
H1153Y-KCNH2 Mutation Identified in a Sudden Arrhythmic Death Syndrome Case Alters Channel Gating

 , , and
, , and
Abstract
:1. Introduction
2. Results
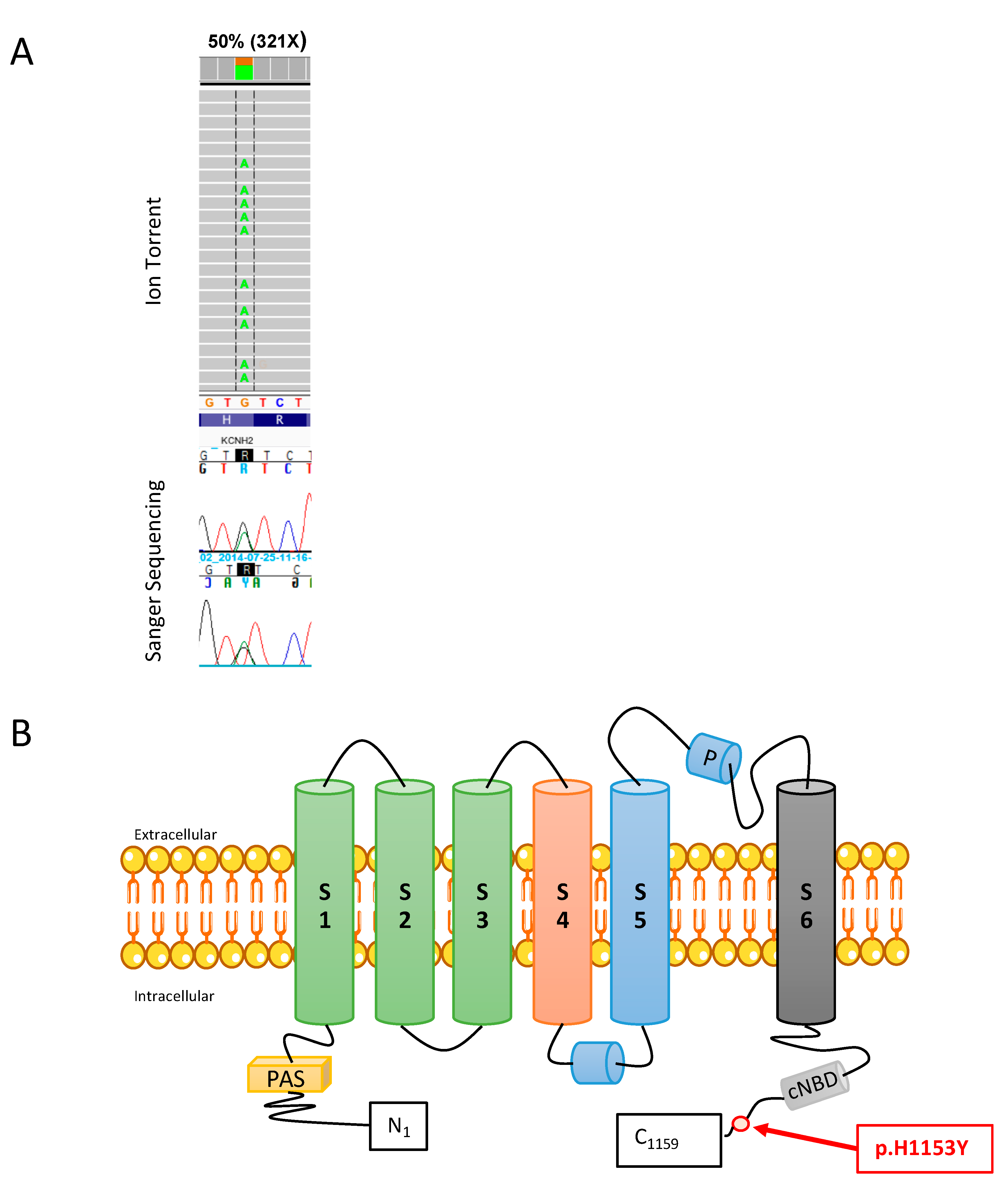
2.1. Mutation Analysis
2.2. H1153Y-Kv11.1 CHANNEL Proteins Have Similar Trafficking Phenotype to WT-Kv11.1 Channel Proteins: Similar Surface Expression and Glycosylation Level
2.3. Electrophysiological Characterization of the Mutation H1153Y in hERG Channel
3. Discussion
4. Material and Methods
4.1. Case Description
4.2. Genetic Analysis
4.3. Plasmid Construct, Cell Cultures, and Transient Transfection in HEK293 Cells
4.4. Immunofluorescence Staining and Microscope Imaging
4.5. Flow Cytometry Assay of Surface Kv11.1 Channel
4.6. Western Blot Analysis and Stain Free Quantification
4.7. Electrophysiological Recording
4.8. Statistical Analysis
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iannetti, P.; Farello, G.; Verrotti, A. Editorial–Channelopathies: A link between brain and heart: The model of epilepsy. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 5523–5526. [Google Scholar] [CrossRef]
- Garcia-Elias, A.; Benito, B. Ion Channel Disorders and Sudden Cardiac Death. Int. J. Mol. Sci. 2018, 19, 692. [Google Scholar] [CrossRef] [Green Version]
- Coll, M.; Ferrer-Costa, C.; Pich, S.; Allegue, C.; Rodrigo, E.; Fernandez-Fresnedo, G.; Barreda, P.; Mates, J.; Martinez de Francisco, A.L.; Ortega, I.; et al. Role of genetic and electrolyte abnormalities in prolonged QTc interval and sudden cardiac death in end-stage renal disease patients. PLoS ONE 2018, 13, e0200756. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Burke, M.; Fornes, P.; Gallagher, P.J.; de Gouveia, R.H.; Sheppard, M.; Thiene, G.; van der Wal, A. Association for European Cardiovascular P. Guidelines for autopsy investigation of sudden cardiac death. Virchows Arch. 2018, 452, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Blomstrom-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. Task Force for the Management of Patients with Ventricular A, the Prevention of Sudden Cardiac Death of the European Society of C 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC)Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Europace 2015, 17, 1601–1687. [Google Scholar] [CrossRef] [PubMed]
- Bagnall, R.D.; Weintraub, R.G.; Ingles, J.; Duflou, J.; Yeates, L.; Lam, L.; Davis, A.M.; Thompson, T.; Connell, V.; Wallace, J.; et al. A Prospective Study of Sudden Cardiac Death among Children and Young Adults. N. Engl. J. Med. 2016, 374, 2441–2452. [Google Scholar] [CrossRef] [PubMed]
- Hertz, C.L.; Christiansen, S.L.; Ferrero-Miliani, L.; Fordyce, S.L.; Dahl, M.; Holst, A.G.; Ottesen, G.L.; Frank-Hansen, R.; Bundgaard, H.; Morling, N. Next-generation sequencing of 34 genes in sudden unexplained death victims in forensics and in patients with channelopathic cardiac diseases. Int. J. Legal Med. 2015, 129, 793–800. [Google Scholar] [CrossRef]
