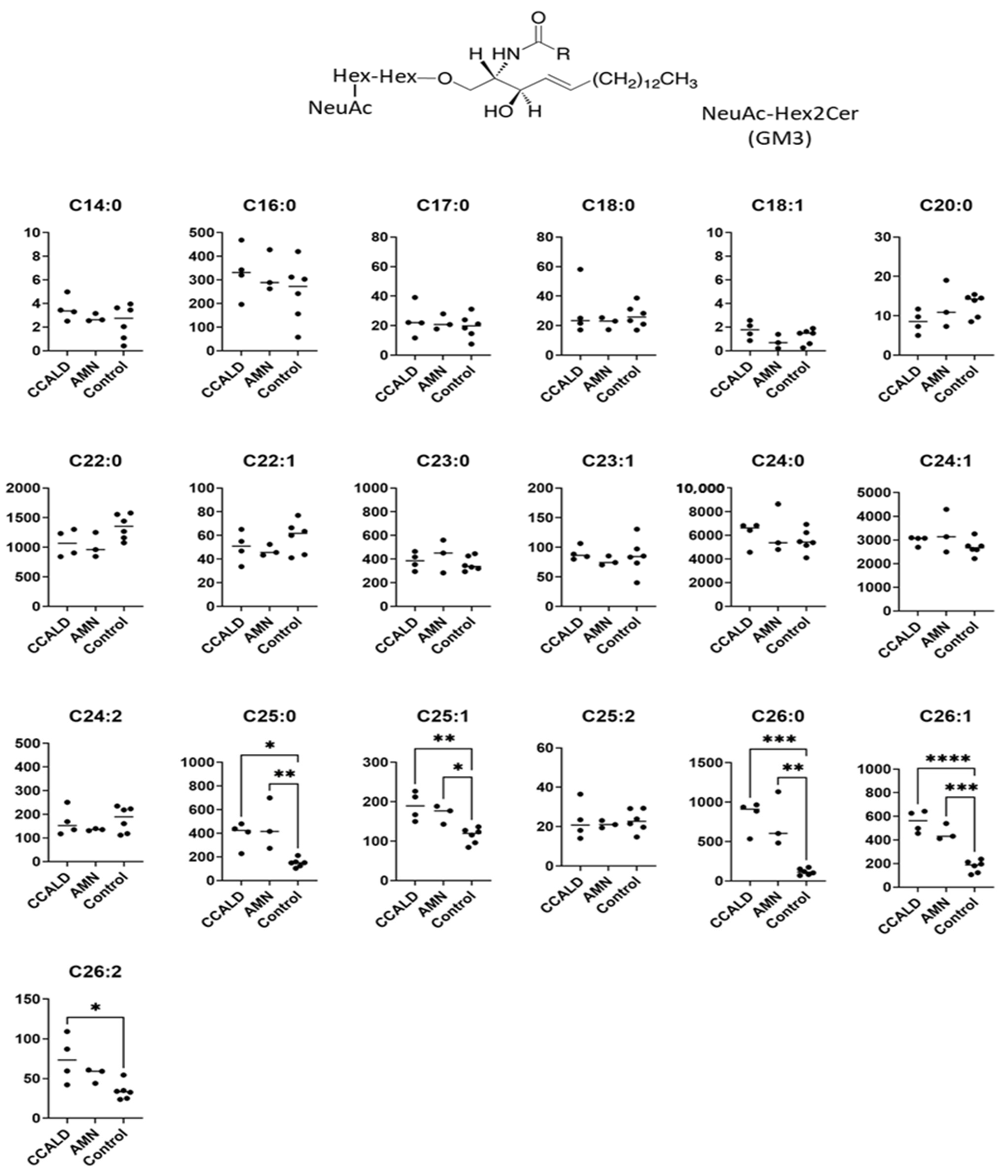
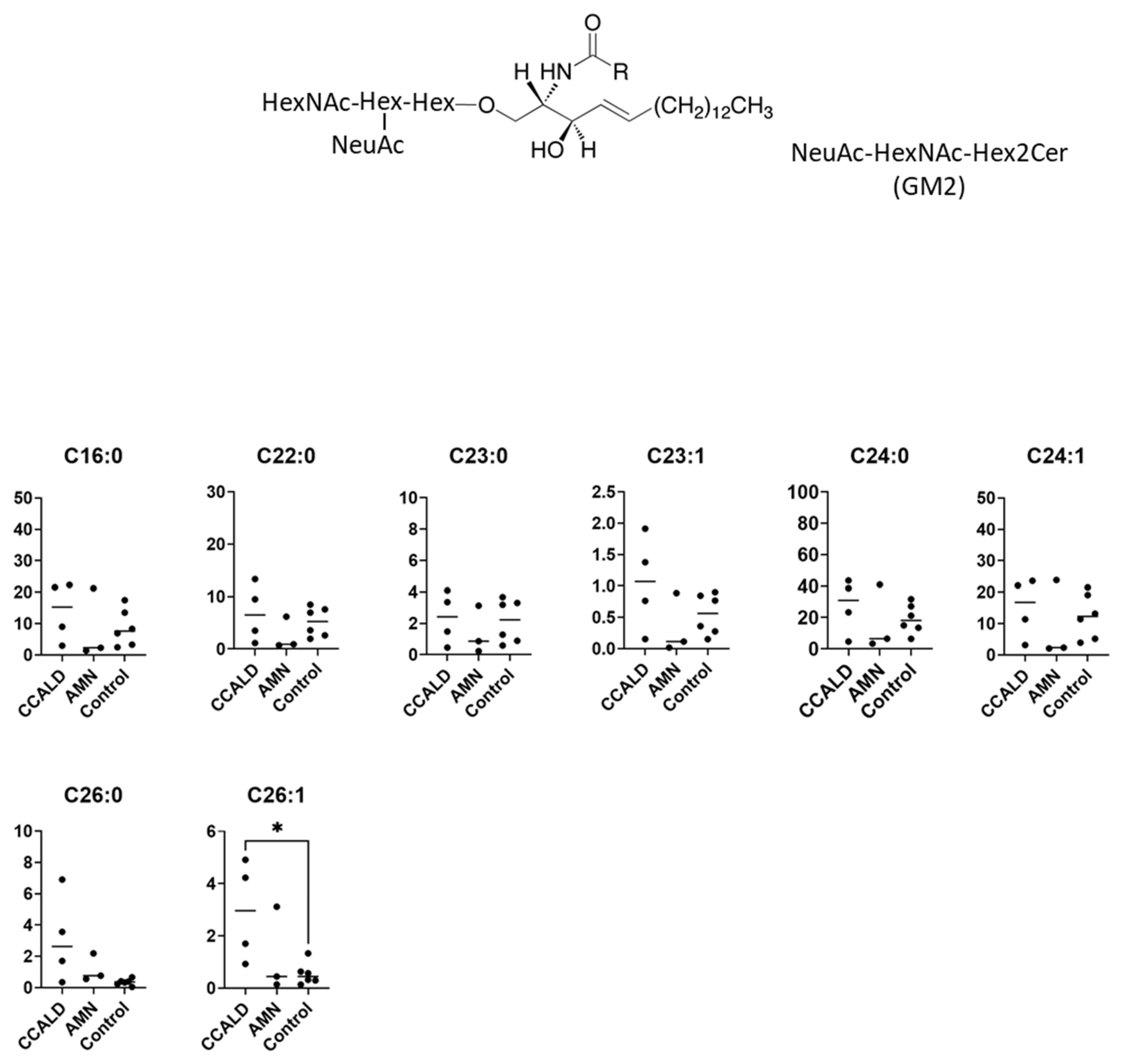
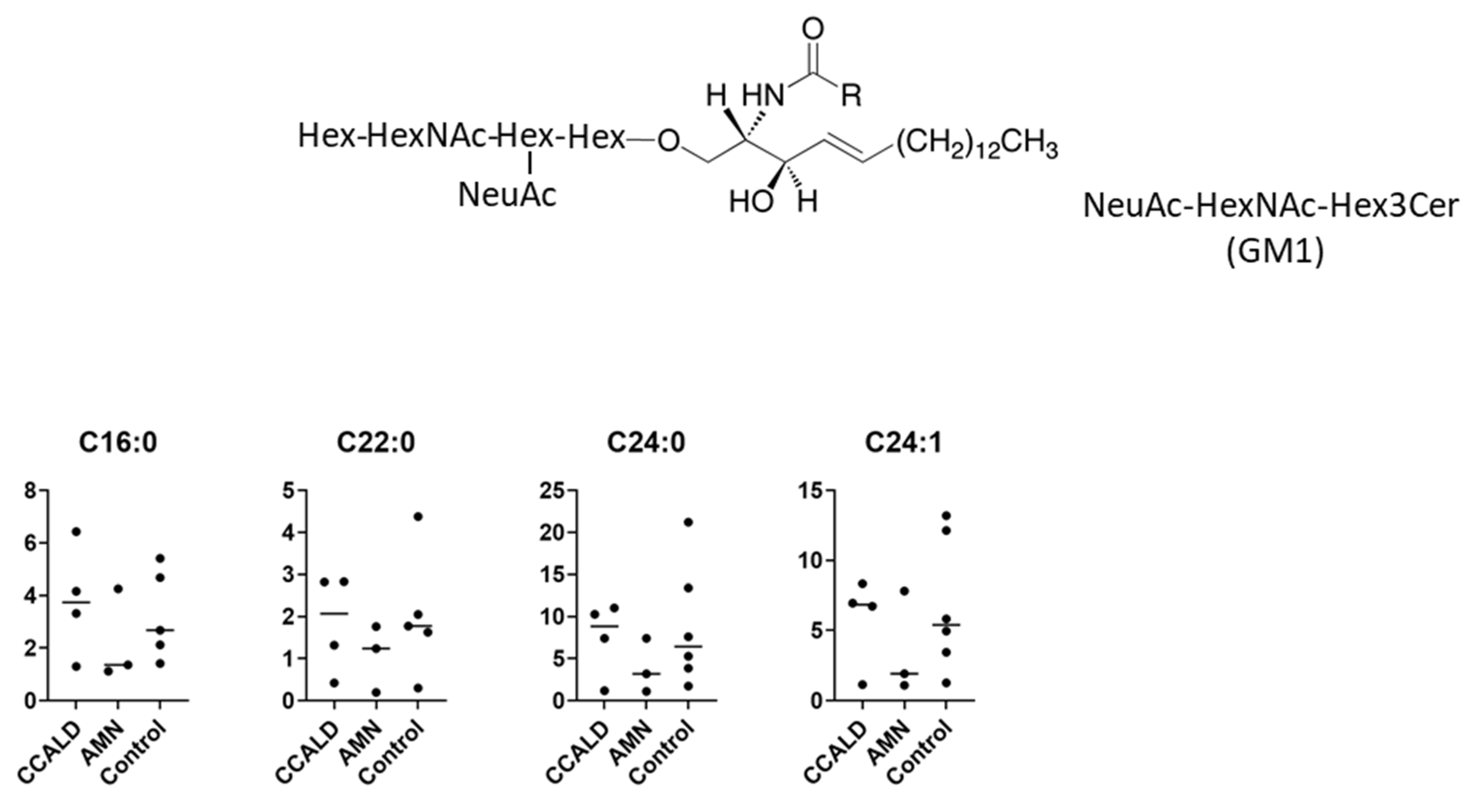
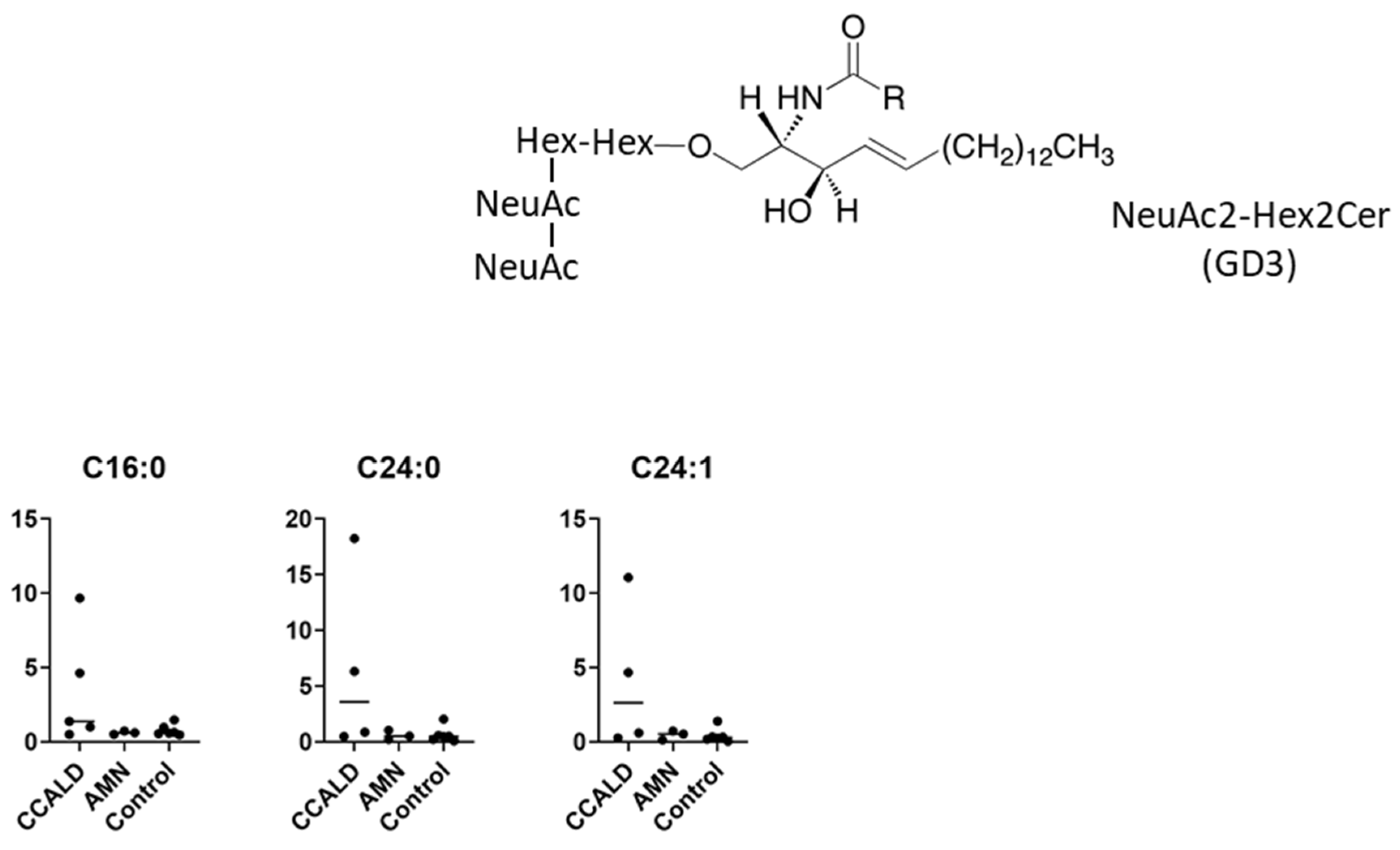
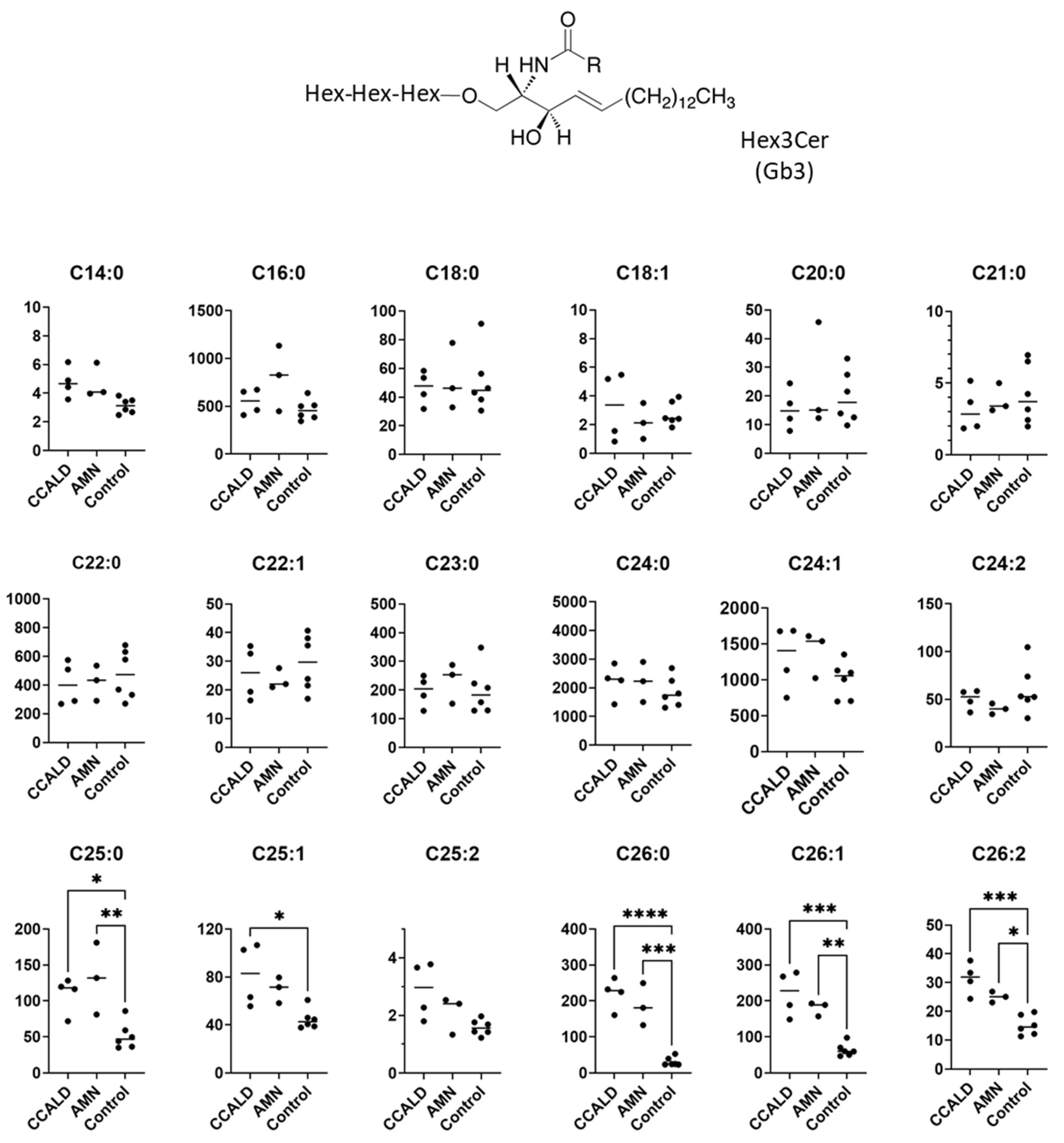
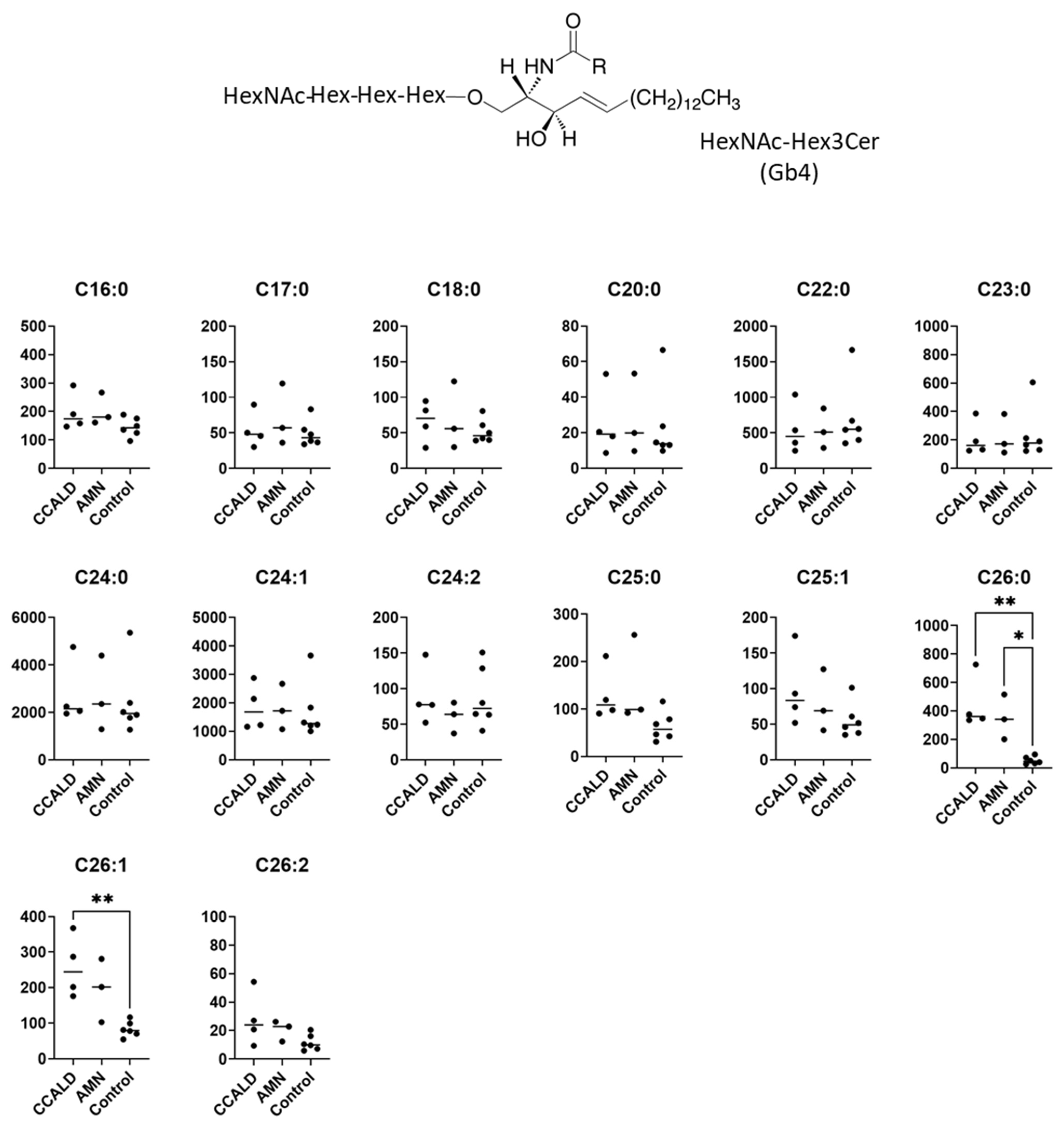
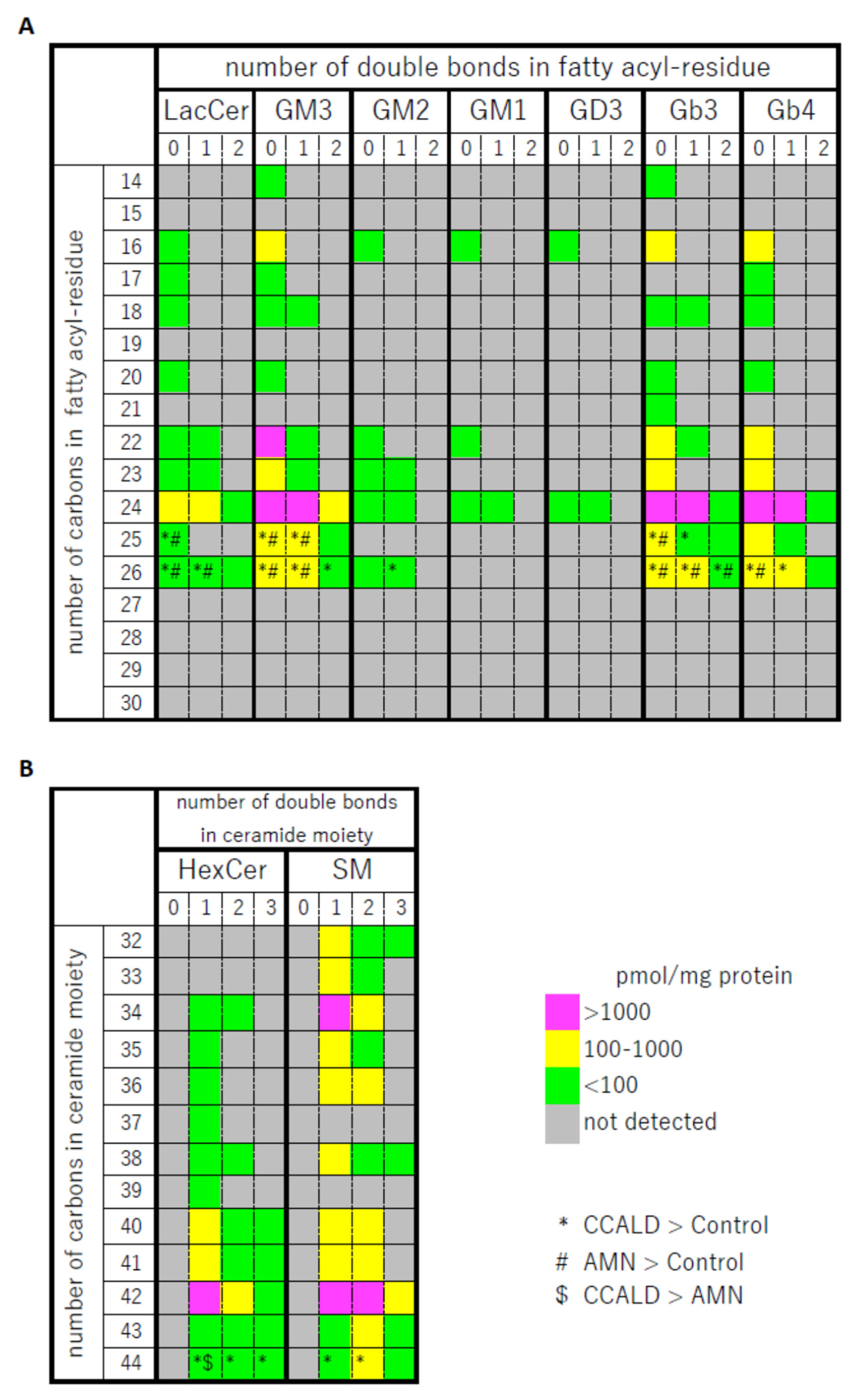
2.1. Quantitative Analysis of GSLs in Fibroblasts from X-ALD Patients
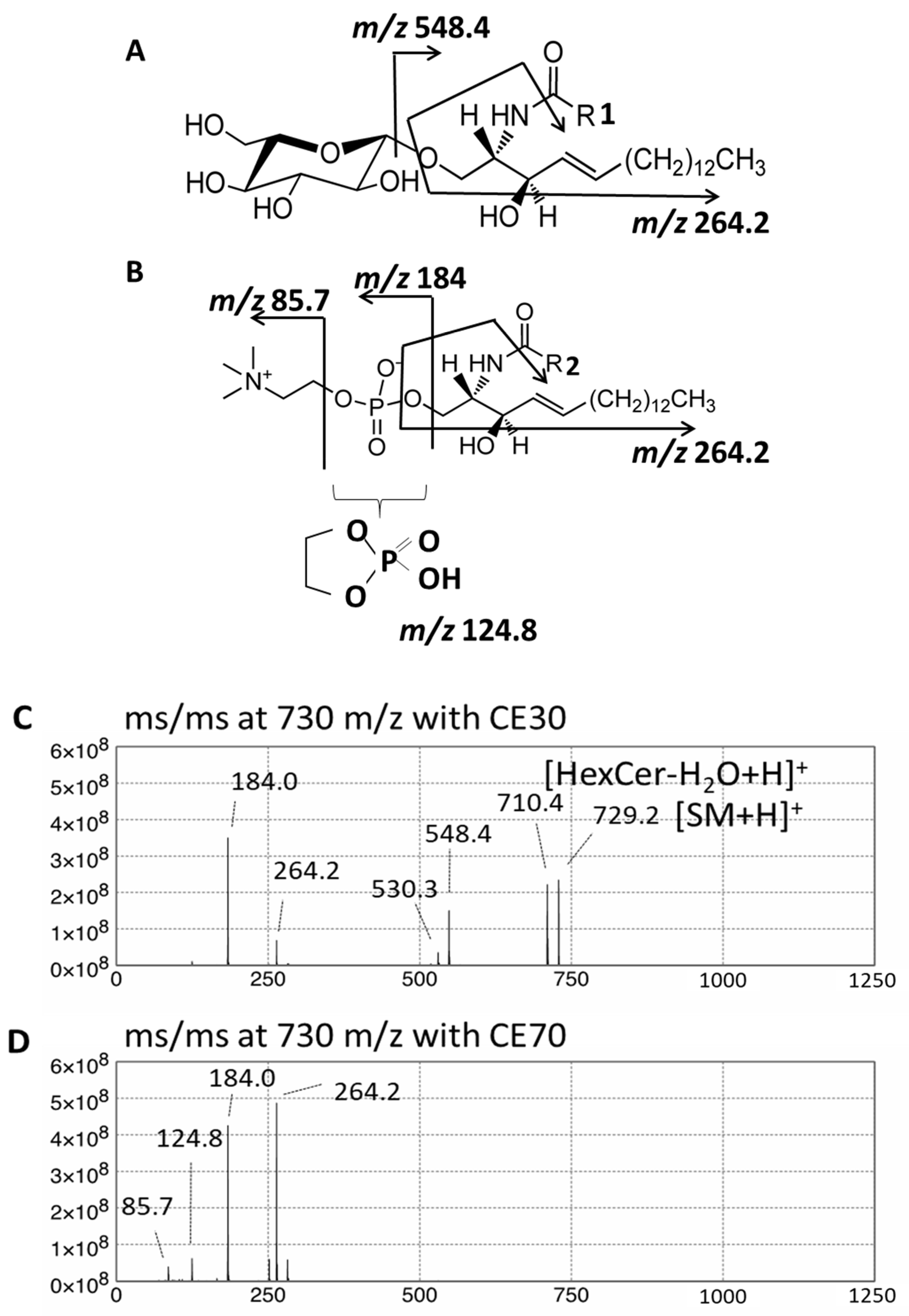
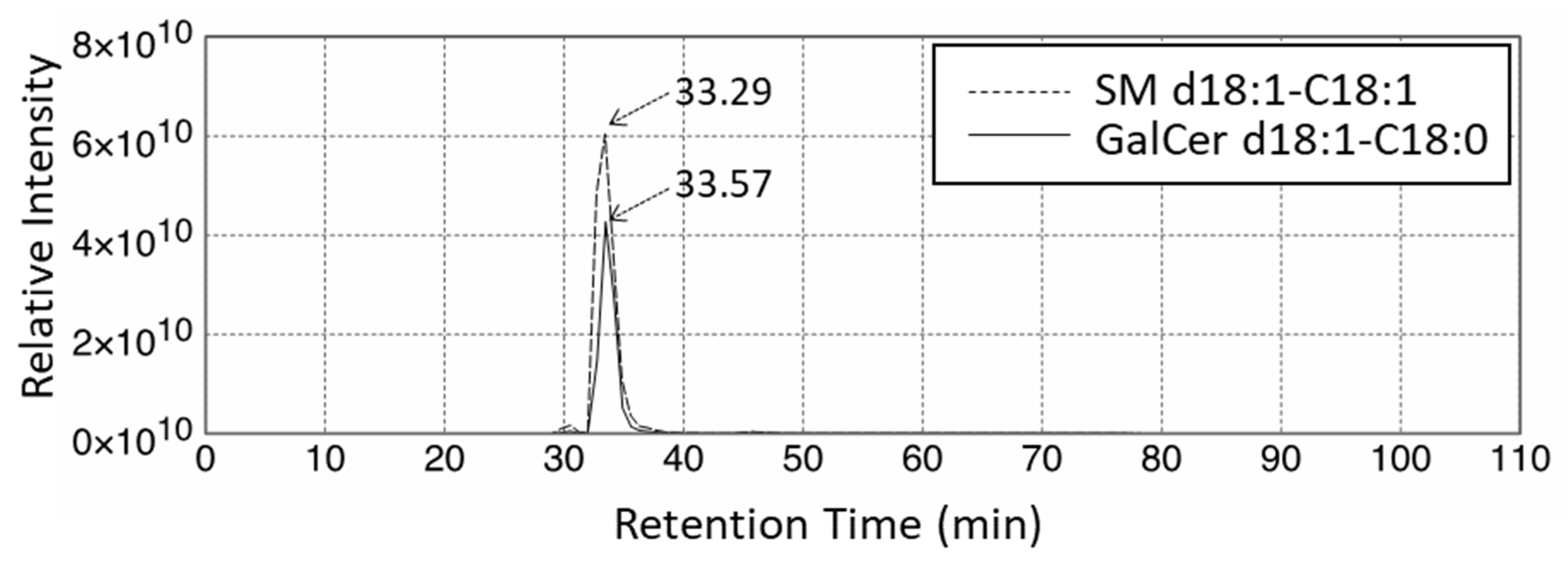
2.2. Quantitative Analysis of Hexosylceramide (HexCer) and Sphingomyelin (SM) in Fibroblasts from X-ALD Patients
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines and Cell Culture
4.3. Sample Preparation
4.4. LC–MS/MS Analysis
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shimozawa, N.; Imanaka, T. Peroxisomes: Biogenesis, Function, and Role in Human Disease; Springer: London, UK, 2019; pp. 107–136. [Google Scholar]
- Shimozawa, N.; Honda, A.; Kajiwara, N.; Kozawa, S.; Nagase, T.; Takemoto, Y.; Suzuki, Y. X-linked adrenoleukodystrophy: Diagnostic and follow-up system in Japan. J. Hum. Genet. 2011, 56, 106–109. [Google Scholar] [CrossRef]
- Suzuki, Y.; Takemoto, Y.; Shimozawa, N.; Imanaka, T.; Kato, S.; Furuya, H.; Kaga, M.; Kato, K.; Hashimoto, N.; Onodera, O.; et al. Natural history of X-linked adrenoleukodystrophy in Japan. Brain Dev. 2005, 27, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Budka, H.; Sluga, E.; Heiss, W.D. Spastic paraplegia associated with Addison’s disease: Adult variant of adreno-leukodystrophy. J. Neurol. 1976, 213, 237–250. [Google Scholar] [CrossRef]
