
Vitamin D Deficiency, Osteoporosis and Effect on Autoimmune Diseases and Hematopoiesis: A Review

 ,
,  ,
,  ,
,  ,
, 
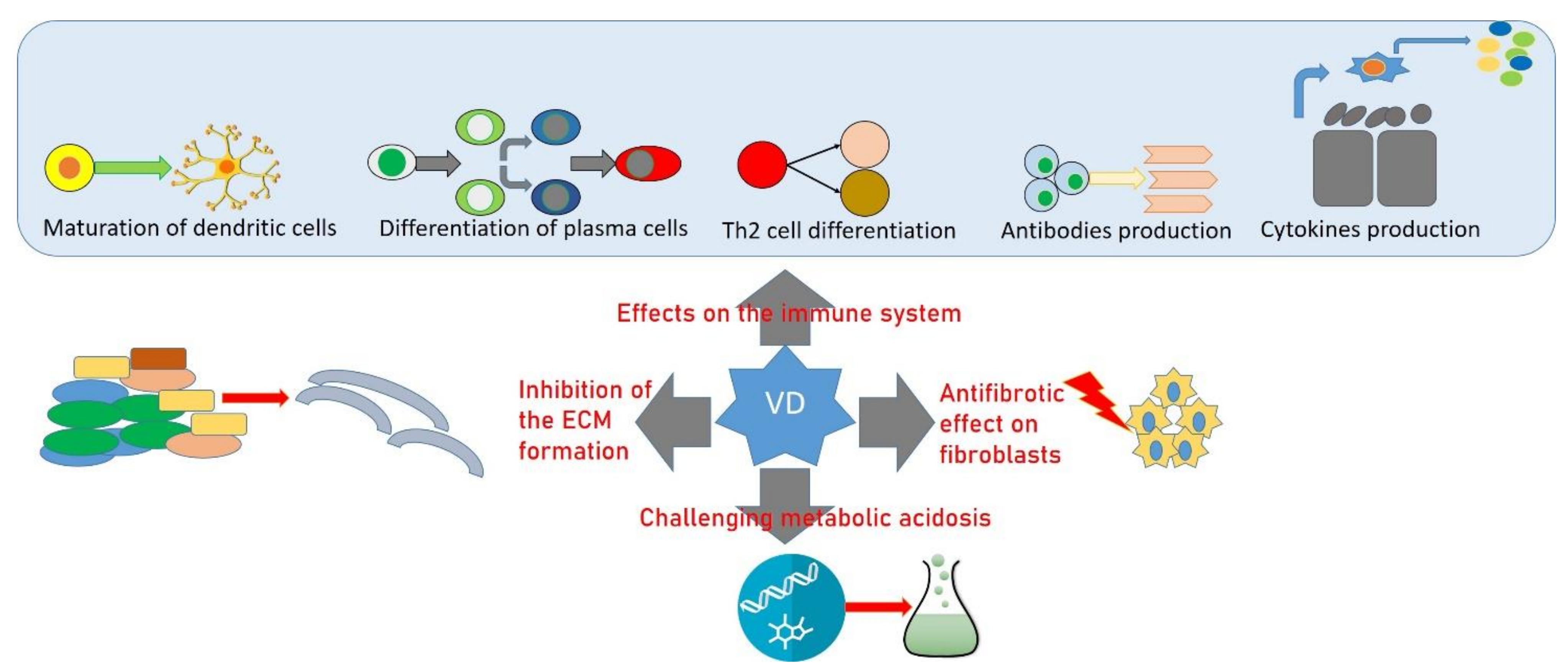{kind=link}
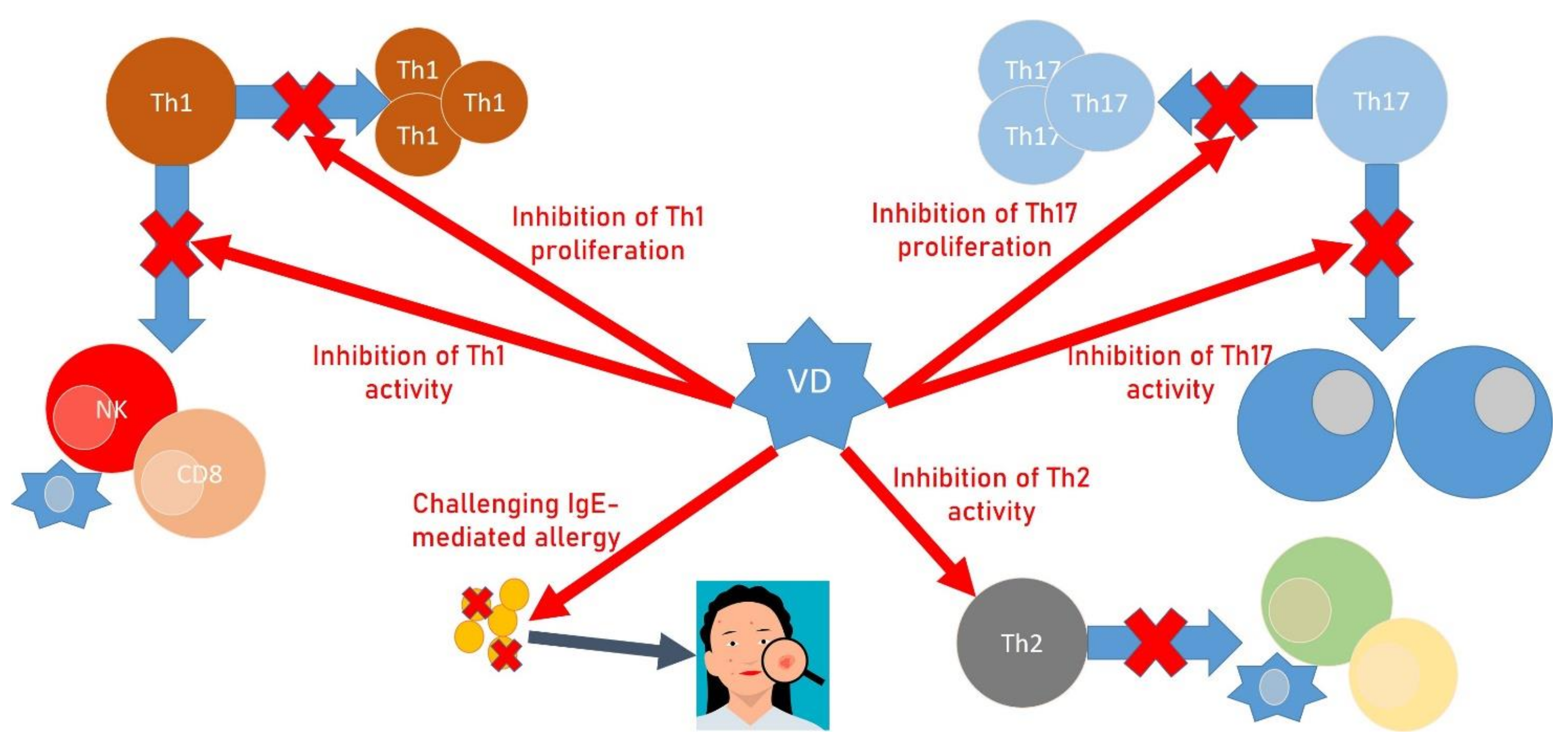{kind=link}
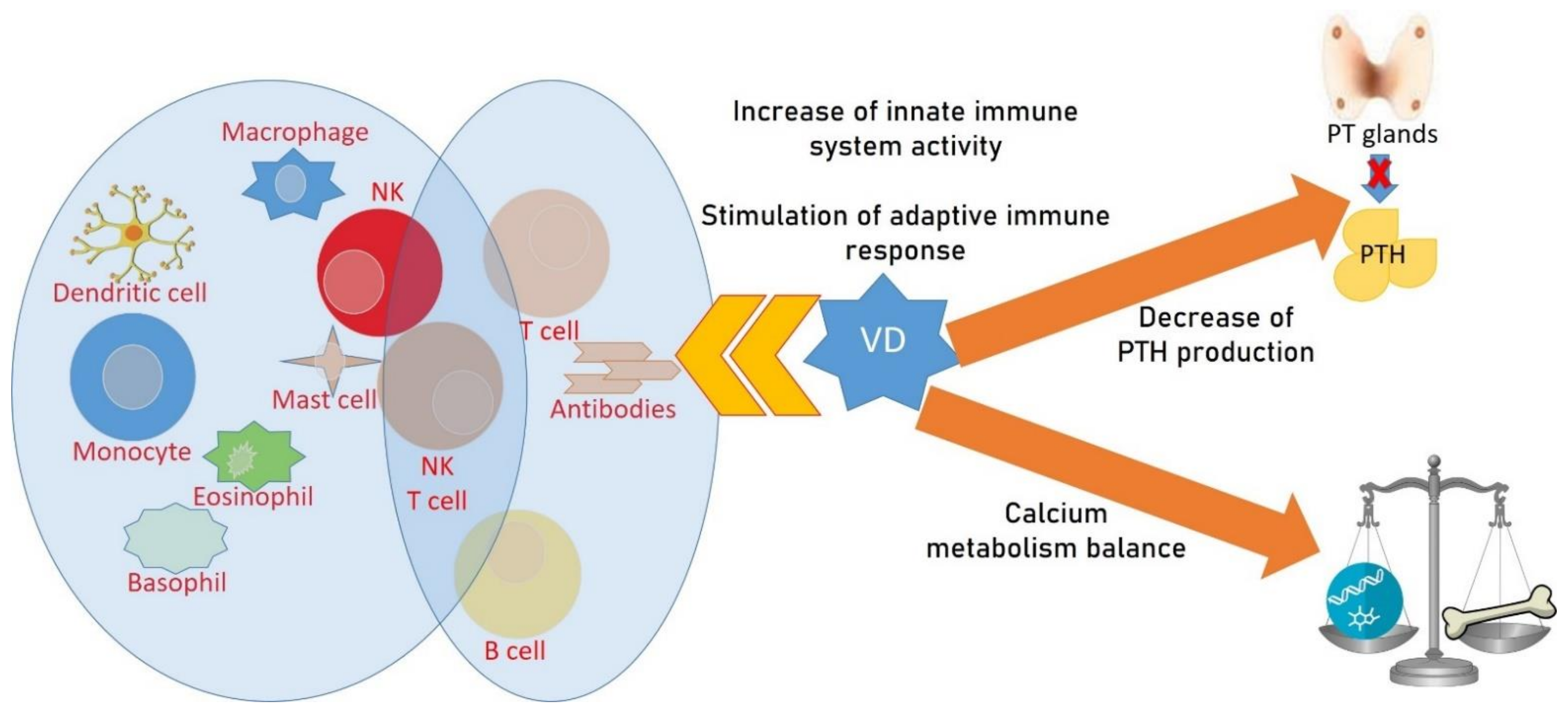{kind=link}
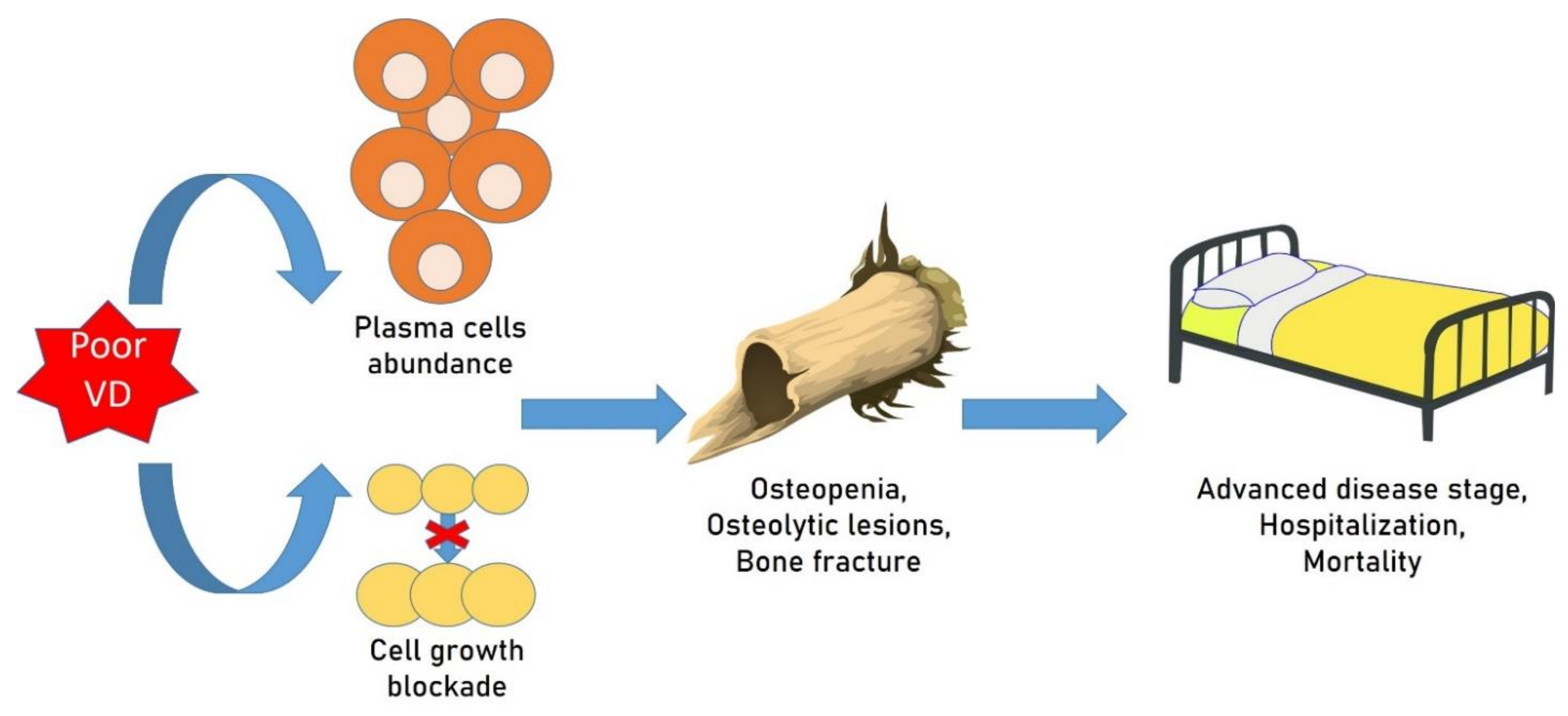{kind=link}
Abstract
:1. General Considerations on Vitamin D
1.1. Vitamin D and Osteoporosis
1.2. Vitamin D and Systemic Diseases
2. Autoimmune Diseases, VD Deficiency and OP
3. Allergy, Vitamin D, and OP
4. Endocrinological Diseases, Vitamin D and OP
5. Osteoporosis, Vitamin D and Monoclonal Gammopathies
6. Bone Marrow Transplantation
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Calvo, M.S.; Whiting, S.J.; Barton, C.N. Vitamin D intake: A global perspective of current status. J. Nutr. 2005, 135, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Cashman, K.D. Vitamin D in childhood and adolescence. Postgrad. Med. J. 2007, 83, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Zanuy, V.M.; Carranza, F.H. Metabolismo, fuentes endógenas y exógenasde vitamina D. Span. J. Metab. Bone Dis. 2007, 16, 63–70. [Google Scholar]
- Bendik, I.; Friedel, A.; Roos, F.F.; Weber, P.; Eggersdorfer, M. Vitamin D: A critical andessential micronutrient for human health. Front. Physiol. 2014, 5, 248. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F.; Binkley, N.C.; Bischoff-Ferrari, H.A.; Gordon, C.M.; Hanley, D.A.; Heaney, R.P.; Murad, M.H.; Weaver, C.M. Guidelines for preventing and treatingvitamin D deficiency and insufficiency revisited. J. Clin. Endocrinol. Metab. 2012, 97, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- De Pergola, G.; Martino, T.; Zupo, R.; Caccavo, D.; Pecorella, C.; Paradiso, S.; Silvestris, F.; Triggiani, V. 25 Hydroxyvitamin D levels are negatively and in-dependently associated with fat mass in a cohort of healthy overweight and obesesubjects. Endocr. Metab. Immune Disord. Drug Targets 2019, 19, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Muscogiuri, G.; Mitri, J.; Mathieu, C.; Badenhoop, K.; Tamer, G.; Orio, F.; Mezza, T.; Vieth, R.; Colao, A.; Pittas, A. Mechanisms in endocrinology: Vitamin D as a potentialcontributor in endocrine health and disease. Eur. J. Endocrinol. 2014, 171, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Pludowski, M.; Holick, W.; Grant, J.; Konstantynowicz, M.; Mascarenhas, A.; Hag, V.; Povoroznyuk, N.; Balatska, N.; Barbosa, A.P.; Karonova, T.; et al. Vitamin D supplementation guidelines. J. Steroid Biochem. Mol. Biol. 2018, 175, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Wimalawansa, J.; Razzaque, M.S.; Al-Daghri, N.M. Calcium and vitamin D inhuman health: Hype or real? J. Steroid Biochem. Mol. Biol. 2018, 180, 4–14. [Google Scholar] [CrossRef]
- Institute of Medicine. Dietary Reference Intakes for Calcium and Vitamin D; The National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Ross, A.C.; Manson, J.E.; Abrams, S.A.; Aloia, J.F.; Brannon, P.M.; Clinton, S.K.; Durazo-Arvizu, R.A.; Gallagher, J.C.; Gallo, R.L.; Jones, G.; et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: What clinicians need to know. J. Clin. Endocrinol. Metab. 2011, 96, 53–58. [Google Scholar] [CrossRef]
- Sempos, C.T.; Durazo-Arvizu, R.A.; Binkley, N.; Jones, J.; Merkel, J.M.; Carter, G.D. Developing vitamin D dietary guidelines and the lack of 25-hydroxyvitamin D assay standardization: The ever-present past. J. Steroid Biochem Mol. Biol. 2016, 164, 115–119. [Google Scholar] [CrossRef]
- Sempos, C.T.; Heijboer, A.C.; Bikle, D.D.; Bollerslev, J.; Bouillon, R.; Brannon, P.M.; DeLuca, H.F.; Jones, G.; Munns, C.F.; Bilezikian, J.P.; et al. Vitamin D assays and the definition of hypovitaminosis D: Results from the First International Conference on Controversies in Vitamin D. Br. J. Clin. Pharmacol. 2018, 84, 2194–2207. [Google Scholar] [CrossRef] [PubMed]
- Giustina, A.; Adler, R.A.; Binkley, N.; Bollerslev, J.; Bouillon, R.; Dawson-Hughes, B.; Ebeling, P.R.; Feldman, D.; Formenti, A.M.; Lazaretti-Castro, M.; et al. Consensus statement from 2nd International Conference on Controversies in Vitamin D. Rev. Endocr. Metab. Disord. 2020, 21, 89–116. [Google Scholar] [CrossRef] [Green Version]
- Nowson, C.A.; McGrath, J.J.; Ebeling, P.R.; Haikerwal, A.; Daly, R.M.; Sanders, K.M.; Seibel, M.J.; Mason, R.S.; Working Group of Australian and New Zealand Bone and Mineral Society; Endocrine Society of Australia and Osteoporosis Australia. Vitamin D and health in adults in Australia and New Zealand: A position statement. Med. J. Aust. 2012, 196, 686–687. [Google Scholar] [CrossRef]
- Bischoff-Ferrari, H.A.; Willett, W.C.; Wong, J.B.; Stuck, A.E.; Staehelin, H.B.; Orav, E.J.; Thoma, A.; Kiel, D.P.; Henschkowski, J. Prevention of nonvertebral fractures with oral vitamin D and dose dependency: A meta-analysis of randomized controlled trials. Arch. Intern. Med. 2009, 169, 551–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouillon, R.; Marcocci, C.; Carmeliet, G.; Bikle, D.; White, J.H.; Dawson-Hughes, B.; Lips, P.; Munns, C.F.; Lazaretti-Castro, M.; Giustina, A.; et al. Skeletal and Extraskeletal Actions of Vitamin D: Current Evidence and Outstanding Questions. Endocr. Rev. 2019, 40, 1109–1151. [Google Scholar] [CrossRef] [Green Version]
- Hewison, M. An update on vitamin D and human immunity. Clin. Endocrinol. 2012, 76, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.M.; Eslick, G.D.; Nowson, C.; Smith, C.; Bensoussan, A. Use of calcium or calcium in combination with vitamin D supplementation to prevent fractures and bone loss in people aged 50 years and older: A meta-analysis. Lancet 2007, 370, 657–666. [Google Scholar] [CrossRef]
- Weaver, C.M.; Alexander, D.D.; Boushey, C.J.; Dawson-Hughes, B.; Lappe, J.M.; LeBoff, M.S.; Liu, S.; Looker, A.C.; Wallace, T.C.; Wang, D.D. Calcium plus vitamin D supplementation and risk of fractures: An updated meta-analysis from the National Osteoporosis Foundation. Osteoporos. Int. 2016, 27, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Yao, P.; Bennett, D.; Mafham, M.; Lin, X.; Chen, Z.; Armitage, J.; Clarke, R. Vitamin D and Calcium for the Prevention of Fracture: A Systematic Review and Meta-analysis. JAMA Netw. Open 2019, 2, e1917789. [Google Scholar] [CrossRef]
- Avenell, A.; Mak, J.C.; O’Connell, D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst. Rev. 2014, 2014, CD000227. [Google Scholar] [CrossRef]
- Bolland, M.J.; Grey, A.; Avenell, A. Effects of vitamin D supplementation on musculoskeletal health: A systematic review, meta-analysis, and trial sequential analysis. Lancet Diabetes Endocrinol. 2018, 6, 847–858. [Google Scholar] [CrossRef] [Green Version]
- Khaw, K.T.; Stewart, A.W.; Waayer, D.; Lawes, C.M.M.; Toop, L.; Camargo, C.A., Jr.; Scragg, R. Effect of monthly high-dose vitamin D supplementation on falls and non-vertebral fractures: Secondary and post-hoc outcomes from the randomised, double-blind, placebo-controlled ViDA trial. Lancet Diabetes Endocrinol. 2017, 5, 438–447. [Google Scholar] [CrossRef] [Green Version]
