
Quantum Calculations of VX Ammonolysis and Hydrolysis Pathways via Hydrated Lithium Nitride

 ,
,
Abstract
:1. Introduction
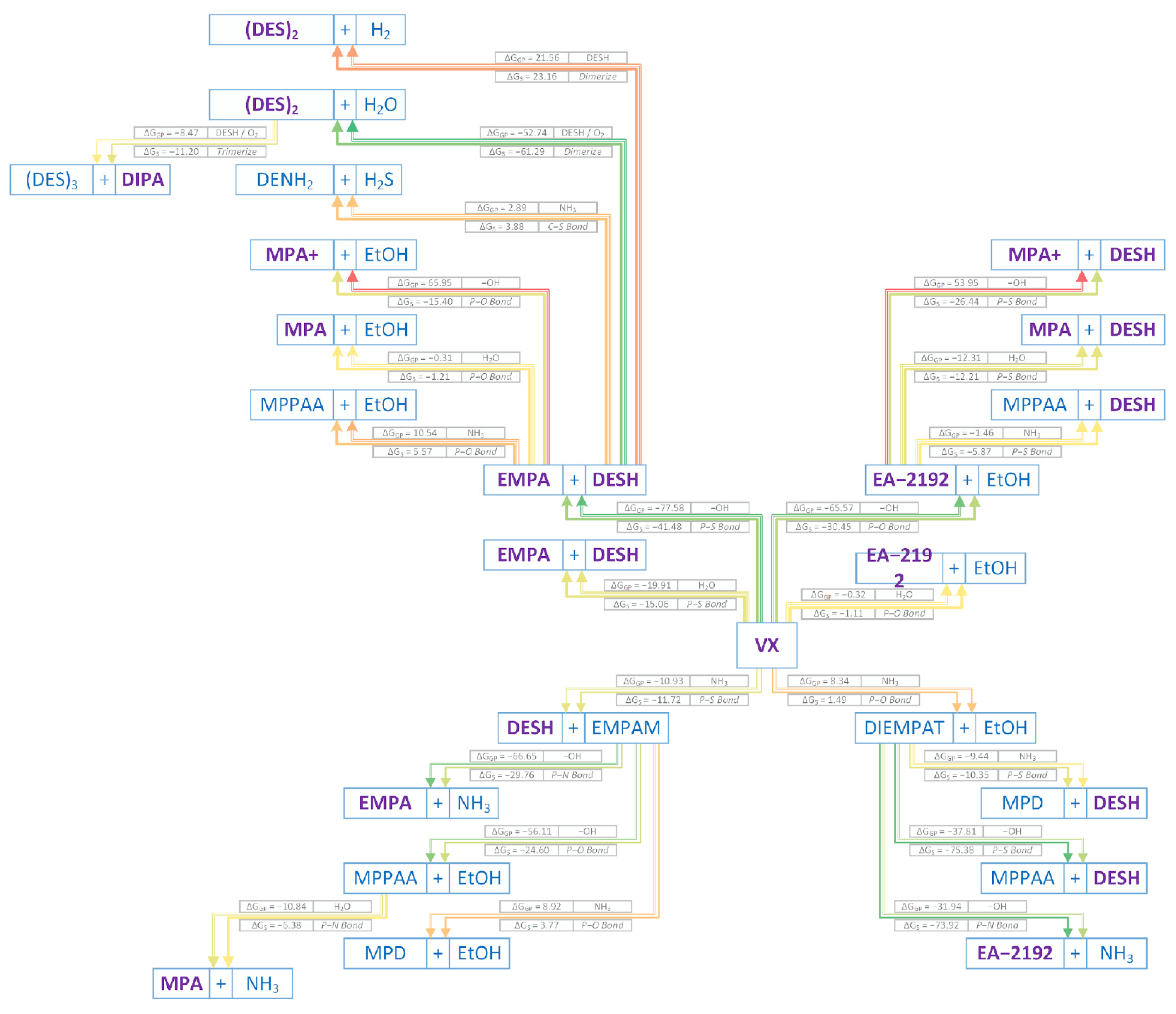
2. Results and Discussions
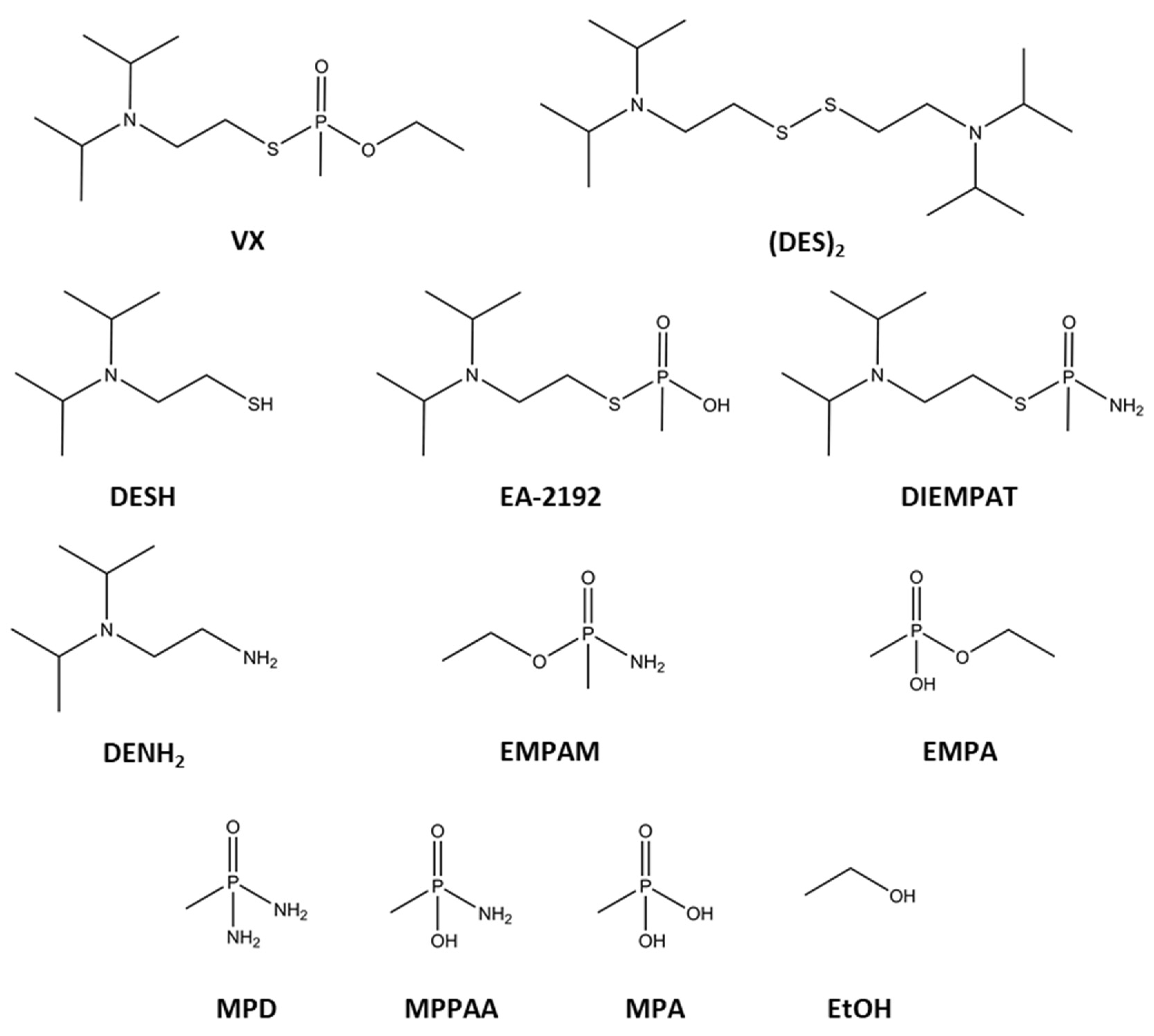
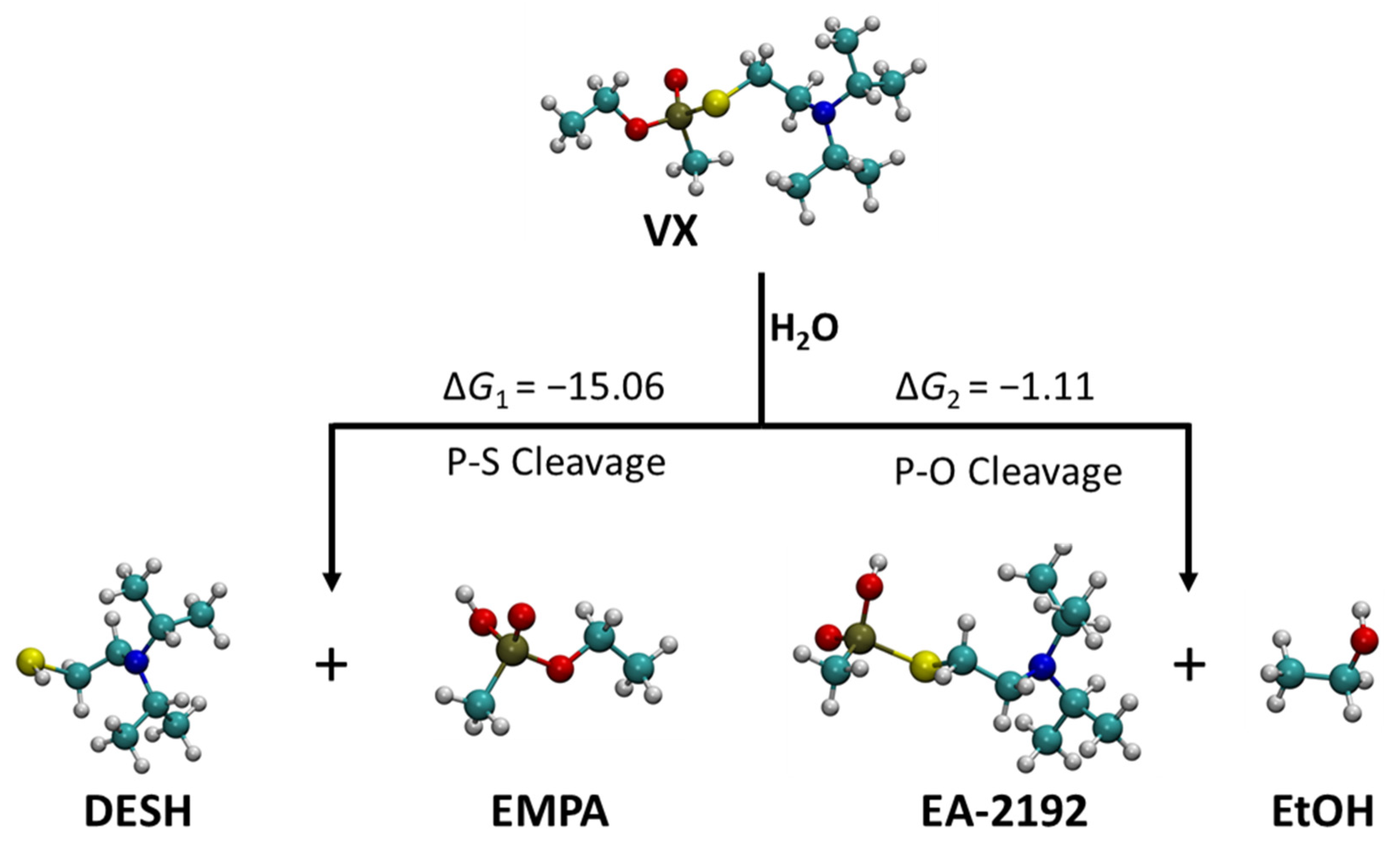
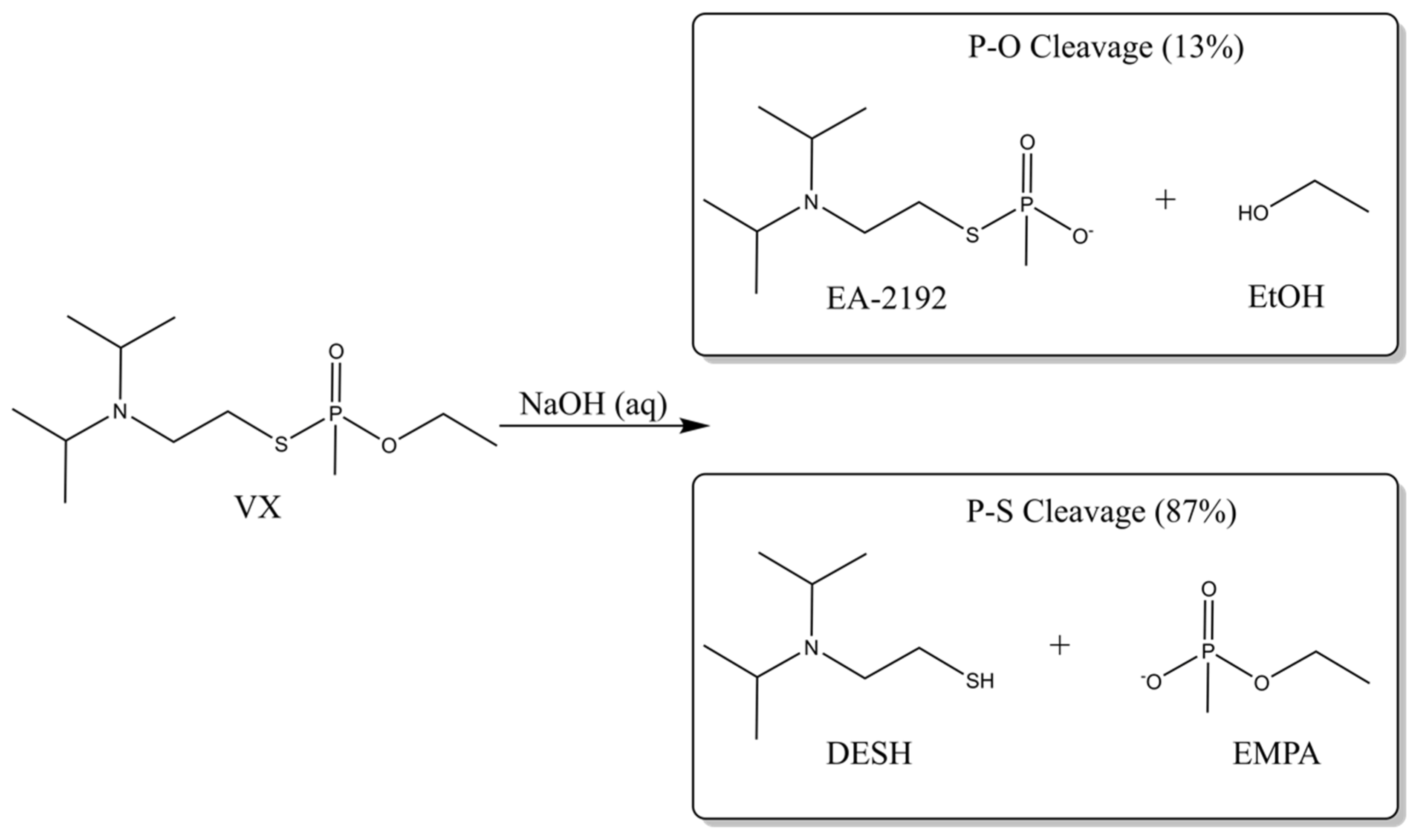
2.1. Neutral Hydrolysis
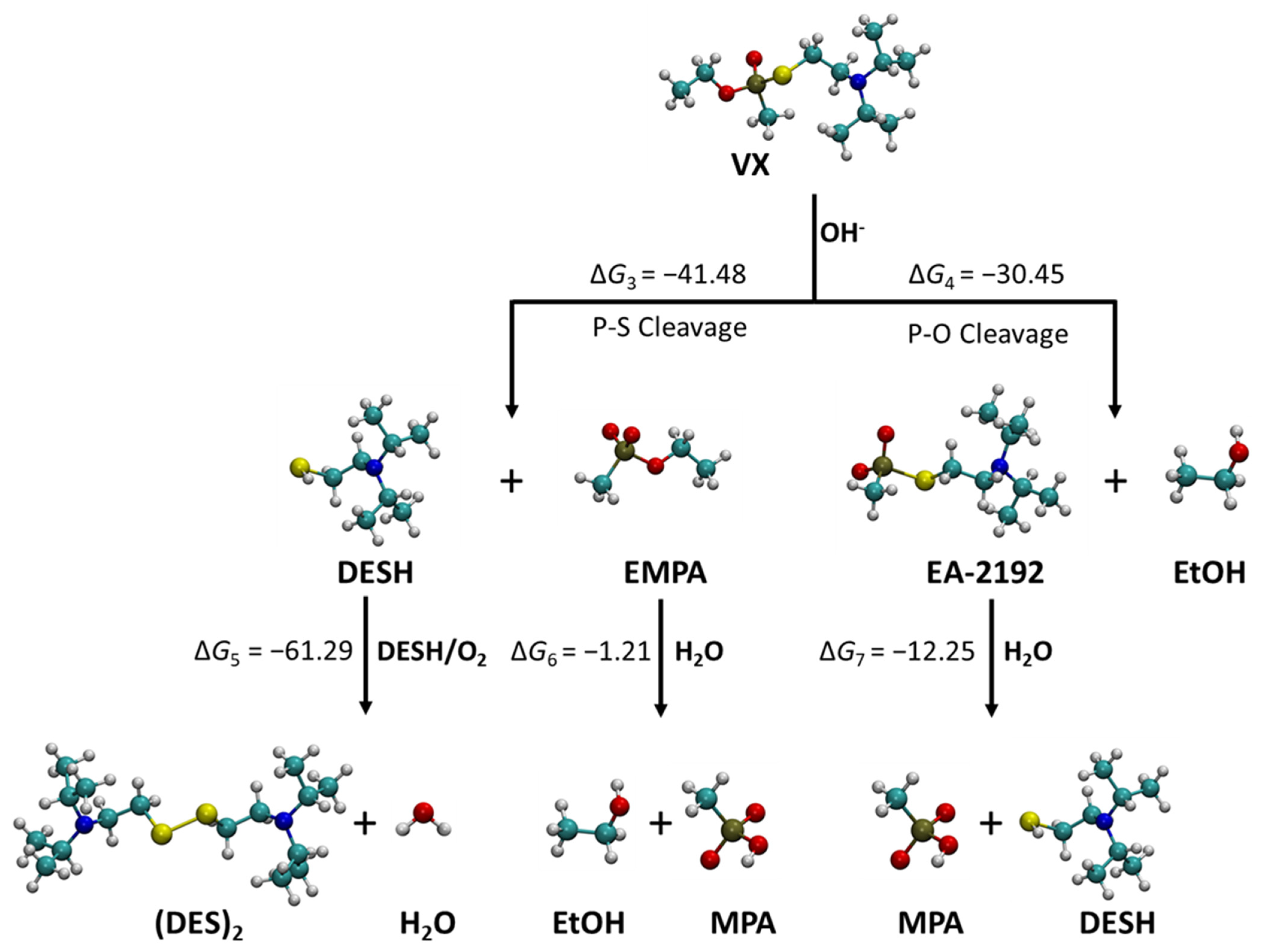
2.2. Alkaline Hydrolysis
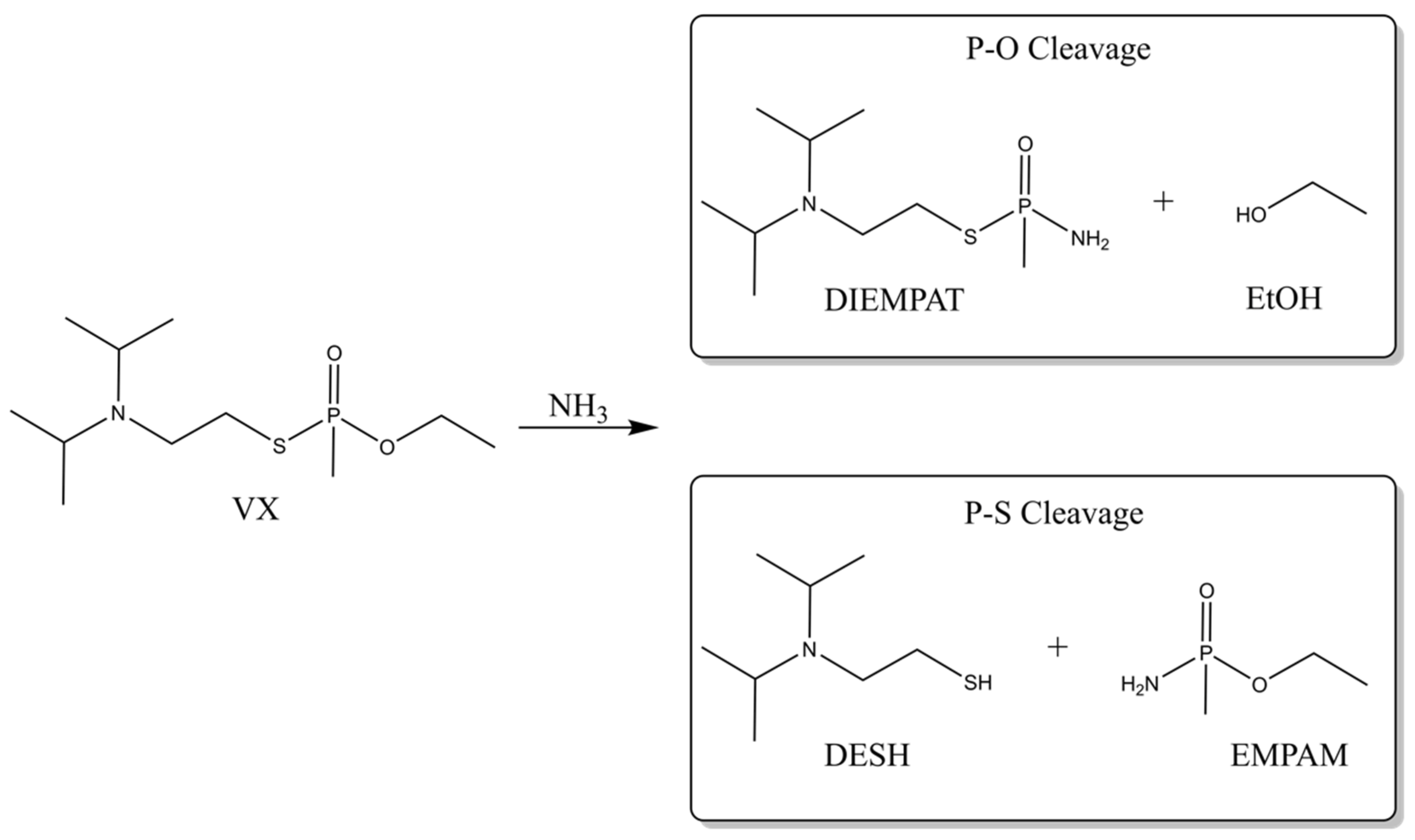
2.3. Ammonolysis
2.4. Computational and Experimental Agreement
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Costero, A.M.; Parra, M.; Gil, S.; Gotor, R.; Martínez-Mañez, R.; Sancenón, F.; Royo, S. Selective Detection of Nerve Agent Simulants by Using Triarylmethanol-Based Chromogenic Chemodosimeters. Eur. J. Org. Chem. 2012, 2012, 4937–4946. [Google Scholar] [CrossRef]
- Friboulet, A.; Rieger, F.; Goudou, D.; Amitai, G.; Taylor, P. Interaction of an Organophosphate with a Peripheral Site on Acetylcholinesterase. Biochemistry 1990, 29, 914–920. [Google Scholar] [CrossRef]
- Shih, T.M.; Kan, R.K.; McDonough, J.H. In Vivo Cholinesterase Inhibitory Specificity of Organophosphorus Nerve Agents. Chem. Biol. Interact. 2005, 157–158, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Tai, K.; Henchman, R.H.; McCammon, J.A. Molecular Dynamics of Acetylcholinesterase. Acc. Chem. Res. 2002, 35, 332–340. [Google Scholar] [CrossRef]
