
Increasing Angiogenesis Factors in Hypoxic Diabetic Wound Conditions by siRNA Delivery: Additive Effect of LbL-Gold Nanocarriers and Desloratadine-Induced Lysosomal Escape

 ,
, 
 and
and 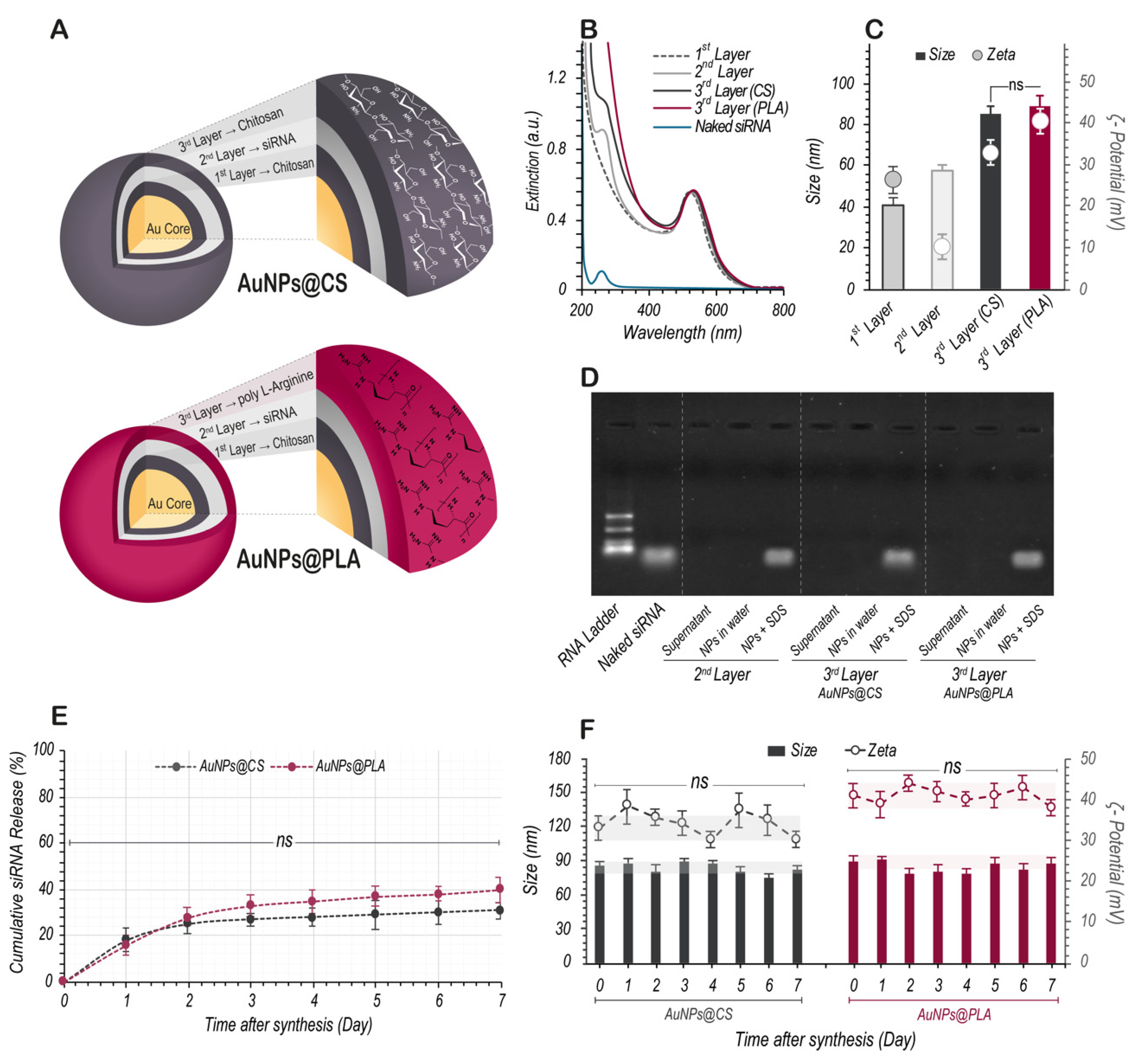{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Synthesis and Physicochemical Evaluation of Nanoformulations
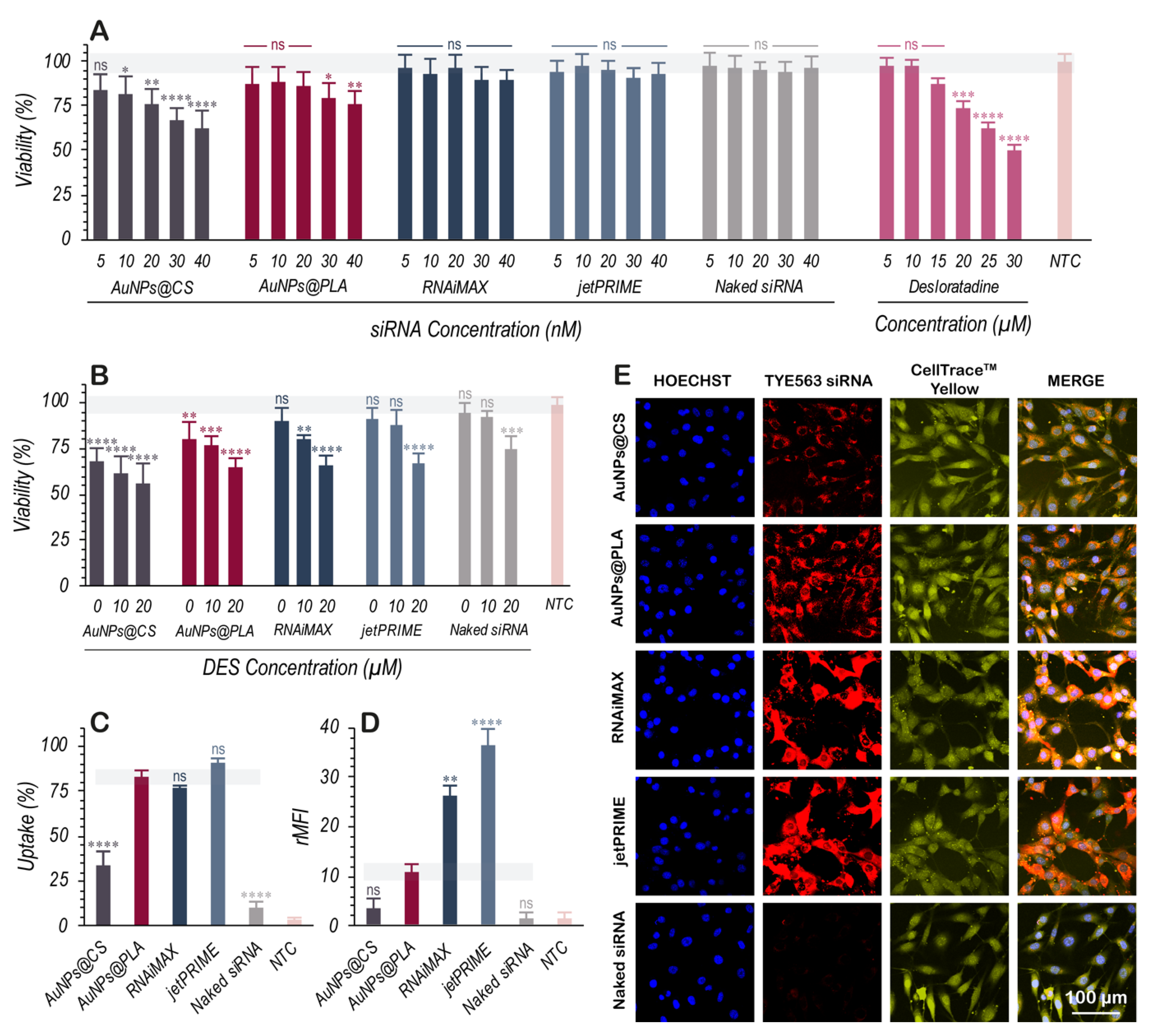
2.2. Cellular Toxicity and Internalization of Nanoformulations
2.3. Lysosomal Swelling by DES and Its Effect on Endosomal Escape in NIH-3T3 Cells
2.4. Forced siRNA Dissociation from the Nanoformulations
2.5. Downregulation of PHD-2 and Its Effect on HIF-1α Expression for Promoting Upregulation of Angiogenesis Factors
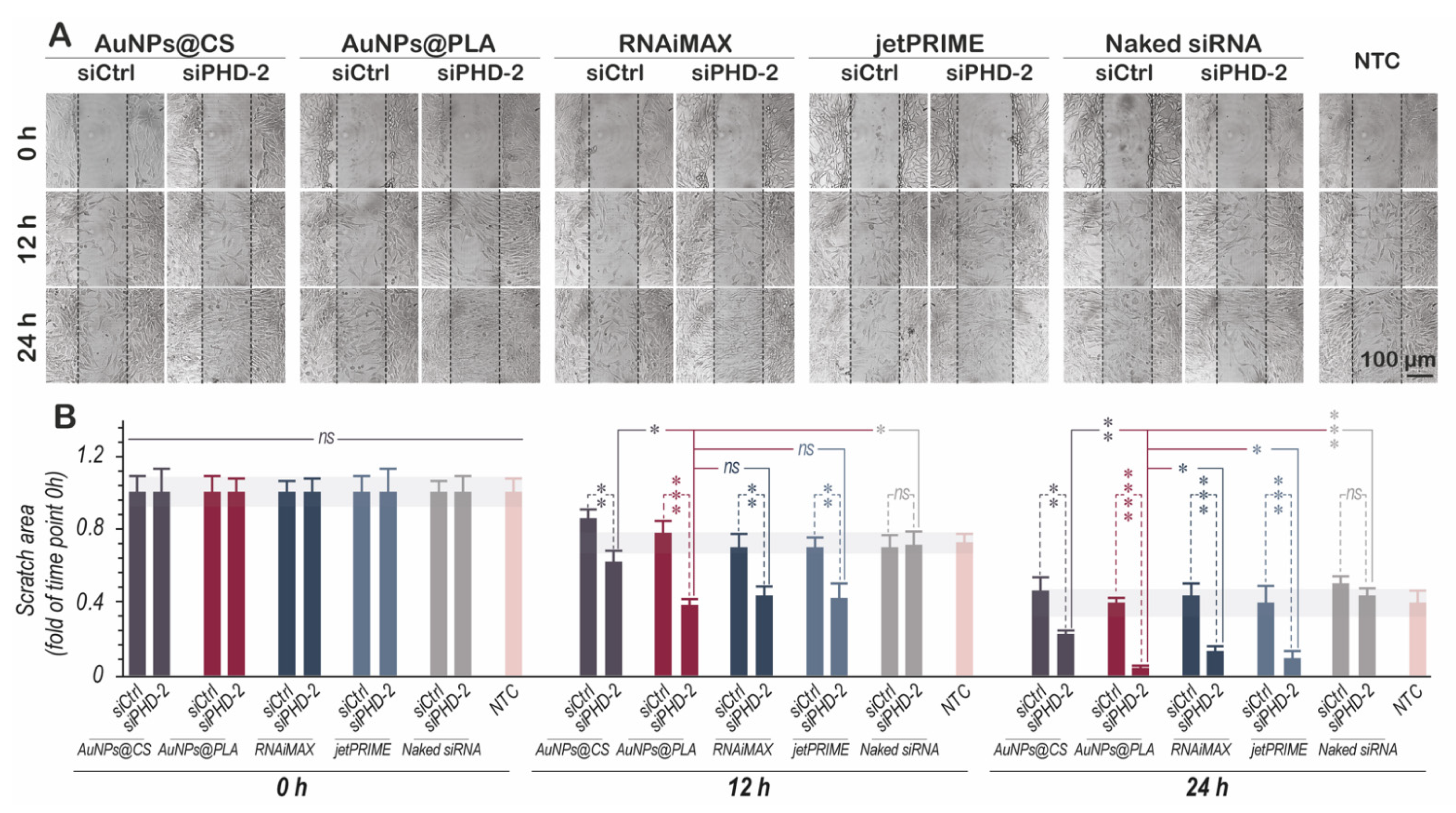
2.6. Enhanced HIF-1α Expression Stimulates Migration and Proliferation In Vitro
3. Discussion
3.1. Nanoformulation Characterization
3.2. Nanoformulation Cytotoxicity and Uptake
3.3. Endosomal Escape Efficiency in Combination with DES Treatment

3.4. Effect of PHD-2 Silencing on the Expression of Angiogenesis Factors
4. Materials and Methods
4.1. Materials
4.2. Nanoformulations Synthesis
4.3. Characterization of Nanoformulations
- −
- UV–vis absorbance spectroscopy
- −
- Dynamic light scattering (DLS) measurements
- −
- Fourier transform infrared (FTIR) spectroscopy
Gel Electrophoresis Assay
4.4. siRNA Release from the Nanoformulations
4.5. Lipofectamine® RNAiMAX and jetPRIME® Preparation
4.6. Dissociation Degree of Nanoformulations by Gel Electrophoresis and Fluorescence Fluctuation Spectroscopy (FFS)
4.7. Cell Culture
4.8. Cell Viability Assay
4.9. Quantification of Nanoformulation Internalization by Flow Cytometry
4.10. Visualizing Nanoformulation Internalization by Confocal Microscopy
4.11. Quantification of Lysosomal Volume by Flow Cytometry
4.12. Visualization of Lysosomes by Confocal Microscopy
4.13. Visualization and Quantification of the Cytosolic Release of AF647 ONs
4.14. Transfection Efficiency Analysis by Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
4.15. In Vitro Scratch Wound and Cell Migration Assays
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mellitus, D. Diagnosis and classification of diabetes mellitus. Diabetes Care 2005, 28, S5–S10. [Google Scholar]
- Zou, Q.; Qu, K.; Luo, Y.; Yin, D.; Ju, Y.; Tang, H. Predicting diabetes mellitus with machine learning techniques. Front. Genet. 2018, 9, 515. [Google Scholar] [CrossRef]
- Mane, K.; Chaluvaraju, K.; Niranjan, M.; Zaranappa, T.; Manjuthej, T. Review of insulin and its analogues in diabetes mellitus. J. Basic Clin. Pharm. 2012, 3, 283. [Google Scholar]
- Verrotti, A.; Prezioso, G.; Scattoni, R.; Chiarelli, F. Autonomic neuropathy in diabetes mellitus. Front. Endocrinol. 2014, 5, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juster-Switlyk, K.; Smith, A.G. Updates in diabetic peripheral neuropathy. Research 2016, 5, 738. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kishore, L.; Kaur, N. Diabetic peripheral neuropathy: Current perspective and future directions. Pharmacol. Res. 2014, 80, 21–35. [Google Scholar] [CrossRef]