- Cesareo, R.; Iozzino, M.; D’onofrio, L.; Terrinoni, I.; Maddaloni, E.; Casini, A.; Campagna, G.; Santonati, A.; Palermo, A. Effectiveness and safety of calcium and vitamin D treatment for postmenopausal osteoporosis. Minerva Endocrinol. 2015, 40, 231–237. [Google Scholar]
- Cranney, A.; Horsley, T.; O’Donnell, S.; Weiler, H.; Puil, L.; Ooi, D.; Atkinson, S.; Ward, L.; Moher, D.; Hanley, D.; et al. Effectiveness and safety of vitamin D in relation to bone health. Evid. Rep. Technol. Assess. 2007, 158, 1–235. [Google Scholar]
- Kahwati, L.C.; Weber, R.P.; Pan, H.; Gourlay, M.; LeBlanc, E.; Coker-Schwimmer, M.; Viswanathan, M. Vitamin D, Calcium, or Combined Supplementation for the Primary Prevention of Fractures in Community-Dwelling Adults: Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA 2018, 319, 1600–1612. [Google Scholar] [CrossRef]
- Burt, L.A.; Billington, E.O.; Rose, M.S.; Raymond, D.A.; Hanley, D.A.; Boyd, S.K. Effect of High-Dose Vitamin D Supplementation on Volumetric Bone Density and Bone Strength: A Randomized Clinical Trial. JAMA 2019, 322, 736–745. [Google Scholar] [CrossRef]
- Bischoff-Ferrari, H.A.; Dawson-Hughes, B.; Orav, E.J.; Staehelin, H.B.; Meyer, O.W.; Theiler, R.; Dick, W.; Willett, W.C.; Egli, A. Monthly High-Dose Vitamin D Treatment for the Prevention of Functional Decline: A Randomized Clinical Trial. JAMA Intern. Med. 2016, 176, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Billington, E.O.; Burt, L.A.; Rose, M.S.; Davison, E.M.; Gaudet, S.; Kan, M.; Boyd, S.K.; Hanley, D.A. Safety of High-Dose Vitamin D Supplementation: Secondary Analysis of a Randomized Controlled Trial. J. Clin. Endocrinol. Metab. 2020, 105, dgz212. [Google Scholar] [CrossRef] [PubMed]
- Malihi, Z.; Wu, Z.; Lawes, C.M.M.; Scragg, R. Adverse events from large dose vitamin D supplementation taken for one year or longer. J. Steroid Biochem. Mol. Biology 2019, 188, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Rizzoli, R. Vitamin D supplementation: Upper limit for safety revisited? Aging Clin. Exp. Res. 2021, 33, 19–24. [Google Scholar] [CrossRef]
- Armas, L.A.G.; Recker, R.R. Pathophysiology of osteoporosis: New mechanistic insights. Endocrinol. Metab. Clin. North. Am. 2014, 41, 475–486. [Google Scholar] [CrossRef]
- Lane, J.M.; Russell, L.; Khan, S.N. Osteoporosis. Clin. Orthop. Relat. Research. 2000, 372, 139–150. [Google Scholar] [CrossRef]
- Moon, S.; Ahn, I.; Jung, H.; Yi, H.; Kim, J.; Kim, Y.; Kwok, S.; Park, K.S.; Min, J.K.; Park, S.H.; et al. Temporal differential effects of proinflammatory cytokines on osteoclastogenesis. Int. J. Mol. Med. 2013, 31, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Montiel, B.Z.; Suárez, L.F.F. Ruta y retos diagnosticos en vasculitis primarias. Reum. Clin. 2011, 7, 1–6. [Google Scholar] [CrossRef]
- Kanis, J.A. Assessment of fracture risk and its application to screening for postmenopausal osteoporosis: Synopsis of a WHO report. Osteoporos. Int. 1994, 4, 368–381. [Google Scholar] [CrossRef]
- Lim, S.Y.; Bolster, M.B. Profile of romosozumab and its potential in the management of osteoporosis. Drug Des. Dev. Ther. 2017, 11, 1221–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirufo, M.M.; De Pietro, F.; Bassino, E.M.; Ginaldi, L.; De Martinis, M. Osteoporosis in Skin Diseases. Int. J. Mol. Sci. 2020, 21, 4749. [Google Scholar] [CrossRef] [PubMed]
- Fleet, J.C.; Desmet, M.; Johnson, R.; Li, Y. Vitamin D and cancer: A review of molecular mechanisms. Biochem. J. 2011, 441, 61–76. [Google Scholar] [CrossRef] [Green Version]
- Shanmugalingam, T.; Crawley, D.; Bosco, C.; Melvin, J.; Rohrmann, S.; Chowdhury, S.; Holmberg, L.; Van Hemelrijck, M. Obesity and cancer: The role of vitamin D. BMC Cancer 2014, 14, 712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wacker, M.; Holick, M.F. Vitamin D-Effects on skeletal and extraskeletal health and the need for supplementation. Nutrients 2013, 5, 111. [Google Scholar] [CrossRef] [Green Version]
- Zittermann, A. Vitamin D in preventive medicine: Are we ignoring the evidence? Br. J. Nutr. 2003, 89, 552–572. [Google Scholar] [CrossRef] [PubMed]
- Cantorna, M.T.; Zhu, Y.; Froicu, M.; Wittke, A. Vitamin D status, 1,25-dihydroxyvitamin D3, and the immune system. Am. J. Clin. Nutr. 2004, 80, 1717–1720. [Google Scholar] [CrossRef] [PubMed]
- Bock, G.; Pieber, T.R.; Prietl, B. Vitamin D: Role in autoimmunity. CAB Rev. 2012, 7, 1–7. [Google Scholar] [CrossRef]
- Antico, A.; Tampoia, M.; Tozzoli, R.; Bizzaro, N. Can supplementation with vitamin D reduce the risk or modify the course of autoimmune diseases? A systematic review of the literature. Autoimmun. Rev. 2012, 12, 127–136. [Google Scholar] [CrossRef]
- Gaudio, A.; Xourafa, A.; Rapisarda, R.; Zanoli, L.; Signorelli, S.S.; Castellino, P. Hematological Diseases and Osteoporosis. Int. J. Mol. Sci. 2020, 21, 3538. [Google Scholar] [CrossRef]
- Valderrábano, R.J.; Wu, J.Y. Bone and blood interactions in human health and disease. Bone 2019, 119, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Hiram-Bab, S.; Liron, T.; Deshet-Unger, N.; Mittelman, M.; Gassmann, M.; Rauner, M.; Franke, K.; Wielockx, B.; Neumann, D.; Gabet, Y. Erythropoietin directly stimulates osteoclast precursors and induces bone loss. FASEB J. 2015, 29, 1890–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toxqui, L.; Vaquero, M.P. Chronic iron deficiency as an emerging risk factor for osteoporosis: A hypothesis. Nutrients 2015, 7, 2324–2344. [Google Scholar] [CrossRef] [Green Version]
- Lemire, J.M.; Archer, D.C. 1,25-dihydroxyvitamin D3 prevents the in vivo induction of murine experimental autoimmune encephalomyelitis. JCI 1991, 87, 1103–1107. [Google Scholar] [CrossRef] [Green Version]
- Cantorna, M.T.; Hayes, C.E.; DeLuca, H.F. 1,25-Dihydroxyvitamin D3 reversibly blocks the progression of relapsing encephalomyelitis, a model of multiple sclerosis. PNAS 1996, 93, 7861–7864. [Google Scholar] [CrossRef] [Green Version]
- Cantorna, M.T.; Hayes, C.E.; DeLuca, H.F. 1,25-Dihydroxycholecalciferol inhibits the progression of arthritis in murine models of human arthritis. J. Nutr. 1998, 128, 68–72. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, H.A.; Mirshafiey, A.; Vahedi, H.; Hemmasi, G.; Khameneh, A.M.N.; Parastouei, K.; Saboor-Yaraghi, A.A. Immunoregulation of inflammatory and inhibitory cytokines by vitamin D3 in patients with inflammatory bowel diseases. Scand. J. Immunol. 2017, 85, 386–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murdaca, G.; Tonacci, A.; Negrini, S.; Greco, M.; Borro, M.; Puppo, F.; Gangemi, S. Emerging role of vitamin D in autoimmune diseases: An update on evidence and therapeutic implications. Autoimmun. Rev. 2019, 18, 102350. [Google Scholar] [CrossRef] [PubMed]
- Zipitis, C.S.; Akobeng, A.K. Vitamin D supplementation in early childhood and risk of type 1 diabetes: A systematic review and meta-analysis. Arch. Dis. Child. 2008, 93, 512–517. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.-Y.; Zhang, W.-G.; Chen, J.J.; Zhang, Z.-L.; Han, S.-F.; Qin, L.-Q. Vitamin D intake and risk of type 1 diabetes: A meta-analysis of observational studies. Nutrients 2013, 5, 3551–3562. [Google Scholar] [CrossRef] [PubMed]
