
NOP53 Suppresses Autophagy through ZKSCAN3-Dependent and -Independent Pathways



Abstract
:1. Introduction
2. Results
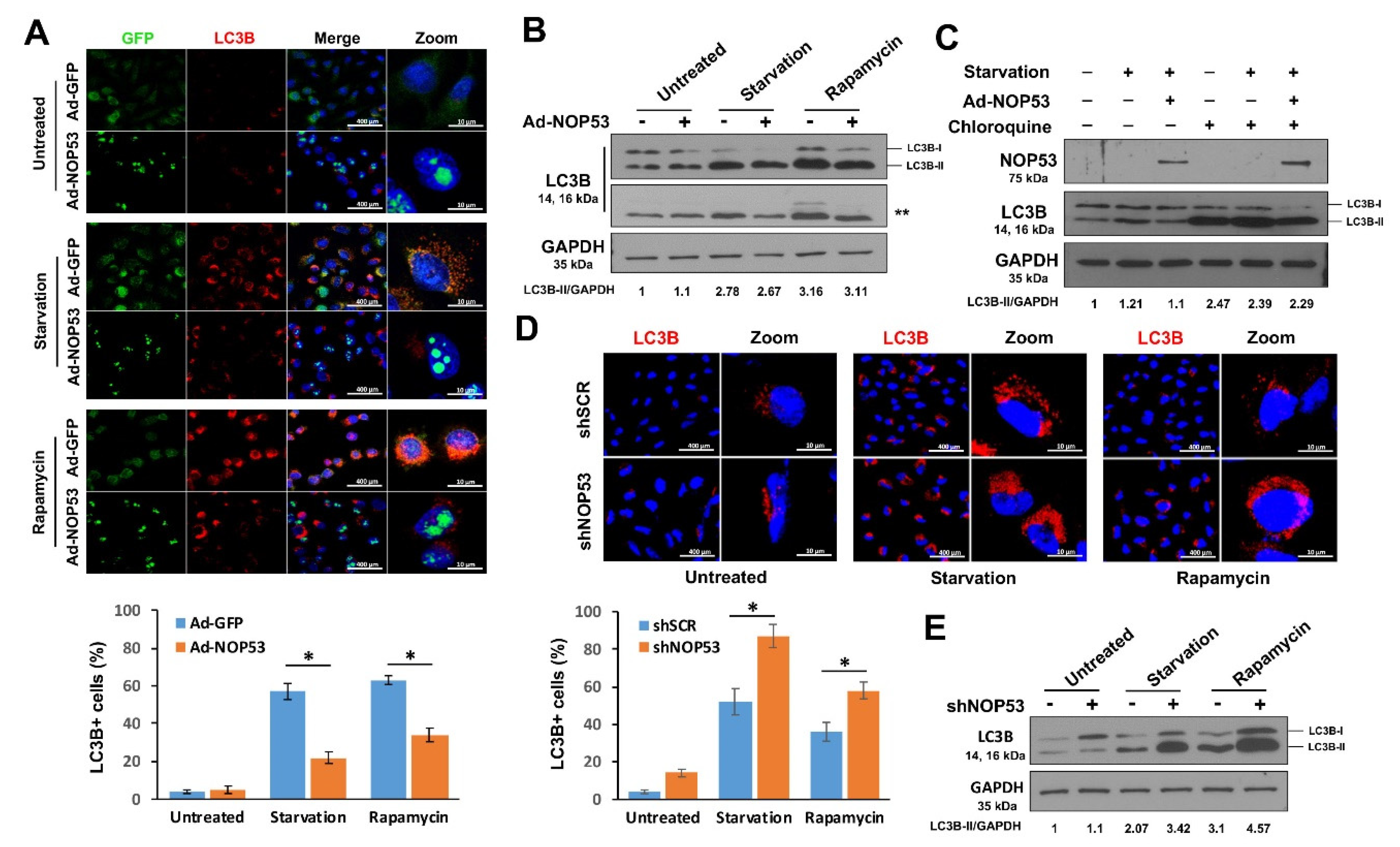
2.1. NOP53 Suppresses Autophagy
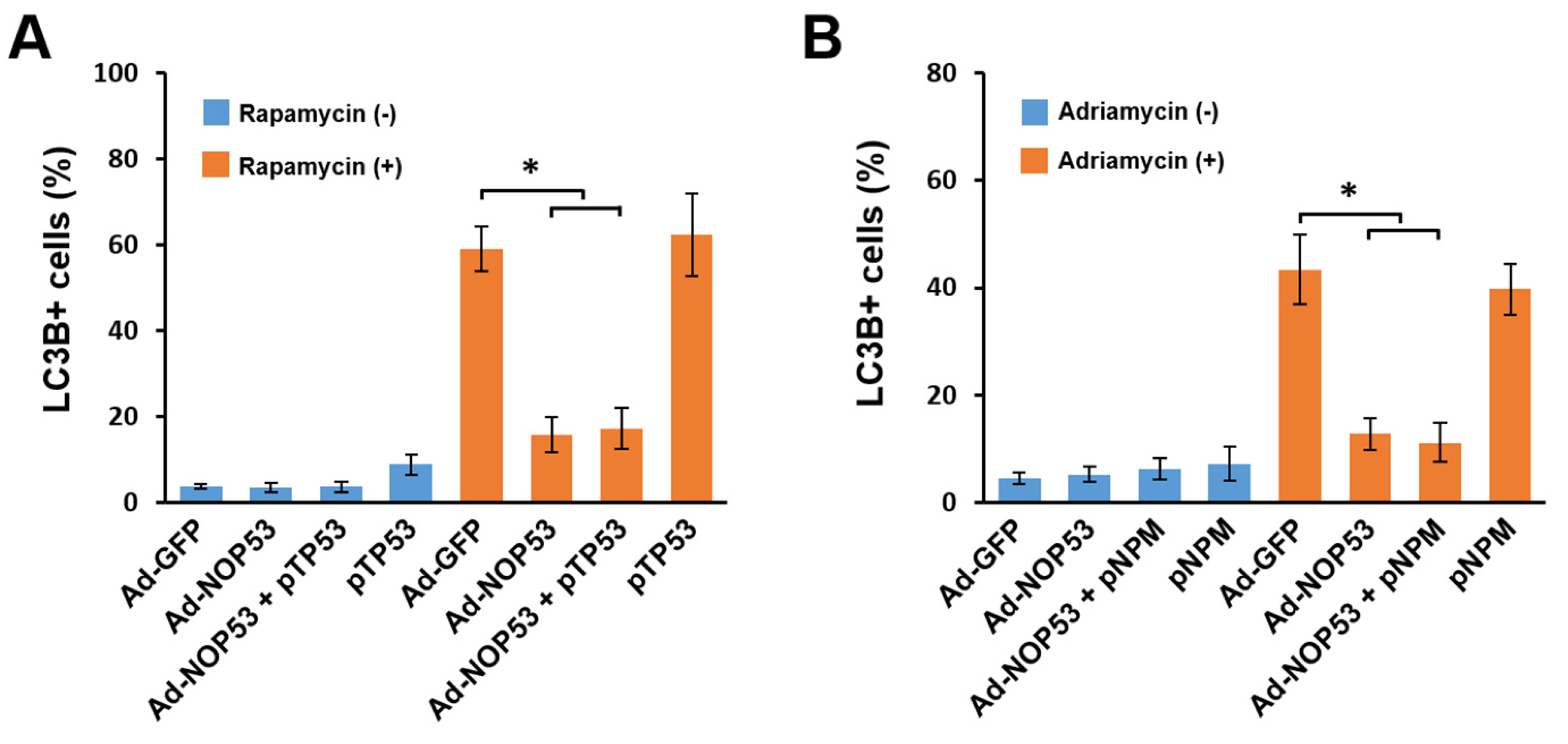
2.2. Suppression of Autophagy by NOP53 Is Independent of p53 or NPM
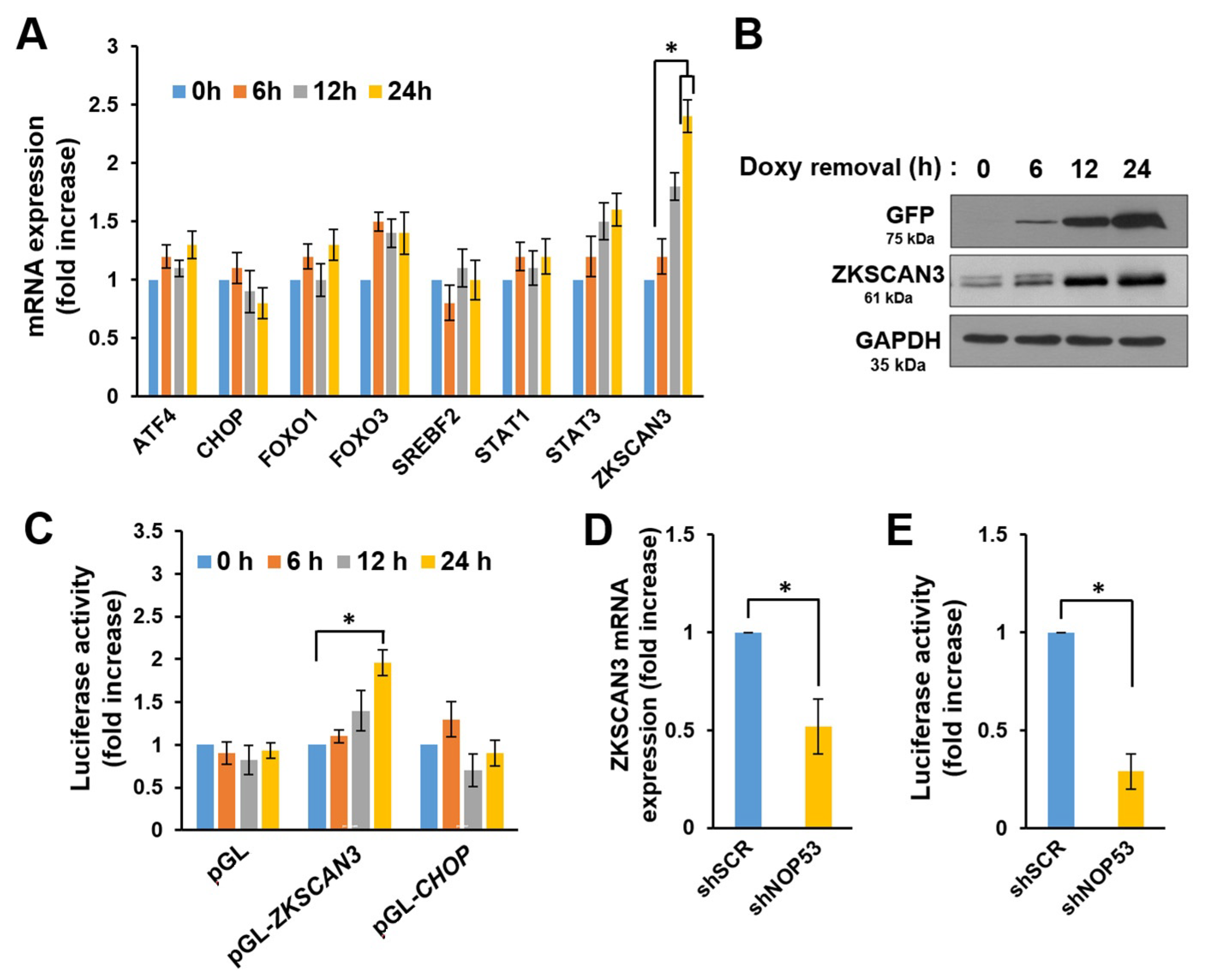
2.3. NOP53 Transcriptionally Upregulates the Expression of Autophagy Repressor ZKSCAN3
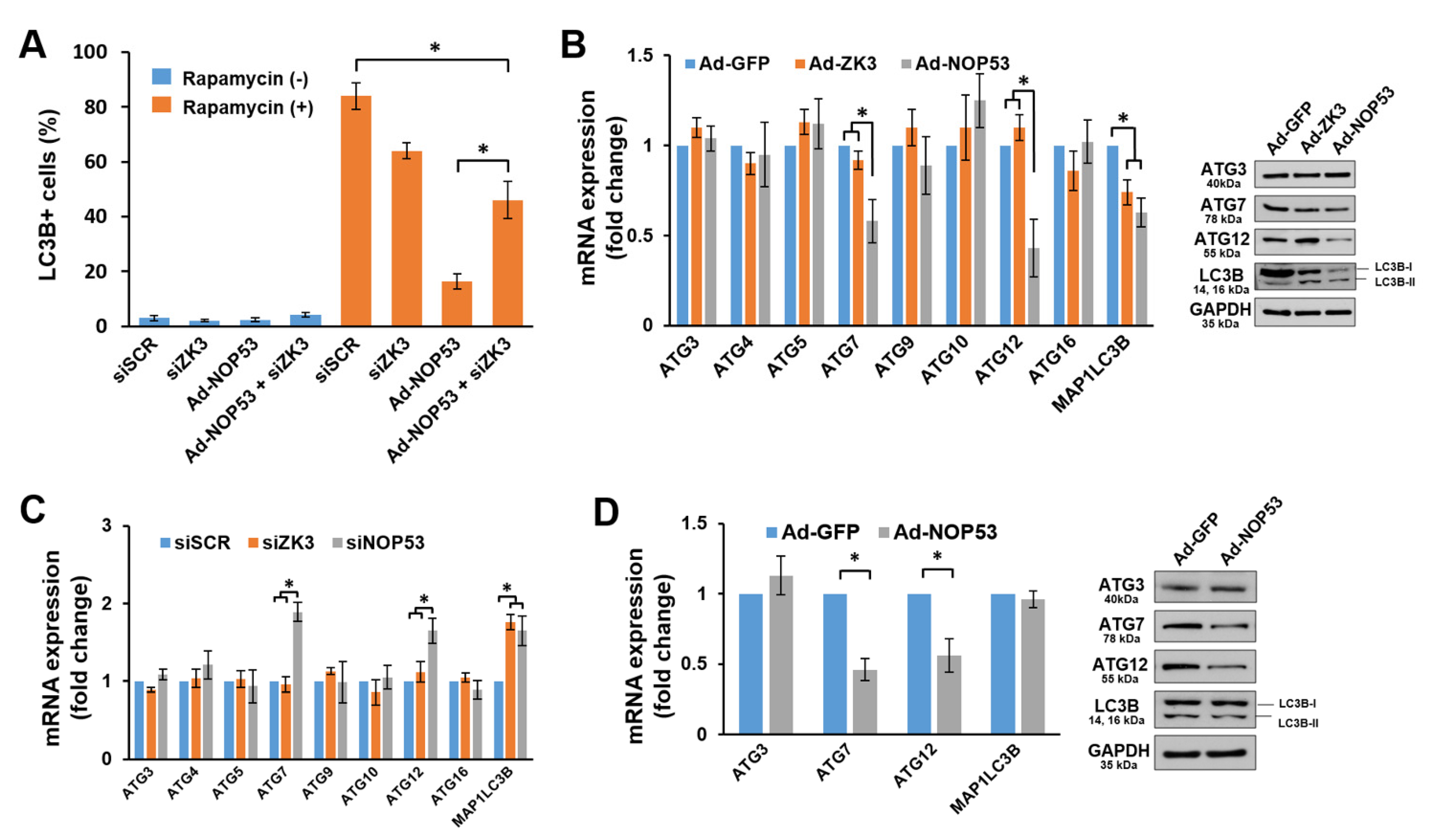
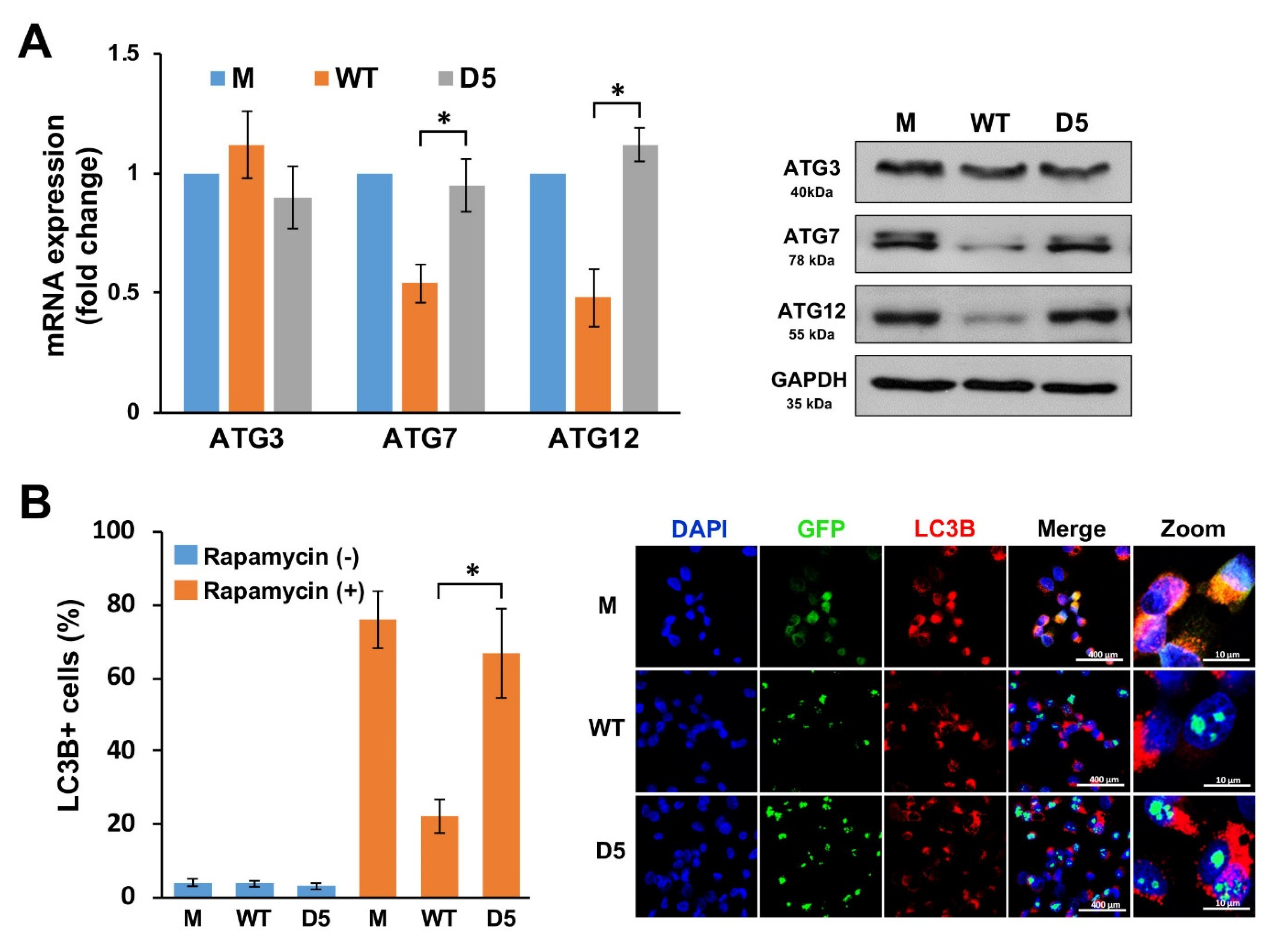
2.4. NOP53 Suppresses Autophagy by Downregulating Autophagy-Related (ATG) Genes and Proteins Both through ZKSCAN3-Dependent and -Independent Pathways
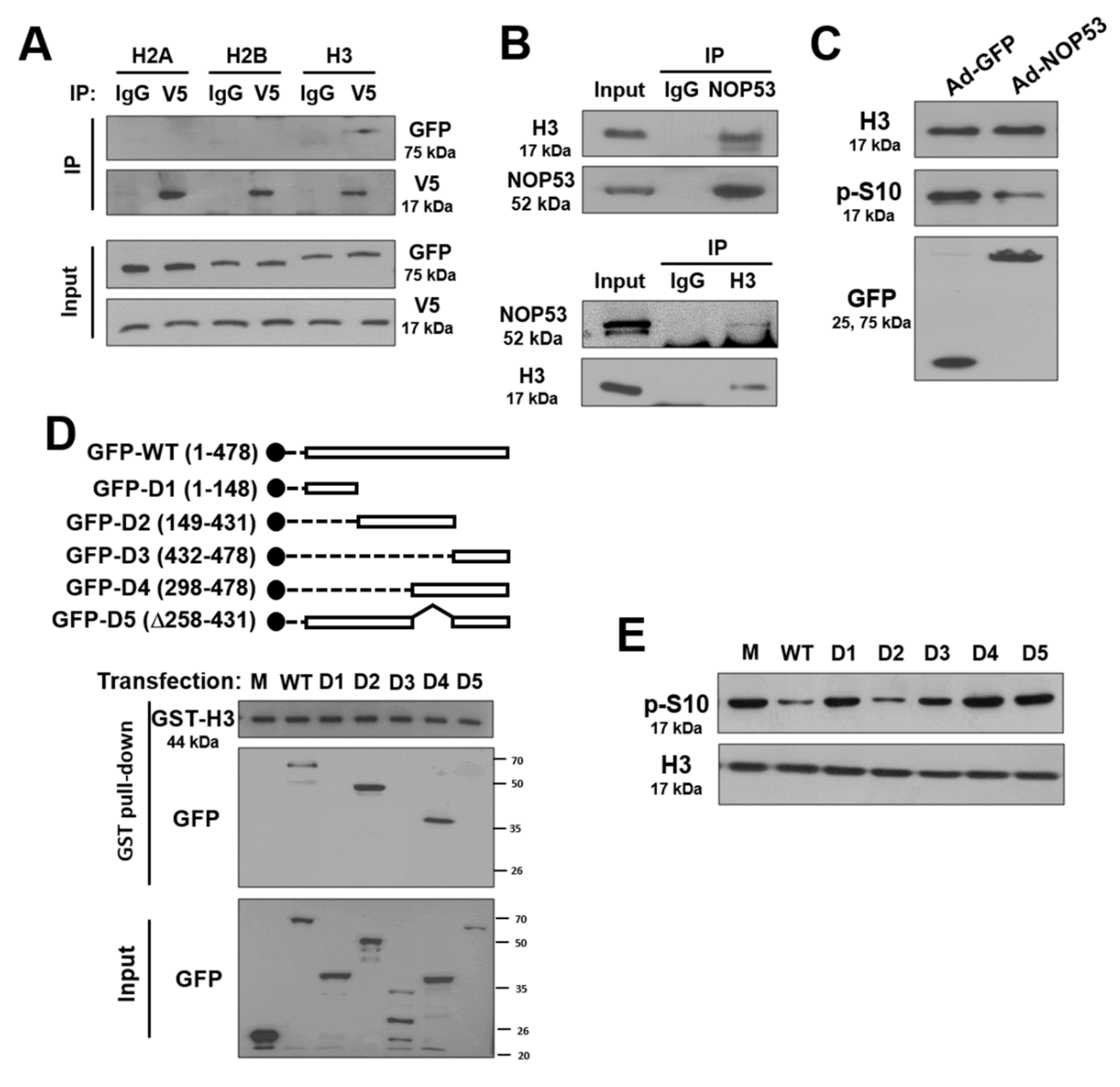
2.5. NOP53 Interacts with Histones and Suppresses Phosphorylation of Histone H3 at S10
2.6. Histone H3 Dephosphorylation at S10 Is Crucial for ATG Suppression by NOP53
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Antibodies, and Reagents
4.2. Plasmid and Viral Constructions and Transduction
4.3. Real-Time qPCR and Promoter Assays
4.4. Immunofluorescence Microscopy and Quantitative Analysis
4.5. Western Blotting and Immunoprecipitation
4.6. Recombinant Proteins and Pull-Down Assays