- van Geel, B.M.; Bezman, L.; Loes, D.J.; Moser, H.W.; Raymond, G.V. Evolution of phenotypes in adult male patients with X-linked adrenoleukodystrophy. Ann. Neurol. 2001, 49, 186–194. [Google Scholar] [CrossRef]
- Wanders, R.; Aubourg, P. Chapter 163—Peroxisomal disorders. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2013; pp. 1593–1609. [Google Scholar]
- Kemper, A.; Brosco, J.; Comeau, A.M.; Green, N.S.; Grosse, S.; Kwon, J.; Prosser, L.A.; Lam, K.K. Newborn Screening for X-Linked Adrenoleukodystrophy (X-ALD): A Systematic Review of Evidence Final Report. Maternal and Child Health Bureau 2015. Available online: https://www.hrsa.gov/sites/default/files/advisorycommittees/mchbadvisory/heritabledisorders/nominatecondition/reviews/alddecisionletter.pdf (accessed on 1 August 2021).
- Berger, J.; Wiesinger, C.; Eichler, F. The genetic landscape of X-linked adrenoleukodystrophy: Inheritance, mutations, modifier genes, and diagnosis. Appl. Clin. Genet. 2015, 8, 109–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, H.W.; Raymond, G.V.; Dubey, P. Adrenoleukodystrophy. JAMA 2005, 294, 3131–3134. [Google Scholar] [CrossRef]
- Pan, H.; Xiong, H.; Wu, Y.; Zhang, Y.; Bao, X.-H.; Jiang, Y.-W.; Wu, X.-R. ABCD1 gene mutations in chinese patients with X-linked adrenoleukodystrophy. Pediatr. Neurol. 2005, 33, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Raymond, G.V.; Moser, A.B.; Fatemi, A. X-linked adrenoleukodystrophy. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2018. [Google Scholar]