- Gunasekar, P.; Swier, V.J.; Fleegel, J.P.; Boosani, C.S.; Radwan, M.M.; Agrawal, D.K. Vitamin D and macrophage polarization in epicardial adipose tissue of atherosclerotic swine. PLoS ONE 2018, 13, e0199411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, K.; You, Y.; Swier, V.; Tang, L.; Radwan, M.M.; Pandya, A.N.; Agrawal, D.K. Vitamin D protects against atherosclerosis via regulation of cholesterol efflux and macrophage polarization in hypercholesterolemic swine. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2432–2442. [Google Scholar] [CrossRef] [Green Version]
- Rosen, Y.; Daich, J.; Soliman, I.; Brathwaite, E.J.; Shoenfeld, Y. Vitamin D and auto-immunity. Scand. J. Rheumatol. 2016, 45, 439–447. [Google Scholar] [CrossRef]
- Vieth, R. Vitamin D toxicity, policy, and science. J. Bone Miner. Res. 2007, 22, 64–68. [Google Scholar] [CrossRef] [Green Version]
- Zittermann, A.; Prokop, S.; Gummert, J.F.; Borgermann, J. Safety issues of vitamin D supplementation. Anticancer Agents Med. Chem. 2013, 13, 4–10. [Google Scholar] [CrossRef]
- Vieth, R. The mechanisms of vitamin D toxicity. Bone Miner. 1990, 11, 267–272. [Google Scholar] [CrossRef]
- Wen, H.; Baker, J.F. Vitamin D: Immunoregulation, and rheumatoid arthritis. J. Clin. Rheumatol. 2011, 17, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Linker-Israeli, M.; Elstner, E.; Klinenberg, J.R.; Wallace, D.J.; Koeffler, H.P. Vitamin D(3) and its syntetic analogs inhibit the spontaneous in vitro immunoglobulin production by SLE-derived PBMC. Clin. Immunol. 2001, 99, 82–93. [Google Scholar] [CrossRef]
- Gatenby, P.; Lucas, R.; Swaminathan, A. Vitamin D deficiency and risk for rheumatic disease: An update. Cur. Opin. Rheumatol. 2013, 25, 148–191. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.R.; Xiao, J.-P.; Zhang, J.J.; Wu, Y.G. Decreased serum/plasma vitamin Dlevels in SLE patients: A meta-analysis. Curr. Pharm. Des. 2018, 24, 4466–4473. [Google Scholar] [CrossRef]
- Salman-Monte, T.C.; Torrente-Segarra, V.; Vega-Vidal, A.; Corzo, P.; Castro Dominguez, F.; Ojeda, F.; Carbonell-Abelló, J. Bone mineral density and vitamin D status in systemic lupus erythematosus (SLE): A systematic review. Autoimmun. Rev. 2017, 16, 1155–1159. [Google Scholar] [CrossRef]
- Ruaro, B.; Casabella, A.; Paolino, S.; Alessandri, E.; Patané, M.; Gotelli, E.; Sulli, A.; Cutolo, M. Trabecular Bone Score and Bone Quality in Systemic Lupus Erythematosus Patients. Front. Med. 2020, 7, 574842. [Google Scholar] [CrossRef] [PubMed]
- Almehed, K.; d’Elia, H.F.; Kvist, G.; Ohlss, C.; Carlsten, H. Prevalence and risk factors of osteoporosis in female SLE patients—extended report. Rheumatology 2007, 46, 1185–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.N.; Feng, X.Y.; He, L.; Zeng, L.X.; Hao, Z.M.; Lv, X.H.; Pu, D. Prevalence and possible risk factors of low bone mineral density in untreated female patients with systemic lupus erythematosus. BioMed Res. International. 2015, 7, 510514. [Google Scholar] [CrossRef]
- Salman-Monte, T.C.; Torrente-Segarra, V.; Almirall, M.; Corzo, P.; Moial, S.; Carbonell-Abell’o, J. Prevalence and predic tors of vitamin D insufciency in supplemented and non supplemented women with systemic lupus erythematosus in the Mediterranean region. Rheumatol. International. 2016, 36, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Bultink, I.E.M.; Lems, W.F.; Kostense, P.J.; Dijkmans, B.A.C.; Voskuyl, A.E. Prevalence of and risk factors for low bone mineral density and vertebral fractures in patients with systemic upus erythematosus. Arthritis Rheumatism. 2005, 52, 2044–2050. [Google Scholar] [CrossRef] [PubMed]
- Veale, D.J.; Fearon, U. The pathogenesis of psoriatic arthritis. Lancet 2018, 391, 2273–2284. [Google Scholar] [CrossRef]
- Ocampo, D.V.; Gladman, D. Psoriatic arthritis. F1000Research 2019, 20, 1665. [Google Scholar] [CrossRef] [PubMed]
- Ogdie, A.; Harter, L.; Shin, D.; Baker, J.; Takeshita, J.; Choi, H.K.; Love, T.J.; Gelfand, J.M. The risk of fracture among patients with psoriatic arthritis and psoriasis: A population-based study. Ann. Rheum. Dis. 2017, 76, 882–888. [Google Scholar] [CrossRef] [Green Version]
- Kathuria, P.; Gordon, K.B.; Silverberg, J.I. Association of psoriasis and psoriatic arthritis with osteoporosis and pathological fractures. J. Am. Acad. Dermatol. 2017, 76, 1045–1053. [Google Scholar] [CrossRef]
- Chen, T.L.; Lu, J.W.; Huang, Y.W.; Wang, J.H.; Su, K.Y. Bone Mineral Density, Osteoporosis, and Fracture Risk in Adult Patients with Psoriasis or Psoriatic Arthritis: A Systematic Review and Meta-Analysis of Observational Studies. J. Clin. Med. 2020, 9, 3712. [Google Scholar] [CrossRef]
- Mohammed, A.; Pagnoux, C.; McDo, H. Low Bone Density in Systemic Sclerosis. A Systematic Review. J. Rheumatol. 2013, 40, 1881–1890. [Google Scholar]
- Loucks, J.; Pope, J.E. Osteoporosis in scleroderma. Semin. Arthritis Rheum. 2005, 34, 678–682. [Google Scholar] [CrossRef]
- Antonelli, A.; Ferri, C.; Fallahi, P.; Cazzato, M.; Ferrari, S.M.; Sebastiani, M.; Ferrannini, E. Clinical and subclinical autoimmune thyroid disorders in systemic sclerosis. Eur. J. Endocrinol. 2007, 156, 431–437. [Google Scholar] [CrossRef]
- Kucharz, E.J. Thyroid disorders in patients with progressive systemic sclerosis: A review. Clin. Rheumatol. 1993, 12, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Leung, P.S.; Adamopoulos, I.E.; Gershwin, M.E. The implication of vitamin D and autoimmunity: A comprehensive review. Clin. Rev. Allergy Immunol. 2013, 45, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Groseanu, L.; Bojinca, V.; Gudu, T.; Saulescu, I.; Predeteanu, D.; Balanescu, A.; Berghea, F.; Opris, D.; Borangiu, A.; Constantinescu, C.; et al. Low vitamin D status in systemic sclerosis and the impact on disease phenotype. Eur. J. Rheumatol. 2016, 3, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Cormier, C.; Piras, M.; Mathieu, A.; Kahan, A.; Allanore, Y. Vitamin D deficiency and insufficiency in 2 independent cohorts of patients. J. Rheumatol. 2009, 36, 1924–1929. [Google Scholar] [CrossRef]
- Robberecht, E.; Vandewalle, S.; Wehlou, C.; Kaufman, J.M.; De Schepper, J. Sunlight is an important determinant of vitamin D serum concentrations in cystic fibrosis. Eur. J. Clin. Nutr. 2011, 65, 574–579. [Google Scholar] [CrossRef] [Green Version]
- Rischmuellerab, M.; Tieua, J.; Lester, S. Primary Sjögren’s syndrome. Best Pract. Res. Clin. Rheumatol. 2013, 30, 189–220. [Google Scholar] [CrossRef]
- Yang, C.; Albietz, J.; Harkin, D.; Kimlin, M.; Schmid, K.L. Impact of oral vitamin D supplementation on the ocular surface in people with dry eye and/or low serum vitamin D. Cont Lens Anterior Eye 2018, 41, 69–76. [Google Scholar] [CrossRef]
- Yin, Z.; Pintea, V.; Lin, Y.; Hammock, B.; Watsky, M.A. Vitamin D enhances corneal epithelial barrier function. Invstig. Ophthalmol. Vis. Sci. 2011, 52, 7359–7364. [Google Scholar] [CrossRef] [Green Version]
- Kizilgul, M.; Kan, S.; Ozcelik, O.; Beysel, S.; Apaydin, M.; Ucan, B.; Cakal, E. Vitamin D replacememnt improves tear osmolarity in patients with vitamin D deficiency. Semin. Ophthalmol. 2018, 33, 589–594. [Google Scholar] [CrossRef]
