
Nitro-Oleic Acid (NO2-OA) Improves Systolic Function in Dilated Cardiomyopathy by Attenuating Myocardial Fibrosis

 , , , , and
, , , , and 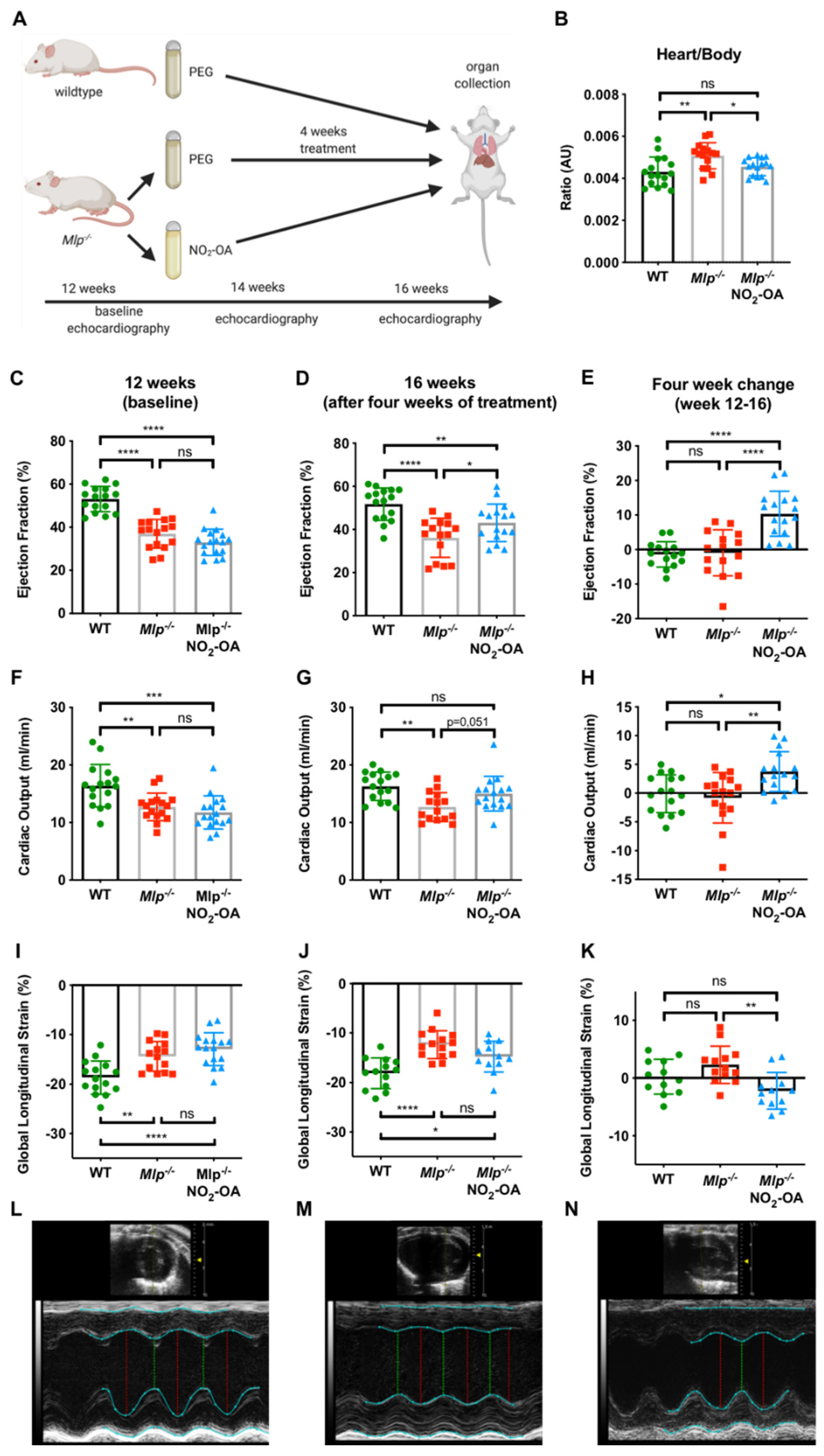{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. NO2-OA Improves Left Ventricular Systolic Function in Mlp−/− Mice
2.2. NO2-OA Attenuates Myocardial Fibrosis in DCM
2.3. Mlp−/− Mice Demonstrate Increased TGFβ Signaling
2.4. NO2-OA Modulates TGFβ Signaling in Isolated Primary Cardiac Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Animal Conditions and Experimental Design
4.2. Echocardiography
4.3. Histology and Total Collagen Assay
4.4. Isolation, Cultivation and Stimulation of Cardiac Fibroblasts
4.5. Quantitative Real-Time PCR
4.6. Immunoblot
4.7. Immunofluorescene Staining
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Towbin, J.A.; Lowe, A.M.; Colan, S.D.; Sleeper, L.A.; Orav, E.J.; Clunie, S.; Messere, J.; Cox, G.F.; Lurie, P.R.; Hsu, D.; et al. Incidence, Causes, and Outcomes of Dilated Cardiomyopathy in Children. JAMA 2006, 296, 1867–1876. [Google Scholar] [CrossRef]
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B. Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006, 113, 1807–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayvanpour, E.; Sedaghat-Hamedani, F.; Amr, A.; Lai, A.; Haas, J.; Holzer, D.B.; Frese, K.S.; Keller, A.; Jensen, K.; Katus, H.A.; et al. Genotype-phenotype associations in dilated cardiomyopathy: Meta-analysis on more than 8000 individuals. Clin. Res. Cardiol. 2016, 106, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Arbustini, E.; Favalli, V.; Narula, N. Extracellular Volume in Dilated Cardiomyopathy. JACC Cardiovasc. Imaging 2018, 11, 60–63. [Google Scholar] [CrossRef]
- Ma, Z.-G.; Yuan, Y.-P.; Wu, H.-M.; Zhang, X.; Tang, Q.-Z. Cardiac fibrosis: New insights into the pathogenesis. Int. J. Biol. Sci. 2018, 14, 1645–1657. [Google Scholar] [CrossRef] [Green Version]
- Watkins, H.; Ashrafian, H.; Redwood, C. Inherited Cardiomyopathies. N. Engl. J. Med. 2011, 364, 1643–1656. [Google Scholar] [CrossRef]
- Györfi, A.H.; Matei, A.-E.; Distler, J.H. Targeting TGF-β signaling for the treatment of fibrosis. Matrix Biol. 2018, 68-69, 8–27. [Google Scholar] [CrossRef]
- Zhang, Y. Non-Smad pathways in TGF-β signaling. Cell Res. 2008, 19, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Ma, C.; Yang, H.; Zhang, P.-Y. Transforming growth factor β and its role in heart disease. Exp. Ther. Med. 2017, 13, 2123–2128. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M.; Strek, M.E. Antifibrotic therapy for idiopathic pulmonary fibrosis: Time to treat. Respir. Res. 2019, 20, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Brilla, C.G.; Funck, R.C.; Rupp, H. Lisinopril-Mediated Regression of Myocardial Fibrosis in Patients with Hypertensive Heart Disease. Circulation 2000, 102, 1388–1393. [Google Scholar] [CrossRef] [Green Version]