- Schmidt, A.M. Diabetes mellitus and cardiovascular disease: Emerging therapeutic approaches. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 558–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haffner, S.M. The metabolic syndrome: Inflammation, diabetes mellitus, and cardiovascular disease. Am. J. Cardiol. 2006, 97, 3–11. [Google Scholar] [CrossRef]
- Malik, V.S.; Popkin, B.M.; Bray, G.A.; Després, J.-P.; Hu, F.B. Sugar-sweetened beverages, obesity, type 2 diabetes mellitus, and cardiovascular disease risk. Circulation 2010, 121, 1356–1364. [Google Scholar] [CrossRef]
- Fineberg, D.; Jandeleit-Dahm, K.A.; Cooper, M.E. Diabetic nephropathy: Diagnosis and treatment. Nat. Rev. Endocrinol. 2013, 9, 713. [Google Scholar] [CrossRef]
- Heyman, S.N.; Rosenberger, C.; Rosen, S.; Khamaisi, M. Why is diabetes mellitus a risk factor for contrast-induced nephropathy? BioMed Res. Int. 2013, 2013, 123589. [Google Scholar] [CrossRef] [Green Version]
- Satirapoj, B. Nephropathy in diabetes. In Diabetes; Springer: Berlin/Heidelberg, Germany, 2013; pp. 107–122. [Google Scholar]
- Hartnett, M.E.; Baehr, W.; Le, Y.Z. Diabetic Retinopathy, an Overview; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Pearce, I.; Simó, R.; Lövestam-Adrian, M.; Wong, D.T.; Evans, M. Association between diabetic eye disease and other complications of diabetes: Implications for care. A systematic review. Diabetes Obes. Metab. 2019, 21, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Saluja, S.; Anderson, S.; Hambleton, I.; Shoo, H.; Livingston, M.; Jude, E.; Lunt, M.; Dunn, G.; Heald, A. Foot ulceration and its association with mortality in diabetes mellitus: A meta-analysis. Diabet. Med. 2020, 37, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setacci, C.; Benevento, D.; De Donato, G.; Viviani, E.; Bracale, U.M.; Del Guercio, L.; Palasciano, G.; Setacci, F. Focusing on diabetic ulcers. Transl. Med. UniSa 2020, 21, 7. [Google Scholar]
- Armstrong, D.G.; Boulton, A.J.; Bus, S.A. Diabetic foot ulcers and their recurrence. N. Engl. J. Med. 2017, 376, 2367–2375. [Google Scholar] [CrossRef] [PubMed]
- Noor, S.; Zubair, M.; Ahmad, J. Diabetic foot ulcer—A review on pathophysiology, classification and microbial etiology. Diabetes Metab. Syndr. Clin. Res. Rev. 2015, 9, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.A.; Eid, M.A.; Creager, M.A.; Goodney, P.P. Epidemiology and risk of amputation in patients with diabetes mellitus and peripheral artery disease. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1808–1817. [Google Scholar] [CrossRef] [PubMed]
- Hasan, R.; Firwana, B.; Elraiyah, T.; Domecq, J.P.; Prutsky, G.; Nabhan, M.; Prokop, L.J.; Henke, P.; Tsapas, A.; Montori, V.M. A systematic review and meta-analysis of glycemic control for the prevention of diabetic foot syndrome. J. Vasc. Surg. 2016, 63, 22S–28S.e22. [Google Scholar] [CrossRef] [Green Version]
- Fortington, L.V.; Geertzen, J.H.; van Netten, J.J.; Postema, K.; Rommers, G.M.; Dijkstra, P.U. Short and long term mortality rates after a lower limb amputation. Eur. J. Vasc. Endovasc. Surg. 2013, 46, 124–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brem, H.; Tomic-Canic, M. Cellular and molecular basis of wound healing in diabetes. J. Clin. Investig. 2007, 117, 1219–1222. [Google Scholar] [CrossRef] [Green Version]