- Bron, A.; De Paiva, C.; Chauhan, S.; Bonini, S.; Gabison, E.; Jain, S.; Knop, E.; Markoulli, M.; Ogawa, Y.; Perez, V.; et al. TFOS DEWS II, pahophysiology report. Ocul. Surf. 2017, 15, 438–510. [Google Scholar] [CrossRef]
- Johnell, O.; Kanis, J. Epidemiology of osteoporotic fractures. Osteoporos. Int. 2005, 16, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Segal, B.; Carpenter, A.; Walk, D. Involvement of nervous system pathways in primary Sjögren’s syndrome. Rheum. Dis. Clin. North. Am. 2008, 34, 885–906. [Google Scholar] [CrossRef] [PubMed]
- Hofbauer, L.C.; Hamann, C.; Ebeling, P.R. Approach to the patient with secondary osteoporosis. Eur. J. Endocrinol. 2010, 162, 1009–1020. [Google Scholar] [CrossRef] [Green Version]
- Silverman, S.L.; Lane, N.E. Glucocorticoid-induced osteoporosis. Curr. Osteoporos. Rep. 2009, 7, 23–26. [Google Scholar] [CrossRef]
- Baldini, C.; Delle Sedie, A.; Luciano, N.; Pepe, P.; Ferro, F.; Talarico, R.; Tani, C.; Mosca, M. Vitamin D in "early" primary Sjögren’s syndrome: Does it play a role in influencing disease phenotypes? Rheumatol. Int. 2014, 34, 1159–1164. [Google Scholar] [CrossRef]
- Kobayashi, T.; Muto, S.; Nemoto, J.; Miyata, Y.; Ishiharajima, S.; Hironaka, M.; Asano, Y.; Kusano, E. Fanconi’s syndrome and distal (type 1) renal tubular acidosis in a patient with primary Sjogren’s syndrome with monoclonal gammopathy of undetermined significance. Clin. Nephrol. 2016, 65, 427–432. [Google Scholar] [CrossRef]
- Weger, W.; Kotanko, P.; Weger, M.; Deutschmann, H.; Scrabal, F. Prevalence and characterization of renal tubular acidosisin patients with osteopenia and osteoporosis and in non-porotic controls. Nephrol. Dial. Transplant. 2000, 15, 975–980. [Google Scholar] [CrossRef] [Green Version]
- Selmi, C.; Meroni, P.L.; Gershwin, M.E. Primary biliary cirrhosis and Sjogren’s syndrome: autoimmune epithelitis. J. Autoimmun. 2012, 39, 34–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, K.; Oxholm, P.; Sorenses, O.; Thymann, M.; Høier-Madsen, M.; Bendtzen, K. Abnormal vitamin D3 metabolism in patients with primary Sjogren’s syndrome. Ann. Rheum. Dis. 1990, 49, 682–684. [Google Scholar] [CrossRef] [Green Version]
- Enestrom, S.; Denneberg, T.; Eriksson, P. Histopathology of renal biopsies with correlation to clinical findings in primari Sjoegren’s syndrome. Clin. Exp. Rheumatol. 1995, 13, 697–703. [Google Scholar] [PubMed]
- Zilahi, E.; Chen, J.Q.; Papp, G.; Szántó, A.; Zeher, M. Lack of association of vitamin D receptor gene polymorphisms/haplotypes in Sjögren’s syndrome. Clin. Rheumatol. 2015, 34, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Agmon-Levin, N.; Kivity, S.; Tzioufas, A.G.; López Hoyos, M.; Rozman, B.; Efes, I.; Shapira, Y.; Shamis, A.; Amital, H.; Youinou, P.; et al. Low levels of vitamin-D are associated with neuropathy and lymphoma amongpatients with Sjögren’s syndrome. J. Autoimmun. 2012, 39, 234–239. [Google Scholar] [CrossRef]
- Fulop, M.; Mackay, M. Renal tubular acidosis, Sjögren’s syndrome, and bone disease. Arch. Intern. Med. 2004, 164, 905–909. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.G.; Ochalek, J.T.; Kaufmann, M.; Jones, G.; Deluca, H.F. CYP2R1 is a major, but not exclusive, contributor to 25-hydroxyvitamin D production in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 15650–15655. [Google Scholar] [CrossRef] [Green Version]
- Rubin, D.C.; Shaker, A.; Levin, M.S. Chronic intestinal inflammation: Inflammatory bowel disease and colitis-associated colon cancer. Front. Immunol. 2012, 3, 107. [Google Scholar] [CrossRef] [Green Version]
- Hewison, M. Vitamin D and the intracrinology of innate immunity. Mol. Cell. Endocrinol. 2010, 321, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.Z.; Li, Y.Y. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. 2014, 20, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Sgambato, D.; Gimigliano, F.; De Musis, C.; Moretti, A.; Toro, G.; Ferrante, E.; Miranda, A.; De Mauro, D.; Romano, L.; Iolascon, G.; et al. Bone alterations in inflammatory bowel diseases. World J. Clin. Cases 2019, 7, 1908–1925. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J.; Tsai, M. Mast cells: Versatile regulators of inflammation, tissue remodeling, host defense and homeostasis. J. Dermatol. Sci. 2008, 49, 7–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobigny, C.; Saffar, J.L. H1 and H2 histamine receptors modulate osteoclastic resorption by different pathways: Evidence obtained by using receptor antagonists in a rat synchronized resorption model. J. Cell. Physiol. 1997, 173, 10–18. [Google Scholar] [CrossRef]
- Rossini, M.; Zanotti, R.; Viapiana, O.; Tripi, G.; Orsolini, G.; Idolazzi, L.; Bonadonna, P.; Schena, D.; Escribano, L.; Adami, S.; et al. Bone Involvement and Osteoporosis in Mastocytosis. Immunol. Allergy Clin. North. Am. 2014, 34, 383–396. [Google Scholar] [CrossRef]
- Gatti, D.; Senna, G.; Viapiana, O.; Rossini, M.; Passalacqua, G.; Adami, S. Allergy and the bone: Unexpected relationships. Ann. Allergy Asthma Immunol. 2011, 107, 202–206. [Google Scholar] [CrossRef]
- Kang, J.; Lim, H.; Lee, D.; Yim, M. Montelukast inhibits RANKL-induced osteoclast formation and bone loss via CysLTR1 and P2Y12. Mol. Med. Rep. 2018, 18, 2387–2398. [Google Scholar] [CrossRef]
- Nachshon, L.; Goldberg, M.R.; Schwartz, N.; Sinai, T.; Amitzur-Levy, R.; Elizur, A.; Eisenberg, E.; Katz, Y. Decreased bone mineral density in young adult IgE-mediated cow’s milk–allergic patients. J. Allergy Clin. Immunol. 2014, 134, 1108–1113. [Google Scholar] [CrossRef]
- Bivona, G.; Agnello, L.; Ciaccio, M. The immunological implication of the new vitamin D metabolism. Central Eur. J. Immunol. 2018, 43, 331–334. [Google Scholar] [CrossRef]
- Arkwright, P.D.; Mughal, M.Z. Vertebral, pelvic, and hip fracture risk in adults with severe atopic dermatitis. J. Allergy Clin. Immunol. 2019, 145, 487–488. [Google Scholar]
- Sirufo, M.M.; Suppa, M.; Ginaldi, L.; De Martinis, M. Does Allergy Break Bones? Osteoporosis and Its Connection to Allergy. Int. J. Mol. Sci. 2020, 21, 712. [Google Scholar] [CrossRef] [Green Version]
- Saini, S.S. Chronic spontaneous urticaria. Immunol. Allergy Clin. N. Am. 2014, 34, 33–52. [Google Scholar]
- De Martinis, M.; Sirufo, M.M.; Ginaldi, L. A “stadium” urticaria, cold urticaria is still a mostly unknown disease, with a wide spectrum of severity degrees and few therapeutic certainties: Is omalizumab one of these? Reflections from a clinical case report. Iran. Red Crescent Med. J. 2019, 21, 84250. [Google Scholar] [CrossRef]
- Heine, G.; Anton, K.; Henz, B.M.; Worm, M. 1α,25-Dihydroxyvitamin D3 inhibits anti-CD40 plus IL-4-mediated IgE production in vitro. Eur. J. Immunol. 2002, 32, 3395–3404. [Google Scholar] [CrossRef]
- Reddy, P.A.; Harinarayan, C.V.; Sachan, A.; Suresh, V.; Rajagopal, G. Bone disease in thyrotoxicosis. Indian J. Med. Res. 2012, 135, 277–286. [Google Scholar] [PubMed]
- Baliram, R.; Latif, R.; Zaidi, M.; Davies, T.F. Expanding the Role of Thyroid-Stimulating Hormone in Skeletal Physiology. Front. Endocrinol. 2017, 8, 252. [Google Scholar] [CrossRef] [Green Version]
