
Mice Harboring a Non-Functional CILK1/ICK Allele Fail to Model the Epileptic Phenotype in Patients Carrying Variant CILK1/ICK

 , , and
, , and 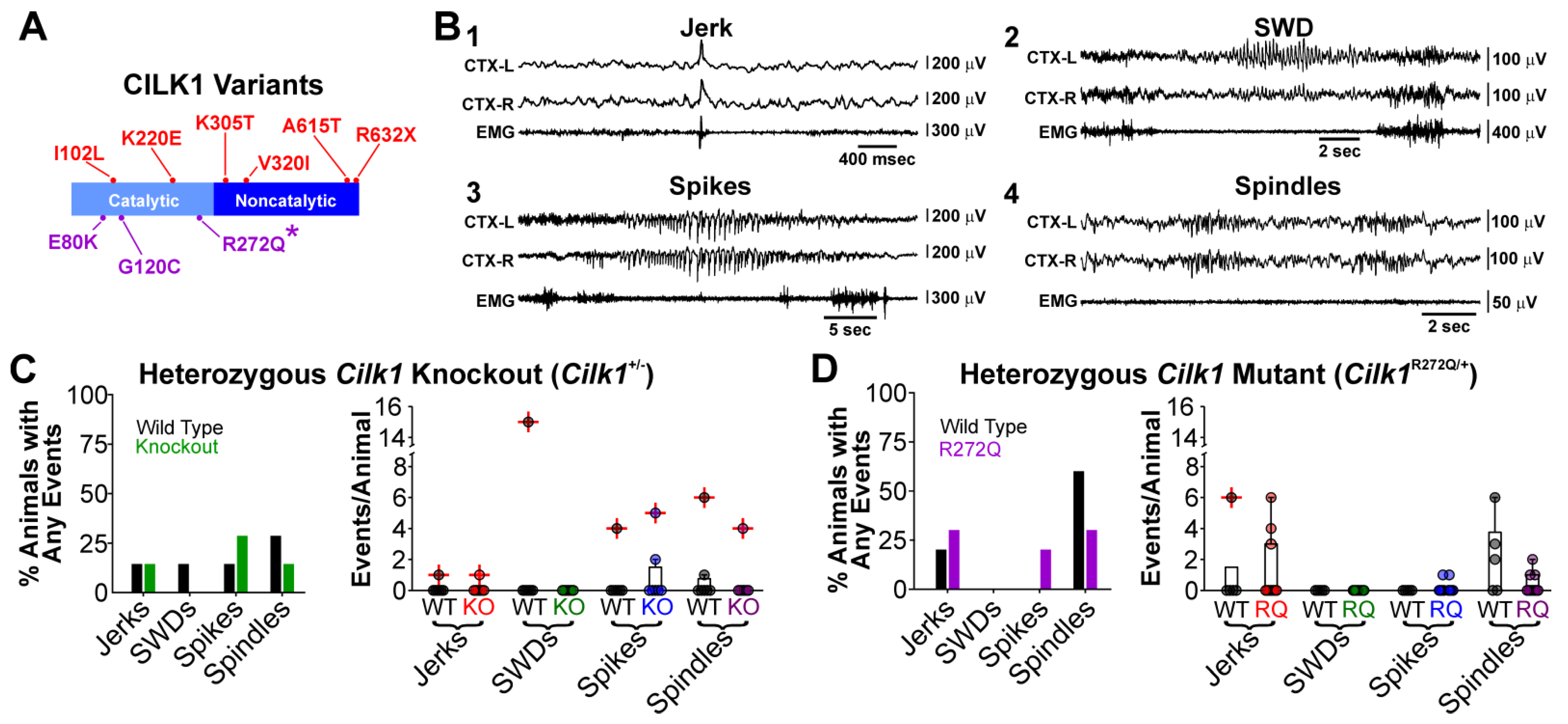{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Cilk1+/− and Cilk1R272Q/+ Mice do Not Exhibit Electrographic Seizures
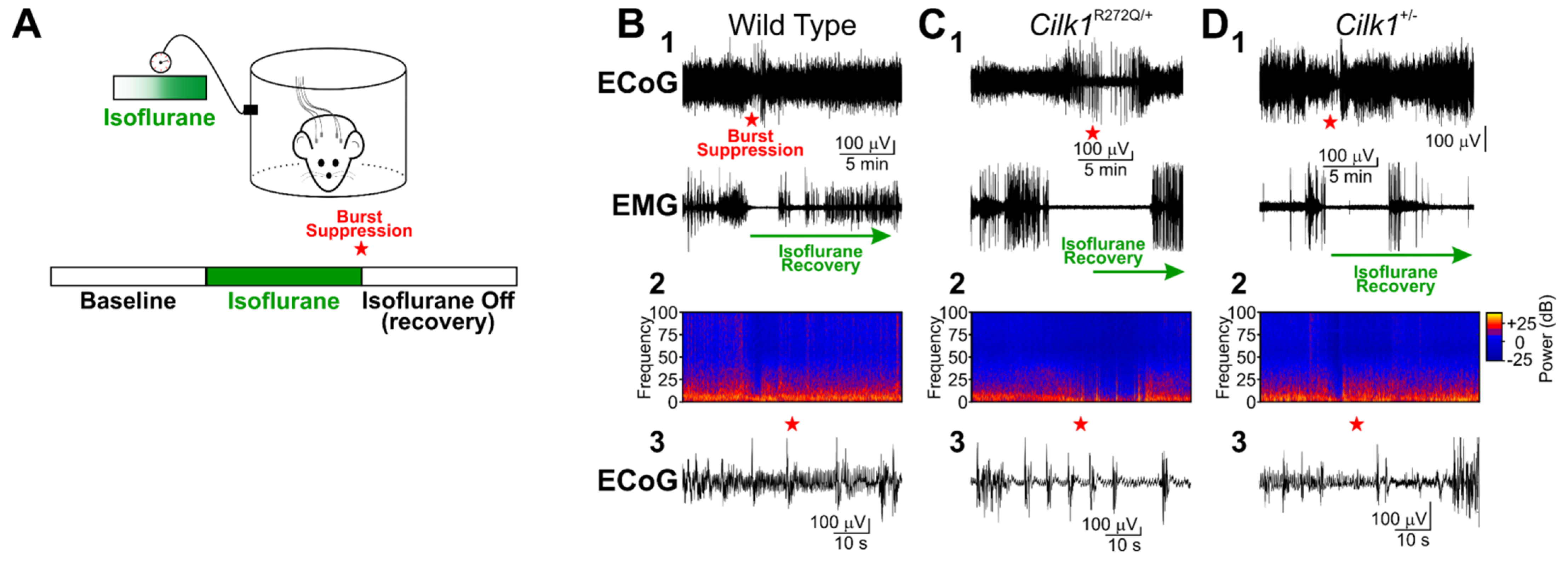
2.2. Isoflurane Does Not Induce Generalized Tonic–Clonic Seizures in Cilk1+/− and Cilk1R272Q/+ Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Electrocorticography (ECoG)/Electromyographic (EMG) Surgery
4.3. Chronic Electrocorticography (ECoG)/Electromyographic (EMG) Recordings
4.4. Electrocorticography (ECoG)/Electromyographic (EMG) Recording with Isoflurane
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gerdes, J.M.; Davis, E.E.; Katsanis, N. The vertebrate primary cilium in development, homeostasis, and disease. Cell 2009, 137, 32–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef]
- Fu, Z.; Gailey, C.D.; Wang, E.J.; Brautigan, D.L. Ciliogenesis associated kinase 1: Targets and functions in various organ systems. FEBS Lett. 2019, 593, 2990–3002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahiry, P.; Wang, J.; Robinson, J.F.; Turowec, J.P.; Litchfield, D.W.; Lanktree, M.B.; Gloor, G.B.; Puffenberger, E.G.; Strauss, K.A.; Martens, M.B.; et al. A multiplex human syndrome implicates a key role for intestinal cell kinase in development of central nervous, skeletal, and endocrine systems. Am. J. Hum. Genet. 2009, 84, 134–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paige Taylor, S.; Kunova Bosakova, M.; Varecha, M.; Balek, L.; Barta, T.; Trantirek, L.; Jelinkova, I.; Duran, I.; Vesela, I.; Forlenza, K.N.; et al. An inactivating mutation in intestinal cell kinase, ICK, impairs hedgehog signalling and causes short rib-polydactyly syndrome. Hum. Mol. Genet. 2016, 25, 3998–4011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oud, M.M.; Bonnard, C.; Mans, D.A.; Altunoglu, U.; Tohari, S.; Ng, A.Y.; Eskin, A.; Lee, H.; Rupar, C.A.; de Wagenaar, N.P.; et al. A novel ICK mutation causes ciliary disruption and lethal endocrine-cerebro-osteodysplasia syndrome. Cilia 2016, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.; Song, J.; Shin, J.O.; Lee, H.; Kim, H.K.; Eggenschwiller, J.T.; Bok, J.; Ko, H.W. Intestinal cell kinase, a protein associated with endocrine-cerebro-osteodysplasia syndrome, is a key regulator of cilia length and Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 8541–8546. [Google Scholar] [CrossRef] [Green Version]