- Simanenko, Y.S.; Savelova, V.A.; Prokop’eva, T.M.; Mikhailov, V.A.; Turovskaya, M.K.; Karpichev, E.A.; Popov, A.F.; Gillitt, N.D.; Bunton, C.A. Bis(Dialkylamide)Hydrogen Dibromobromate Precursors of Hypobromite Ion in Reactions with Nerve and Blister Agent Simulants. J. Org. Chem. 2004, 69, 9238–9240. [Google Scholar] [CrossRef]
- Yang, Y.-C. Chemical Detoxification of Nerve Agent VX. Acc. Chem. Res. 1999, 32, 109–115. [Google Scholar] [CrossRef]
- Bigley, A.N.; Xu, C.; Henderson, T.J.; Harvey, S.P.; Raushel, F.M. Enzymatic Neutralization of the Chemical Warfare Agent VX: Evolution of Phosphotriesterase for Phosphorothiolate Hydrolysis. J. Am. Chem. Soc. 2013, 135, 10426–10432. [Google Scholar] [CrossRef] [Green Version]
- Tsai, P.C.; Fox, N.; Bigley, A.N.; Harvey, S.P.; Barondeau, D.P.; Raushel, F.M. Enzymes for the Homeland Defense: Optimizing Phosphotriesterase for the Hydrolysis of Organophosphate Nerve Agents. Biochemistry 2012, 51, 6463–6475. [Google Scholar] [CrossRef]
- LeJeune, K.E.; Wild, J.R.; Russell, A.J. Nerve Agents Degraded by Enzymatic Foams. Nature 1998, 395, 27–28. [Google Scholar] [CrossRef] [PubMed]