- Bassett, J.H.D.; Williams, G.R. Role of thyroid hormones in skeletal development and bone maintenance. Endocr. Rev. 2016, 37, 135–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, S.; Bruun, N.; Pedersen, K.M.; Laurberg, P. Biologic variation is important for interpretation of Thyroid function tests. Thyroid 2003, 13, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Bassett, J.H.D.; Williams, G.R. Critical role of the hypothalamic–pituitary–thyroid axis in bone. Bone 2008, 43, 418–426. [Google Scholar] [CrossRef]
- Kim, D. The Role of Vitamin D in Thyroid Diseases. Int. J. Mol. Sci. 2017, 18, 1949. [Google Scholar] [CrossRef] [Green Version]
- Muscogiuri, G.; Tirabassi, G.; Bizzaro, G.; Orio, F.; Paschou, S.A.; Vryonidou, A.; Balercia, G.; Shoenfeld, Y.; Colao, A. Vitamin D and thyroid disease: To D or not to D? Eur. J. Clin. Nutr. 2015, 69, 291–296. [Google Scholar] [CrossRef]
- Vondra, K.; Stárka, L.; Hampl, R. Vitamin D and thyroid diseases. Physiol. Res. 2015, 64, S95–S100. [Google Scholar] [CrossRef]
- Tamer, G.; Mesçi, B. Role of vitamin D in the immune system. Turk. J. Endocrinol. Metab. 2013, 17, 5–7. [Google Scholar] [CrossRef] [Green Version]
- Nettore, I.C.; Albano, L.; Ungaro, P.; Colao, A.; Macchia, P.E. Sunshine vitamin and thyroid. Rev. Endocr. Metab. Disord. 2017, 18, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandeira, F.; Cusano, N.E.; Silva, B.C.; Cassibba, S.; Almeida, C.B.; Machado, V.C.; Bilezikian, J.P. Bone disease in primary hyperparathyroidism. Arq. Bras. Endocrinol. Metabol. 2014, 58, 553–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christiansen, P. The skeleton in primary hyperparathyroidism: A review focusing on bone remodeling, structure, mass, and fracture. APMIS Suppl. 2001, 102, 1–52. [Google Scholar]
- Khosla, S.; Melton, L.J., 3rd.; Wermers, R.A.; Crowson, C.S.; O’Fallon, W.; Riggs, B. Primary hyperparathyroidism and the risk of fracture: A population-based study. J. Bone Miner. Res. 1999, 14, 1700–1707. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, P.; Mollerup, C.L.; Frokjaer, V.G.; Christiansen, P.; Blichert-Toft, M.; Mosekilde, L. Cohort study of risk of fracture before and after surgery for primary hyperparathyroidism. BMJ 2000, 321, 598–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.; Grey, A.; Shoback, D. Medical management of asymptomatic primary hyperparathyroidism: Proceedings of the third international workshop. J. Clin. Endocrinol. Metab. 2009, 94, 373–381. [Google Scholar] [CrossRef]
- Christiansen, P.; Steiniche, T.; Brixen, K.; Hessov, I.; Melsen, F.; Heickendorff, L.; Mosekilde, L. Primary hyperparathyroidism: Effect of parathyroidectomy on regional bone mineral density in Danish patients. A three-year follow-up study. Bone 1999, 25, 589–595. [Google Scholar] [CrossRef]
- Camozzi, V.; Luisetto, G.; Basso, S.M.; Cappelletti, P.; Tozzoli, R.; Lumachi, F. Treatment of chronic hypercalcemia. Med. Chem. 2012, 8, 556–563. [Google Scholar] [CrossRef]
- Yendt, E.R.; Kovacs, K.A.; Jones, G. Secondary hyperparathyroidism in primary osteoporosis and osteopenia: Optimizing calcium and vitamin D intakes to levels recommended by expert panels may not be sufficient for correction. Clin. Endocrinol. 2008, 69, 855–863. [Google Scholar] [CrossRef]
- Song, A.; Zhao, H.; Yang, Y.; Liu, S.; Nie, M.; Wang, O.; Xing, X. Safety and efficacy of common vitamin D supplementation in primary hyperparathyroidism and coexistent vitamin D deficiency and insufficiency: A systematic review and meta-analysis. J. Endocrinol. Investig. 2021, 44, 1667–1677. [Google Scholar] [CrossRef]
- Zini, M.; Attanasio, R.; Cesareo, R.; Emmolo, I.; Frasoldati, A.; Gianotti, L.; Guglielmi, R.; Piovesan, A.; Procopio, M.; Scillitani, A.; et al. Italian Association of Clinical Endocrinologists. AME position statement: Primary hyperparathyroidism in clinical practice. J. Endocrinol. Investig. 2012, 35 (Suppl. S7), 2–21. [Google Scholar] [PubMed]
- Janghorbani, M.; Van Dam, R.M.; Willett, W.C.; Hu, F.B. Systematic review of type 1 and type 2 diabetes mellitus and risk of fracture. Am. J. Epidemiol. 2007, 166, 495–505. [Google Scholar] [CrossRef]
- Melton, L.J., 3rd; Leibson, C.L.; Achenbach, S.J.; Therneau, T.M.; Khosla, S. Fracture risk in type 2 diabetes: Update of a population-based study. J. Bone Miner. Res. 2008, 23, 1334–1342. [Google Scholar] [CrossRef]
- Vestergaard, P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes—A meta-analysis. Osteoporos. Int. 2007, 18, 427–444. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Sugimoto, T. Bone metabolism and fracture risk in type 2 diabetes mellitus. Bonekey Rep. 2012, 7, 1–36. [Google Scholar] [CrossRef]
- Sanguineti, R.; Puddu, A.; Mach, F.; Montecucco, F.; Viviani, G.L. Advanced glycation end products play adverse proinflammatory activities in osteoporosis. Mediators Inflamm. 2014, 2014, 975872. [Google Scholar] [CrossRef] [Green Version]
- Lontchi-Yimagou, E.; Sobngwi, E.; Matsha, T.E.; Kengne, A.P. Diabetes mellitus and inflammation. Curr. Diab. Rep. 2013, 13, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Karstoft, K.; Pedersen, B.K. Exercise and type 2 diabetes: Focus on metabolism and inflammation. Immunol. Cell Biol. 2016, 94, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.E.; Coleman, C.M. Impact of Diabetes Mellitus on Bone Health. Int. J. Mol. Sci. 2019, 20, 4873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paschou, A.; Anastasia, D.; Dede, D.; Panagiotis, G. Type 2 Diabetes and Osteoporosis: A Guide to Optimal Management. J. Clin. Endocrinol. Metab. 2017, 102, 3621–3634. [Google Scholar] [CrossRef] [PubMed]
- Pappachan, J.M.; Hariman, C.; Edavalath, M.; Waldron, J.; Hanna, F.W. Cushing’s syndrome: A practical approach to diagnosis and differential diagnoses. J. Clin. Pathol. 2017, 70, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Cushing, H. The basophil adenomas of the pituitary body and their clinical manifestation (pituitary basophilism). Bull. Johns. Hopkins Hosp. 1932, 50, 137–195. [Google Scholar]
- Kaltsas, G.; Manetti, L.; Grossman, A.B. Osteoporosis in Cushing’s syndrome. Front. Horm. Res. 2002, 30, 60–72. [Google Scholar]
- Vestergaard, P.; Lindholm, J.; Jørgensen, J. Increased risk of osteoporotic fractures in patients with Cushing’s syndrome. Eur. J. Endocrinol. 2002, 146, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Sato, A.Y.; Peacock, M.; Bellido, T. Glucocorticoid Excess in Bone and Muscle. Clin. Rev. Bone Miner. Metab. 2018, 16, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Arnaldi, G.; Angeli, A.; Atkinson, A.B.; Bertagna, X.; Cavagnini, F.; Chrousos, G.P.; Fava, G.A.; Findling, J.W.; Gaillard, R.C.; Grossman, A.B.; et al. Diagnosis and complications of Cushing’s syndrome: A consensus statement. J. Clin. Endocrinol. Metab. 2003, 88, 5593–5602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, F.; Canalis, E. Management of endocrine disease: Secondary osteoporosis: Pathophysiology and management. Eur. J. Endocrinol. 2015, 173, 131–151. [Google Scholar] [CrossRef] [Green Version]
- Ritz, E.; Kreusser, W.; Rambausek, M. Effects of glucocorticoids on calcium and phosphate excretion. Adv. Exp. Med. Biol. 1984, 171, 381–397. [Google Scholar] [PubMed]