- University of Washington. GeneReviews; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Takano, H.; Koike, R.; Onodera, O.; Sasaki, R.; Tsuji, S. Mutational analysis and genotype-phenotype correlation of 29 unrelated Japanese patients with X-linked adrenoleukodystrophy. Arch. Neurol. 1999, 56, 295–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elrington, G.M.; Bateman, D.; Jeffrey, M.J.; Lawton, N.F. Adrenoleukodystrophy: Heterogeneity in two brothers. J. Neurol. Neurosurg. Psychiatry 1989, 52, 310–313. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.; Dompas, B.; Ceuterick, C.; Jacobs, K. Adrenomyeloneuropathy and adrenoleukodystrophy in two brothers. Eur. Neurol. 1980, 19, 281–287. [Google Scholar] [CrossRef]
- O’Neill, B.P.; Mannion, L.C.; Feringa, E.R. The adrenoleukomyeloneuropathy complex: Expression in four generations. Neurology 1981, 31, 151. [Google Scholar] [CrossRef]
- Moser, H.W. X-linked adrenoleukodystrophy. In The Metabolic and Molecular Bases of Inherited Disease; McGraw Hill: New York, NY, USA, 2002; pp. 3257–3301. [Google Scholar]
- Abe, Y.; Honsho, M.; Nakanishi, H.; Taguchi, R.; Fujiki, Y. Very-long-chain polyunsaturated fatty acids accumulate in phosphatidylcholine of fibroblasts from patients with Zellweger syndrome and acyl-CoA oxidase1 deficiency. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-K.; Long, N.P.; Jung, J.; Kim, T.J.; Na, E.; Kang, Y.P.; Kwon, S.W.; Jang, J. Integrative lipidomic and transcriptomic analysis of X-linked adrenoleukodystrophy reveals distinct lipidome signatures between adrenomyeloneuropathy and childhood cerebral adrenoleukodystrophy. Biochem. Biophys. Res. Commun. 