
PYTHIA: Deep Learning Approach for Local Protein Conformation Prediction

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Results
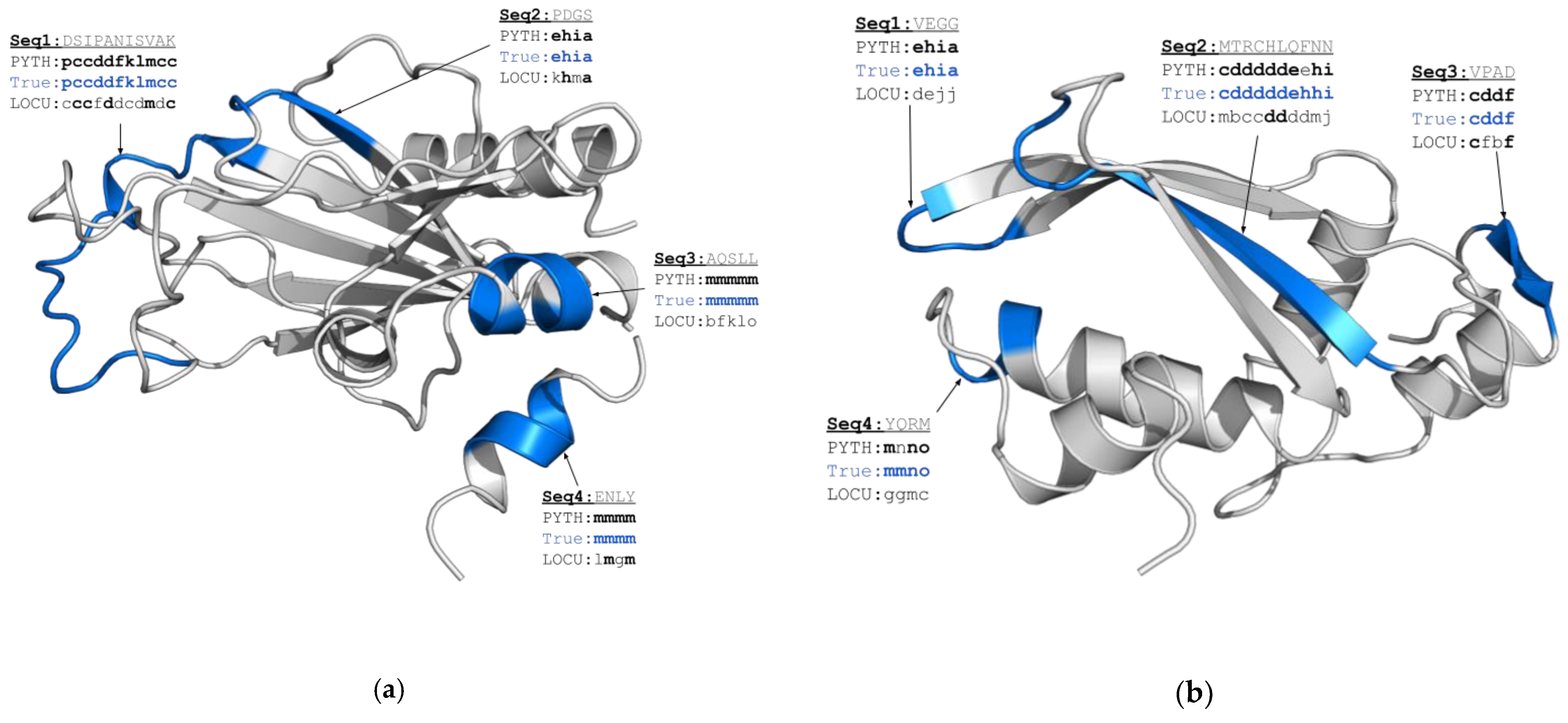
2.1. PYTHIA Performance on the CASP14 Free Modelling Targets
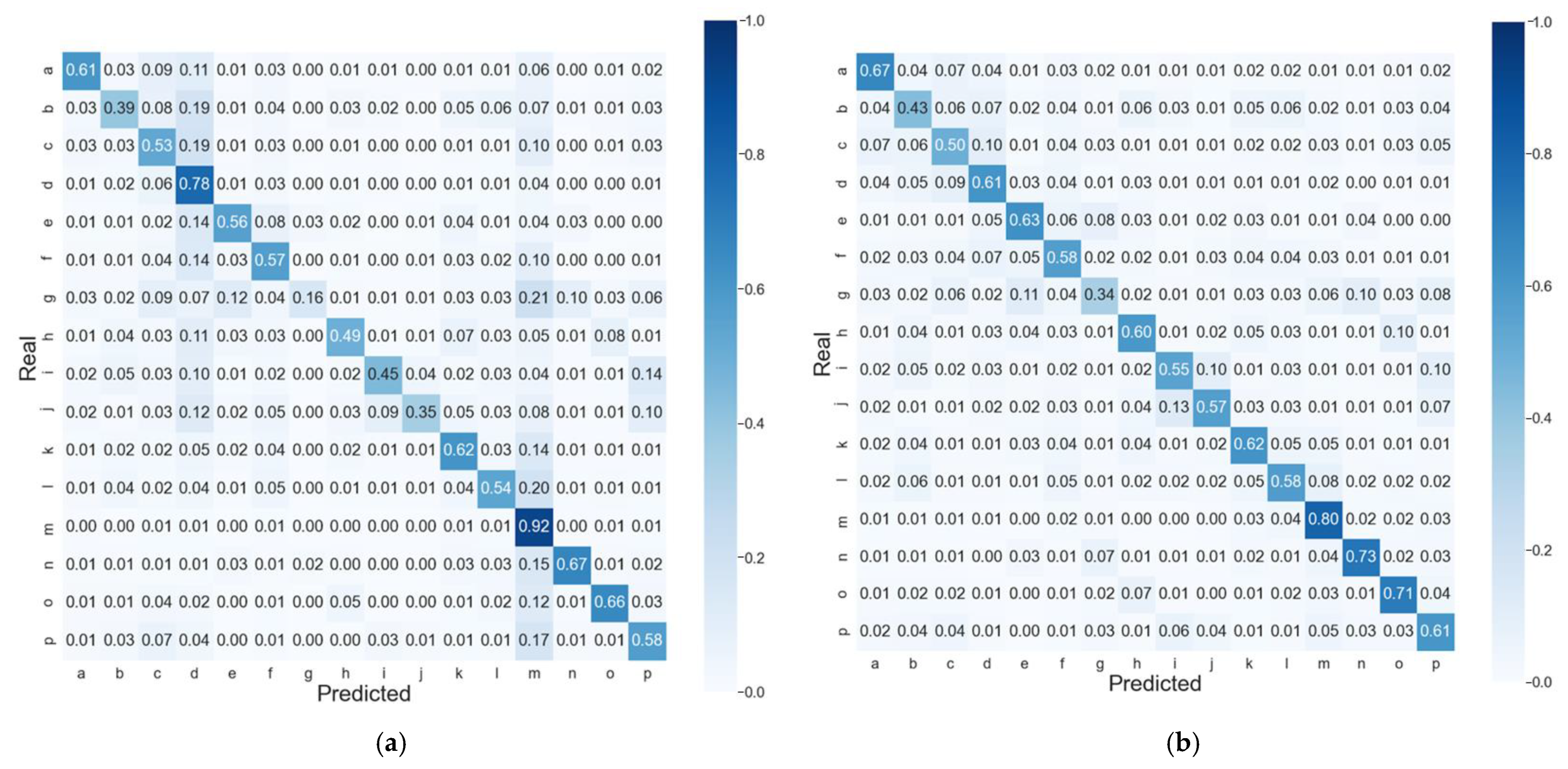
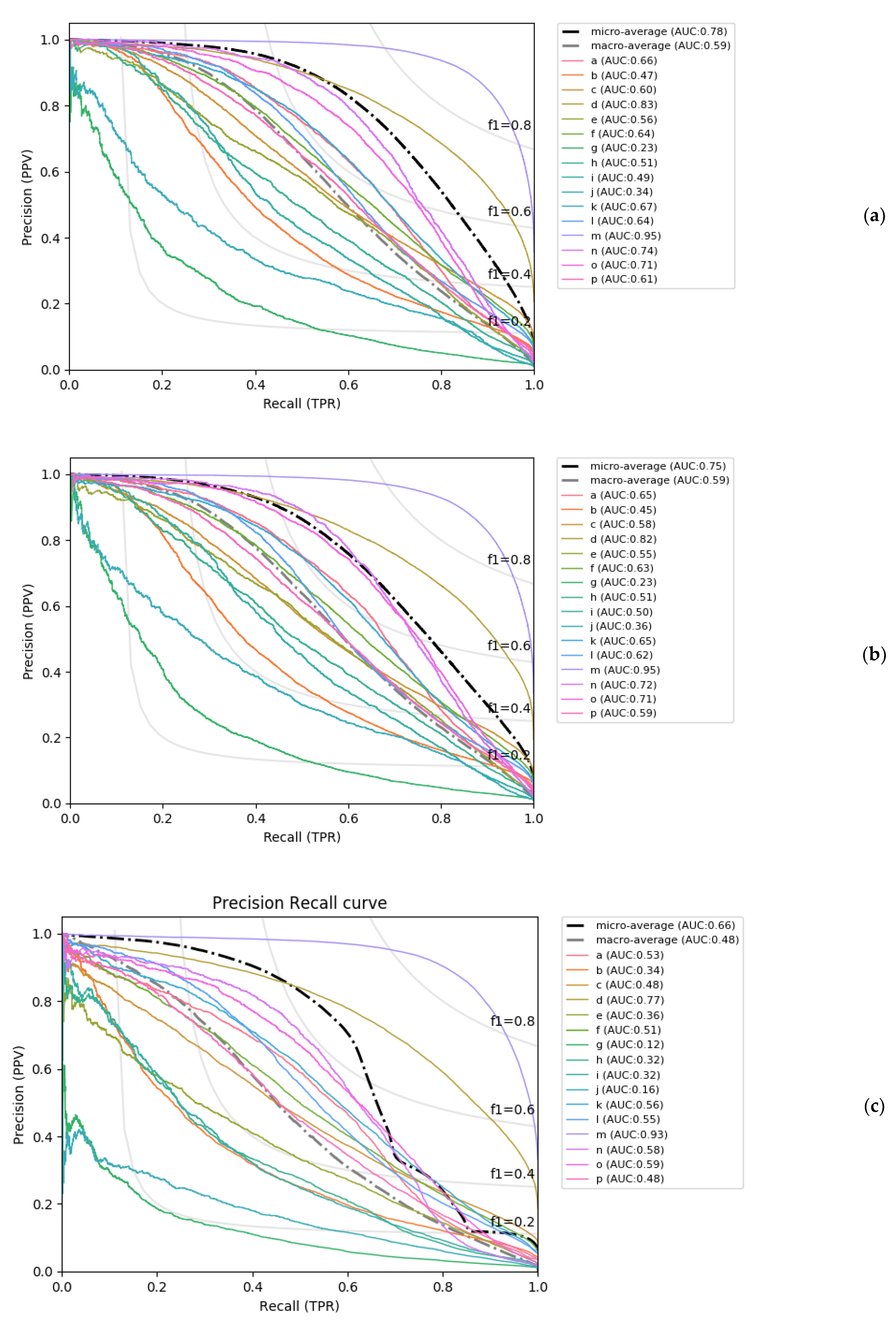
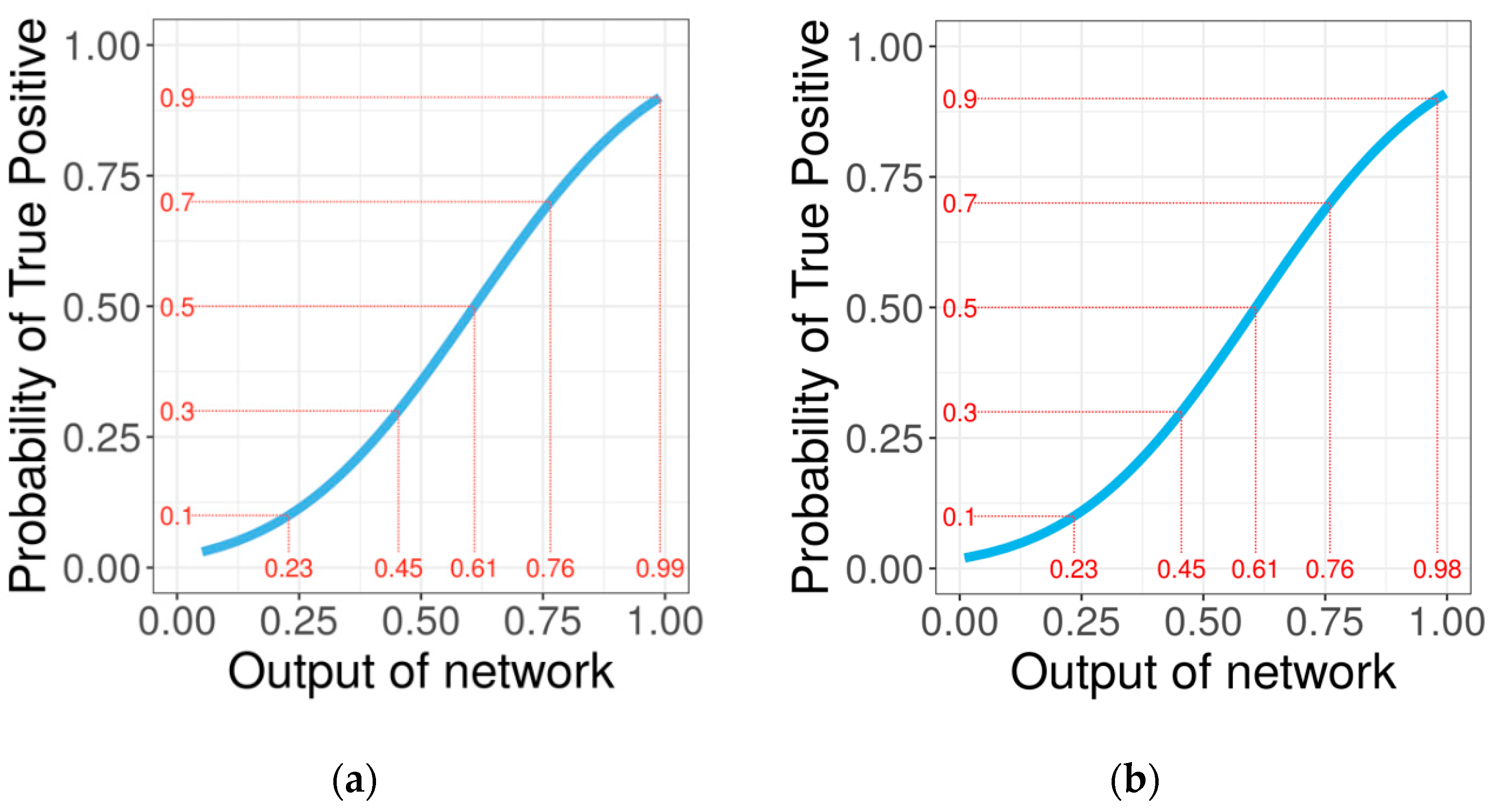
2.2. Confidence of Predictions
3. Discussion
4. Materials and Methods
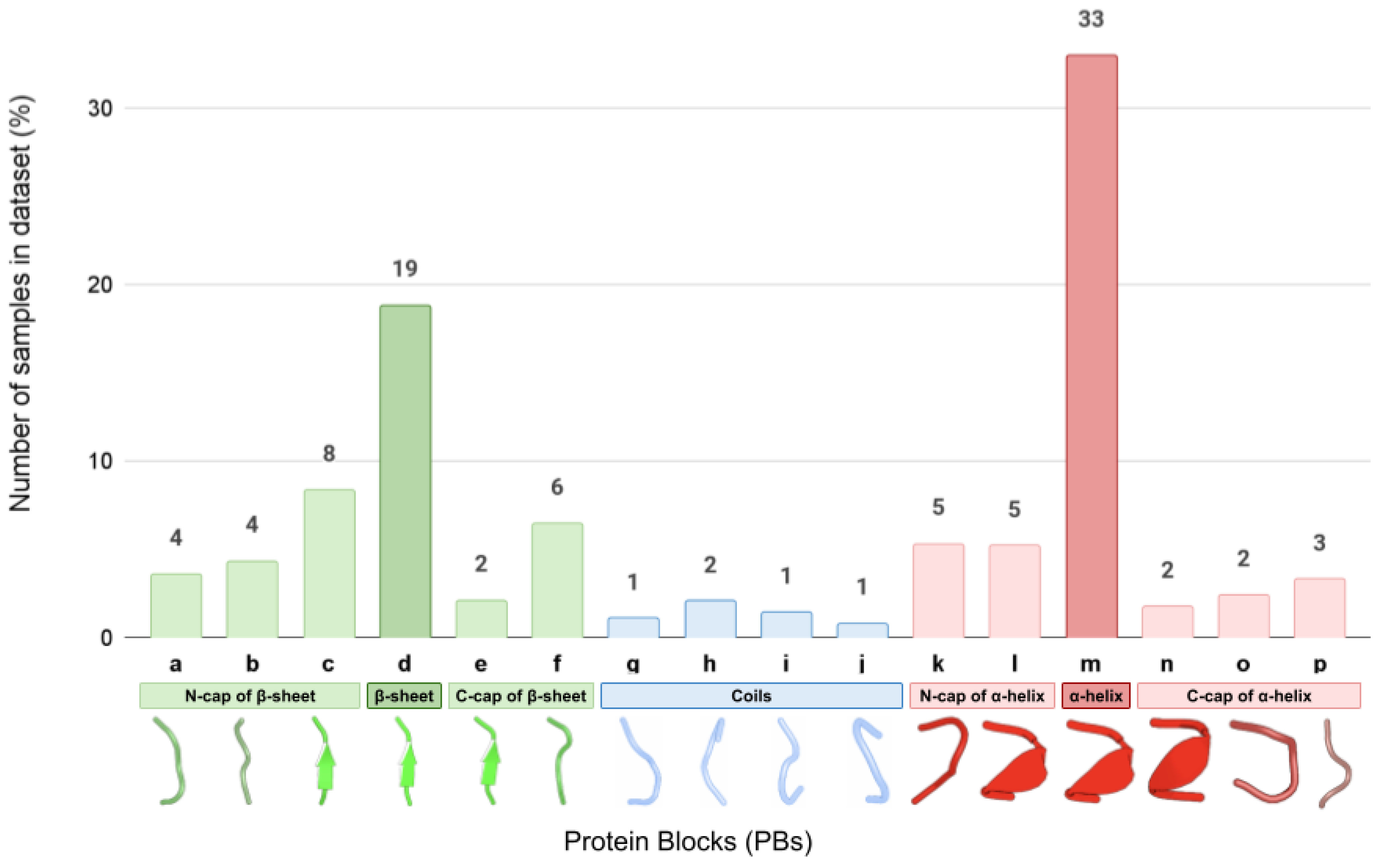
4.1. Dataset Preparation
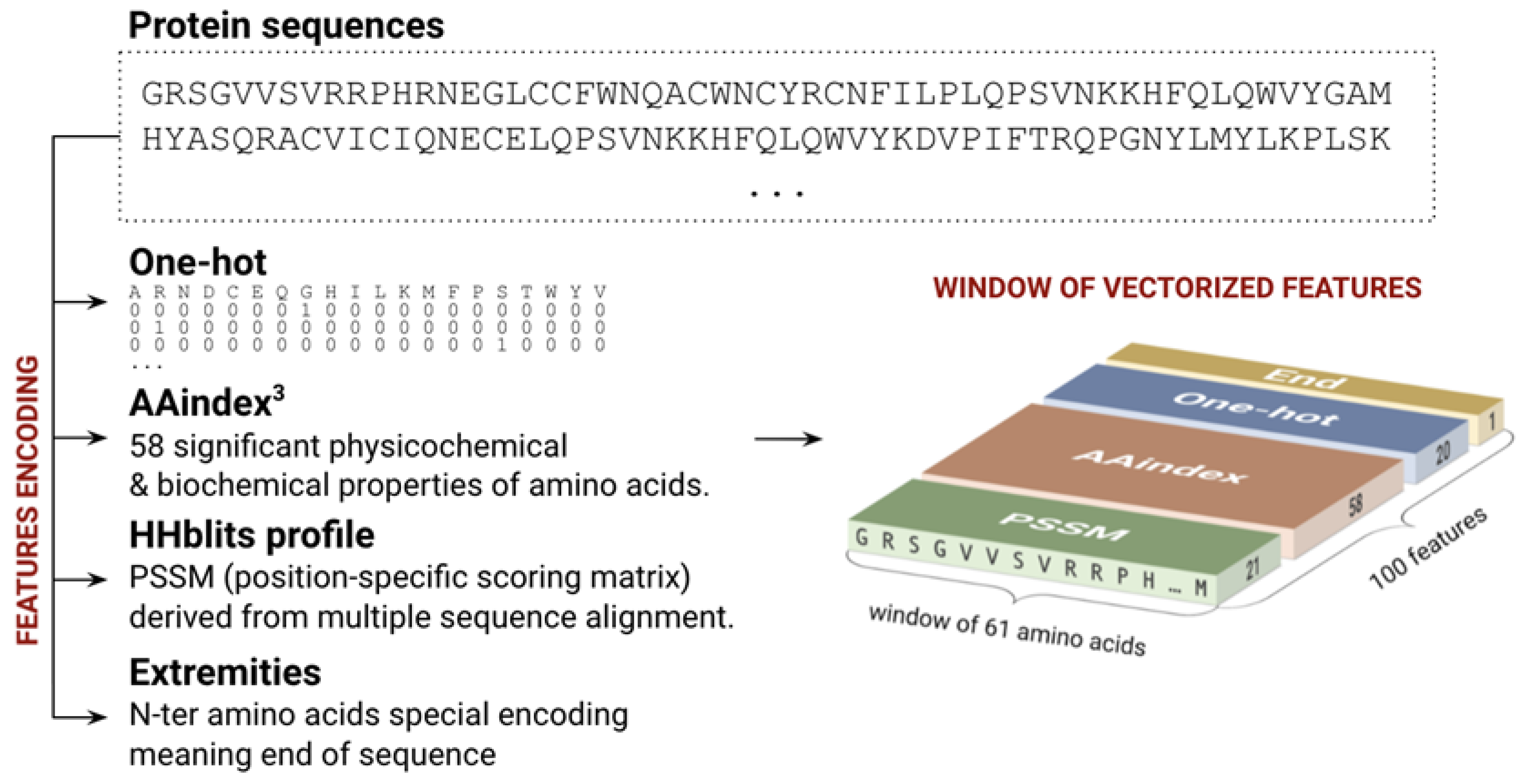
4.2. Features Encoding
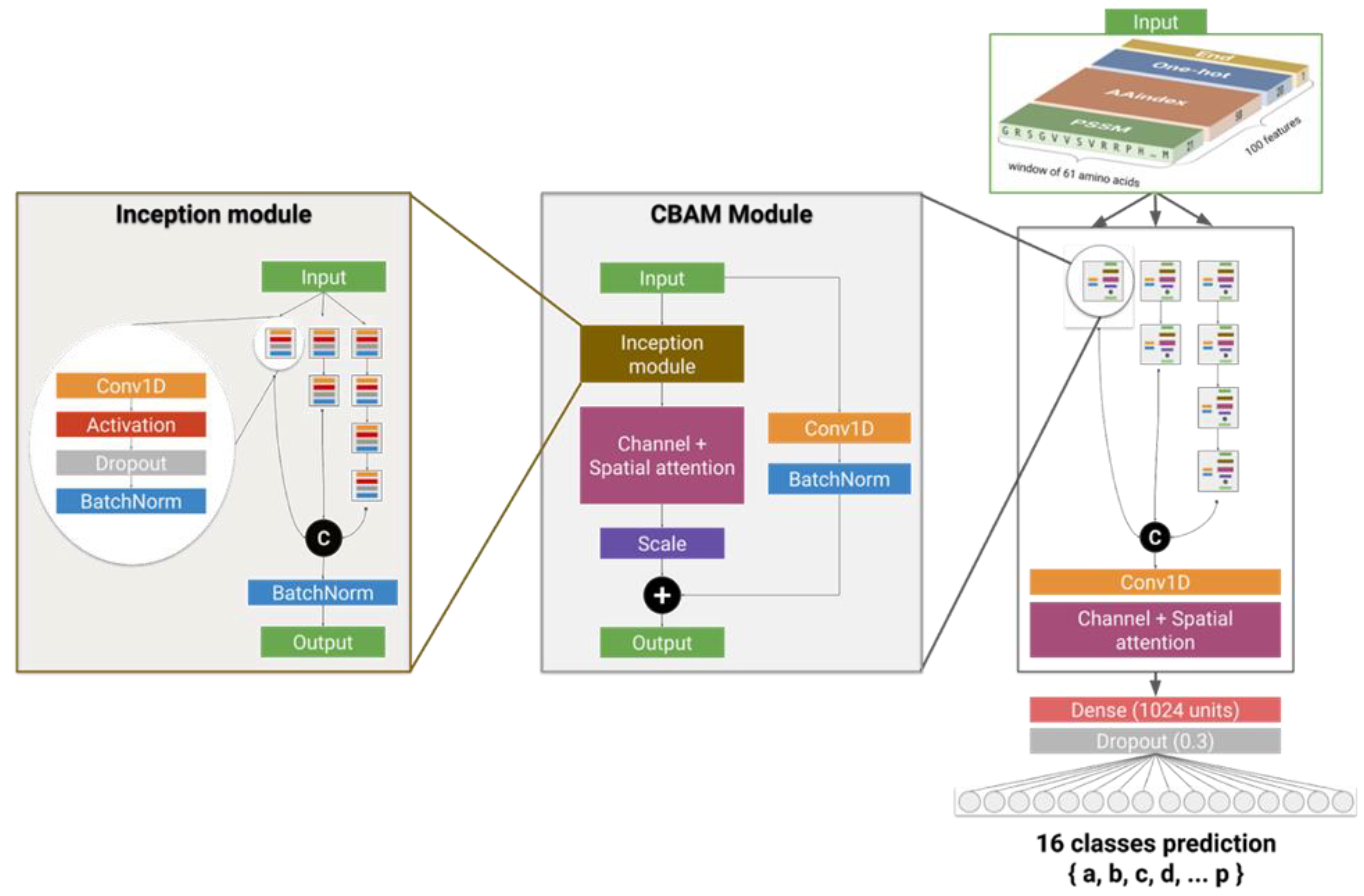
4.3. Deep Neural Network
4.4. Network Training
4.4.1. Hyperparameters
4.4.2. Class Weighting
4.5. Evaluation of Model Performances
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.D.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- De Brevern, A.; Etchebest, C.; Hazout, S. Bayesian probabilistic approach for predicting backbone structures in terms of protein blocks. Proteins Struct. Funct. Bioinform. 2000, 41, 271–287. [Google Scholar] [CrossRef] [Green Version]
- De Brevern, A.G. New Assessment of a Structural Alphabet. In Silico Biol. 2005, 5, 283–289. [Google Scholar]