- Rafehi, H.; El-Osta, A.; Karagiannis, T.C. Genetic and epigenetic events in diabetic wound healing. Int. Wound J. 2011, 8, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Falanga, V. Wound healing and its impairment in the diabetic foot. Lancet 2005, 366, 1736–1743. [Google Scholar] [CrossRef]
- Costa, P.Z.; Soares, R. Neovascularization in diabetes and its complications. Unraveling the angiogenic paradox. Life Sci. 2013, 92, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Okonkwo, U.A.; DiPietro, L.A. Diabetes and wound angiogenesis. Int. J. Mol. Sci. 2017, 18, 1419. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wang, C.; Chen, M.; Xi, Y.; Cheng, W.; Mao, C.; Xu, T.; Zhang, X.; Lin, C.; Gao, W. Efficient angiogenesis-based diabetic wound healing/skin reconstruction through bioactive antibacterial adhesive ultraviolet shielding nanodressing with exosome release. ACS Nano 2019, 13, 10279–10293. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-W.; Bae, S.-H.; Jeong, J.-W.; Kim, S.-H.; Kim, K.-W. Hypoxia-inducible factor (HIF-1) α: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar] [CrossRef]
- Giaccia, A.J.; Simon, M.C.; Johnson, R. The biology of hypoxia: The role of oxygen sensing in development, normal function, and disease. Genes Dev. 2004, 18, 2183–2194. [Google Scholar] [CrossRef] [Green Version]
- Zimna, A.; Kurpisz, M. Hypoxia-inducible factor-1 in physiological and pathophysiological angiogenesis: Applications and therapies. BioMed Res. Int. 2015, 2015, 549412. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFα targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.-M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. Proline hydroxylation and gene expression. Annu. Rev. Biochem. 2005, 74, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.D.; Hackett, S.F.; Hirota, K.; Oshima, Y.; Cai, Z.; Berg-Dixon, S.; Rowan, A.; Yan, Z.; Campochiaro, P.A.; Semenza, G.L. Cell type–specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ. Res. 2003, 93, 1074–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin. Cancer Biol. 2009, 19, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Schultz, K.; Fanburg, B.L.; Beasley, D. Hypoxia and hypoxia-inducible factor-1α promote growth factor-induced proliferation of human vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2528–H2534. [Google Scholar] [CrossRef] [PubMed]
- Kütscher, C.; Lampert, F.M.; Kunze, M.; Markfeld-Erol, F.; Stark, G.B.; Finkenzeller, G. Overexpression of hypoxia-inducible factor-1 alpha improves vasculogenesis-related functions of endothelial progenitor cells. Microvasc. Res. 2016, 105, 85–92. [Google Scholar] [CrossRef]
- Hong, W.X.; Hu, M.S.; Esquivel, M.; Liang, G.Y.; Rennert, R.C.; McArdle, A.; Paik, K.J.; Duscher, D.; Gurtner, G.C.; Lorenz, H.P. The role of hypoxia-inducible factor in wound healing. Adv. Wound Care 2014, 3, 390–399. [Google Scholar] [CrossRef] [Green Version]
- Catrina, S.-B.; Okamoto, K.; Pereira, T.; Brismar, K.; Poellinger, L. Hyperglycemia regulates hypoxia-inducible factor-1α protein stability and function. Diabetes 2004, 53, 3226–3232. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Ferguson, G.; Connell, P.; Walshe, T.; Murphy, R.; Birney, Y.A.; O’Brien, C.; Cahill, P.A. High glucose concentrations alter hypoxia-induced control of vascular smooth muscle cell growth via a HIF-1α-dependent pathway. J. Mol. Cell. Cardiol. 2007, 42, 609–619. [Google Scholar] [CrossRef]