2018, 508, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Herzog, K.; Pras-Raves, M.L.; Ferdinandusse, S.; Vervaart, M.A.T.; Luyf, A.C.M.; Van Kampen, A.H.C.; Wanders, R.J.A.; Waterham, H.R.; Vaz, F.M. Functional characterisation of peroxisomal β-oxidation disorders in fibroblasts using lipidomics. J. Inherit. Metab. Dis. 2017, 41, 479–487. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, M.; Schaumburg, H.H.; Powers, J.; Kishimoto, Y.; Koilodny, E.; Suzuki, K. Fatty acid abnormality in adrenoleukodystrophy. J. Neurochem. 1976, 26, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, M.; Belchjs, D.; Suzuki, K. Brain gangliosides in adrenoleukodystrophy. J. Neurochem. 1976, 27, 327–328. [Google Scholar] [CrossRef] [PubMed]
- Miyatake, T.; Ariga, T.; Atsumi, T.; Komiya, Y. Biochemical Study on Myelin in Adrenoleukodystrophy; Springer: Boston, MA, USA, 1978; pp. 585–599. [Google Scholar] [CrossRef]
- Tsuji, S.; Suzuki, M.; Ariga, T.; Sekine, M.; Kuriyama, M.; Miyatake, T. Abnormality of long-chain fatty acids in erythrocyte membrane sphingomyelin from patients with adrenoleukodystrophy. J. Neurochem. 1981, 36, 1046–1049. [Google Scholar] [CrossRef]
- Schaumburg, H.H.; Powers, J.M.; Raine, C.; Spencer, P.; Griffin, J.W.; Prineas, J.W.; Boehme, D.M. Adrenomyeloneuropathy: A probable variant of adrenoleukodystrophy: II. General pathologic, neuropathologic, and biochemical aspects. Neurology 1977, 27, 1114. [Google Scholar] [CrossRef]
- Probst, A.; Ulrich, J.; Heitz, P.U.; Herschkowitz, N. Adrenomyeloneuropathy. Acta Neuropathol. 1980, 49, 105–115. [Google Scholar] [CrossRef]