- Lahrouchi, N.; Raju, H.; Lodder, E.M.; Papatheodorou, S.; Miles, C.; Ware, J.S.; Papadakis, M.; Tadros, R.; Cole, D.; Skinner, J.R.; et al. The yield of postmortem genetic testing in sudden death cases with structural findings at autopsy. Eur. J. Hum. Genet. 2020, 28, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, C.; Mazzanti, A.; Priori, S.G. Genetic risk stratification in cardiac arrhythmias. Curr. Opin. Cardiol. 2018, 33, 298–303. [Google Scholar] [CrossRef]
- Neubauer, J.; Lecca, M.R.; Russo, G.; Bartsch, C.; Medeiros-Domingo, A.; Berger, W.; Haas, C. Post-mortem whole-exome analysis in a large sudden infant death syndrome cohort with a focus on cardiovascular and metabolic genetic diseases. Eur. J. Hum. Genet. 2017, 25, 404–409. [Google Scholar] [CrossRef] [Green Version]
- Campuzano, O.; Allegue, C.; Fernandez, A.; Iglesias, A.; Brugada, R. Determining the pathogenicity of genetic variants associated with cardiac channelopathies. Sci. Rep. 2015, 5, 7953. [Google Scholar] [CrossRef] [Green Version]
- Refsgaard, L.; Holst, A.G.; Sadjadieh, G.; Haunso, S.; Nielsen, J.B.; Olesen, M.S. High prevalence of genetic variants previously associated with LQT syndrome in new exome data. Eur. J. Hum. Genet. 2012, 20, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Boycott, K.M.; Vanstone, M.R.; Bulman, D.E.; MacKenzie, A.E. Rare-disease genetics in the era of next-generation sequencing: Discovery to translation. Nat. Rev. Genet. 2013, 14, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, A.; Keyser, C.; Hollard, C.; Raul, J.S.; Muller, J.; Ludes, B. Targeted next generation sequencing application in cardiac channelopathies: Analysis of a cohort of autopsy-negative sudden unexplained deaths. Forensic Sci. Int. 2015, 254, 5–11. [Google Scholar] [CrossRef]
- Sanguinetti, M.C.; Jiang, C.; Curran, M.E.; Keating, M.T. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 1995, 81, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Trudeau, M.C.; Warmke, J.W.; Ganetzky, B.; Robertson, G.A. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science 1995, 269, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.E.; Splawski, I.; Timothy, K.W.; Vincent, G.M.; Green, E.D.; Keating, M.T. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995, 80, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.L.; Kuzmicki, C.E.; Childs, R.R.; Hintz, C.J.; Delisle, B.P. January CT Large-scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome. Nat. Commun. 2014, 5, 5535. [Google Scholar] [CrossRef]
- Smith, J.L.; Anderson, C.L.; Burgess, D.E.; Elayi, C.S.; January, C.T.; Delisle, B.P. Molecular pathogenesis of long QT syndrome type 2. J. Arrhythm. 2016, 32, 373–380. [Google Scholar] [CrossRef] [Green Version]