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mizushima, N.; Klionsky, D.J. Protein turnover via autophagy: Implications for metabolism. Annu. Rev. Nutr. 2007, 27, 19–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niedergang, F.; Grinstein, S. How to build a phagosome. new concepts for an old process. Curr. Opin. Cel. Biol. 2018, 50, 57–63. [Google Scholar] [CrossRef]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Rusten, T.E.; Lindmo, K.; Juhász, G.; Sass, M.; Seglen, P.O.; Brech, A.; Stenmark, H. Programmed autophagy in the Drosophila fat body is induced by ecdysone through regulation of the PI3K pathway. Dev. Cell 2004, 7, 179–192. [Google Scholar] [CrossRef] [Green Version]
- Panda, P.K.; Mukhopadhyay, S.; Das, D.N.; Sinha, N.; Naik, P.P.; Bhutia, S.K. Mechanism of autophagic regulation in carcinogenesis and cancer therapeutics. Semin. Cell Dev. Biol. 2015, 39, 43–55. [Google Scholar] [CrossRef]
- Füllgrabe, J.; Klionsky, D.J.; Joseph, B. The return of the nucleus: Transcriptional and epigenetic control of autophagy. Nat. Rev. Mol. Cell Biol. 2014, 15, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.F.; Bertram, P.G.; Ai, W.; Zheng, X.F. Regulation of APG14 expression by the GATA-type transcription factor Gln3p. J. Biol. Chem. 2001, 276, 6463–6467. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Das, M.; Reilly, J.; Davis, R.J. JNK regulates FoxO-dependent autophagy in neurons. Genes. Dev. 2011, 25, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zhou, Y.; Sun, Q.; Zhou, J.; Pan, H.; Sui, X. Regulation of Autophagy by MiRNAs and Their Emerging Roles in Tumorigenesis and Cancer Treatment. Int. Rev. Cell Mol. Biol. 2017, 334, 1–26. [Google Scholar] [CrossRef]
- Gozuacik, D.; Akkoc, Y.; Ozturk, D.G.; Kocak, M. Autophagy-regulating microRNAs and cancer. Front. Oncol. 2017, 18, 65. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wu, H.; Liu, X.; Li, B.; Chen, Y.; Ren, Z.; Liu, C.G.; Yang, J.M. Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy 2009, 5, 816–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comincini, S.; Allavena, G.; Palumbo, S.; Morini, M.; Durando, F.; Angeletti, F.; Pirtli, L.; Miracco, C. microRNA-17 regulates the expression of ATG7 and modulates the autophagy process, improving the sensitivity to temozolomide and low-dose ionizing radiation treatments in human glioblastoma cells. Cancer Biol. Ther. 2013, 14, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Emmott, E.; Hiscox, J.A. Nucleolar targeting: The hub of the matter. EMBO Rep. 2009, 10, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Katagiri, N.; Kuroda, T.; Kishimoto, H.; Hayashi, Y.; Kumazawa, T.; Kimura, K. The nucleolar protein nucleophosmin is essential for autophagy induced by inhibiting Pol I transcription. Sci. Rep. 2015, 5, 8903. [Google Scholar] [CrossRef] [PubMed]
