
Hydration of Simple Model Peptides in Aqueous Osmolyte Solutions

 , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
2. Material and Methods
2.1. Chemicals and Solutions
2.2. NAGMA Synthesis

2.3. FTIR Measurements
2.4. Spectral Data Analysis
2.5. Molecular Dynamics Simulations
2.5.1. Investigated Systems
- -
- one diglycine molecule in water and one NAGMA molecule in water—these systems were used to find the most frequently occurring conformations of the solutes (through temperature-replica exchange molecular dynamics simulations) and to obtain trajectories (through molecular dynamics simulations) to study properties of solutes’ solvation shells (these included hydrogen bond analysis—described in more detail in the Supplementary Materials—and weak noncovalent interaction analysis—described below in Section 2.6). The solutes were placed in rectangular cuboid simulation boxes, with minimal distance from the box walls equal to about 20 Å. It ensured that the system remained relatively small but simultaneously big enough for the properties of solvation water to be able to converge to properties of bulk water with increasing distance from the peptide. The number of water molecules in these systems was equal to 4038.
- -
- one diglycine molecule or one NAGMA molecule in water with three molecules of the osmolytes (TMAO or urea)—these systems were used to study interactions of the osmolytes with diglycine and NAGMA. They contained 4035 molecules of water. This corresponds to molal concentration equal to about 0.04 molkg.
- -
- three TMAO or three urea molecules in water—these systems were used for the purposes of the initial assessment of the validity and applicability of the chosen sets of force field parameters. They contained 4040 molecules of water.
2.5.2. Force Field Parameters
2.5.3. Protocols of the Simulations
2.6. Weak Noncovalent Interaction Analysis
3. Results and Discussion
3.1. FTIR Investigation of Water Structure
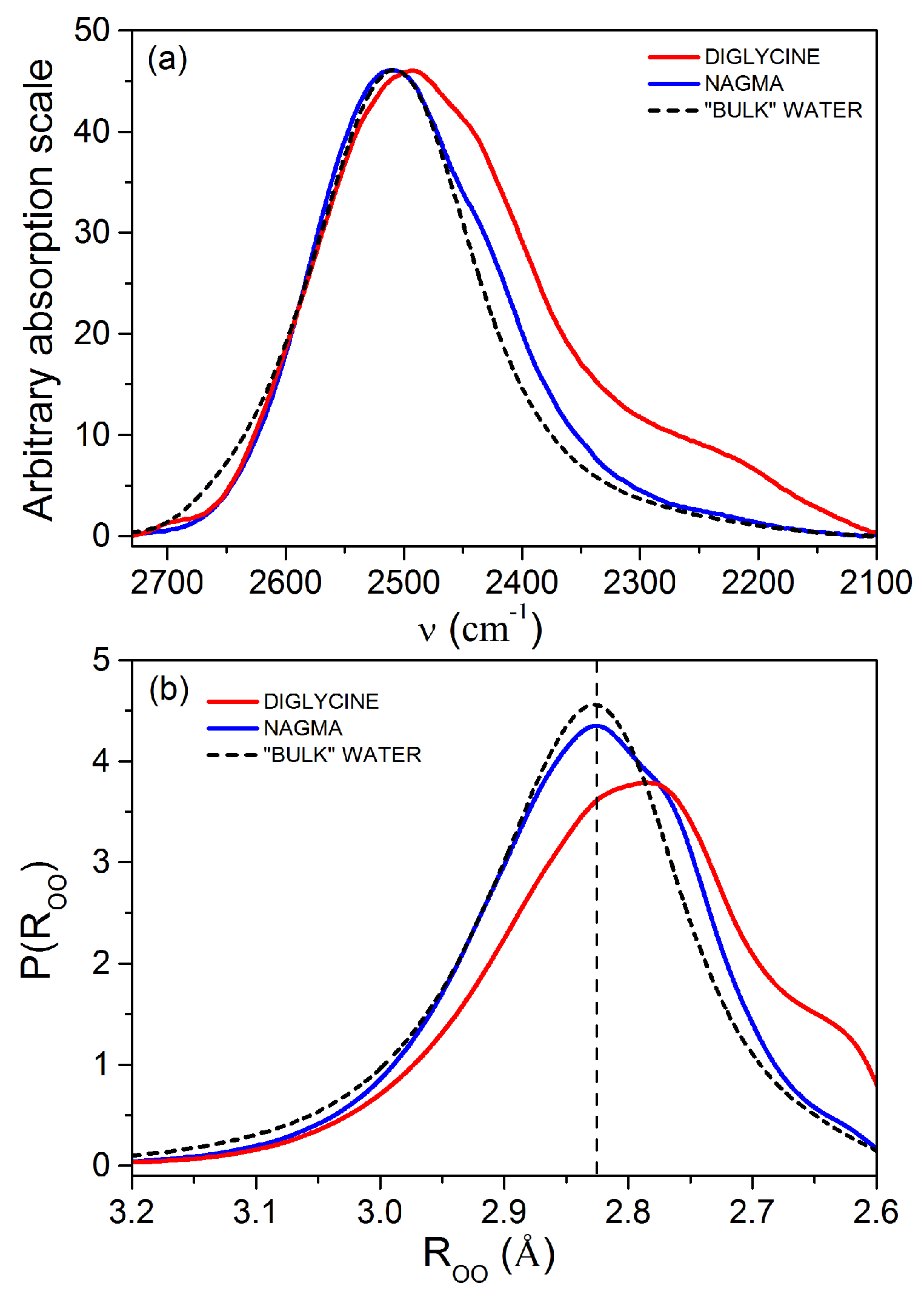
3.1.1. Characteristics of the Hydration Shell of Diglycine and NAGMA
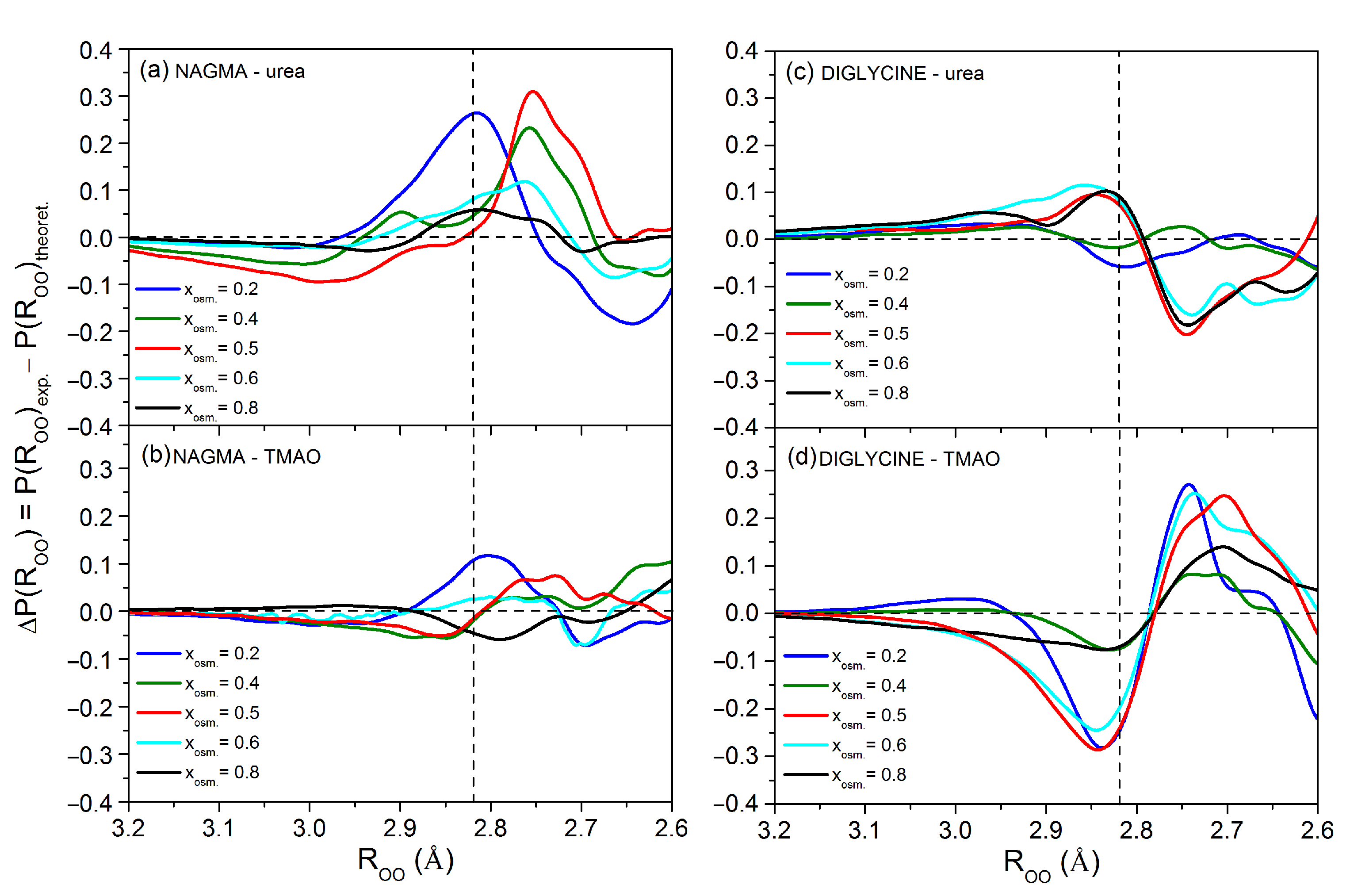
3.1.2. Analysis of the Water Structure around Peptides in the Presence of Osmolytes
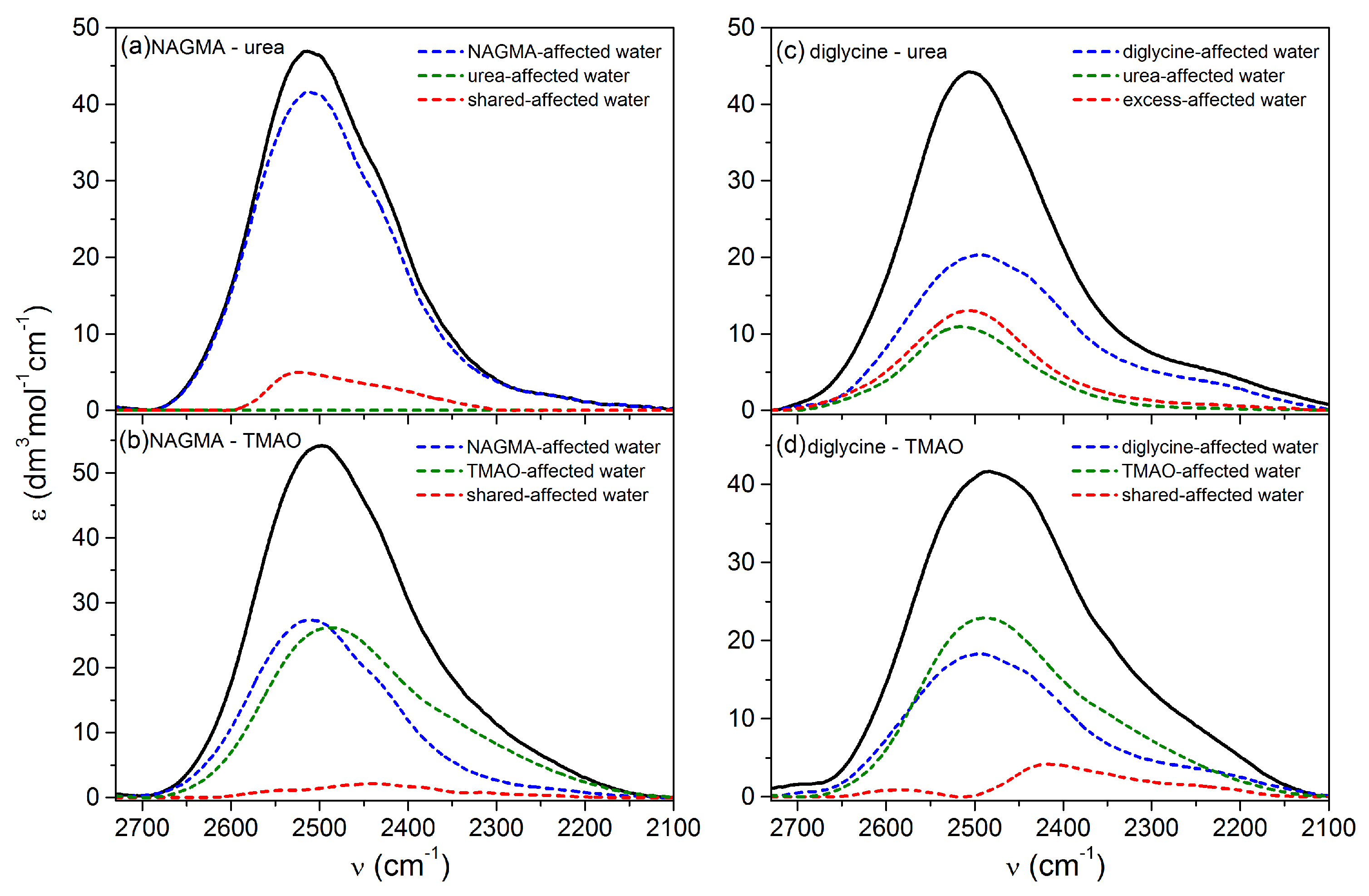
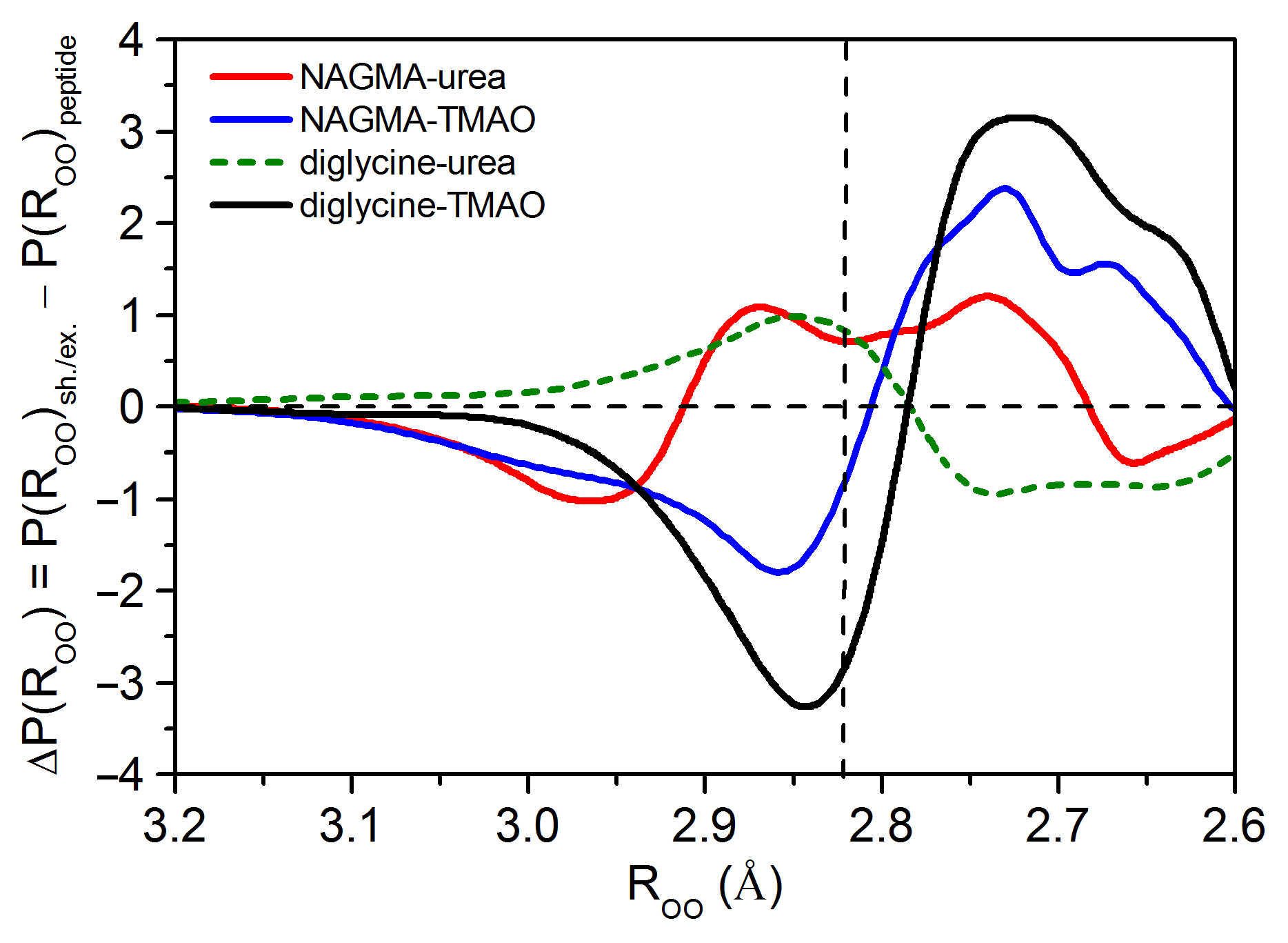
3.1.3. Water Spectra Changed Simultaneously by Peptide and Osmolyte
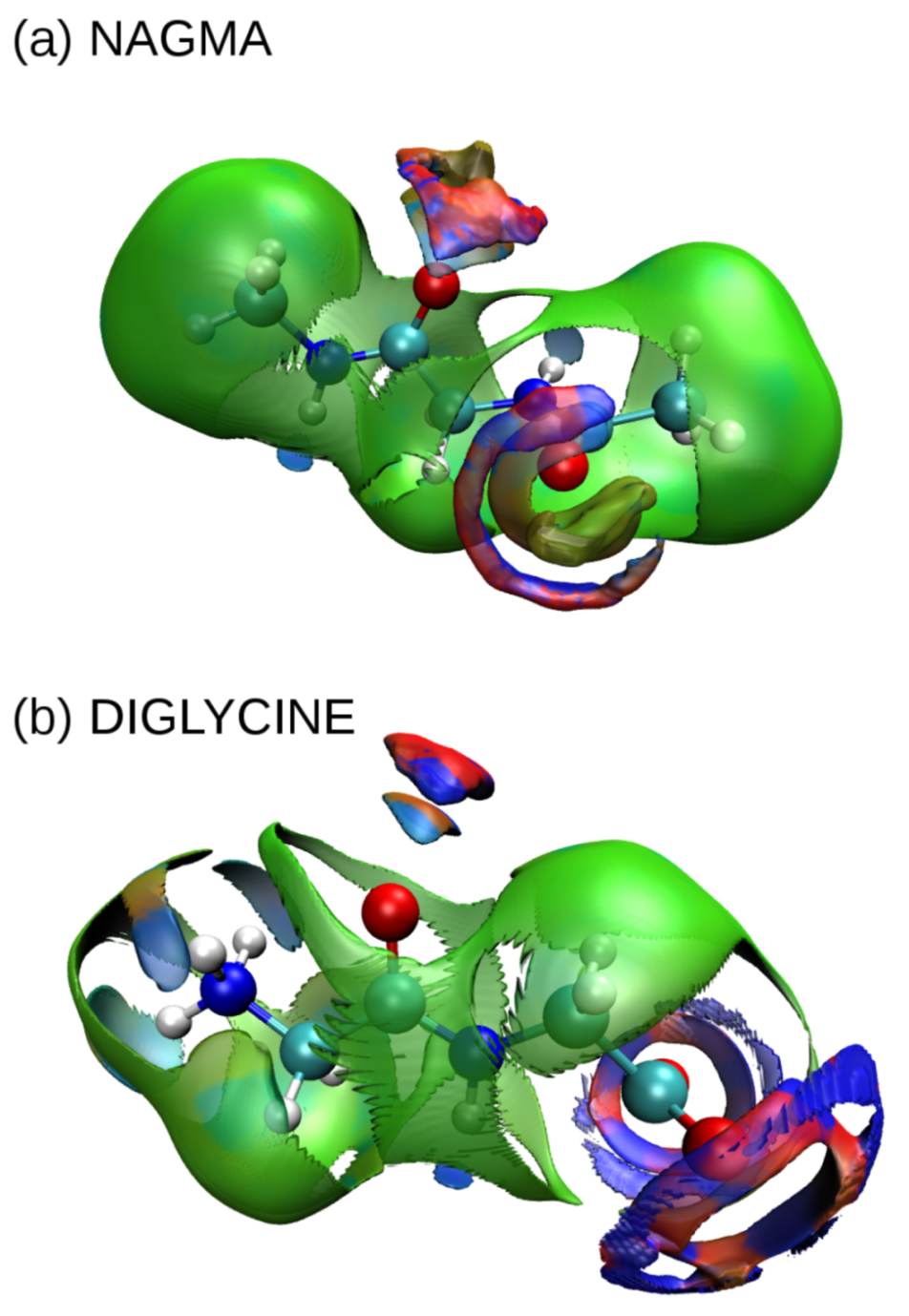
3.2. Noncovalent Interaction Analysis in Peptide–Water Systems
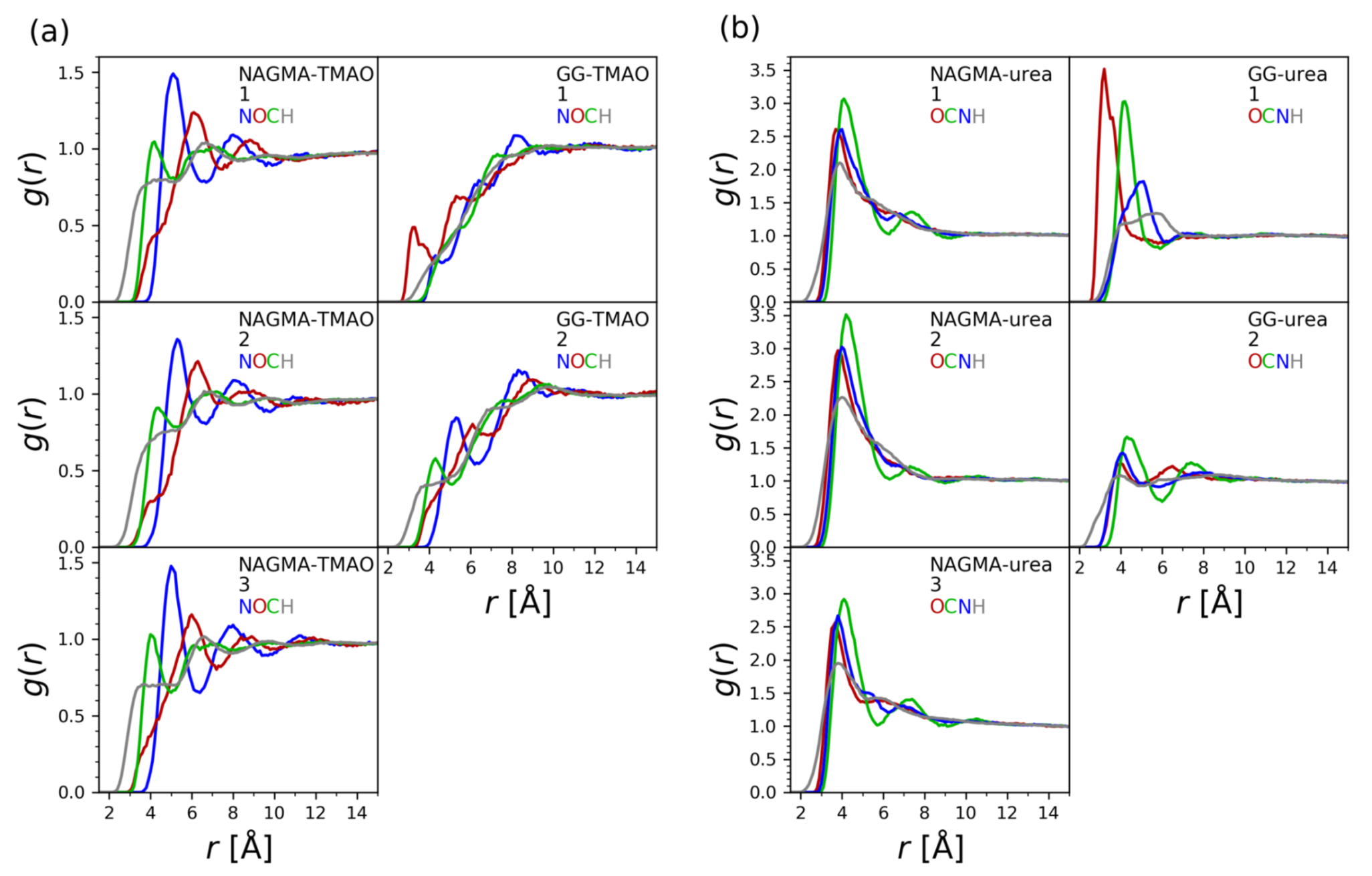
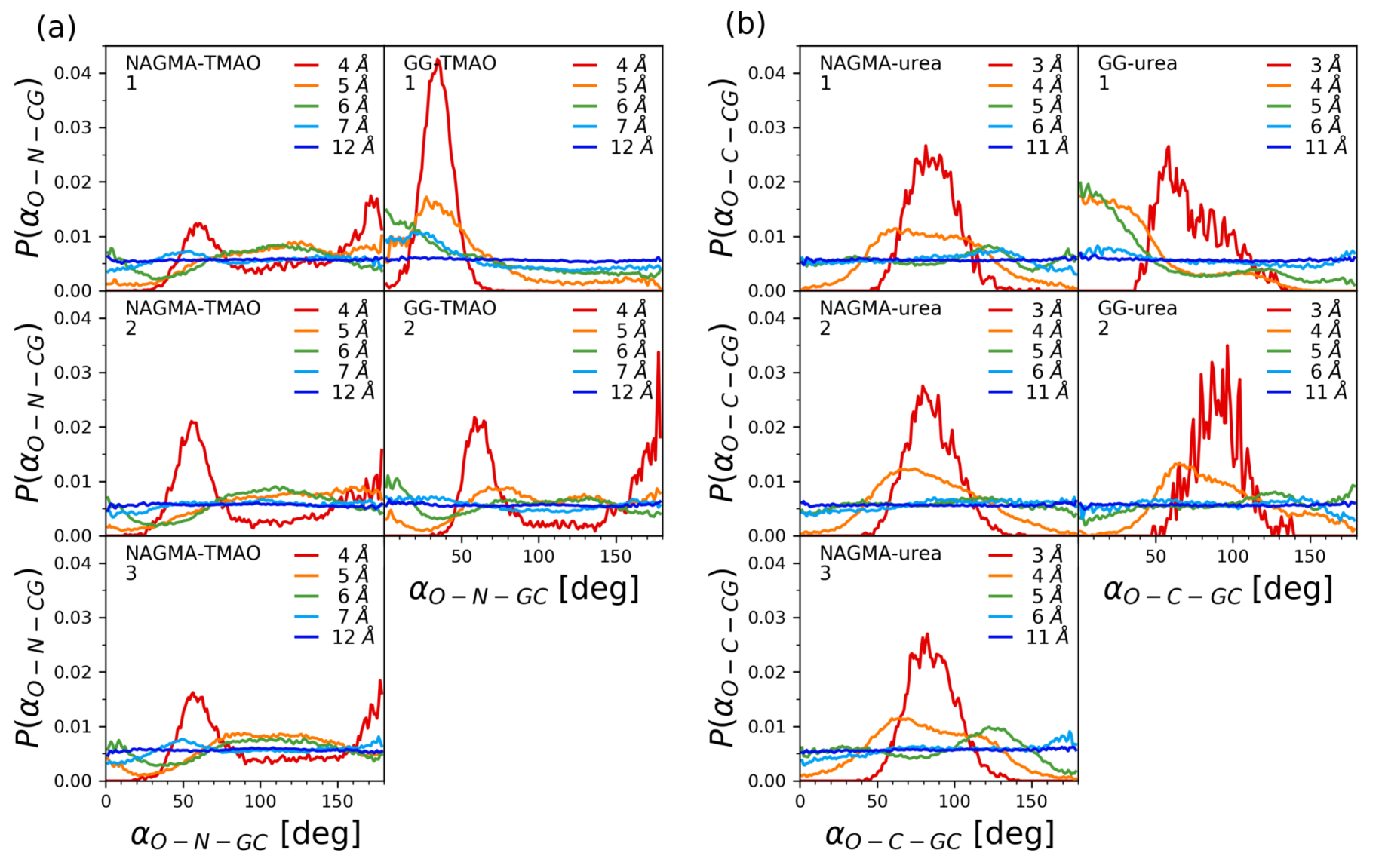
3.3. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AcOEt | ethyl acetate |
| aNCI | average noncovalent interactions |
| CDI | 1,1-carbonyldiimidazole |
| DMF | dimethylformamide |
| DMSO | dimethyl sulfoxide |
| FTIR | Fourier transform infrared |
| GC | geometric center |
| GdmCl | guanidinium chloride |