- Biliczki, P.; Girmatsion, Z.; Harenkamp, S.; Anneken, L.; Brandes, R.P.; Varro, A.; Marschall, C.; Herrera, D.; Hohnloser, S.H.; Nattel, S.; et al. Cellular properties of C-terminal KCNH2 long QT syndrome mutations: Description and divergence from clinical phenotypes. Heart Rhythm 2008, 5, 1159–1167. [Google Scholar] [CrossRef]
- Vandenberg, J.I.; Perry, M.D.; Perrin, M.J.; Mann, S.A.; Ke, Y.; Hill, A.P. hERG K(+) channels: Structure, function, and clinical significance. Physiol. Rev. 2012, 92, 1393–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, C.; Priori, S.G.; Schwartz, P.J.; Bloise, R.; Ronchetti, E.; Nastoli, J.; Bottelli, G.; Cerrone, M.; Leonardi, S. Genetic testing in the long QT syndrome: Development and validation of an efficient approach to genotyping in clinical practice. JAMA 2005, 294, 2975–2980. [Google Scholar] [CrossRef] [Green Version]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shaw, K.; Cooper, D.N. The Human Gene Mutation Database (HGMD) and its exploitation in the fields of personalized genomics and molecular evolution. Curr. Protoc. Bioinform. 2012, 39, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Diebold, I.; Schon, U.; Scharf, F.; Benet-Pages, A.; Laner, A.; Holinski-Feder, E. Abicht A Critical assessment of secondary findings in genes linked to primary arrhythmia syndromes. Hum. Mutat. 2020, 41, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Kanner, S.A.; Jain, A.; Colecraft, H.M. Development of a High-Throughput Flow Cytometry Assay to Monitor Defective Trafficking and Rescue of Long QT2 Mutant hERG Channels. Front. Physiol. 2018, 9, 397. [Google Scholar] [CrossRef]
- Rajamani, S.; Anderson, C.L.; Valdivia, C.R.; Eckhardt, L.L.; Foell, J.D.; Robertson, G.A.; Kamp, T.J.; Makielski, J.C.; Anson, B.D.; January, C.T. Specific serine proteases selectively damage KCNH2 (hERG1) potassium channels and I(Kr). Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1278–H1288. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Gong, Q.; Epstein, M.L.; January, C.T. HERG channel dysfunction in human long QT syndrome. Intracellular transport and functional defects. J. Biol. Chem. 1998, 273, 21061–21066. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.L.; Delisle, B.P.; Anson, B.D.; Kilby, J.A.; Will, M.L.; Tester, D.J.; Gong, Q.; Zhou, Z.; Ackerman, M.J.; January, C.T. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation 2006, 113, 365–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergeev, V.; Perry, F.; Roston, T.M.; Sanatani, S.; Tibbits, G.F.; Claydon, T.W. Functional characterization of a novel hERG variant in a family with recurrent sudden infant death syndrome: Retracting a genetic diagnosis. Forensic Sci. Int. 2018, 284, 39–45. [Google Scholar] [CrossRef]
- Smith, J.L.; Tester, D.J.; Hall, A.R.; Burgess, D.E.; Hsu, C.C.; Claude Elayi, S.; Anderson, C.L.; January, C.T.; Luo, J.Z.; Hartzel, D.N. Functional Invalidation of Putative Sudden Infant Death Syndrome-Associated Variants in the KCNH2-Encoded Kv11.1 Channel. Circ. Arrhythm. Electrophysiol. 2018, 11, e005859. [Google Scholar] [CrossRef]