- Daniel, K.A.; Kopff, L.A.; Patterson, E.V. Computational Studies on the Solvolysis of the Chemical Warfare Agent VX. J. Phys. Org. Chem. 2008, 21, 321–328. [Google Scholar] [CrossRef]
- Menke, J.L.; Patterson, E.V. Quantum Mechanical Calculations on the Reaction of Ethoxide Anion with O,S-Dimethyl Methylphosphonothiolate. J. Mol. Struct. THEOCHEM 2007, 811, 281–291. [Google Scholar] [CrossRef]
- Seckute, J.; Menke, J.L.; Emnett, R.J.; Patterson, E.V.; Cramer, C.J. Ab Initio Molecular Orbital and Density Functional Studies on the Solvolysis of Sarin and O,S-Dimethyl Methylphosphonothiolate, a VX-Like Compound. J. Org. Chem. 2005, 70, 8649–8660. [Google Scholar] [CrossRef] [PubMed]
- Glaude, P.A.; Melius, C.; Pitz, W.J.; Westbrook, C.K. Detailed Chemical Kinetic Reaction Mechanisms for Incineration of Organophosphorus and Fluoroorganophosphorus Compounds. Proc. Combust. Inst. 2002, 29, 2469–2476. [Google Scholar] [CrossRef] [Green Version]
- Picard, B.; Chataigner, I.; Maddaluno, J.; Legros, J. Introduction to Chemical Warfare Agents, Relevant Simulants and Modern Neutralisation Methods. Org. Biomol. Chem. 2019, 17, 6528–6537. [Google Scholar] [CrossRef] [PubMed]