- Botusan, I.R.; Sunkari, V.G.; Savu, O.; Catrina, A.I.; Grünler, J.; Lindberg, S.; Pereira, T.; Ylä-Herttuala, S.; Poellinger, L.; Brismar, K. Stabilization of HIF-1α is critical to improve wound healing in diabetic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 19426–19431. [Google Scholar] [CrossRef] [Green Version]
- Fadini, G.; Sartore, S.; Schiavon, M.; Albiero, M.; Baesso, I.; Cabrelle, A.; Agostini, C.; Avogaro, A. Diabetes impairs progenitor cell mobilisation after hindlimb ischaemia–reperfusion injury in rats. Diabetologia 2006, 49, 3075–3084. [Google Scholar] [CrossRef] [Green Version]
- Hirota, K.; Semenza, G.L. Regulation of hypoxia-inducible factor 1 by prolyl and asparaginyl hydroxylases. Biochem. Biophys. Res. Commun. 2005, 338, 610–616. [Google Scholar] [CrossRef]
- Wu, S.; Nishiyama, N.; Kano, M.R.; Morishita, Y.; Miyazono, K.; Itaka, K.; Chung, U.-i.; Kataoka, K. Enhancement of angiogenesis through stabilization of hypoxia-inducible factor-1 by silencing prolyl hydroxylase domain-2 gene. Mol. Ther. 2008, 16, 1227–1234. [Google Scholar] [CrossRef]
- Wetterau, M.; George, F.; Weinstein, A.; Nguyen, P.D.; Tutela, J.P.; Knobel, D.; Oriana, C.B.; Warren, S.M.; Saadeh, P.B. Topical prolyl hydroxylase domain-2 silencing improves diabetic murine wound closure. Wound Repair Regen. 2011, 19, 481–486. [Google Scholar] [CrossRef] [Green Version]
- Dallas, A.; Trotsyuk, A.; Ilves, H.; Bonham, C.A.; Rodrigues, M.; Engel, K.; Barrera, J.A.; Kosaric, N.; Stern-Buchbinder, Z.A.; White, A. Acceleration of diabetic wound healing with PHD2-and miR-210-targeting oligonucleotides. Tissue Eng. Part A 2019, 25, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.R.; Nelson, C.E.; Gupta, M.K.; Yu, F.; Sarett, S.M.; Hocking, K.M.; Pollins, A.C.; Nanney, L.B.; Davidson, J.M.; Guelcher, S.A. Local delivery of PHD2 siRNA from ROS-degradable scaffolds to promote diabetic wound healing. Adv. Healthc. Mater. 2016, 5, 2751–2757. [Google Scholar] [CrossRef] [Green Version]
- Nelson, C.E.; Kim, A.J.; Adolph, E.J.; Gupta, M.K.; Yu, F.; Hocking, K.M.; Davidson, J.M.; Guelcher, S.A.; Duvall, C.L. Tunable delivery of siRNA from a biodegradable scaffold to promote angiogenesis in vivo. Adv. Mater. 2014, 26, 607–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, K.J.; Maan, Z.N.; Zielins, E.R.; Duscher, D.; Whittam, A.J.; Morrison, S.D.; Brett, E.A.; Ransom, R.C.; Hu, M.S.; Wu, J.C. Short hairpin RNA silencing of PHD-2 improves neovascularization and functional outcomes in diabetic wounds and ischemic limbs. PLoS ONE 2016, 11, e0150927. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, J.C.; Kowalski, P.S.; Anderson, D.G. Advances in the delivery of RNA therapeutics: From concept to clinical reality. Genome Med. 2017, 9, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittrup, A.; Lieberman, J. Knocking down disease: A progress report on siRNA therapeutics. Nat. Rev. Genet. 2015, 16, 543–552. [Google Scholar] [CrossRef]