- Chaya, T.; Omori, Y.; Kuwahara, R.; Furukawa, T. ICK is essential for cell type-specific ciliogenesis and the regulation of ciliary transport. EMBO J. 2014, 33, 1227–1242. [Google Scholar] [CrossRef] [Green Version]
- Tong, Y.; Park, S.H.; Wu, D.; Xu, W.; Guillot, S.J.; Jin, L.; Li, X.; Wang, Y.; Lin, C.S.; Fu, Z. An essential role of intestinal cell kinase in lung development is linked to the perinatal lethality of human ECO syndrome. FEBS Lett. 2017, 591, 1247–1257. [Google Scholar] [CrossRef] [Green Version]
- Youn, Y.H.; Han, Y.G. Primary Cilia in Brain Development and Diseases. Am. J. Pathol. 2018, 188, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.N.; de Nijs, L.; Bai, D.; Suzuki, T.; Miyamoto, H.; Tanaka, M.; Patterson, C.; Lin, Y.C.; Medina, M.T.; Alonso, M.E.; et al. Variant Intestinal-Cell Kinase in Juvenile Myoclonic Epilepsy. N. Engl. J. Med. 2018, 378, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Jin, L.; Xie, L.; Park, S.H.; Tong, Y.; Wu, D.; Chhabra, A.B.; Fu, Z.; Li, X. A Murine Model for Human ECO Syndrome Reveals a Critical Role of Intestinal Cell Kinase in Skeletal Development. Calcif. Tissue Int. 2018, 102, 348–357. [Google Scholar] [CrossRef]
- Lerche, H.; Berkovic, S.F.; Lowenstein, D.H. Intestinal-Cell Kinase and Juvenile Myoclonic Epilepsy. N. Engl. J. Med. 2019, 380, e24. [Google Scholar] [PubMed] [Green Version]
- Uygun, D.S.; Katsuki, F.; Bolortuya, Y.; Aguilar, D.D.; McKenna, J.T.; Thankachan, S.; McCarley, R.W.; Basheer, R.; Brown, R.E.; Strecker, R.E.; et al. Validation of an automated sleep spindle detection method for mouse electroencephalography. Sleep 2019, 42, zsy218. [Google Scholar] [CrossRef]
- Purdon, P.L.; Sampson, A.; Pavone, K.J.; Brown, E.N. Clinical Electroencephalography for Anesthesiologists: Part I: Background and Basic Signatures. Anesthesiology 2015, 123, 937–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Stierer, T.; Zuckerman, R.; Sakima, N.; Parker, S.D.; Fleisher, L.A. Comparison of recovery profile after ambulatory anesthesia with propofol, isoflurane, sevoflurane and desflurane: A systematic review. Anesth Analg. 2004, 98, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Stokes, E.L.; Flecknell, P.A.; Richardson, C.A. Reported analgesic and anaesthetic administration to rodents undergoing experimental surgical procedures. Lab. Anim. 2009, 43, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, H.; Ogli, K. Opisthotonus during exposure to isoflurane, enflurane, and halothane in mice. Anesthesiology 1987, 67, 771–774. [Google Scholar] [CrossRef]
- Komatsu, H.; Izumikawa, N.; Yoda, K.; Morita, J.; Chujo, K.; Endo, S.; Nogaya, J.; Ueki, M.; Ogli, K. Clonic convulsive movements during and on emergence from sevoflurance anesthesia. J. Anesth. 1996, 10, 72–75. [Google Scholar] [CrossRef]