- Nawała, J.; Jóźwik, P.; Popiel, S. Thermal and Catalytic Methods Used for Destruction of Chemical Warfare Agents. Int. J. Environ. Sci. Technol. 2019, 16, 3899–3912. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, S.; Chauhan, S.; D’Cruz, R.; Faruqi, S.; Singh, K.K.; Varma, S.; Singh, M.; Karthik, V. Chemical Warfare Agents. Environ. Toxicol. Pharmacol. 2008, 26, 113–122. [Google Scholar] [CrossRef]
- Priest, C.W.; Greathouse, J.A.; Kinnan, M.K.; Burton, P.D.; Rempe, S.B. Ab Initio and Force Field Molecular Dynamics Study of Bulk Organophosphorus and Organochlorine Liquid Structures. J. Chem. Phys. 2021, 154, 084503. [Google Scholar] [CrossRef]
- Emelianova, A.; Basharova, E.A.; Kolesnikov, A.L.; Arribas, E.V.; Ivanova, E.V.; Gor, G.Y. Force Fields for Molecular Modeling of Sarin and Its Simulants: Dmmp and Dimp. J. Phys. Chem. B 2021, 125, 4086–4098. [Google Scholar] [CrossRef]
- Harvey, J.A.; McEntee, M.L.; Garibay, S.J.; Durke, E.M.; DeCoste, J.B.; Greathouse, J.A.; Sava Gallis, D.F. Spectroscopically Resolved Binding Sites for the Adsorption of Sarin Gas in a Metal-Organic Framework: Insights Beyond Lewis Acidity. J. Phys. Chem. Lett. 2019, 10, 5142–5147. [Google Scholar] [CrossRef]
- Bermudez, V.M. Computational Study of the Adsorption of Trichlorophosphate, Dimethyl Methylphosphonate, and Sarin on Amorphous Sio2. J. Phys. Chem. C 2007, 111, 9314–9323. [Google Scholar] [CrossRef]