- Reef, S.; Zalckvar, E.; Shifman, O.; Bialik, S.; Sabanay, H.; Oren, M.; Kimchi, A. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol. Cell 2006, 22, 463–475. [Google Scholar] [CrossRef]
- Budina-Kolomets, A.; Hontz, R.D.; Pimkina, J.; Murphy, M.E. A conserved domain in exon 2 coding for the human and murine ARF tumor suppressor protein is required for autophagy induction. Autophagy 2013, 9, 1553–1565. [Google Scholar] [CrossRef] [Green Version]
- Okahara, F.; Itoh, K.; Nakagawara, A.; Murakami, M.; Kanaho, Y.; Maehama, T. Critical role of PICT-1, a tumor suppressor candidate, in phosphatidylinositol 3,4,5-trisphosphate signals and tumorigenic transformation. Mol. Biol. Cell 2006, 17, 4888–4895. [Google Scholar] [CrossRef] [Green Version]
- Yim, J.H.; Kim, Y.J.; Ko, J.H.; Cho, Y.E.; Kim, S.M.; Kim, J.Y.; Lee, S.; Park, J.H. The putative tumor suppressor gene GLTSCR2 induces PTEN-modulated cell death. Cell Death Differ. 2007, 14, 1872–1879. [Google Scholar] [CrossRef]