- Perez, O.; Garvin, A.; Hale, T. Transient ACE-Inhibitor Treatment Produces Persistent Change in Cardiac Fibroblast Physiology. FASEB J. 2018, 32, 867.4. [Google Scholar] [CrossRef]
- Rudolph, T.K.; Ravekes, T.; Klinke, A.; Friedrichs, K.; Mollenhauer, M.; Pekarova, M.; Ambrozova, G.; Martiskova, H.; Kaur, J.-J.; Matthes, B.; et al. Nitrated fatty acids suppress angiotensin II-mediated fibrotic remodelling and atrial fibrillation. Cardiovasc. Res. 2015, 109, 174–184. [Google Scholar] [CrossRef] [Green Version]
- Khoo, N.K.; Freeman, B.A. Electrophilic nitro-fatty acids: Anti-inflammatory mediators in the vascular compartment. Curr. Opin. Pharmacol. 2010, 10, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsikas, D.; Zoerner, A.A.; Mitschke, A.; Gutzki, F.-M. Nitro-fatty Acids Occur in Human Plasma in the Picomolar Range: A Targeted Nitro-lipidomics GC–MS/MS Study. Lipids 2009, 44, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, S.; Vitturi, D.; Baker, P.R.; Bonacci, G.; Koenitzer, J.R.; Woodcock, S.R.; Freeman, B.A.; Schopfer, F.J. Characterization and quantification of endogenous fatty acid nitroalkene metabolites in human urine. J. Lipid Res. 2013, 54, 1998–2009. [Google Scholar] [CrossRef] [Green Version]
- Cole, M.P.; Rudolph, T.K.; Khoo, N.K.H.; Motanya, U.N.; Golin-Bisello, F.; Wertz, J.W.; Schopfer, F.J.; Rudolph, V.; Woodcock, S.R.; Bolisetty, S.; et al. Nitro-Fatty Acid Inhibition of Neointima Formation After Endoluminal Vessel Injury. Circ. Res. 2009, 105, 965–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolph, V.; Rudolph, T.K.; Schopfer, F.J.; Bonacci, G.; Woodcock, S.R.; Cole, M.P.; Baker, P.R.; Ramani, R.; Freeman, B.A. Endogenous generation and protective effects of nitro-fatty acids in a murine model of focal cardiac ischaemia and reperfusion. Cardiovasc. Res. 2009, 85, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Mollenhauer, M.; Mehrkens, D.; Rudolph, V. Nitrated fatty acids in cardiovascular diseases. Nitric Oxide 2018, 78, 146–153. [Google Scholar] [CrossRef]
- Klinke, A.; Berghausen, E.; Friedrichs, K.; Molz, S.; Lau, D.; Remane, L.; Berlin, M.; Kaltwasser, C.; Adam, M.; Mehrkens, D.; et al. Myeloperoxidase aggravates pulmonary arterial hypertension by activation of vascular Rho-kinase. JCI Insight 2018, 3, e97530. [Google Scholar] [CrossRef]
- Schopfer, F.J.; Vitturi, D.A.; Jorkasky, D.K.; Freeman, B.A. Nitro-fatty acids: New drug candidates for chronic inflammatory and fibrotic diseases. Nitric Oxide 2018, 79, 31–37. [Google Scholar] [CrossRef]