- Jouypazadeh, H.; Farrokhpour, H. DFT and TD-DFT Study of the Adsorption and Detection of Sulfur Mustard Chemical Warfare Agent by the C24, C12Si12, Al12N12, Al12P12, Be12O12, B12N12 and Mg12O12 Nanocages. J. Mol. Struct. 2018, 1164, 227–238. [Google Scholar] [CrossRef]
- Khan, M.A.; Kesharwani, M.K.; Bandyopadhyay, T.; Ganguly, B. Solvolysis of Chemical Warfare Agent VX Is More Efficient with Hydroxylamine Anion: A Computational Study. J. Mol. Graph. Model. 2009, 28, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.J.; Kim, K.; Tsay, O.G.; Atwood, D.A.; Churchill, D.G. Update 1 of: Destruction and Detection of Chemical Warfare Agents. Chem. Rev. 2015, 115, PR1–PR76. [Google Scholar] [CrossRef] [PubMed]
- Mandal, D.; Sen, K.; Das, A.K. Aminolysis of a Model Nerve Agent: A Computational Reaction Mechanism Study of O,S-Dimethyl Methylphosphonothiolate. J. Phys. Chem. A 2012, 116, 8382–8396. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Sambrook, M.R.; Clary, D.C. Theoretical Study of Gas-Phase Unimolecular Decomposition of Simulants of the Nerve Agent VX. J. Phys. Chem. A 2019, 123, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-C.; Szafraniec, L.L.; Beaudry, W.T.; Bunton, C.A. Perhydrolysis of Nerve Agent VX. J. Org. Chem. 1993, 58, 6964–6965. [Google Scholar] [CrossRef]