- Sasaki, M.; Kawahara, K.; Nishio, M.; Mimori, K.; Kogo, R.; Hamada, K.; Itoh, B.; Wang, J.; Komatsu, Y.; Yang, Y.R.; et al. Regulation of the MDM2-P53 pathway and tumor growth by PICT1 via nucleolar RPL11. Nat. Med. 2011, 17, 944–951. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Kim, J.Y.; Kim, Y.J.; Seok, K.O.; Kim, J.H.; Chang, Y.J.; Kang, H.Y.; Park, J.H. Nucleolar protein GLTSCR2 stabilizes p53 in response to ribosomal stresses. Cell Death. Differ. 2012, 19, 1613–1622. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Cho, Y.E.; An, Y.M.; Kim, S.H.; Lee, Y.G.; Park, J.H.; Lee, S. GLTSCR2 is an upstream negative regulator of nucleophosmin in cervical cancer. J. Cell Mol. Med. 2015, 19, 1245–1252. [Google Scholar] [CrossRef]
- Lee, S.; Cho, Y.E.; Kim, S.H.; Kim, Y.J.; Park, J.H. GLTSCR2 promotes the nucleoplasmic translocation and subsequent degradation of nucleolar ARF. Oncotarget 2017, 8, 16293–16302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, S.; Goodwin, J.G.; Chauhan, S.; Manyam, G.; Wang, J.; Kamat, A.M.; Boyd, D.D. ZKSCAN3 is a master transcriptional repressor of autophagy. Mol. Cell 2013, 50, 16–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdone, L.; Agricola, E.; Caserta, M.; Di Mauro, E. Histone acetylation in gene regulation. Brief. Funct. Genomic. Proteomic. 2006, 5, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Rikiishi, H. Autophagic and apoptotic effects of HDAC inhibitors on cancer cells. J. Biomed. Biotechnol. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Duo, Y.; Hu, B.; Wang, Z.; Zhang, F.; Tsai, H.; Zhang, J.; Zhou, L.; Wang, L.; Wang, X.; et al. PICT-1 triggers a pro-death autophagy through inhibiting rRNA transcription and AKT/mTOR/p70S6K signaling pathway. Oncotarget 2016, 7, 78747–78763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in ATG7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef]
- Eisenberg, T.; Schroeder, S.; Büttner, S.; Carmona-Gutierrez, D.; Pendl, T.; Andryushkova, A.; Marino, G.; Pietrocola, F.; Harger, A.; Zimmermann, A.; et al. A histone point mutation that switches on autophagy. Autophagy 2014, 10, 1143–1145. [Google Scholar] [CrossRef] [Green Version]
- Koeneke, E.; Witt, O.; Oehme, I. HDAC Family Members Intertwined in the Regulation of Autophagy. A Druggable Vulnerability in Aggressive Tumor Entities. Cells 2015, 4, 135–168. [Google Scholar] [CrossRef] [PubMed]
- N’Diaye, E.N.; Kajihara, K.K.; Hsieh, I.; Morisaki, H.; Debnath, J.; Brown, E.J. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep. 2009, 10, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Renna, M.; Schaffner, C.; Winslow, A.R.; Menzies, F.M.; Peden, A.A.; Floto, R.A.; Rubinsztein, D.C. Autophagic substrate clearance requires activity