- Kanasty, R.; Dorkin, J.R.; Vegas, A.; Anderson, D. Delivery materials for siRNA therapeutics. Nat. Mater. 2013, 12, 967–977. [Google Scholar] [CrossRef]
- Pack, D.W.; Hoffman, A.S.; Pun, S.; Stayton, P.S. Design and development of polymers for gene delivery. Nat. Rev. Drug Discov. 2005, 4, 581–593. [Google Scholar] [CrossRef]
- Chuan, D.; Jin, T.; Fan, R.; Zhou, L.; Guo, G. Chitosan for gene delivery: Methods for improvement and applications. Adv. Colloid Interface Sci. 2019, 268, 25–38. [Google Scholar] [CrossRef]
- Zhou, Y.; Han, S.; Liang, Z.; Zhao, M.; Liu, G.; Wu, J. Progress in arginine-based gene delivery systems. J. Mater. Chem. B 2020, 8, 5564–5577. [Google Scholar] [CrossRef]
- Wang, Y.; Ye, M.; Xie, R.; Gong, S. Enhancing the in vitro and in vivo stabilities of polymeric nucleic acid delivery nanosystems. Bioconjug. Chem. 2018, 30, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Sahay, G.; Querbes, W.; Alabi, C.; Eltoukhy, A.; Sarkar, S.; Zurenko, C.; Karagiannis, E.; Love, K.; Chen, D.; Zoncu, R. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat. Biotechnol. 2013, 31, 653–658. [Google Scholar] [CrossRef] [Green Version]
- Joris, F.; De Backer, L.; Van de Vyver, T.; Bastiancich, C.; De Smedt, S.C.; Raemdonck, K. Repurposing cationic amphiphilic drugs as adjuvants to induce lysosomal siRNA escape in nanogel transfected cells. J. Control. Release 2018, 269, 266–276. [Google Scholar] [CrossRef] [Green Version]
- Van de Vyver, T.; Bogaert, B.; De Backer, L.; Joris, F.; Guagliardo, R.; Van Hoeck, J.; Merckx, P.; Van Calenbergh, S.; Ramishetti, S.; Peer, D. Cationic amphiphilic drugs boost the lysosomal escape of small nucleic acid therapeutics in a nanocarrier-dependent manner. ACS Nano 2020, 14, 4774–4791. [Google Scholar] [CrossRef] [PubMed]
- Villamil, G.A.M.; Appelqvist, H.; Ederth, T.; Öllinger, K. Lysosomotropic agents: Impact on lysosomal membrane permeabilization and cell death. Biochem. Soc. Trans. 2014, 42, 1460–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, N.H.; Olsen, O.D.; Groth-Pedersen, L.; Ellegaard, A.-M.; Bilgin, M.; Redmer, S.; Ostenfeld, M.S.; Ulanet, D.; Dovmark, T.H.; Lønborg, A. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell 2013, 24, 379–393. [Google Scholar] [CrossRef] [Green Version]
- Shaabani, E.; Sharifiaghdam, M.; De Keersmaecker, H.; De Rycke, R.; De Smedt, S.; Faridi-Majidi, R.; Braeckmans, K.; Fraire, J.C. Layer by Layer Assembled Chitosan-Coated Gold Nanoparticles for Enhanced siRNA Delivery and Silencing. Int. J. Mol. Sci. 2021, 22, 831. [Google Scholar] [CrossRef]
- Richard, I.; Thibault, M.; De Crescenzo, G.; Buschmann, M.D.; Lavertu, M. Ionization behavior of chitosan and chitosan–DNA polyplexes indicate that chitosan has a similar capability to induce a proton-sponge effect as PEI. Biomacromolecules 2013, 14, 1732–1740. [Google Scholar] [CrossRef]