- Gelly, J.-C.; Joseph, A.P.; Srinivasan, N.; De Brevern, A.G. iPBA: A tool for protein structure comparison using sequence alignment strategies. Nucleic Acids Res. 2011, 39, W18–W23. [Google Scholar] [CrossRef] [Green Version]
- Ghouzam, Y.; Postic, G.; Guerin, P.-E.; De Brevern, A.G.; Gelly, J.-C. ORION: A web server for protein fold recognition and structure prediction using evolutionary hybrid profiles. Sci. Rep. 2016, 6, 28268. [Google Scholar] [CrossRef] [PubMed]
- Akhila, M.V.; Narwani, T.J.; Floch, A.; Maljković, M.; Bisoo, S.; Shinada, N.K.; Kranjc, A.; Gelly, J.-C.; Srinivasan, N.; Mitić, N.; et al. A structural entropy index to analyse local conformations in intrinsically disordered proteins. J. Struct. Biol. 2020, 210, 107464. [Google Scholar] [CrossRef] [PubMed]
- Meersche, Y.V.; Cretin, G.; de Brevern, A.G.; Gelly, J.-C.; Galochkina, T. MEDUSA: Prediction of Protein Flexibility from Sequence. J. Mol. Biol. 2021, 433, 166882. [Google Scholar] [CrossRef] [PubMed]
- De Brevern, A.G.; Bornot, A.; Craveur, P.; Etchebest, C.; Gelly, J.-C. PredyFlexy: Flexibility and local structure prediction from sequence. Nucleic Acids Res. 2012, 