| MD | molecular dynamics |
| MeCN | acetonitrile |
| NAAMA | N-acetyl-alanine-methylamide |
| NAGMA | N-acetyl-glycine-methylamide |
| NATA | N-acetyl-tryptophan-amide |
| NCI | noncovalent interactions |
| PTFE | polytetrafluoroethylene |
| REMD | replica exchange molecular dynamics |
| TMAO | trimethylamine N-oxide |
References
- Irvine, G.B.; El-Agnaf, O.M.; Shankar, G.M.; Walsh, D.M. Protein aggregation in the brain: The molecular basis for Alzheimer’s and Parkinson’s diseases. Mol. Med. 2008, 14, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Marzban, L.; Park, K.; Verchere, C.B. Islet amyloid polypeptide and type 2 diabetes. Exp. Gerontol. 2003, 38, 347–351. [Google Scholar] [CrossRef]
- Sabate, R.; Rousseau, F.; Schymkowitz, J.; Batlle, C.; Ventura, S. Amyloids or prions? That is the question. Prion 2015, 9, 200–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Stephens, A.D.; Kaminski Schierle, G.S. The role of water in amyloid aggregation kinetics. Curr. Opin. Struct. Biol. 2019, 58, 115–123. [Google Scholar] [CrossRef]
- Chaplin, M. Do we underestimate the importance of water in cell biology? Nat. Rev. Mol. Cell Biol. 2006, 7, 861–866. [Google Scholar] [CrossRef]
- Bellissent-Funel, M.C.; Hassanali, A.; Havenith, M.; Henchman, R.; Pohl, P.; Sterpone, F.; Van Der Spoel, D.; Xu, Y.; Garcia, A.E. Water Determines the Structure and Dynamics of Proteins. Chem. Rev. 2016, 116, 7673–7697. [Google Scholar] [CrossRef]
- Levy, Y.; Onuchic, J.N. Water Mediation in Protein Folding and Molecular Recognition. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 389–415. [Google Scholar] [CrossRef] [Green Version]
- Dielmann-Gessner, J.; Grossman, M.; Nibali, V.C.; Born, B.; Solomonov, I.; Fields, G.B.; Havenith, M.; Sagi, I. Enzymatic turnover of macromolecules generates long-lasting protein-water-coupled motions beyond reaction steady state. Proc. Natl. Acad. Sci. USA 2014, 111, 17857–17862. [Google Scholar] [CrossRef] [Green Version]
- Pizzitutti, F.; Marchi, M.; Sterpone, F.; Rossky, P.J. How protein surfaces induce anomalous dynamics of hydration water. J. Phys. Chem. B 2007, 111, 7584–7590. [Google Scholar] [CrossRef]
- Sterpone, F.; Stirnemann, G.; Laage, D. Magnitude and molecular origin of water slowdown next to a protein. J. Am. Chem. Soc. 2012, 134, 4116–4119. [Google Scholar] [CrossRef]
- Fogarty, A.C.; Laage, D. Water dynamics in protein hydration shells: The molecular origins of the dynamical perturbation. J. Phys. Chem. B 2014, 118, 7715–7729. [Google Scholar] [CrossRef]
- Gavrilov, Y.; Leuchter, J.D.; Levy, Y. On the coupling between the dynamics of protein and water. Phys. Chem. Chem. Phys. 2017, 19, 8243–8257. [Google Scholar] [CrossRef]