- Arber, S.; Hunter, J.J.; Ross, J.; Hongo, M.; Sansig, G.; Borg, J.; Perriard, J.-C.; Chien, K.R.; Caroni, P. MLP-Deficient Mice Exhibit a Disruption of Cardiac Cytoarchitectural Organization, Dilated Cardiomyopathy, and Heart Failure. Cell 1997, 88, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Knöll, R.; Kostin, S.; Klede, S.; Savvatis, K.; Klinge, L.; Stehle, I.; Gunkel, S.; Kötter, S.; Babicz, K.; Sohns, M.; et al. A Common MLP (Muscle LIM Protein) Variant Is Associated with Cardiomyopathy. Circ. Res. 2010, 106, 695–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Repetti, G.G.; Toepfer, C.N.; Seidman, J.G.; Seidman, C.E. Novel Therapies for Prevention and Early Treatment of Cardiomyopathies. Circ. Res. 2019, 124, 1536–1550. [Google Scholar] [CrossRef] [PubMed]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; John, G.F.C.; Andrew, J.S.C.; Falk, V.; Juanatey, J.R.G.; Harjola, V.-P.; Jankowska, E.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Hear. Fail. 2016, 18, 891–975. [Google Scholar] [CrossRef]
- Taniguchi, H.; Ebina, M.; Kondoh, Y.; Ogura, T.; Azuma, A.; Suga, M.; Taguchi, Y.; Takahashi, H.; Nakata, K.; Sato, A.; et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur. Respir. J. 2009, 35, 821–829. [Google Scholar] [CrossRef]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolph, V.; Schopfer, F.J.; Khoo, N.K.H.; Rudolph, T.K.; Cole, M.P.; Woodcock, S.R.; Bonacci, G.; Groeger, A.L.; Golin-Bisello, F.; Chen, C.-S.; et al. Nitro-fatty Acid Metabolome: Saturation, Desaturation, β-Oxidation, and Protein Adduction. J. Biol. Chem. 2009, 284, 1461–1473. [Google Scholar] [CrossRef] [Green Version]
- Rea, D.; Coppola, C.; Barbieri, A.; Monti, M.G.; Misso, G.; Palma, G.; Bimonte, S.; Zarone, M.R.; Luciano, A.; Liccardo, D.; et al. Strain Analysis in the Assessment of a Mouse Model of Cardiotoxicity due to Chemotherapy: Sample for Preclinical Research. In Vivo 2016, 30, 279–290. [Google Scholar] [PubMed]
- De Lucia, C.; Wallner, M.; Eaton, D.M.; Zhao, H.; Houser, S.R.; Koch, W.J. Echocardiographic Strain Analysis for the Early Detection of Left Ventricular Systolic/Diastolic Dysfunction and Dyssynchrony in a Mouse Model of Physiological Aging. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2018, 74, 455–461. [Google Scholar] [CrossRef]
- Hoffman, M.; Kyriazis, I.; Lucchese, A.M.; De Lucia, C.; Piedepalumbo, M.; Bauer, M.; Schulze, P.C.; Bonios, M.J.; Koch, W.J.; Drosatos, K. Myocardial Strain and Cardiac Output are Preferable Measurements for Cardiac Dysfunction and Can Predict Mortality in Septic Mice. J. Am. Hear. Assoc. 2019, 8, e012260. [Google Scholar] [CrossRef] [PubMed]
- Pauschinger, M.; Knopf, D.; Petschauer, S.; Doerner, A.; Poller, W.; Schwimmbeck, P.L.; Kühl, U.; Schultheiss, H.-P. Dilated Cardiomyopathy Is Associated with Significant Changes in Collagen Type I/III ratio. Circulation 1999, 99, 2750–2756. [Google Scholar] [CrossRef] [Green Version]