- Gong, Q.; Anderson, C.L.; January, C.T.; Zhou, Z. Role of glycosylation in cell surface expression and stability of HERG potassium channels. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H77–H84. [Google Scholar] [CrossRef] [Green Version]
- Limberg, M. Genetische Ursachen des Langen-QT-Syndroms. Doctoral Thesis, Philipps-Universität Marburg, Marburg, Germany, 2011. [Google Scholar] [CrossRef]
- Anson, B.D.; Ackerman, M.J.; Tester, D.J.; Will, M.L.; Delisle, B.P.; Anderson, C.L.; January, C.T. Molecular and functional characterization of common polymorphisms in HERG (KCNH2) potassium channels. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H2434–H2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordeiro, J.M.; Brugada, R.; Wu, Y.S.; Hong, K.; Dumaine, R. Modulation of I(Kr) inactivation by mutation N588K in KCNH2: A link to arrhythmogenesis in short QT syndrome. Cardiovasc. Res. 2005, 67, 498–509. [Google Scholar] [CrossRef]
- McPate, M.J.; Duncan, R.S.; Milnes, J.T.; Witchel, H.J.; Hancox, J.C. The N588K-HERG K+ channel mutation in the ‘short QT syndrome’: Mechanism of gain-in-function determined at 37 degrees C. Biochem. Biophys. Res. Commun. 2005, 334, 441–449. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Priori, S.G.; Spazzolini, C.; Moss, A.J.; Vincent, G.M.; Napolitano, C.; Denjoy, I.; Guicheney, P.; Breithardt, G.; Keating, M.T.; et al. Genotype-phenotype correlation in the long-QT syndrome: Gene-specific triggers for life-threatening arrhythmias. Circulation 2001, 103, 89–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenewein, T.; Neumann, T.; Erkapic, D.; Kuniss, M.; Verhoff, M.A.; Thiel, G.; Kauferstein, S. Influence of genetic modifiers on sudden cardiac death cases. Int. J. Legal Med. 2018, 132, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Du, C.Y.; El Harchi, A.; Zhang, Y.H.; Orchard, C.H.; Hancox, J.C. Pharmacological inhibition of the hERG potassium channel is modulated by extracellular but not intracellular acidosis. J. Cardiovasc. Electrophysiol. 2011, 22, 1163–1170. [Google Scholar] [CrossRef]
- Liu, L.; Tian, J.; Lu, C.; Chen, X.; Fu, Y.; Xu, B.; Zhu, C.; Sun, Y.; Zhang, Y.; Zhao, Y.; et al. Electrophysiological Characteristics of the LQT2 Syndrome Mutation KCNH2-G572S and Regulation by Accessory Protein KCNE2. Front. Physiol. 2016, 7, 650. [Google Scholar] [CrossRef]
- Cui, J.; Melman, Y.; Palma, E.; Fishman, G.I. McDonald TV Cyclic AMP regulates the HERG K(+) channel by dual pathways. Curr. Biol. 2000, 10, 671–674. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Committee ALQA Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Thomas, P.; Smart, T.G. HEK293 cell line: A vehicle for the expression of recombinant proteins. J. Pharmacol. Toxicol. Methods 2005, 51, 187–200. [Google Scholar] [CrossRef]