- Jana, B.; Pal, S.; Bagchi, B. Hydration dynamics of protein molecules in aqueous solution: Unity among diversity. J. Chem. Sci. 2012, 124, 317–325. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Mondal, S.; Bagchi, B. Distinguishing dynamical features of water inside protein hydration layer: Distribution reveals what is hidden behind the average. J. Chem. Phys. 2017, 147, 24901. [Google Scholar] [CrossRef] [PubMed]
- Russo, D.; Hura, G.; Head-Gordon, T. Hydration Dynamics Near a Model Protein Surface. Biophys. J. 2004, 86, 1852–1862. [Google Scholar] [CrossRef] [Green Version]
- Russo, D.; Copley, J.R.D.; Ollivier, J.; Teixeira, J. On the behaviour of water hydrogen bonds at biomolecular sites: Dependences on temperature and on network dimensionality. J. Mol. Struct. 2010, 972, 81–86. [Google Scholar] [CrossRef]
- Russo, D.; Ollivier, J.; Teixeira, J. Water hydrogen bond analysis on hydrophilic and hydrophobic biomolecule sites. Phys. Chem. Chem. Phys. 2008, 10, 4968–4974. [Google Scholar] [CrossRef] [PubMed]
- Russo, D.; Murarka, R.K.; Copley, J.R.; Head-Gordon, T. Molecular view of water dynamics near model peptides. J. Phys. Chem. B 2005, 109, 12966–12975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazur, K.; Heisler, I.A.; Meech, S.R. Ultrafast dynamics and hydrogen-bond structure in aqueous solutions of model peptides. J. Phys. Chem. B 2010, 114, 10684–10691. [Google Scholar] [CrossRef]
- Bolen, D.W.; Baskakov, I.V. The osmophobic effect: Natural selection of a thermodynamic force in protein folding. J. Mol. Biol. 2001, 310, 955–963. [Google Scholar] [CrossRef]
- Auton, M.; Bolen, D.W. Predicting the energetics of osmolyte-induced protein folding/unfolding. Proc. Natl. Acad. Sci. USA 2005, 102, 15065–15068. [Google Scholar] [CrossRef] [Green Version]
- Street, T.O.; Bolen, D.W.; Rose, G.D. A molecular mechanism for osmolyte-induced protein stability. Proc. Natl. Acad. Sci. USA 2006, 103, 13997–14002. [Google Scholar] [CrossRef] [Green Version]
- Auton, M.; Rösgen, J.; Sinev, M.; Holthauzen, L.M.F.; Bolen, D.W. Osmolyte effects on protein stability and solubility: A balancing act between backbone and side-chains. Biophys. Chem. 2011, 159, 90–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, G.; Muttathukattil, A.N.; Mondal, B. Cosolvent effects on the growth of amyloid fibrils. Curr. Opin. Struct. Biol. 2020, 60, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Yancey, P.H. Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. J. Exp. Biol. 2005, 208, 2819–2830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, A.T.; Rösgen, J.; Bolen, D.W. Osmolyte effects on kinetics of FKBP12 C22A folding coupled with prolyl isomerization. J. Mol. Biol. 2003, 330, 851–866. [Google Scholar] [CrossRef]
- Ortiz-Costa, S.; Sorenson, M.M.; Sola-Penna, M. Counteracting effects of urea and methylamines in function and structure of skeletal muscle myosin. Arch. Biochem. Biophys. 2002, 408, 272–278. [Google Scholar] [CrossRef]