- Yang, Y.-C.; Szafraniec, L.L.; Beaudry, W.T.; Rohrbaugh, D.K. Oxidative Detoxification of Phosphonothiolates. J. Am. Chem. Soc. 1990, 112, 6621–6627. [Google Scholar] [CrossRef]
- McGarvey, D.J.; Creasy, W.R.; Kinnan, M.K. Characterization of Solid Reaction Products from the Reaction of VX with Li3N+H2O for the Tactical Disablement Project; CCDC CBC-TR-1635; U.S. Army Combat Capabilities Development Command Chemical Biological Center: Aberdeen, MD, USA, 2020. [Google Scholar]
- McGarvey, D.J.; Creasy, W.R.; Kinnan, M.K. Reaction of QL with Li3N+H2O for the Tactical Disablement Project; CCDC CBC-TR-1722; U.S. Army Combat Capabilities Development Command Chemical Biological Center: Aberdeen, MD, USA, 2020. [Google Scholar]
- Yang, Y.-C.; Szafraniec, L.L.; Beaudry, W.T.; Rohrbaugh, D.K.; Procell, L.R.; Samuel, J.B. Autocatalytic Hydrolysis of V-Type Nerve Agents. J. Org. Chem. 1996, 61, 8407–8413. [Google Scholar] [CrossRef]
- Smith, B.M. Catalytic Methods for the Destruction of Chemical Warfare Agents under Ambient Conditions. Chem. Soc. Rev. 2008, 37, 470–478. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Baker, J.; Andzelm, J.; Muir, M.; Taylor, P.R. OH + H2 → H2O + H. The Importance of ‘Exact Exchange’ in Density Functional Theory. Chem. Phys. Lett. 1995, 237, 53–60. [Google Scholar] [CrossRef]