- Askanas, V.; McLaughlin, J.; Engel, W.K.; Adornato, B.T. Abnormalities in cultured muscle and peripheral nerve of a patient with adrenomyeloneuropathy. N. Engl. J. Med. 1979, 301, 588–590. [Google Scholar] [CrossRef]
- Calvano, C.D.; Ventura, G.; Sardanelli, A.M.; Losito, I.; Palmisano, F.; Cataldi, T.R. Identification of neutral and acidic glycosphingolipids in the human dermal fibroblasts. Anal. Biochem. 2019, 581, 113348. [Google Scholar] [CrossRef]
- Dawson, G.; Matalon, R.; Dorfman, A. Glycosphingolipids in cultured human skin fibroblasts. J. Biol. Chem. 1972, 247, 5944–5950. [Google Scholar] [CrossRef]
- Sawada, R.; Hotta, H.; Chung, Y.-S.; Sowa, M.; Tai, T.; Yano, I. Globotriaosyl ceramide and globoside as major glycolipid components of fibroblasts in scirrhous gastric carcinoma tissues. Jpn. J. Cancer Res. 1998, 89, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Fujiwaki, T.; Yamaguchi, S.; Sukegawa, K.; Taketomi, T. Application of delayed extraction matrix-assisted laser desorption ionization time-of-flight mass spectrometry for analysis of sphingolipids in cultured skin fibroblasts from sphingolipidosis patients. Brain Dev. 2002, 24, 170–173. [Google Scholar] [CrossRef]
- Scherer, M.; Leuthäuser-Jaschinski, K.; Ecker, J.; Schmitz, G.; Liebisch, G. A rapid and quantitative LC-MS/MS method to profile sphingolipids. J. Lipid Res. 2010, 51, 2001–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacques, I.P. Distribution of gangliosides in human epidermis, dermis and whole skin. J. Clin. Exp. Dermatol. Res. 2015, 6, 282. [Google Scholar] [CrossRef]