- Venkatesu, P.; mu Lin, H.; Lee, M.J. Counteracting effects of trimethylamine N-oxide and betaine on the interactions of urea with zwitterionic glycine peptides. Thermochim. Acta 2009, 491, 20–28. [Google Scholar] [CrossRef]
- Zeman, J.; Holm, C.; Smiatek, J. The Effect of Small Organic Cosolutes on Water Structure and Dynamics. J. Chem. Eng. Data 2020, 65, 1197–1210. [Google Scholar] [CrossRef]
- Oprzeska-Zingrebe, E.A.; Smiatek, J. Interactions of a DNA G-quadruplex with TMAO and urea: A molecular dynamics study on co-solute compensation mechanisms. Phys. Chem. Chem. Phys. 2021, 23, 1254–1264. [Google Scholar] [CrossRef]
- Felitsky, D.J.; Cannon, J.G.; Capp, M.W.; Hong, J.; Van Wynsberghe, A.W.; Anderson, C.F.; Record, M.T. The exclusion of glycine betaine from anionic biopolymer surface: Why glycine betaine is an effective osmoprotectant but also a compatible solute. Biochemistry 2004, 43, 14732–14743. [Google Scholar] [CrossRef]
- Canchi, D.R.; Paschek, D.; Garcia, A.E. Equilibrium study of protein denaturation by urea. J. Am. Chem. Soc. 2010, 132, 2338–2344. [Google Scholar] [CrossRef]
- Canchi, D.R.; Jayasimha, P.; Rau, D.C.; Makhatadze, G.I.; Garcia, A.E. Molecular mechanism for the preferential exclusion of TMAO from protein surfaces. J. Phys. Chem. B 2012, 116, 12095–12104. [Google Scholar] [CrossRef] [Green Version]
- Saladino, G.; Marenchino, M.; Pieraccini, S.; Campos-Olivas, R.; Sironi, M.; Gervasio, F.L. A simple mechanism underlying the effect of protecting osmolytes on protein folding. J. Chem. Theory Comput. 2011, 7, 3846–3852. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Bennion, B.J.; Daggett, V.; Murphy, K.P. The molecular mechanism of stabilization of proteins by TMAO and its ability to counteract the effects of urea. J. Am. Chem. Soc. 2002, 124, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Bennion, B.J.; Daggett, V. The molecular basis for the chemical denaturation of proteins by urea. Proc. Natl. Acad. Sci. USA 2003, 100, 5142–5147. [Google Scholar] [CrossRef] [Green Version]
- Bennion, B.J.; Daggett, V. Counteraction of urea-induced protein denaturation by trimethylamine N-oxide: A chemical chaperone at atomic resolution. Proc. Natl. Acad. Sci. USA 2004, 101, 6433–6438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, S.; Patey, G.N. Structure and interaction in aqueous urea-Trimethylamine-N-oxide solutions. J. Am. Chem. Soc. 2007, 129, 4476–4482. [Google Scholar] [CrossRef] [PubMed]
- Russo, D. The impact of kosmotropes and chaotropes on bulk and hydration shell water dynamics in a model peptide solution. Chem. Phys. 2008, 345, 200–211. [Google Scholar] [CrossRef]
- Panuszko, A.; Bruździak, P.; Kaczkowska, E.; Stangret, J. General Mechanism of Osmolytes’ Influence on Protein Stability Irrespective of the Type of Osmolyte Cosolvent. J. Phys. Chem. B 2016, 120, 11159–11169. [Google Scholar] [CrossRef]