- Komatsu, H.; Yokono, S.; Ogli, K. The central nervous stimulating effect of four different halogenated ether anesthetics and halothane in mice. J. Anesth. 1988, 2, 115–117. [Google Scholar] [CrossRef]
- Sadan, O.; Neufeld, M.Y.; Parmet, Y.; Rozenberg, A.; Kipervasser, S. Psychogenic seizures: Long-term outcome in patients with and without epilepsy. Acta Neurol Scand. 2016, 133, 145–151. [Google Scholar] [CrossRef]
- Kholi, H.; Bellier, A.; Vercueil, L. PNESSE 1: Psychogenic status and status epilepticus: Could they be distinguished retrospectively? A survey among neurologists. Epilepsy Behav. 2020, 102, 106665. [Google Scholar] [CrossRef]
- Delgado-Escueta, A.V. Advances in genetics of juvenile myoclonic epilepsies. Epilepsy Curr. 2007, 7, 61–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vulliemoz, S.; Vollmar, C.; Koepp, M.J.; Yogarajah, M.; O’Muircheartaigh, J.; Carmichael, D.W.; Stretton, J.; Richardson, M.P.; Symms, M.R.; Duncan, J.S. Connectivity of the supplementary motor area in juvenile myoclonic epilepsy and frontal lobe epilepsy. Epilepsia 2011, 52, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Woermann, F.G.; Free, S.L.; Koepp, M.J.; Sisodiya, S.M.; Duncan, J.S. Abnormal cerebral structure in juvenile myoclonic epilepsy demonstrated with voxel-based analysis of MRI. Brain 1999, 122 Pt 11, 2101–2108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caciagli, L.; Wandschneider, B.; Xiao, F.; Vollmar, C.; Centeno, M.; Vos, S.B.; Trimmel, K.; Sidhu, M.K.; Thompson, P.J.; Winston, G.P.; et al. Abnormal hippocampal structure and function in juvenile myoclonic epilepsy and unaffected siblings. Brain 2019, 142, 2670–2687. [Google Scholar] [CrossRef]
- Medina, M.T.; Suzuki, T.; Alonso, M.E.; Duron, R.M.; Martinez-Juarez, I.E.; Bailey, J.N.; Bai, D.; Inoue, Y.; Yoshimura, I.; Kaneko, S.; et al. Novel mutations in Myoclonin1/EFHC1 in sporadic and familial juvenile myoclonic epilepsy. Neurology 2008, 70 Pt 2, 2137–2144. [Google Scholar] [CrossRef]
- Suzuki, T.; Delgado-Escueta, A.V.; Aguan, K.; Alonso, M.E.; Shi, J.; Hara, Y.; Nishida, M.; Numata, T.; Medina, M.T.; Takeuchi, T.; et al. Mutations in EFHC1 cause juvenile myoclonic epilepsy. Nat. Genet. 2004, 36, 842–849. [Google Scholar] [CrossRef]