40, W317–W322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craveur, P.; Joseph, A.P.; Esque, J.; Narwani, T.J.; Noël, F.; Shinada, N.; Goguet, M.; Leonard, S.; Poulain, P.; Bertrand, O.; et al. Protein flexibility in the light of structural alphabets. Front. Mol. Biosci. 2015, 2, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnoud, J.; Santuz, H.; Craveur, P.; Joseph, A.P.; Jallu, V.; De Brevern, A.G.; Poulain, P. PBxplore: A tool to analyze local protein structure and deformability with Protein Blocks. PeerJ 2017, 5, e4013. [Google Scholar] [CrossRef] [Green Version]
- Goguet, M.; Narwani, T.J.; Petermann, R.; Jallu, V.; De Brevern, A.G. In silico analysis of Glanzmann variants of Calf-1 domain of αIIbβ3 integrin revealed dynamic allosteric effect. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Jallu, V.; Poulain, P.; Fuchs, P.F.; Kaplan, C.; de Brevern, A.G. Modeling and molecular dynamics simulations of the V33 variant of the integrin subunit β3: Structural comparison with the L33 (HPA-1a) and P33 (HPA-1b) variants. Biochimie 2014, 105, 84–90. [Google Scholar] [CrossRef]
- Ladislav, M.; Černý, J.; Krusek, J.; Horak, M.; Balik, A.; Vyklicky, L. The LILI Motif of M3-S2 Linkers Is a Component of the NMDA Receptor Channel Gate. Front. Mol. Neurosci. 