- Bruździak, P.; Panuszko, A.; Stangret, J. Influence of Osmolytes on Protein and Water Structure: A Step To Understanding the Mechanism of Protein Stabilization. J. Phys. Chem. B 2013, 117, 11502–11508. [Google Scholar] [CrossRef]
- Bruździak, P.; Panuszko, A.; Kaczkowska, E.; Piotrowski, B.; Daghir, A.; Demkowicz, S.; Stangret, J. Taurine as a water structure breaker and protein stabilizer. Amino Acids 2018, 50, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Rezus, Y.L.A.; Bakker, H.J. Effect of urea on the structural dynamics of water. Proc. Natl. Acad. Sci. USA 2006, 103, 18417–18420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandyopadhyay, D.; Mohan, S.; Ghosh, S.K.; Choudhury, N. Molecular Dynamics Simulation of Aqueous Urea Solution: Is Urea a Structure Breaker? J. Phys. Chem. B 2014, 118, 11757–11768. [Google Scholar] [CrossRef]
- Pazos, I.M.; Gai, F. Solute’s Perspective on How Trimethylamine Oxide, Urea, and Guanidine Hydrochloride Affect Water’s Hydrogen Bonding Ability. J. Phys. Chem. B 2012, 116, 12473–12478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oprzeska-Zingrebe, E.A.; Smiatek, J. Aqueous Mixtures of Urea and Trimethylamine-N-oxide: Evidence for Kosmotropic or Chaotropic Behavior? J. Phys. Chem. B 2019, 123, 4415–4424. [Google Scholar] [CrossRef]
- Lim, W.K.; Rösgen, J.; Englander, S.W. Urea, but not guanidinium, destabilizes proteins by forming hydrogen bonds to the peptide group. Proc. Natl. Acad. Sci. USA 2009, 106, 2595–2600. [Google Scholar] [CrossRef] [Green Version]
- Jas, G.S.; Rentchler, E.C.; Słowicka, A.M.; Hermansen, J.R.; Johnson, C.K.; Middaugh, C.R.; Kuczera, K. Reorientation Motion and Preferential Interactions of a Peptide in Denaturants and Osmolyte. J. Phys. Chem. B 2016, 120, 3089–3099. [Google Scholar] [CrossRef]
- Choudhary, S.; Kishore, N. Interactions of some short peptides with the osmolyte trimethylamine N-oxide in aqueous solution: Physico-chemical insights. J. Chem. Thermodyn. 2012, 52, 36–42. [Google Scholar] [CrossRef]
- Johnson, M.E.; Malardier-Jugroot, C.; Head-Gordon, T. Effects of co-solvents on peptide hydration water structure and dynamics. Phys. Chem. Chem. Phys. 2010, 12, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Malardier-Jugroot, C.; Bowron, D.T.; Soper, A.K.; Johnson, M.E.; Head-Gordon, T. Structure and water dynamics of aqueous peptide solutions in the presence of co-solvents. Phys. Chem. Chem. Phys. 2010, 12, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Panuszko, A.; Stangret, J.; Nowosielski, B.; Bruździak, P. Interactions between hydration spheres of two different solutes in solution: The least squares fitting with constraints as a tool to determine water properties in ternary systems. J. Mol. Liq. 2020, 310. [Google Scholar] [CrossRef]