- Zhao, Y.; Pu, J.; Lynch, B.J.; Truhlar, D.G. Tests of Second-Generation and Third-Generation Density Functionals for Thermochemical Kinetics. Phys. Chem. Chem. Phys. 2004, 6, 673–676. [Google Scholar] [CrossRef]
- Easton, R.E.; Giesen, D.J.; Welch, A.; Cramer, C.J.; Truhlar, D.G. The Midi! Basis Set for Quantum Mechanical Calculations of Molecular Geometries and Partial Charges. Theor. Chim. Acta 1996, 93, 281–301. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. Vmd: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HF-S a | HF | B3LYP-S a | B3LYP | |
|---|---|---|---|---|
| ⊗G1 | −15.06 | −19.91 | −12.27 | −12.54 |
| ⊗G2 | −1.11 | −0.32 | −1.08 | 0.33 |
| HF-S | HF | B3LYP-S | B3LYP | |
|---|---|---|---|---|
| ⊗G3 | −41.48 | −77.58 | −87.18 | −114.18 |
| ⊗G4 | −30.45 | −65.58 | −78.87 | −112.15 |
| ⊗G5 | −61.29 | −52.74 | −42.84 | −41.28 |
| ⊗G6 | −1.21 | −0.31 | −0.39 | 2.08 |
| ⊗G7 | −12.25 | −12.31 | −8.71 | 0.05 |
| HF-S | HF | B3LYP-S | B3LYP | |
|---|---|---|---|---|
| ⊗G8 | −11.72 | −10.93 | 3.88 | 5.05 |
| ⊗G9 | 1.49 | 8.34 | 14.32 | 16.61 |
| ⊗G10 | −29.76 | −66.65 | −91.06 | −119.22 |