of the syntaxin-5 SNARE complex. J. Cell Sci. 2011, 124, 469–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Sugita, H.; Yoshimori, T.; Ohsumi, Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 1998, 273, 33889–33892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polson, H.E.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbé, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Seok, K.O.; Kim, Y.J.; Bae, W.K.; Lee, S.; Park, J.H. Involvement of GLTSCR2 in the DNA Damage Response. Am. J. Pathol. 2011, 179, 1257–1264. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| ATF4 | CTTACGTTGCCATGATCCCT | TCCCATCTCCAGGTGTTCTC |
| CHOP | TGGAAGCCTGGTATGAGGAC | CAGAACCAGCAGAGGTCACA |
| FOXO1 | TGGACATGCTCAGCAGACATC | TGAACCGCCTGACCCAA |
| FOXO3 | AGAAGTTCCCCAGCGACTTG | GTTGGTTTGAACGTGGGGA |
| SREBF2 | AGGAGAACATGGTGCTGA | TTGACTCTGAGCCAGGAA |
| STAT1 | CGGTTGAACCCTACACGAAG | ACCAGAGCCAATGGAACTTG |
| STAT3 | AGCAGCACCTTCAGGATGTC | AGTGACCAGGCAGAAGATGC |
| ZKSCAN3 | GGTCTCCCTGGGTGATGAAA | GCACATGTAGGAATCTGGGC |
| MAPILC3B | ACGATACAAGGGTGAGAAGCA | GACCATGCTGTGTCCGTTC |
| ATG3 | GTTGGAAACAGATGAGGCTACC | TAGCCAAACAACCATAATCGTG |
| ATG4A | GTGCTCGTCTATGGTTTACATAC | AATACCAACGCATCCTACAGTGC |
| ATG5 | ACCAGTTTTGGGCCATCAAT | GTGTGTGCAACTGTCCATCTG |