- Giustina, A.; Mazziotti, G.; Canalis, E. Growth hormone, insulin-like growth factors, and the skeleton. Endocr. Rev. 2008, 29, 535–559. [Google Scholar] [CrossRef] [Green Version]
- Snow-Harter, C.; Bouxsein, M.; Lewis, B.; Charette, S.; Weinstein, P.; Marcus, R. Muscle strength as a predictor of bone mineral density in young women. J. Bone Miner. Res. 1990, 5, 589–595. [Google Scholar] [CrossRef]
- Löfberg, E.; Gutierrez, A.; Wernerman, J.; Anderstam, B.; Mitch, W.E.; Price, S.R.; Bergström, J.; Alvestrand, A. Effects of high doses of glucocorticoids on free amino acids, ribosomes and protein turnover in human muscle. Eur. J. Clin. Investig. 2002, 32, 345–353. [Google Scholar] [CrossRef]
- Tomas, F.M.; Munro, H.N.; Young, V.R. Effect of glucocorticoid administration on the rate of muscle protein breakdown in vivo in rats, as measured by urinary excretion of NT-methylhistidine. Biochem. J. 1979, 178, 139–146. [Google Scholar] [CrossRef]
- De Paula, F.J.; Rosen, C.J. Bone Remodeling and Energy Metabolism: New Perspectives. Bone Research. 2013, 1, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Russell, M.; Mendes, N.; Miller, K.K.; Rosen, C.J.; Lee, H.; Klibanski, A.; Misra, M. Visceral fat is a negative predictor of bone density measures in obese adolescent girls. J. Clin. Endocrinol. Metab. 2010, 95, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Rozhinskaia, L.; Ermakova, I.P.; Marova, E.I.; Bukhman, A.I.; Pronchenko, I.A. Phosphorus-calcium metabolism and calcium-regulating hormones in endogenous hypercorticism. Probl. Endokrinol. 1986, 32, 13–18. [Google Scholar]
- Chiodini, I.; Carnevale, V.; Torlontano, V.M.; Fusilli, S.; Guglielmi, G.; Pileri, M.; Modoni, S.; Di Giorgio, A.; Liuzzi, A.; Minisola, S.; et al. Alterations of bone turnover and bone mass at different skeletal sites due to pure glucocorticoid excess: Study in eumenorrheic patients with Cushing’s syndrome. J. Clin. Endocrinol. Metab. 1998, 83, 1863–1867. [Google Scholar] [PubMed]
- Meng, X.W.; Liu, S.Q.; Zhang, K.Q. Metabolism of calcium and phosphorus in Cushing syndrome with osteoporosis. Zhonghua Nei Ke Za Zhi 1989, 28, 548–552. [Google Scholar] [PubMed]
- Kyle, R.A.; Benson, J.; Larson, D.; Therneau, T.; Dispenzieri, A.; Iii, L.J.M.; Rajkumar, S.V.; Melton, L.J. IgM monoclonal gammopathy of undetermined significance and smoldering Waldenström’s macroglobulinemia. Clin. Lymphoma Myeloma 2009, 9, 17–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyle, R.A.; Larson, D.R.; Therneau, T.M.; Dispenzieri, A.; Kumar, S.; Cerhan, J.R.; Rajkumar, S.V. Long-term follow-up of monoclonal gammopathy of undetermined significance. N. Engl. J. Med. 2018, 378, 241–249. [Google Scholar] [CrossRef]
- DisPenzieri, A.; Katzmann, J.A.; Kyle, R.A.; Larson, D.R.; Melton, L.J.; Colby, C.L.; Therneau, T.M.; Clark, R.; Kumar, S.K.; Bradwell, A.R.; et al. Prevalence and risk of progression of light-chain monoclonal gammopathy of undetermined significance: A retrospective population-based cohort study. Lancet 2010, 375, 1721–1728. [Google Scholar] [CrossRef] [Green Version]
- Therneau, T.M.; Kyle, R.A.; Melton, L.J.; Larson, D.R.; Benson, J.T.; Colby, C.L.; Dispenzieri, A.; Kumar, S.; Katzman, J.A.; Cerhan, J.R.; et al. Incidence of monoclonal gammopathy of undetermined signifiance and estimation of duration before first clinical recognition. Mayo Clin. Proc. 2012, 87, 1071–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Drake, M.T. Unveiling skeletal fragility in patients diagnosed with MGUS: No longer a condition of undetermined significance? J. Bone Miner. Res. 2014, 29, 2529–2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golombick, T.; Diamond, T. Prevalence of monoclonal gammopathy of undetermined significance/myeloma in patients with acute osteoporotic vertebral fractures. Acta Haematol. 2008, 120, 87–90. [Google Scholar] [CrossRef]
- Melton, L.J., 3rd.; Rajkumar, S.V.; Khosla, S.; Achenbach, S.J.; Oberg, A.L.; Kyle, R.A. Fracture risk in monoclonal gammopathy of undetermined significance. J. Bone Miner. Res. 2004, 19, 25–30. [Google Scholar] [CrossRef]
- Pepe, J.; Petrucci, M.T.; Nofroni, I.; Fassino, V.; Diacinti, D.; Romagnoli, E.; Minisola, S. Lumbar bone mineral density as the major factor determining increased prevalence of vertebral fractures in monoclonal gammopathy of undetermined significance. Br. J. Haematol. 2006, 134, 485–490. [Google Scholar] [CrossRef]
- Veronese, N.; Luchini, C.; Solmi, M.; Sergi, G.; Manzato, E.; Stubbs, B. Monoclonal gammopathy of undetermined significance and bone health outcomes: A systematic review and exploratory meta-analysis. J. Bone Miner. Metab. 2018, 36, 128–132. [Google Scholar] [CrossRef] [Green Version]
- Gregersen, H.; Ibsen, J.S.; Mellemkjoer, L.; Dahlerup, J.F.; Olsen, J.H.; Soerensen, H.T. Mortality and causes of death in patients with monoclonal gammopathy of undetermined significance. Br. J. Haematol. 2001, 112, 353–357. [Google Scholar] [CrossRef]
- Kristinsson, S.Y.; Tang, M.; Pfeiffer, R.M.; Björkholm, M.; Blimark, C.; Mellqvist, U.-H.; Wahlin, A.; Turesson, I.; Landgren, O. Monoclonal gammopathy of undetermined significance and risk of skeletal fractures: A population-based study. Blood 2010, 116, 2651–2655. [Google Scholar] [CrossRef] [Green Version]
- Abrahamsen, B.; Andersen, I.; Christensen, S.S.; Madsen, J.S.; Brixen, K. Utility of testing for monoclonal bands in serum of patients with suspected osteoporosis: Retrospective, cross sectional study. BMJ 2005, 330, 818. [Google Scholar] [CrossRef] [Green Version]
- Farr, J.N.; Zhang, W.; Kumar, S.K.; Jacques, R.M.; Ng, A.C.; McCready, L.K.; Rajkumar, S.V.; Drake, M.T. Altered cortical microarchitecture in patients with monoclonal gammopathy of undetermined significance. Blood 2014, 123, 647–649. [Google Scholar] [CrossRef] [Green Version]
- Stein, E.M.; Dash, A.; Bucovsky, M.; Agarwal, S.; Fu, J.; Lentzsch, S.; Shane, E. Disrupted radial and tibial microarchitecture in patients with monoclonal gammopathy of undetermined significance. Osteoporos. Int. 2019, 30, 629–635. [Google Scholar] [CrossRef]
- Piot, J.M.; Royer, M.; Schmidt-Tanguy, A.; Hoppé, E.; Gardembas, M.; Bourrée, T.; Hunault, M.; Francois, S.; Boyer, F.; Ifrah, N.; et al. Factors associated with an increased risk of vertebral fracture in monoclonal gammopathies of undetermined significance. Blood Cancer J. 2015, 5, e345. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.C.; Khosla, S.; Charatcharoenwitthaya, N.; Kumar, S.K.; Achenbach, S.J.; Holets, M.F.; McCready, L.K.; Melton, L.J.; Kyle, R.A.; Raikumar, S.V.; et al. Bone microstructural changes revealed by high-resolution peripheral quantitative computed tomography imaging and elevated DKK1 and MIP-1a levels i patients with MGUS. Blood 2011, 118, 6529–6534. [Google Scholar] [CrossRef]
- Orduna, G.; Mellibovsky, L.; Abella, E.; Nogués, X.; Granero, R.; García-Giralt, N.; Pineda-Moncusí, M.; Güerri-Fernández, R.; Prieto-Alhambra, D.; Díez-Pérez, A. Bone tissue quality in patients with monoclonal gammopathy of uncertain significance. J. Bone Miner. Metab. 2020, 38, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Kristinsson, S.Y.; Minter, A.R.; Korde, N.; Tan, E.; Landgren, O. Bone disease in multiple myeloma and precursor disease: Novel diagnostic approaches and implications on clinical management. Expert Rev. Mol. Diagn. 2011, 11, 593–603. [Google Scholar] [CrossRef]