- Stangret, J. Solute-affected vibrational spectra of water in Ca(ClO4)2 aqueous solutions. Spectrosc. Lett. 1988, 21, 369–381. [Google Scholar] [CrossRef]
- Stangret, J.; Gampe, T. Hydration sphere of tetrabutylammonium cation. FTIR studies of HDO spectra. J. Phys. Chem. B 1999, 103, 3778–3783. [Google Scholar] [CrossRef]
- Śmiechowski, M.; Stangret, J. Vibrational spectroscopy of semiheavy water (HDO) as a probe of solute hydration. Pure Appl. Chem. 2010, 82, 1869–1887. [Google Scholar] [CrossRef]
- Raskar, T.; Khavnekar, S.; Hosur, M. Time-Dependent X-Ray Diffraction Studies on Urea/Hen Egg White Lysozyme Complexes Reveal Structural Changes that Indicate Onset of Denaturation. Sci. Rep. 2016, 6, 32277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastings, J.; Owen, G.; Dekker, A.; Ennis, M.; Kale, N.; Muthukrishnan, V.; Turner, S.; Swainston, N.; Mendes, P.; Steinbeck, C. ChEBI in 2016: Improved services and an expanding collection of metabolites. Nucleic Acids Res. 2015, 44, D1214–D1219. [Google Scholar] [CrossRef]
- Patriksson, A.; van der Spoel, D. A Temperature Predictor for Parallel Tempering Simulations. Phys. Chem. Chem. Phys. 2008, 10, 2073–2077. [Google Scholar] [CrossRef]
- Wernet, P.; Nordlund, D.; Bergmann, U.; Cavalleri, M.; Odelius, N.; Ogasawara, H.; Naslund, L.Å.; Hirsch, T.K.; Ojamae, L.; Glatzel, P.; et al. The structure of the first coordination shell in liquid water. Science 2004, 304, 995–999. [Google Scholar] [CrossRef] [Green Version]
- Czapiewski, D.; Zielkiewicz, J. Structural properties of hydration shell around various conformations of simple polypeptides. J. Phys. Chem. B 2010, 114, 4536–4550. [Google Scholar] [CrossRef]
- Kuffel, A.; Zielkiewicz, J. Why the solvation water around proteins is more dense than bulk water. J. Phys. Chem. B 2012, 116, 12113–12124. [Google Scholar] [CrossRef]
- Best, R.B.; Mittal, J. Protein Simulations with an Optimized Water Model: Cooperative Helix Formation and Temperature-Induced Unfolded State Collapse. J. Phys. Chem. B 2010, 114, 14916–14923. [Google Scholar] [CrossRef]
- Abascal, J.L.F.; Vega, C. A General Purpose Model for the Condensed Phases of Water: TIP4P/2005. J. Chem. Phys. 2005, 123, 234505. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A Point-Charge Force Field for Molecular Mechanics Simulations of Proteins Based on Condensed-Phase Quantum Mechanical Calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Camilloni, C.; Bonetti, D.; Morrone, A.; Giri, R.; Dobson, C.M.; Brunori, M.; Gianni, S.; Vendruscolo, M. Towards a Structural Biology of the Hydrophobic Effect in Protein Folding. Sci. Rep. 2016, 6, 28285. [Google Scholar] [CrossRef] [PubMed]