- Herce, H.; Garcia, A.; Litt, J.; Kane, R.; Martín, P.; Enrique, N.; Rebolledo, A.; Milesi, V. Arginine-rich peptides destabilize the plasma membrane, consistent with a pore formation translocation mechanism of cell-penetrating peptides. Biophys. J. 2009, 97, 1917–1925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allolio, C.; Magarkar, A.; Jurkiewicz, P.; Baxová, K.; Javanainen, M.; Mason, P.E.; Šachl, R.; Cebecauer, M.; Hof, M.; Horinek, D. Arginine-rich cell-penetrating peptides induce membrane multilamellarity and subsequently enter via formation of a fusion pore. Proc. Natl. Acad. Sci. USA 2018, 115, 11923–11928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbeek, S.F.; Awasthi, N.; Teiwes, N.K.; Mey, I.; Hub, J.S.; Janshoff, A. How arginine derivatives alter the stability of lipid membranes: Dissecting the roles of side chains, backbone and termini. Eur. Biophys. J. 2021, 50, 127–142. [Google Scholar] [CrossRef]
- Tang, H.; Yin, L.; Kim, K.H.; Cheng, J. Helical poly (arginine) mimics with superior cell-penetrating and molecular transporting properties. Chem. Sci. 2013, 4, 3839–3844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilfinger, W.; Mackey, K.; Chomczynski, P. DNA sequencing II optimizing preparation and cleanup. In Assessing the Quantity, Purity and Integrity of RNA and DNA Following Nucleic Acid Purification; Jones and Bartlett Publishers: Burlington, MA, USA, 2006; pp. 291–312. [Google Scholar]
- Prasad, S.; Mandal, I.; Singh, S.; Paul, A.; Mandal, B.; Venkatramani, R.; Swaminathan, R. Near UV-Visible electronic absorption originating from charged amino acids in a monomeric protein. Chem. Sci. 2017, 8, 5416–5433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anicuta, S.-G.; Dobre, L.; Stroescu, M.; Jipa, I. Fourier transform infrared (FTIR) spectroscopy for characterization of antimicrobial films containing chitosan. An. Univ. Din. Oradea Fasc. Ecotoxicol. Zooteh. Ind. Aliment. 2010, 65, 1234–1240. [Google Scholar]
- Szyk-Warszyńska, L.; Raszka, K.; Warszyński, P. Interactions of casein and polypeptides in multilayer films studied by FTIR and molecular dynamics. Polymers 2019, 11, 920. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Wang, Z.; Zhang, Y.; Zheng, Z.; Wang, C.; Du, Y.; Ye, W. Simultaneous electrochemical determination of uric acid, xanthine and hypoxanthine based on poly (l-arginine)/graphene composite film modified electrode. Talanta 2012, 93, 320–325. [Google Scholar] [CrossRef]
- Addis, R.; Cruciani, S.; Santaniello, S.; Bellu, E.; Sarais, G.; Ventura, C.; Maioli, M.; Pintore, G. Fibroblast proliferation and migration in wound healing by phytochemicals: Evidence for a novel synergic outcome. Int. J. Med. Sci. 2020, 17, 1030. [Google Scholar] [CrossRef] [Green Version]
- Bainbridge, P. Wound healing and the role of fibroblasts. J. Wound Care 2013, 22, 407–411. [Google Scholar]
- Ciortan, L.; Macarie, R.D.; Cecoltan, S.; Vadana, M.; Tucureanu, M.M.; Mihaila, A.C.; Droc, I.; Butoi, E.; Manduteanu, I. Chronic High Glucose Concentration Induces Inflammatory and Remodeling Changes in Valvular Endothelial Cells and Valvular Interstitial Cells in a Gelatin Methacrylate 3D Model of the Human Aortic Valve. Polymers 2020, 12, 2786. [Google Scholar] [CrossRef]
- Wu, W.-y.; Yan, H.; Wang, X.-b.; Gui, Y.-z.; Gao, F.; Tang, X.-l.; Qin, Y.-l.; Su, M.; Chen, T.; Wang, Y.-p. Sodium tanshinone IIA silate inhibits high glucose-induced vascular smooth muscle cell proliferation and migration through activation of AMP-activated protein kinase. PLoS ONE 2014, 9, e94957. [Google Scholar] [CrossRef]