- Brun, S.; Schall, N.; Jeltsch-David, H.; Seze, J.; Muller, S. Assessing Autophagy in Sciatic Nerves of a Rat Model that Develops Inflammatory Autoimmune Peripheral Neuropathies. Cells 2017, 6, 30. [Google Scholar] [CrossRef] [Green Version]
- Gilda, J.E.; Gomes, A.V. Stain-Free total protein staining is a superior loading control to beta-actin for Western blots. Anal. Biochem. 2013, 440, 186–188. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA Change (cDNA) | Protein | Frequency of Variant (GnomAD) | Mutation Tester | SIFT Prediction | Polyphen-2 | Previously Reported in Patients (HGMD) and Phenotype | ACMG |
|---|---|---|---|---|---|---|---|
| c.3457C > T | P.H1153Y | 0.0048% | Disease causing | Deleterious | Probably damaging (0.981) | Yes/LQTs [22] | 3 [24] |
| Study | Phenotype | Variant | Current Density |
|---|---|---|---|
| pA/pF | |||
| This study | SADS | WT | 58.3 ± 14.6 (19) |
| H1153Y | 18.7 ± 5.1 * (23) | ||
| WT + H153Y | 55.7 ± 20.4 (8) | ||
| Anderson et al., 2014 [18] | WT | 97.7 ± 6.2 (9) | |
| LQT2 patient (0–90 y.o) | R1005Q | 97.7 ± 6.2 (7) | |
| LQT2 patient (0–90 y.o) | L1049P | 34.6 ± 4.5 (7) | |
| LQT2 patient (0–90 y.o) | L1066V | 74.3 ± 8.7 (6) | |
| Biliczki et al., 2008 [20] | WT | 8.0 ± 1.0(13) | |
| Macrolide-induced QT prolongation, asymptomatic | R954C | 4.2 ± 1.2 * (5) | |
| Survived SCD, no syncope | G1036D | 6.3 ± 1.4 * (7) | |
| Anson et al., 2004 [33] | WT | 94.4 ± 18.3 (19) | |
| SNP in SIDS cohort | K897T | 57.3 ± 7.2 (22) | |
| SNP in SIDS cohort | P967L | 116.9 ± 18.3 (20) | |
| SNP in SIDS cohort | R1047L | 129.5 ± 14.5 (20) | |
| SNP in SIDS cohort | Q1068R | 126.1 ± 13.9 (20) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farrugia, A.; Rollet, K.; Sinniger, J.; Brun, S.; Spenle, C.; Ludes, B.; Taleb, O.; Mensah-Nyagan, A.G. H1153Y-KCNH2 Mutation Identified in a Sudden Arrhythmic Death Syndrome Case Alters Channel Gating. Int. J. Mol. Sci. 2021, 22, 9235. https://doi.org/10.3390/ijms22179235
Farrugia A, Rollet K, Sinniger J, Brun S, Spenle C, Ludes B, Taleb O, Mensah-Nyagan AG. H1153Y-KCNH2 Mutation Identified in a Sudden Arrhythmic Death Syndrome Case Alters Channel Gating. International Journal of Molecular Sciences. 2021; 22(17):9235. https://doi.org/10.3390/ijms22179235
Chicago/Turabian StyleFarrugia, Audrey, Kevin Rollet, Jérome Sinniger, Susana Brun, Caroline Spenle, Bertrand Ludes, Omar Taleb, and Ayikoe Guy Mensah-Nyagan. 2021. "H1153Y-KCNH2 Mutation Identified in a Sudden Arrhythmic Death Syndrome Case Alters Channel Gating" International Journal of Molecular Sciences 22, no. 17: 9235. https://doi.org/10.3390/ijms22179235
APA StyleFarrugia, A., Rollet, K., Sinniger, J., Brun, S., Spenle, C., Ludes, B., Taleb, O., & Mensah-Nyagan, A. G. (2021). H1153Y-KCNH2 Mutation Identified in a Sudden Arrhythmic Death Syndrome Case Alters Channel Gating. International Journal of Molecular Sciences, 22(17), 9235. https://doi.org/10.3390/ijms22179235