- Pi, H.L.; Aragones, J.L.; Vega, C.; Noya, E.G.; Abascal, J.L.; Gonzalez, M.A.; McBride, C. Anomalies in Water as Obtained from Computer Simulations of the TIP4P/2005 Model: Density Maxima, and Density, Isothermal Compressibility and Heat Capacity Minima. Mol. Phys. 2009, 107, 365–374. [Google Scholar] [CrossRef] [Green Version]
- González, M.A.; Abascal, J.L.F. The Shear Viscosity of Rigid Water Models. J. Chem. Phys. 2010, 132, 096101. [Google Scholar] [CrossRef] [PubMed]
- Vega, C.; Abascal, J.L.F.; Nezbeda, I. Vapor-Liquid Equilibria from the Triple Point up to the Critical Point for the New Generation of TIP4P-Like Models: TIP4P/Ew, TIP4P/2005, and TIP4P/ice. J. Chem. Phys. 2006, 125, 034503. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 2016; ;University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Hölzl, C.; Kibies, P.; Imoto, S.; Frach, R.; Suladze, S.; Winter, R.; Marx, D.; Horinek, D.; Kast, S.M. Design Principles for High–Pressure Force Fields: Aqueous TMAO Solutions from Ambient to Kilobar Pressures. J. Chem. Phys. 2016, 144, 144104. [Google Scholar] [CrossRef]
- Markthaler, D.; Zeman, J.; Baz, J.; Smiatek, J.; Hansen, N. Validation of Trimethylamine-N-oxide (TMAO) Force Fields Based on Thermophysical Properties of Aqueous TMAO Solutions. J. Phys. Chem. B 2017, 121, 10674–10688. [Google Scholar] [CrossRef]
- Boopathi, S.; Kolandaivel, P. Molecular dynamics simulations and density functional theory studies of NALMA and NAGMA dipeptides. J. Biomol. Struct. Dyn. 2013, 31, 158–173. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Chaudret, R.; Hu, X.; Yang, W. Noncovalent Interaction Analysis in Fluctuating Environments. J. Chem. Theory Comput. 2013, 9, 2226–2234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, K.A.; Madan, B.; Manas, E.; Vanderkooi, J.M. Water structure changes induced by hydrophobic and polar solutes revealed by simulations and infrared spectroscopy. J. Chem. Phys. 2001, 114, 1791–1796. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.T.; Manson, A.C.; DeLyser, M.R.; Noid, W.G.; Cremer, P.S. Trimethylamine N-oxide stabilizes proteins via a distinct mechanism compared with betaine and glycine. Proc. Natl. Acad. Sci. USA 2017, 114, 2479–2484. [Google Scholar] [CrossRef] [Green Version]
- Bruździak, P.; Adamczak, B.; Kaczkowska, E.; Czub, J.; Stangret, J. Are stabilizing osmolytes preferentially excluded from the protein surface? FTIR and MD studies. Phys. Chem. Chem. Phys. 2015, 17, 23155–23164. [Google Scholar] [CrossRef] [PubMed]
- Stumpe, M.C.; Grubmüller, H. Interaction of Urea with Amino Acids: Implications for Urea-Induced Protein Denaturation. J. Am. Chem. Soc. 2007, 129, 16126–16131. [Google Scholar] [CrossRef] [Green Version]
- Despa, F.; Fernández, A.; Scott, L.R.; Berry, R.S. Hydration profiles of amyloidogenic molecular structures. J. Biol. Phys. 2008, 34, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Munroe, K.L.; Magers, D.H.; Hammer, N.I. Raman Spectroscopic Signatures of Noncovalent Interactions Between Trimethylamine N-oxide (TMAO) and Water. J. Phys. Chem. B 2011, 115, 7699–7707. [Google Scholar] [CrossRef]