- Cozma, C.; Iurașcu, M.-I.; Eichler, S.; Hovakimyan, M.; Brandau, O.; Zielke, S.; Böttcher, T.; Giese, A.-K.; Lukas, J.; Rolfs, A. C26-Ceramide as highly sensitive biomarker for the diagnosis of Farber Disease. Sci. Rep. 2017, 7, 6149. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, Y.; Hama, K.; Yokoyama, K. Mass spectrometry in combination with a chiral column and multichannel-MRM allows comprehensive analysis of glycosphingolipid molecular species from mouse brain. Carbohydr. Res. 2020, 490, 107959. [Google Scholar] [CrossRef]
- Lago, J.C.; Puzzi, M.B. The effect of aging in primary human dermal fibroblasts. PLoS ONE 2019, 14, e0219165. [Google Scholar] [CrossRef] [Green Version]
- Vukelić, Z.; Kalanj-Bognar, S. Cell density-dependent changes of glycosphingolipid biosynthesis in cultured human skin fibroblasts. Glycoconj. J. 2001, 18, 429–437. [Google Scholar] [CrossRef]
- Valsecchi, M.; Mauri, L.; Casellato, R.; Prioni, S.; Loberto, N.; Prinetti, A.; Chigorno, V.; Sonnino, S. Ceramide and sphingomyelin species of fibroblasts and neurons in culture. J. Lipid Res. 2007, 48, 417–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hama, K.; Fujiwara, Y.; Takashima, S.; Hayashi, Y.; Yamashita, A.; Shimozawa, N.; Yokoyama, K. Hexacosenoyl-CoA is the most abundant very long-chain acyl-CoA in ATP binding cassette transporter D1-deficient cells. J. Lipid Res. 2020, 61, 523–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huitema, K.; Dikkenberg, J.V.D.; Brouwers, J.F.H.M.; Holthuis, J.C.M. Identification of a family of animal sphingomyelin synthases. EMBO J. 2003, 23, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, S.; Sakiyama, H.; Suzuki, G.; Hidari, K.I.; Hirabayashi, Y. Expression cloning of a cDNA for human ceramide glucosyltransferase that catalyzes the first glycosylation step of glycosphingolipid synthesis. Proc. Natl. Acad. Sci. USA 1996, 93, 4638–4643. [Google Scholar] [CrossRef] [Green Version]
- Novgorodov, S.; I Gudz, T. Ceramide and mitochondria in ischemic brain injury. Int. J. Biochem. Mol. Boil. 2011, 2, 347–361. [Google Scholar]
- Moser, A.B.; Kreiter, N.; Bezman, L.; Lu, S.; Raymond, G.V.; Naidu, S.; Moser, H.W. Plasma very long chain fatty acids in 3000 peroxisome disease patients and 29,000 controls. Ann. Neurol. 1999, 45, 100–110. [Google Scholar] [CrossRef]
- Yamashita, T.; Mitsui, J.; Shimozawa, N.; Takashima, S.; Umemura, H.; Sato, K.; Takemoto, M.; Hishikawa, N.; Ohta, Y.; Matsukawa, T.; et al. Ataxic form of autosomal recessive PEX10-related peroxisome biogenesis disorders with a novel compound heterozygous gene mutation and characteristic clinical phenotype. J. Neurol. Sci. 2017, 375, 424–429. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
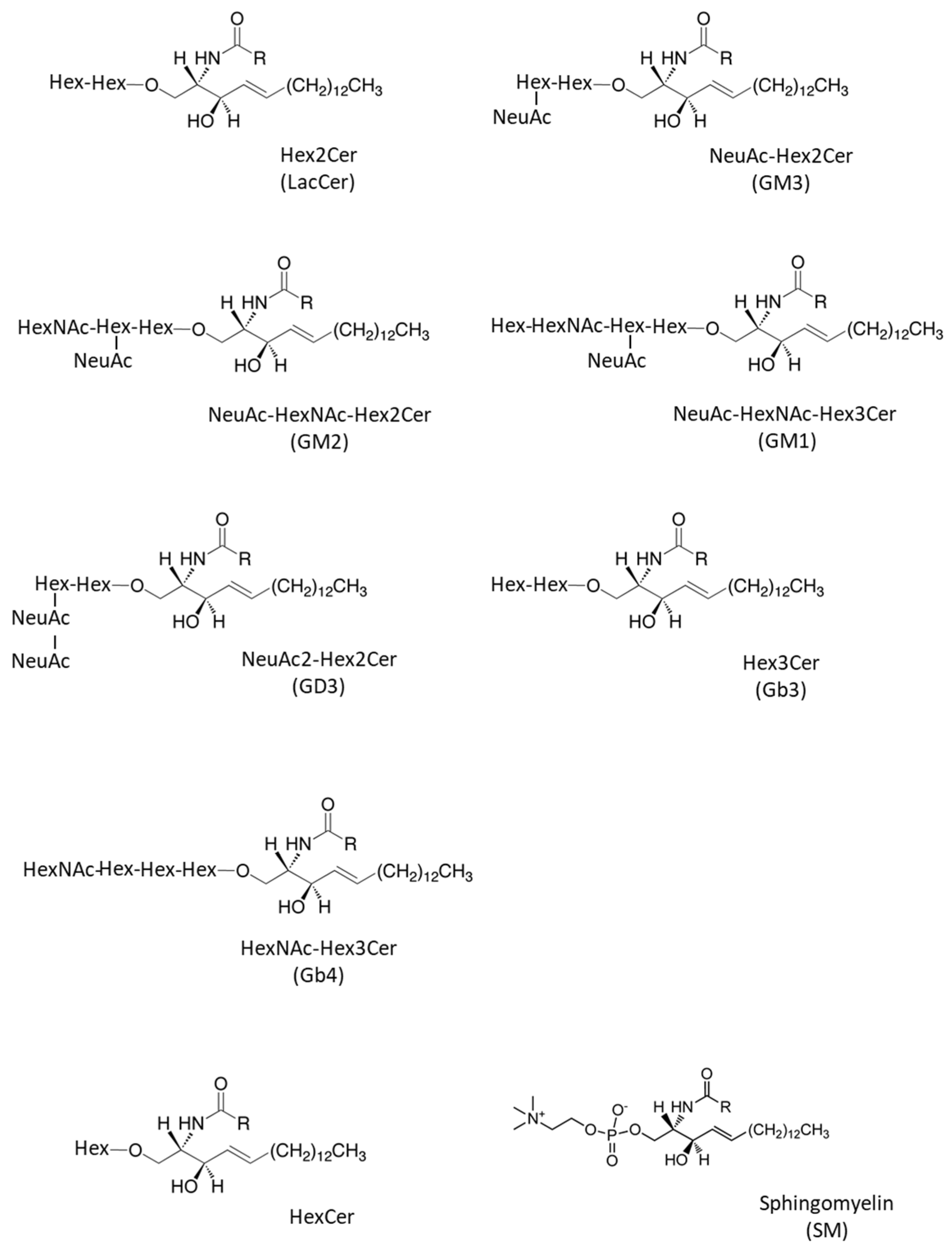
| Abbreviated Name | Systematic Name | Common Name | Ref |
|---|---|---|---|
| HexCer | Glc/Galβ-Cer | HexCer | [27,28,29,30,31] |
| Hex2Cer | Galβ1-4Glcβ-Cer | LacCer | [27,28,29,31] |
| NeuAc-Hex2Cer | NeuAcα2-3Galβ1-4Glcβ-Cer | GM3 | [27,29,32] |
| NeuAc-HexNAc-Hex2Cer | GalNAcβ1-4(NeuAcα2-3)Galβ1-4Glcβ-Cer | GM2 | [27,32] |
| NeuAc-HexNAc-Hex3Cer | Galβ1-3GalNAcβ1-4(NeuAcα2-3)Galβ1-4Glcβ-Cer | GM1 | [27] |
| NeuAc2-Hex2Cer | NeuAcα2-8NeuAcα2-3Galβ1-4Glcβ-Cer | GD3 | [28,29,32] |