- Schalla, S.; Bekkers, S.C.; Dennert, R.; Van Suylen, R.J.; Waltenberger, J.; Leiner, T.; Wildberger, J.; Crijns, H.J.; Heymans, S. Replacement and reactive myocardial fibrosis in idiopathic dilated cardiomyopathy: Comparison of magnetic resonance imaging with right ventricular biopsy. Eur. J. Hear. Fail. 2010, 12, 227–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Jia, Z.; Liu, S.; Downton, M.; Liu, G.; Du, Y.; Yang, T. Combined losartan and nitro-oleic acid remarkably improves diabetic nephropathy in mice. Am. J. Physiol. Physiol. 2013, 305, F1555–F1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaka, Y. Targeting TGF-β Signaling in Kidney Fibrosis. Int. J. Mol. Sci. 2018, 19, 2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabregat, I.; Moreno-Caceres, J.; Sánchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; Dijke, P.T. The IT-LIVER Consortium TGF-β signalling and liver disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef] [Green Version]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Zhang, Y. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nat. Cell Biol. 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Klinke, A.; Möller, A.; Pekarova, M.; Ravekes, T.; Friedrichs, K.; Berlin, M.; Scheu, K.M.; Kubala, L.; Kolarova, H.; Ambrozova, G.; et al. Protective Effects of 10-nitro-oleic Acid in a Hypoxia-Induced Murine Model of Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2014, 51, 155–162. [Google Scholar] [CrossRef] [Green Version]
- Su, W.; Wang, H.; Feng, Z.; Sun, J. Nitro-oleic acid inhibits the high glucose-induced epithelial-mesenchymal transition in peritoneal mesothelial cells and attenuates peritoneal fibrosis. Am. J. Physiol. Physiol. 2020, 318, F457–F467. [Google Scholar] [CrossRef]
- Chakraborty, D.; Šumová, B.; Mallano, T.; Chen, C.-W.; Distler, A.; Bergmann, C.; Ludolph, I.; Horch, R.E.; Gelse, K.; Ramming, A.; et al. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.-A.; Yang, D.; Wu, Y.; Xie, Y.; Zhu, W.; Cai, Z.; Shen, J.; Fu, Z.; Wang, Y.; Jia, L.; et al. EphrinB2 Regulates Cardiac Fibrosis Through Modulating the Interaction of Stat3 and TGF-β/Smad3 Signaling. Circ. Res. 2017, 121, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Killeen, M.E.; Sumpter, T.L.; Ferris, L.K.; Falo, L.D.; Freeman, B.A.; Schopfer, F.J.; Mathers, A.R. Electrophilic nitro-fatty acids suppress psoriasiform dermatitis: STAT3 inhibition as a contributory mechanism. Redox Biol. 2021, 43, 101987. [Google Scholar] [CrossRef] [PubMed]
- Nettersheim, F.S.; Lemties, J.; Braumann, S.; Geißen, S.; Bokredenghel, S.; Nies, R.; Hof, A.; Winkels, H.; Freeman, B.; Klinke, A.; et al. Nitro-oleic acid (NO2-OA) reduces thoracic aortic aneurysm progression in a mouse model of Marfan syndrome. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef]