2018, 11, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etchebest, C.; Benros, C.; Hazout, S.; de Brevern, A.G. A structural alphabet for local protein structures: Improved prediction methods. Proteins Struct. Funct. Bioinform. 2005, 59, 810–827. [Google Scholar] [CrossRef] [PubMed]
- De Brevern, A.G.; Etchebest, C.; Benros, C.; Hazout, S.; De Brevern, A. “Pinning strategy”: A novel approach for predicting the backbone structure in terms of protein blocks from sequence. J. Biosci. 2007, 32, 51–70. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetrivel, I.; Mahajan, S.; Tyagi, M.; Hoffmann, L.; Sanejouand, Y.-H.; Srinivasan, N.; De Brevern, A.G.; Cadet, F.; Offmann, B. Knowledge-based prediction of protein backbone conformation using a structural alphabet. PLoS ONE 2017, 12, e0186215. [Google Scholar] [CrossRef] [Green Version]
- Suresh, V.; Ganesan, K.; Parthasarathy, S. A Protein Block Based Fold Recognition Method for the Annotation of Twilight Zone Sequences. Protein Pept. Lett. 2013, 20, 249–254. [Google Scholar] [CrossRef]
- Dong, Q.W.; Wang, X.L.; Lin, L. Methods for optimizing the structure alphabet sequences of proteins. Comput. Biol. Med. 2007, 37, 1610–1616. [Google Scholar] [CrossRef]