| Hex3Cer | Galα1-4Galβ1-4Glcβ-Cer | Gb3 | [27,28,29] |
| HexNAc-Hex3Cer | GalNAcβ1-3Galα1-4Galβ1-4Glcβ-Cer | Gb4 | [27,29,33] |
| Analytes | MRM Transitions | CE (V) |
|---|---|---|
| LacCer | M + H → 264.2 | 500 m/z~; 70, 700 m/z~; 80, 1000 m/z~; 90 |
| GM3 | ||
| GM2 | ||
| GM1 | ||
| GD3 | ||
| Gb3 | ||
| Gb4 | ||
| HexCer | Neutral loss of 180 | 500 m/z~; 30, 900 m/z~; 40, 1000 m/z~; 50 |
| SM | M + H → 184 | 70 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujiwara, Y.; Hama, K.; Shimozawa, N.; Yokoyama, K. Glycosphingolipids with Very Long-Chain Fatty Acids Accumulate in Fibroblasts from Adrenoleukodystrophy Patients. Int. J. Mol. Sci. 2021, 22, 8645. https://doi.org/10.3390/ijms22168645
Fujiwara Y, Hama K, Shimozawa N, Yokoyama K. Glycosphingolipids with Very Long-Chain Fatty Acids Accumulate in Fibroblasts from Adrenoleukodystrophy Patients. International Journal of Molecular Sciences. 2021; 22(16):8645. https://doi.org/10.3390/ijms22168645
Chicago/Turabian StyleFujiwara, Yuko, Kotaro Hama, Nobuyuki Shimozawa, and Kazuaki Yokoyama. 2021. "Glycosphingolipids with Very Long-Chain Fatty Acids Accumulate in Fibroblasts from Adrenoleukodystrophy Patients" International Journal of Molecular Sciences 22, no. 16: 8645. https://doi.org/10.3390/ijms22168645
APA StyleFujiwara, Y., Hama, K., Shimozawa, N., & Yokoyama, K. (2021). Glycosphingolipids with Very Long-Chain Fatty Acids Accumulate in Fibroblasts from Adrenoleukodystrophy Patients. International Journal of Molecular Sciences, 22(16), 8645. https://doi.org/10.3390/ijms22168645