- Cojan-Minzat, B.O.; Zlibut, A.; Agoston-Coldea, L. Non-ischemic dilated cardiomyopathy and cardiac fibrosis. Hear. Fail. Rev. 2020, 26, 1081–1101. [Google Scholar] [CrossRef]
- Czubryt, M.P. Cardiac Fibroblast to Myofibroblast Phenotype Conversion—An Unexploited Therapeutic Target. J. Cardiovasc. Dev. Dis. 2019, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Ford, D.A. A BOSSS platform: Using functionalized lipids and click chemistry for new discoveries in lipid research. J. Lipid Res. 2021, 62, 100025. [Google Scholar] [CrossRef]
- Fang, M.; Huang, K.H.; Tu, W.-J.; Chen, Y.-T.; Pan, P.-Y.; Hsiao, W.-C.; Ke, Y.-Y.; Tsou, L.K.; Zhang, M.M. Chemoproteomic profiling reveals cellular targets of nitro-fatty acids. bioRxiv 2021. [Google Scholar] [CrossRef]
- Mollenhauer, M.; Mehrkens, D.; Klinke, A.; Lange, M.; Remane, L.; Friedrichs, K.; Braumann, S.; Geißen, S.; Simsekyilmaz, S.; Nettersheim, F.S.; et al. Nitro-fatty acids suppress ischemic ventricular arrhythmias by preserving calcium homeostasis. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braumann, S.; Schumacher, W.; Im, N.G.; Nettersheim, F.S.; Mehrkens, D.; Bokredenghel, S.; Hof, A.; Nies, R.J.; Adler, C.; Winkels, H.; et al. Nitro-Oleic Acid (NO2-OA) Improves Systolic Function in Dilated Cardiomyopathy by Attenuating Myocardial Fibrosis. Int. J. Mol. Sci. 2021, 22, 9052. https://doi.org/10.3390/ijms22169052
Braumann S, Schumacher W, Im NG, Nettersheim FS, Mehrkens D, Bokredenghel S, Hof A, Nies RJ, Adler C, Winkels H, et al. Nitro-Oleic Acid (NO2-OA) Improves Systolic Function in Dilated Cardiomyopathy by Attenuating Myocardial Fibrosis. International Journal of Molecular Sciences. 2021; 22(16):9052. https://doi.org/10.3390/ijms22169052
Chicago/Turabian StyleBraumann, Simon, Wibke Schumacher, Nam Gyu Im, Felix Sebastian Nettersheim, Dennis Mehrkens, Senai Bokredenghel, Alexander Hof, Richard Julius Nies, Christoph Adler, Holger Winkels, and et al. 2021. "Nitro-Oleic Acid (NO2-OA) Improves Systolic Function in Dilated Cardiomyopathy by Attenuating Myocardial Fibrosis" International Journal of Molecular Sciences 22, no. 16: 9052. https://doi.org/10.3390/ijms22169052
APA StyleBraumann, S., Schumacher, W., Im, N. G., Nettersheim, F. S., Mehrkens, D., Bokredenghel, S., Hof, A., Nies, R. J., Adler, C., Winkels, H., Knöll, R., Freeman, B. A., Rudolph, V., Klinke, A., Adam, M., Baldus, S., Mollenhauer, M., & Geißen, S. (2021). Nitro-Oleic Acid (NO2-OA) Improves Systolic Function in Dilated Cardiomyopathy by Attenuating Myocardial Fibrosis. International Journal of Molecular Sciences, 22(16), 9052. https://doi.org/10.3390/ijms22169052