- Suresh, V. SVM-PB-Pred: SVM Based Protein Block Prediction Method Using Sequence Profiles and Secondary Structures. Protein Pept. Lett. 2013, 21, 736–742. [Google Scholar] [CrossRef]
- Zimmermann, O.; Hansmann, U.H.E. LOCUSTRA: Accurate Prediction of Local Protein Structure Using a Two-Layer Support Vector Machine Approach. J. Chem. Inf. Model. 2008, 48, 1903–1908. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, H.; Kauffman, C.; Karypis, G. svm PRAT: SVM-based Protein Residue Annotation Toolkit. BMC Bioinform. 2009, 10, 439. [Google Scholar] [CrossRef] [Green Version]
- Kinch, L.N.; Pei, J.; Kryshtafovych, A.; Schaeffer, R.D.; Grishin, N.V. Topology evaluation of models for difficult targets in the 14th round of the critical assessment of protein structure prediction. Proteins Struct. Funct. Bioinform. 2021. [Google Scholar] [CrossRef]
- Pereira, J.; Simpkin, A.J.; Hartmann, M.D.; Rigden, D.J.; Keegan, R.M.; Lupas, A.N. High-accuracy protein structure prediction in CASP14. Proteins Struct. Funct. Bioinform. 2021. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly Accurate Protein Structure Prediction for the Human Proteome. Nature 2021, 1–9. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, eabj8754. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 1–11. [Google Scholar] [CrossRef]
- Shapovalov, M.; Dunbrack, R.L., Jr.; Vucetic, S. Multifaceted analysis of training and testing convolutional neural networks for protein secondary structure prediction. PLoS ONE 2020, 15, e0232528. [Google Scholar] [CrossRef]
- Fang, C.; Shang, Y.; Xu, D. MUFOLD-SS: New deep inception-inside-inception networks for protein secondary structure prediction. Proteins 2018, 86, 592–598. [Google Scholar] [CrossRef]
- Uddin, M.R.; Mahbub, S.; Rahman, M.S.; Bayzid, S. SAINT: Self-attention augmented inception-inside-inception network improves protein secondary structure prediction. Bioinformatics 2020, 36, 4599–4608. [Google Scholar] [CrossRef]
- Fang, C.; Shang, Y.; Xu, D. A deep dense inception network for protein beta-turn prediction. Proteins 2020, 88, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Dunbrack, R.L. PISCES: A protein sequence culling server. Bioinformatics 2003, 19, 1589–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bank, R.P.D. PDB Statistics: Overall Growth of Released Structures per Year. 2021. Available online: https://www.rcsb.org/stats/growth/growth-released-structures (accessed on 6 August 2021).