| ATG7 | AAGCAAGAGAAAGCTGGTCATC | AGTAGCAGCCAAGCTTGTAACC |
| ATG9A | CTCATGCAGTTCCTCTTTGTGGT | GTGCCAGGATTCAGGAAAATGG |
| ATG10 | CTGAAGGACATATGGGAAGGAG | GAGGTAGATTCAGCCCAACAAC |
| ATG12 | GCAGCTTCCTACTTCAATTGCT | ATTGCAGAATGTTTGCAGACTA |
| ATG16L1 | AAATGGCCCAACTGAGGATTAA | ATTGCAGAATGTTTGCAGACTA |
| GAPDH | GGCATGGACTGTGGTCATGAG | GGCATGGACTGTGGTCATGAG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, Y.-E.; Kim, Y.-J.; Lee, S.; Park, J.-H. NOP53 Suppresses Autophagy through ZKSCAN3-Dependent and -Independent Pathways. Int. J. Mol. Sci. 2021, 22, 9318. https://doi.org/10.3390/ijms22179318
Cho Y-E, Kim Y-J, Lee S, Park J-H. NOP53 Suppresses Autophagy through ZKSCAN3-Dependent and -Independent Pathways. International Journal of Molecular Sciences. 2021; 22(17):9318. https://doi.org/10.3390/ijms22179318
Chicago/Turabian StyleCho, Young-Eun, Yong-Jun Kim, Sun Lee, and Jae-Hoon Park. 2021. "NOP53 Suppresses Autophagy through ZKSCAN3-Dependent and -Independent Pathways" International Journal of Molecular Sciences 22, no. 17: 9318. https://doi.org/10.3390/ijms22179318
APA StyleCho, Y. -E., Kim, Y. -J., Lee, S., & Park, J. -H. (2021). NOP53 Suppresses Autophagy through ZKSCAN3-Dependent and -Independent Pathways. International Journal of Molecular Sciences, 22(17), 9318. https://doi.org/10.3390/ijms22179318