- Mitchell, D.J.; Steinman, L.; Kim, D.; Fathman, C.; Rothbard, J. Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Pept. Res. 2000, 56, 318–325. [Google Scholar] [CrossRef]
- Vermeulen, L.M.; Brans, T.; Samal, S.K.; Dubruel, P.; Demeester, J.; De Smedt, S.C.; Remaut, K.; Braeckmans, K. Endosomal size and membrane leakiness influence proton sponge-based rupture of endosomal vesicles. ACS Nano 2018, 12, 2332–2345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousselle, P.; Braye, F.; Dayan, G. Re-epithelialization of adult skin wounds: Cellular mechanisms and therapeutic strategies. Adv. Drug Deliv. Rev. 2019, 146, 344–365. [Google Scholar] [CrossRef] [PubMed]
- Hostanska, K.; Rostock, M.; Melzer, J.; Baumgartner, S.; Saller, R. A homeopathic remedy from arnica, marigold, St. John’s wort and comfrey accelerates in vitro wound scratch closure of NIH 3T3 fibroblasts. BMC Complement. Altern. Med. 2012, 12, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.-C.; Park, A.Y.; Guan, J.-L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329. [Google Scholar] [CrossRef] [Green Version]
- Naik, R.J.; Chandra, P.; Mann, A.; Ganguli, M. Exogenous and cell surface glycosaminoglycans alter DNA delivery efficiency of arginine and lysine homopeptides in distinctly different ways. J. Biol. Chem. 2011, 286, 18982–18993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Huang, Y.; Cai, X.; Wang, S. Impact of pH, ionic strength and chitosan charge density on chitosan/casein complexation and phase behavior. Carbohydr. Polym. 2019, 208, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.F.; Mundargi, R.C.; Chen, M.H.A.; Lessig, J.; Neu, B.; Venkatraman, S.S.; Wong, T.T. Layer-by-layer nanoparticles as an efficient siRNA delivery vehicle for SPARC silencing. Small 2014, 10, 1790–1798. [Google Scholar] [CrossRef] [PubMed]
- Raemdonck, K.; Naeye, B.; Høgset, A.; Demeester, J.; De Smedt, S.C. Prolonged gene silencing by combining siRNA nanogels and photochemical internalization. J. Control. Release 2010, 145, 281–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.A.; Selby, L.I.; Johnston, A.P.; Such, G.K. The endosomal escape of nanoparticles: Toward more efficient cellular delivery. Bioconjugate Chem. 2018, 30, 263–272. [Google Scholar] [CrossRef]
- De Backer, L.; Braeckmans, K.; Demeester, J.; De Smedt, S.C.; Raemdonck, K. The influence of natural pulmonary surfactant on the efficacy of siRNA-loaded dextran nanogels. Nanomedicine 2013, 8, 1625–1638. [Google Scholar] [CrossRef] [Green Version]
- De Backer, L.; Braeckmans, K.; Stuart, M.C.; Demeester, J.; De Smedt, S.C.; Raemdonck, K. Bio-inspired pulmonary surfactant-modified nanogels: A promising siRNA delivery system. J. Control. Release 2015, 206, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Lucas, B.; Remaut, K.; Sanders, N.; Braeckmans, K.; De Smedt, S.; Demeester, J. Towards a better understanding of the dissociation behavior of liposome-oligonucleotide complexes in the cytosol of cells. J. Control. Release 2005, 103, 435–450. [Google Scholar] [CrossRef]