- Van Westen, G.J.; Swier, R.F.; Wegner, J.K.; Ijzerman, A.P.; Van Vlijmen, H.W.; Bender, A. Benchmarking of protein descriptor sets in proteochemometric modeling (part 1): Comparative study of 13 amino acid descriptor sets. J. Cheminform. 2013, 5, 41. [Google Scholar] [CrossRef] [Green Version]
- Henikoff, S.; Henikoff, J.G. Position-based sequence weights. J. Mol. Biol. 1994, 243, 574–578. [Google Scholar] [CrossRef]
- Henikoff, J.G.; Henikoff, S. Using substitution probabilities to improve position-specific scoring matrices. Bioinformatics 1996, 12, 135–143. [Google Scholar] [CrossRef]
- Steinegger, M.; Meier, M.; Mirdita, M.; Vöhringer, H.; Haunsberger, S.J.; Söding, J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinform. 2019, 20, 473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szegedy, C.; Ioffe, S.; Vanhoucke, V.; Alemi, A. Inception-v4, Inception-ResNet and the Impact of Residual Connections on Learning. In Proceedings of the Thirty-First AAAI Conference on Artificial Intelligence (AAAI-17), Hilton San Francisco, San Francisco, CA, USA, 4–9 February 2016. [Google Scholar]
- Woo, S.; Park, J.; Lee, J.-Y.; Kweon, I.S. CBAM: Convolutional Block Attention Module. In Proceedings of the Computer Vision—ECCV, Munich, Germany, 8–14 September 2018; Ferrari, V., Hebert, M., Sminchisescu, C., Weiss, Y., Eds.; 2018. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Jiang, H.; He, P.; Chen, W.; Liu, X.; Gao, J.; Han, J. On the Variance of the Adaptive Learning Rate and Beyond. Available online: http://arxiv.org/abs/1908.03265 (accessed on 11 August 2021).
- Misra, D. Mish: A Self Regularized Non-Monotonic Activation Function. Available online: http://arxiv.org/abs/1908.08681 (accessed on 11 August 2021).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TPR/SENS | F1 Score | MCC | |||||||
|---|---|---|---|---|---|---|---|---|---|
| PB | LOCUSTRA | PYTHIA | DIFF | LOCUSTRA | PYTHIA | DIFF | LOCUSTRA | PYTHIA | DIFF |
| a | 47.6 | 60.7 | +13.1 | 53.9 | 62.9 | +9.0 | 52.9 | 61.2 | +8.3 |
| b | 20.6 | 39.2 | +18.6 | 29.2 | 45.1 | +16.0 | 30.2 | 42.9 | +12.7 |
| c | 37.1 | 52.8 | +15.7 | 44.6 | 55.5 | +10.9 | 41.6 | 50.6 | +9.0 |
| d | 69.0 | 78.4 | +9.4 | 69.9 | 75.4 | +5.5 | 63.0 | 67.7 | +4.7 |
| e | 28.1 | 55.9 | +27.8 | 35.9 | 55.7 | +19.9 | 36.3 | 54.4 | +18.1 |
| f | 37.6 | 57.4 | +19.8 | 46.7 | 60.2 | +13.5 | 45.5 | 56.8 | +11.3 |
| g | 3.4 | 16.0 | +12.6 | 6.2 | 22.9 | +16.7 | 10.6 | 24.8 | +14.2 |
| h | 24.6 | 49.4 | +24.8 | 33.1 | 51.6 | +18.5 | 34.4 | 50.3 | +15.9 |
| i | 26.4 | 45.0 | +18.6 | 34.6 | 49.3 | +14.7 | 35.7 | 48.6 | +12.9 |
| j | 2.9 | 35.2 | +32.3 | 5.4 | 37.2 | +31.8 | 10.1 | 36.6 | +26.5 |
| k | 43.5 | 61.7 | +18.2 | 52.7 | 63.0 | +10.3 | 52.0 | 60.3 | +8.3 |
| l | 38.3 | 54.1 | +15.8 | 50.0 | 59.0 | +9.0 | 50.8 | 56.6 | +5.8 |
| m | 93.0 | 91.8 | −1.2 | 71.1 | 87.2 | +16.2 | 56.0 | 77.7 | +21.7 |
| n | 51.0 | 67.2 | +16.2 | 58.8 | 67.1 | +8.3 | 58.9 | 66.3 | +7.4 |
| o | 50.6 | 65.8 | +15.2 | 56.6 | 65.7 | +9.0 | 56.1 | 64.5 | +8.4 |
| p | 37.1 | 57.6 | +20.5 | 46.0 | 57.1 | +11.1 | 46.0 | 55.1 | +9.1 |