- Moeser, B.; Horinek, D. Unified Description of Urea Denaturation: Backbone and Side Chains Contribute Equally in the Transfer Model. J. Phys. Chem. B 2014, 118, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Rossky, P.J. Protein denaturation by urea: Slash and bond. Proc. Natl. Acad. Sci. USA 2008, 105, 16825–16826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.S.; Reddy, G.; Straub, J.E.; Thirumalai, D. Entropic Stabilization of Proteins by TMAO. J. Phys. Chem. B 2011, 115, 13401–13407. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solute | oODa | gODb | fwhh c | I d | RoOO e | RgOO f |
|---|---|---|---|---|---|---|
| pure solutes | ||||||
| diglycine | 2492 ± 2 | 2454 ± 2 | 209 ± 4 | 9100 | 2.777 ± 0.003 | 2.803 ± 0.003 |
| NAGMA | 2511 ± 2 | 2485 ± 2 | 179 ± 4 | 8914 | 2.823 ± 0.003 | 2.833 ± 0.003 |
| urea | 2515 ± 2 | 2496 ± 2 | 153 ± 4 | 7708 | 2.836 ± 0.003 | 2.844 ± 0.003 |
| TMAO | 2486 ± 2 | 2445 ± 2 | 211 ± 4 | 11204 | 2.793 ± 0.003 | 2.795 ± 0.003 |
| bulk water | 2509 ± 2 | 2496 ± 2 | 162 ± 4 | 10053 | 2.823 ± 0.003 | 2.844 ± 0.003 |
| shared or excess affected water | ||||||
| diglycine–urea | 2507 ± 2 | 2487 ± 2 | 162 ± 4 | 2481 | 2.821 ± 0.003 | 2.836 ± 0.003 |
| diglycine–TMAO | 2416 ± 2 | 2381 ± 2 | 153 ± 4 | 786 | 2.747 ± 0.003 | 2.729 ± 0.003 |
| NAGMA–urea | 2522 ± 2 | 2472 ± 2 | 161 ± 4 | 778 | 2.836 ± 0.003 | 2.818 ± 0.003 |
| NAGMA–TMAO | 2438 ± 2 | 2432 ± 2 | 193 ± 4 | 426 | 2.767 ± 0.003 | 2.775 ± 0.003 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panuszko, A.; Pieloszczyk, M.; Kuffel, A.; Jacek, K.; Biernacki, K.A.; Demkowicz, S.; Stangret, J.; Bruździak, P. Hydration of Simple Model Peptides in Aqueous Osmolyte Solutions. Int. J. Mol. Sci. 2021, 22, 9350. https://doi.org/10.3390/ijms22179350
Panuszko A, Pieloszczyk M, Kuffel A, Jacek K, Biernacki KA, Demkowicz S, Stangret J, Bruździak P. Hydration of Simple Model Peptides in Aqueous Osmolyte Solutions. International Journal of Molecular Sciences. 2021; 22(17):9350. https://doi.org/10.3390/ijms22179350
Chicago/Turabian StylePanuszko, Aneta, Maciej Pieloszczyk, Anna Kuffel, Karol Jacek, Karol A. Biernacki, Sebastian Demkowicz, Janusz Stangret, and Piotr Bruździak. 2021. "Hydration of Simple Model Peptides in Aqueous Osmolyte Solutions" International Journal of Molecular Sciences 22, no. 17: 9350. https://doi.org/10.3390/ijms22179350
APA StylePanuszko, A., Pieloszczyk, M., Kuffel, A., Jacek, K., Biernacki, K. A., Demkowicz, S., Stangret, J., & Bruździak, P. (2021). Hydration of Simple Model Peptides in Aqueous Osmolyte Solutions. International Journal of Molecular Sciences, 22(17), 9350. https://doi.org/10.3390/ijms22179350