- Zhang, H.; De Smedt, S.C.; Remaut, K. Fluorescence Correlation Spectroscopy to find the critical balance between extracellular association and intracellular dissociation of mRNA complexes. Acta Biomater. 2018, 75, 358–370. [Google Scholar] [CrossRef] [Green Version]
- Dakwar, G.R.; Zagato, E.; Delanghe, J.; Hobel, S.; Aigner, A.; Denys, H.; Braeckmans, K.; Ceelen, W.; De Smedt, S.C.; Remaut, K. Colloidal stability of nano-sized particles in the peritoneal fluid: Towards optimizing drug delivery systems for intraperitoneal therapy. Acta Biomater. 2014, 10, 2965–2975. [Google Scholar] [CrossRef] [Green Version]
- Naeye, B.; Deschout, H.; Röding, M.; Rudemo, M.; Delanghe, J.; Devreese, K.; Demeester, J.; Braeckmans, K.; De Smedt, S.C.; Raemdonck, K. Hemocompatibility of siRNA loaded dextran nanogels. Biomaterials 2011, 32, 9120–9127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buyens, K.; Lucas, B.; Raemdonck, K.; Braeckmans, K.; Vercammen, J.; Hendrix, J.; Engelborghs, Y.; De Smedt, S.C.; Sanders, N.N. A fast and sensitive method for measuring the integrity of siRNA-carrier complexes in full human serum. J. Control. Release 2008, 126, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Rehman, Z.U.; Hoekstra, D.; Zuhorn, I.S. Mechanism of polyplex-and lipoplex-mediated delivery of nucleic acids: Real-time visualization of transient membrane destabilization without endosomal lysis. ACS Nano 2013, 7, 3767–3777. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaabani, E.; Sharifiaghdam, M.; Lammens, J.; De Keersmaecker, H.; Vervaet, C.; De Beer, T.; Motevaseli, E.; Ghahremani, M.H.; Mansouri, P.; De Smedt, S.; et al. Increasing Angiogenesis Factors in Hypoxic Diabetic Wound Conditions by siRNA Delivery: Additive Effect of LbL-Gold Nanocarriers and Desloratadine-Induced Lysosomal Escape. Int. J. Mol. Sci. 2021, 22, 9216. https://doi.org/10.3390/ijms22179216
Shaabani E, Sharifiaghdam M, Lammens J, De Keersmaecker H, Vervaet C, De Beer T, Motevaseli E, Ghahremani MH, Mansouri P, De Smedt S, et al. Increasing Angiogenesis Factors in Hypoxic Diabetic Wound Conditions by siRNA Delivery: Additive Effect of LbL-Gold Nanocarriers and Desloratadine-Induced Lysosomal Escape. International Journal of Molecular Sciences. 2021; 22(17):9216. https://doi.org/10.3390/ijms22179216
Chicago/Turabian StyleShaabani, Elnaz, Maryam Sharifiaghdam, Joris Lammens, Herlinde De Keersmaecker, Chris Vervaet, Thomas De Beer, Elahe Motevaseli, Mohammad Hossein Ghahremani, Parvin Mansouri, Stefaan De Smedt, and et al. 2021. "Increasing Angiogenesis Factors in Hypoxic Diabetic Wound Conditions by siRNA Delivery: Additive Effect of LbL-Gold Nanocarriers and Desloratadine-Induced Lysosomal Escape" International Journal of Molecular Sciences 22, no. 17: 9216. https://doi.org/10.3390/ijms22179216
APA StyleShaabani, E., Sharifiaghdam, M., Lammens, J., De Keersmaecker, H., Vervaet, C., De Beer, T., Motevaseli, E., Ghahremani, M. H., Mansouri, P., De Smedt, S., Raemdonck, K., Faridi-Majidi, R., Braeckmans, K., & Fraire, J. C. (2021). Increasing Angiogenesis Factors in Hypoxic Diabetic Wound Conditions by siRNA Delivery: Additive Effect of LbL-Gold Nanocarriers and Desloratadine-Induced Lysosomal Escape. International Journal of Molecular Sciences, 22(17), 9216. https://doi.org/10.3390/ijms22179216