| ⊗G11 | −24.60 | −56.11 | −73.08 | −98.64 |
| ⊗G12 | −37.81 | −75.38 | −93.19 | −128.76 |
| ⊗G13 | −31.94 | −73.92 | −83.52 | −110.20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leverant, C.J.; Priest, C.W.; Greathouse, J.A.; Kinnan, M.K.; Rempe, S.B. Quantum Calculations of VX Ammonolysis and Hydrolysis Pathways via Hydrated Lithium Nitride. Int. J. Mol. Sci. 2021, 22, 8653. https://doi.org/10.3390/ijms22168653
Leverant CJ, Priest CW, Greathouse JA, Kinnan MK, Rempe SB. Quantum Calculations of VX Ammonolysis and Hydrolysis Pathways via Hydrated Lithium Nitride. International Journal of Molecular Sciences. 2021; 22(16):8653. https://doi.org/10.3390/ijms22168653
Chicago/Turabian StyleLeverant, Calen J., Chad W. Priest, Jeffery A. Greathouse, Mark K. Kinnan, and Susan B. Rempe. 2021. "Quantum Calculations of VX Ammonolysis and Hydrolysis Pathways via Hydrated Lithium Nitride" International Journal of Molecular Sciences 22, no. 16: 8653. https://doi.org/10.3390/ijms22168653
APA StyleLeverant, C. J., Priest, C. W., Greathouse, J. A., Kinnan, M. K., & Rempe, S. B. (2021). Quantum Calculations of VX Ammonolysis and Hydrolysis Pathways via Hydrated Lithium Nitride. International Journal of Molecular Sciences, 22(16), 8653. https://doi.org/10.3390/ijms22168653