- Graklanov, V.; Popov, V. Vitamin D levels in patients with non-Hodgkin lymphoma/diffuse large B-cell lymphoma, chronic lymphocytic leukemia and multiple myeloma. J. Int. Med. Res. 2020, 48, 1–8. [Google Scholar] [CrossRef]
- Ng, C.A.; Kumar, S.; Rajkumar, S.; Drake, M. Impact of vitamin D deficiency on the clinical presentation and prognosis of patients with newly diagnosed multiple myeloma. Am. J. Hematol. 2009, 84, 397–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauter, B.; Schmidt-Wolf, I. Prevalence, supplementation, and impact of vitamin D deficiency in multiple myeloma patients. Cancer Investig. 2015, 33, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Hudzik, S.; Snoad, B.; Mousa, L.; Sborov, D.W.; Williams, N.; Jones, D.; Hofmeister, C.C. The Majority of Myeloma Patients Are Vitamin D Deficient, Unrelated to Survival or Cytogenetics. Blood 2015, 126, 5336. [Google Scholar] [CrossRef]
- Diamond, T.; Golombrick, T.; Manoharan, A. Vitamin D status may affect the skeletal complications of multiple myeloma. Am. J. Hematol. 2010, 85, 302–303. [Google Scholar] [CrossRef]
- Park, W.H.; Seol, J.G.; Kim, E.S.; Binderup, L.; Koeffler, H.P.; Kim, B.K.; Lee, Y.Y. The induction of apoptosis by a combined 1,25(OH)2D3 analog, EB1089 and TGF-beta1 in NCI-H929 multiple myeloma cells. Int. J. Oncol. 2002, 20, 533–542. [Google Scholar]
- Rossi, J.F.; Durie, B.G.; Duperray, C.; Braich, T.; Marion, S.L.; Pike, J.W.; Haussler, M.R.; Janbon, C.; Bataille, R. Phenotypic and functional analysis of 1, 25- dihydroxyvitamin D3 receptor mediated modulation of the human myeloma cell line RPMI 8226. Cancer Res. 1988, 48, 1213–1216. [Google Scholar]
- Ozdemir, F.; Esen, N.; Ovali, E.; Tekelioglu, Y.; Yilmaz, M.; Aydin, F.; Kavgaci, H.; Boruban, C. Effects of Dexamethasone, All-Trans Retinoic Acid, Vitamin D3 and Interferon-alpha on FO Myeloma Cells. Chemotherapy 2004, 50, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Badros, A.; Goloubeva, O.; Terpos, E.; Milliron, T.; Baer, M.R.; Streeten, E. Prevalence and significance of vitamin D deficiency in multiple myeloma patients. Br. J. Haematol. 2008, 142, 492–494. [Google Scholar] [CrossRef]
- Lipe, B.; Kambhampati, S.; Van Veldhuizen, P.; Yacoub, A.; Aljitawi, O.; Mikhael, J. Correlation between markers of bone metabolism and vitamin D levels in patients with monoclonal gammopathy of undetermined significance (MGUS). Blood Cancer J. 2017, 7, 646. [Google Scholar] [CrossRef]
- Pepe, J.; Petrucci, M.T; Mascia, M.L.; Piemonte, S.; Fassino, V.; Romagnoli, E.; Minisola, S. The effects of alendronate treatment in osteoporotic patients affected by monoclonal gammopathy of undetermined significance. Calcif. Tissue Int. 2008, 82, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Berenson, J.R.; Yellin, O.; Boccia, R.V.; Flam, M.; Wong, S.F.; Batuman, O.; Moezi, M.M.; Woytowitz, D.; Duvivier, H.; Nassir, R.A. Zoledronic acid markedly improves bone mineral density for patients with monoclonal gammopathy of undetermined significance and bone loss. Clin. Cancer Res. 2008, 14, 6289–6295. [Google Scholar] [CrossRef] [Green Version]
- Ebeling, P.R.; Thomas, D.M.; Erbas, B.; Hopper, J.L.; Szer, J.; Grigg, A.P. Mechanisms of bone loss following allogeneic and autologous hemopoietic stem cell transplantation. J. Bone Miner. Res. 1999, 14, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Schulte, C.; Beelen, D.W.; Schaefer, U.W.; Mann, K. Bone loss in long-term survivors after transplantation of hematopoietic stem cells: A prospective study. Osteoporos. Int. 2000, 11, 344–353. [Google Scholar] [CrossRef]
- Weilbaecher, K.N. Mechanisms of osteoporosis after hematopoietic cell transplantation. Biol. Blood Marrow Transpl. 2000, 6, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Schulte, C.M.; Beelen, D.W. Bone loss following hematopoietic stem cell transplantation: A long-term follow-up. Blood 2004, 103, 3635–3643. [Google Scholar] [CrossRef] [Green Version]
- De Laet, C.; Kanis, J.A.; Oden, A.; Johanson, H.; Johnell, O.; Delmas, P.; Eisman, J.A.; Kroger, H.; Fujiwara, S.; Garnero, P.; et al. Body mass index as a predictor of fracture risk: A meta-analysis. Osteoporos. Int. 2005, 16, 1330–1338. [Google Scholar] [CrossRef]
- Van der Voort, D.J.; Geusens, P.P.; Dinant, G.J. Risk factors for osteoporosis related to their outcome: Fractures. Osteoporos. Int. 2001, 12, 630–638. [Google Scholar] [CrossRef]
- Stern, J.M.; Sullivan, K.M.; Ott, S.M.; Seidel, K.; Fink, J.C.; Longton, G. Bone density loss after allogeneic hematopoietic stem cell transplantation: A prospective study. Biol. Blood Marrow Transpl. 2001, 7, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Banfi, A.; Podestà, M.; Fazzuoli, L.; Sertoli, M.R.; Venturini, M.; Santini, G.; Cancedda, R.; Quarto, R. High-dose chemotherapy shows a dose-dependent toxicity to bone marrow osteoprogenitors: A mechanism for post-bone marrow transplantation osteopenia. Cancer 2001, 92, 2419–2428. [Google Scholar] [CrossRef]
- Lee, W.Y.; Cho, S.W.; Oh, E.S.; Oh, K.W.; Lee, J.M.; Yoon, K.H.; Kang, M.I.; Cha, B.Y.; Lee, K.W.; Son, H.Y.; et al. The effect of bone marrow transplantation on the osteoblastic differentiation of human bone marrow stromal cells. J. Clin. Endocrinol. Metab. 2002, 87, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Pundole, X.N.; Barbo, A.G.; Lin, H.; Champlin, R.E.; Lu, H. Increased incidence of fractures in recipients of hematopoietic stem-cell transplantation. J. Clin. Oncol. 2015, 33, 1364–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valimaki, M.J.; Kinnunen, K.; Volin, L.; Tahtela, R.; Loyttyniemi, E.; Laitinen, K.; Makela, P.; Keto, P.; Ruutu, T. A prospective study of bone loss and turnover after allogeneic bone marrow transplantation: Effect of calcium supplementation with or with. Bone Marrow Transpl. 1999, 23, 355–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, M.; Lekamwasam, S.; Inman, I.; Kaptoge, S.; Sizer, L.; Love, S.; Bearcroft, P.; Milligan, T.; Price, C.; Marcus, R.; et al. Significant and persistent loss of bone mineral density in the femoral neck after haematopoietic stem cell transplantation: Long-term follow-up of a prospective study. Br. J. Haematol. 2003, 121, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.N.; Vrooman, L.; Apfelbaum, E.M.; Whitley, K.; Bechard, L.; Lehmann, L.E. 25-hydroxy vitamin D deficiency following pediatric hematopoietic stem cell transplant. Biol. Blood Marrow Transpl. 2011, 17, 749–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faulhaber, G.A.; Premaor, M.O.; Moser Filho, H.L.; Silla, L.M.; Furlanetto, T.W. Low bone mineral density is associated with insulin resistance in bone marrow transplant subjects. Bone Marrow Transpl. 2009, 43, 953–957. [Google Scholar] [CrossRef] [Green Version]
- Baumgartner, A.; Moesch, M.; Zumsteg, M.; Struja, T.; Bernet, S.; Medinger, M.; Mueller, B.; Passweg, J.; Bargetzi, M.; Schuetz, P. Predictors of impaired bone health in long-term survivors after allogeneic stem cell transplantation. Bone Marrow Transplant. 2019, 54, 1651–1661. [Google Scholar] [CrossRef] [PubMed]