- Kang, S.K.; Hawkins, N.A.; Kearney, J.A. C57BL/6J and C57BL/6N substrains differentially influence phenotype severity in the Scn1a (+/−) mouse model of Dravet syndrome. Epilepsia Open 2019, 4, 164–169. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.J.; Gailey, C.D.; Brautigan, D.L.; Fu, Z. Functional Alterations in Ciliogenesis-Associated Kinase 1 (CILK1) that Result from Mutations Linked to Juvenile Myoclonic Epilepsy. Cells 2020, 9, 694. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Otis, J.M.; Higginbotham, H.; Monckton, C.; Cheng, J.; Asokan, A.; Mykytyn, K.; Caspary, T.; Stuber, G.D.; Anton, E.S. Primary Cilia Signaling Shapes the Development of Interneuronal Connectivity. Dev. Cell 2017, 42, 286–300.e4. [Google Scholar] [CrossRef] [PubMed]
- Arkan, S.; Kasap, M.; Akman, O.; Akpinar, G.; Ates, N.; Karson, A. The lower expression of parvalbumin in the primary somatosensory cortex of WAG/Rij rats may facilitate the occurrence of absence seizures. Neurosci. Lett. 2019, 709, 134299. [Google Scholar] [CrossRef] [PubMed]
- Magloire, V.; Mercier, M.S.; Kullmann, D.M.; Pavlov, I. GABAergic Interneurons in Seizures: Investigating Causality with Optogenetics. Neuroscientist 2019, 25, 344–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panthi, S.; Leitch, B. The impact of silencing feed-forward parvalbumin-expressing inhibitory interneurons in the cortico-thalamocortical network on seizure generation and behaviour. Neurobiol. Dis. 2019, 132, 104610. [Google Scholar] [CrossRef]
- Staley, K. Molecular mechanisms of epilepsy. Nat. Neurosci. 2015, 18, 367–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salvati, K.A.; Mason, A.J.; Gailey, C.D.; Wang, E.J.; Fu, Z.; Beenhakker, M.P. Mice Harboring a Non-Functional CILK1/ICK Allele Fail to Model the Epileptic Phenotype in Patients Carrying Variant CILK1/ICK. Int. J. Mol. Sci. 2021, 22, 8875. https://doi.org/10.3390/ijms22168875
Salvati KA, Mason AJ, Gailey CD, Wang EJ, Fu Z, Beenhakker MP. Mice Harboring a Non-Functional CILK1/ICK Allele Fail to Model the Epileptic Phenotype in Patients Carrying Variant CILK1/ICK. International Journal of Molecular Sciences. 2021; 22(16):8875. https://doi.org/10.3390/ijms22168875
Chicago/Turabian StyleSalvati, Kathryn A., Ashley J. Mason, Casey D. Gailey, Eric J. Wang, Zheng Fu, and Mark P. Beenhakker. 2021. "Mice Harboring a Non-Functional CILK1/ICK Allele Fail to Model the Epileptic Phenotype in Patients Carrying Variant CILK1/ICK" International Journal of Molecular Sciences 22, no. 16: 8875. https://doi.org/10.3390/ijms22168875
APA StyleSalvati, K. A., Mason, A. J., Gailey, C. D., Wang, E. J., Fu, Z., & Beenhakker, M. P. (2021). Mice Harboring a Non-Functional CILK1/ICK Allele Fail to Model the Epileptic Phenotype in Patients Carrying Variant CILK1/ICK. International Journal of Molecular Sciences, 22(16), 8875. https://doi.org/10.3390/ijms22168875