| Macro | 38.2 | 55.5 | +17.3 | 43.4 | 57.2 | +13.8 | 42.5 | 54.7 | +12.2 |
| Micro | 60.8 | 71.1 | +10.3 | 60.8 | 71.1 | +10.3 | 58.2 | 68.5 | +10.3 |
| TPR/SENS | F1 Score | MCC | |||||||
|---|---|---|---|---|---|---|---|---|---|
| PB | LOCUSTRA | PYTHIAb | DIFF | LOCUSTRA | PYTHIAb | DIFF | LOCUSTRA | PYTHIAb | DIFF |
| a | 47.6 | 67.0 | +19.4 | 53.9 | 58.5 | +4.6 | 52.9 | 56.4 | +3.5 |
| b | 20.6 | 43.0 | +22.4 | 29.2 | 41.4 | +12.2 | 30.2 | 37.4 | +7.2 |
| c | 37.1 | 49.9 | +12.8 | 44.6 | 52.6 | +8.0 | 41.6 | 46.9 | +5.3 |
| d | 69.0 | 61.5 | −7.5 | 69.9 | 70.4 | +0.5 | 63.0 | 63.4 | +0.4 |
| e | 28.1 | 62.7 | +34.6 | 35.9 | 51.6 | +15.7 | 36.3 | 50.6 | +14.3 |
| f | 37.6 | 57.4 | +19.8 | 46.7 | 57.0 | +10.3 | 45.5 | 52.6 | +7.1 |
| g | 3.4 | 34.1 | +30.7 | 6.2 | 22.7 | +16.5 | 10.6 | 22.4 | +11.8 |
| h | 24.6 | 59.7 | +35.1 | 33.1 | 48.0 | +15.0 | 34.4 | 47.1 | +12.7 |
| i | 26.4 | 55.3 | +28.9 | 34.6 | 46.2 | +11.7 | 35.7 | 45.5 | +9.8 |
| j | 2.9 | 56.5 | +53.6 | 5.4 | 35.9 | +30.5 | 10.1 | 37.5 | +27.4 |
| k | 43.5 | 61.7 | +18.2 | 52.7 | 59.4 | +6.7 | 52.0 | 56.0 | +4.0 |
| l | 38.3 | 57.9 | +19.6 | 50.0 | 54.2 | +4.2 | 50.8 | 50.5 | −0.3 |
| m | 93.0 | 80.3 | −12.7 | 71.1 | 85.9 | +14.9 | 56.0 | 76.7 | +20.7 |
| n | 51.0 | 72.8 | +21.8 | 58.8 | 60.3 | +1.6 | 58.9 | 60.1 | +1.2 |
| o | 50.6 | 71.2 | +20.6 | 56.6 | 59.8 | +3.2 | 56.1 | 59.0 | +2.8 |
| p | 37.1 | 60.5 | +23.4 | 46.0 | 51.4 | +5.5 | 46.0 | 49.2 | +3.2 |
| Macro | 38.2 | 59.5 | +21.3 | 43.4 | 53.5 | +10.1 | 42.5 | 50.7 | +8.2 |
| Micro | 60.8 | 65.4 | +4.6 | 60.8 | 65.4 | +4.6 | 58.2 | 62.0 | +3.8 |
| CASP 14Targets | Length | PYTHIA | LOCUSTRA | |
|---|---|---|---|---|
| Balanced | Global | |||
| 6uf2A | 125 | 69.4 | 69.4 | 47.7 |
| 6xc0C | 105 | 63.9 | 61.1 | 59.5 |
| 6y4fA | 141 | 56.9 | 57.7 | 48.2 |
| 6ya2A | 199 | 43.5 | 47.8 | 45.5 |
| 6zycA | 148 | 66.0 | 68.1 | 57.0 |
| 7d2oA | 174 | 36.6 | 42.1 | 30.4 |
| 7jtlA | 107 | 27.7 | 43.6 | 37.6 |
| 7k7wA | 590 | 66.9 | 69.2 | 56.4 |
| 7m7aA | 197 | 59.1 | 69.6 | 64.5 |
| 7m7aB | 590 | 69.1 | 70.5 | 66.5 |
| Mean | 55.9 | 59.9 | 51.3 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cretin, G.; Galochkina, T.; de Brevern, A.G.; Gelly, J.-C. PYTHIA: Deep Learning Approach for Local Protein Conformation Prediction. Int. J. Mol. Sci. 2021, 22, 8831. https://doi.org/10.3390/ijms22168831
Cretin G, Galochkina T, de Brevern AG, Gelly J-C. PYTHIA: Deep Learning Approach for Local Protein Conformation Prediction. International Journal of Molecular Sciences. 2021; 22(16):8831. https://doi.org/10.3390/ijms22168831
Chicago/Turabian StyleCretin, Gabriel, Tatiana Galochkina, Alexandre G. de Brevern, and Jean-Christophe Gelly. 2021. "PYTHIA: Deep Learning Approach for Local Protein Conformation Prediction" International Journal of Molecular Sciences 22, no. 16: 8831. https://doi.org/10.3390/ijms22168831
APA StyleCretin, G., Galochkina, T., de Brevern, A. G., & Gelly, J. -C. (2021). PYTHIA: Deep Learning Approach for Local Protein Conformation Prediction. International Journal of Molecular Sciences, 22(16), 8831. https://doi.org/10.3390/ijms22168831