- Laroche, M.; Lemaire, O.; Attal, M. Vitamin D deficiency does not alter biochemical markers of bone metabolism before or after autograft in patients with multiple myeloma. Eur. J. Haematol. 2010, 85, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Campos, D.J.; Biagini, G.L.; Funke, V.A.; Bonfim, C.M.; Boguszewski, C.L.; Borba, V.Z. Vitamin D deficiency in children and adolescents submitted to hematopoietic stem cell transplantation. Rev. Bras. Hematol. Hemoter. 2014, 36, 126–131. [Google Scholar] [CrossRef] [Green Version]
- Wallace, G.; Jodele, S.; Howell, J.; Myers, K.C.; Teusink, A.; Zhao, X.; Setchell, K.; Holtzapfel, C.; Lane, A.; Taggart, C.; et al. Vitamin D deficiency and survival in children after hematopoietic stem cell transplant. Biol Blood Marrow Transpl. 2015, 21, 1627–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robien, K.; Strayer, L.G.; Majhail, N.; Lazovich, D.; Baker, K.S.; Smith, A.R.; Mulrooney, D.A.; Burns, L.J. Vitamin D status among long-term survivors of hematopoietic cell transplantation. Bone Marrow Transpl. 2011, 46, 1472–1479. [Google Scholar] [CrossRef] [Green Version]
- Margulies, S.L.; Kurian, D.; Elliott, M.S.; Han, Z. Vitamin D deficiency in patients with intestinal malabsorption syndromes-think in and outside the gut. J. Dig. Dis. 2015, 16, 617–633. [Google Scholar] [CrossRef]
- Prytula, A.; Cransberg, K.; Raes, A. CYP3A4 is a crosslink between vitamin D and calcineurin inhibitors in solid organ transplant recipients: Implications for bone health. Pharm. J. 2017, 17, 481–487. [Google Scholar] [CrossRef]
- Prytula, A.; Cransberg, K.; Raes, A. Drug-metabolizing enzymes CYP3A as a link between tacrolimus and vitamin D in renal transplant recipients: Is it relevant in clinical practice? Pediatr. Nephrol. 2019, 34, 1201–1210. [Google Scholar] [CrossRef]
- Pappa, H.M.; Bern, E.; Kamin, D.; Grand, R.J. Vitamin D status in gastrointestinal and liver disease. Curr. Opin. Gastroenterol. 2008, 24, 176–183. [Google Scholar] [CrossRef]
- Strugnell, S.A.; Sprague, S.M.; Ashfaq, A.; Petkovich, M.; Bishop, C.W. Rationale for raising current clinical practice guideline target for serum 25-hydroxyvitamin D in chronic kidney disease. Am. J. Nephrol. 2019, 49, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Anandi, P.; Jain, N.A.; Tian, X.; Wu, C.O.; Pophali, P.A.; Koklanaris, E.; Ito, S.; Savani, B.N.; Barrett, J.; Battiwalla, M. Factors influencing the late phase of recovery after bone mineral density loss in allogeneic stem cell transplantation survivors. Bone Marrow Transpl. 2016, 51, 1101–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosman, F.; de Beur, S.J.; LeBoff, M.S.; Lewiecki, E.M.; Tanner, B.; Randall, S.; Lindsay, R. Clinician’s guide to prevention and treatment of osteoporosis. Osteoporos. Int. 2014, 25, 2359–2381. [Google Scholar] [CrossRef] [Green Version]
- Tauchmanova, L.; De Simone, G.; Musella, T.; Orio, F.; Ricci, P.; Nappi, C.; Lombardi, G.; Colao, A.; Rotoli, B.; Selleri, C. Effects of various antireabsorptive treatments on bone mineral density in hypogonadal young women after allogeneic stem cell transplantation. Bone Marrow Transpl. 2006, 37, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Kananen, K.; Volin, L.; Laitinen, K.; Alfthan, H.; Ruutu, T.; Valimaki, M.J. Prevention of bone loss after allogeneic stem cell transplantation by calcium, vitamin D, and sex hormone replacement with or without pamidronate. J. Clin. Endocrinol. Metab. 2005, 90, 3877–3885. [Google Scholar] [CrossRef] [Green Version]
- Hermann, M.; Berger, P. Hormonal changes in aging men: A therapeutic indication? Exp. Gerontol. 2001, 36, 1075–1082. [Google Scholar] [CrossRef]
- Bar, M.; Ott, S.M.; Lewiecki, E.M.; Sarafoglou, K.; Wu, J.Y.; Thompson, M.J.; Vaux, J.J.; Dean, D.R.; Saag, K.G.; Hashmi, S.K.; et al. Bone Health Management After Hematopoietic Cell Transplantation: An Expert Panel Opinion from the American Society for Transplantation and Cellular Therapy. Biol. Blood Marrow Transpl. 2020, 26, 1784–1802. [Google Scholar] [CrossRef]
- Campos, D.J.; Boguszewski, C.L.; Funke, V.A.; Bonfim, C.M.; Kulak, C.A.; Pasquini, R.; Borba, V.Z. Bone mineral density, vitamin D, and nutritional status of children submitted to hematopoietic stem cell transplantation. Nutrition 2014, 30, 654–659. [Google Scholar] [CrossRef]
- Caballero-Velazquez, T.; Montero, I.; Sanchez-Guijo, F.; Parody, R.; Saldana, R.; Valcarcel, D.; López-Godino, O.; Ferra I Coll, C.; Cuesta, M.; Carrillo-Vico, A.; et al. Immunomodulatory effect of vitamin D after allogeneic stem cell transplantation: Results of a prospective multicenter clinical trial. Clin. Cancer Res. 2016, 22, 5673–5681. [Google Scholar] [CrossRef] [Green Version]
- Glotzbecker, B.; Ho, V.T.; Aldridge, J.; Kim, H.T.; Horowitz, G.; Ritz, J.; Soiffer, R.; Avigan, D.; Rosenblatt, J. Low levels of 25-hydroxyvitamin D before allogeneic hematopoietic SCT correlate with the development of chronic GVHD. Bone Marrow Transpl. 2013, 48, 593–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Bahr, L.; Blennow, O.; Alm, J.; Bjorklund, A.; Malmberg, K.J.; Mougiakakos, D.; Le Blanc, A.; Oefner, P.J.; Labopin, M.; Ljungman, P.; et al. Increased incidence of chronic GvHD and CMV disease in patients with vitamin D deficiency before allogeneic stem cell transplantation. Bone Marrow Transpl. 2015, 50, 1217–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snegarova, V.; Naydenova, D. Vitamin D: A Review of its Effects on Epigenetics and Gene Regulation. Folia Med. 2020, 62, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Wimalawansa, S.J. Vitamin D Deficiency: Effects on Oxidative Stress, Epigenetics, Gene Regulation, and Aging. Biology 2019, 8, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Martinis, M.; Allegra, A.; Sirufo, M.M.; Tonacci, A.; Pioggia, G.; Raggiunti, M.; Ginaldi, L.; Gangemi, S. Vitamin D Deficiency, Osteoporosis and Effect on Autoimmune Diseases and Hematopoiesis: A Review. Int. J. Mol. Sci. 2021, 22, 8855. https://doi.org/10.3390/ijms22168855
De Martinis M, Allegra A, Sirufo MM, Tonacci A, Pioggia G, Raggiunti M, Ginaldi L, Gangemi S. Vitamin D Deficiency, Osteoporosis and Effect on Autoimmune Diseases and Hematopoiesis: A Review. International Journal of Molecular Sciences. 2021; 22(16):8855. https://doi.org/10.3390/ijms22168855
Chicago/Turabian StyleDe Martinis, Massimo, Alessandro Allegra, Maria Maddalena Sirufo, Alessandro Tonacci, Giovanni Pioggia, Martina Raggiunti, Lia Ginaldi, and Sebastiano Gangemi. 2021. "Vitamin D Deficiency, Osteoporosis and Effect on Autoimmune Diseases and Hematopoiesis: A Review" International Journal of Molecular Sciences 22, no. 16: 8855. https://doi.org/10.3390/ijms22168855
APA StyleDe Martinis, M., Allegra, A., Sirufo, M. M., Tonacci, A., Pioggia, G., Raggiunti, M., Ginaldi, L., & Gangemi, S. (2021). Vitamin D Deficiency, Osteoporosis and Effect on Autoimmune Diseases and Hematopoiesis: A Review. International Journal of Molecular Sciences, 22(16), 8855. https://doi.org/10.3390/ijms22168855