
Metformin Attenuates Postinfarction Myocardial Fibrosis and Inflammation in Mice

 , ,
, ,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
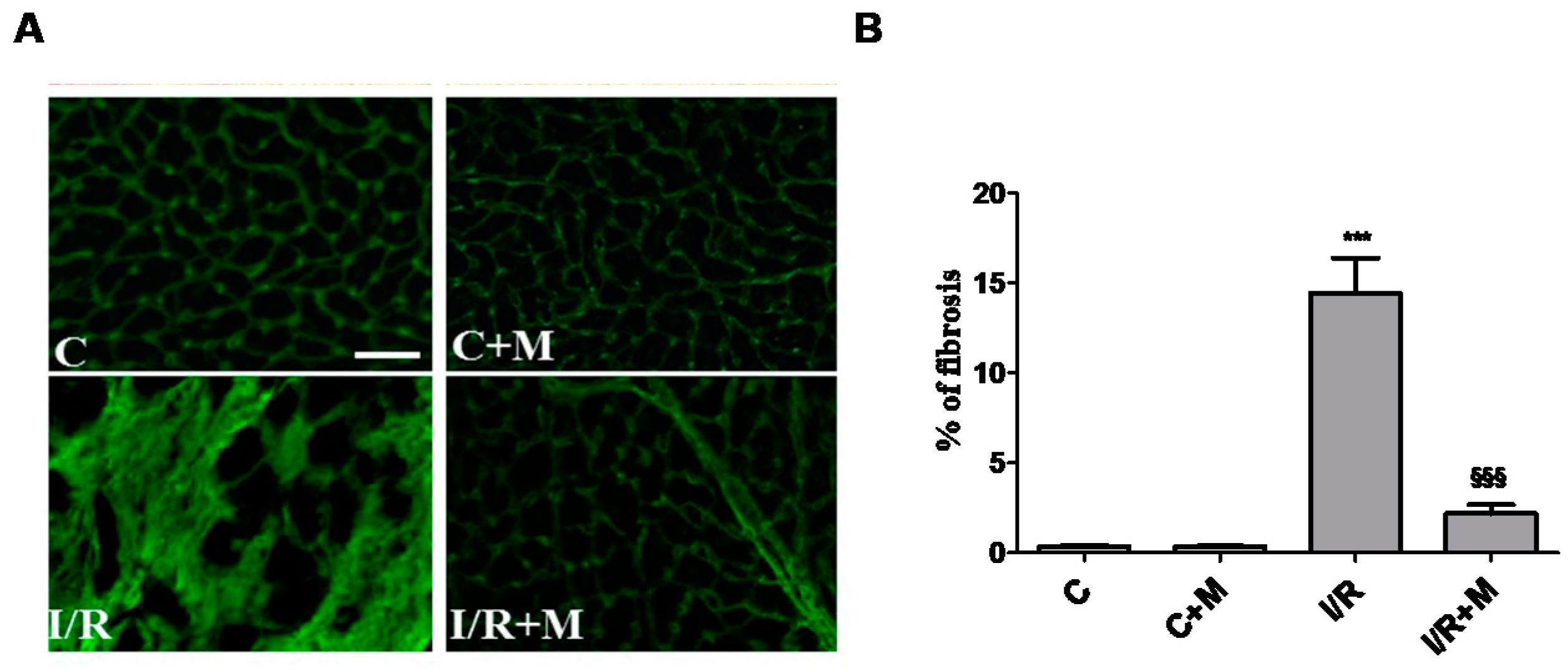
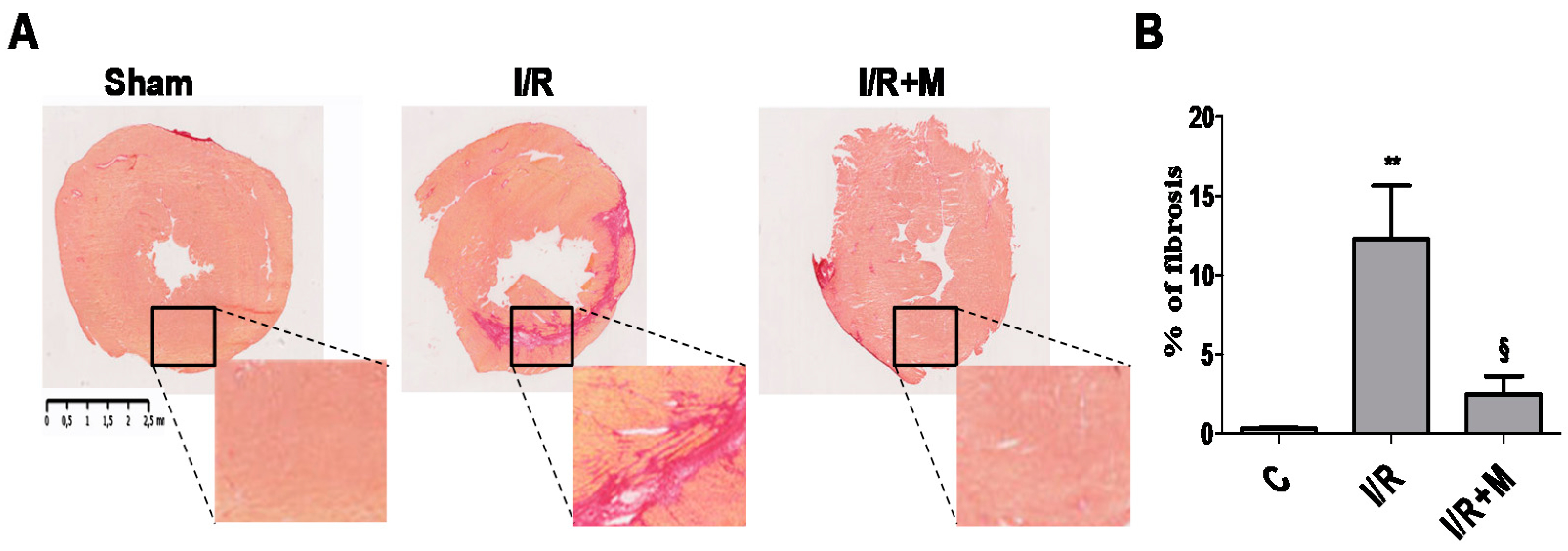
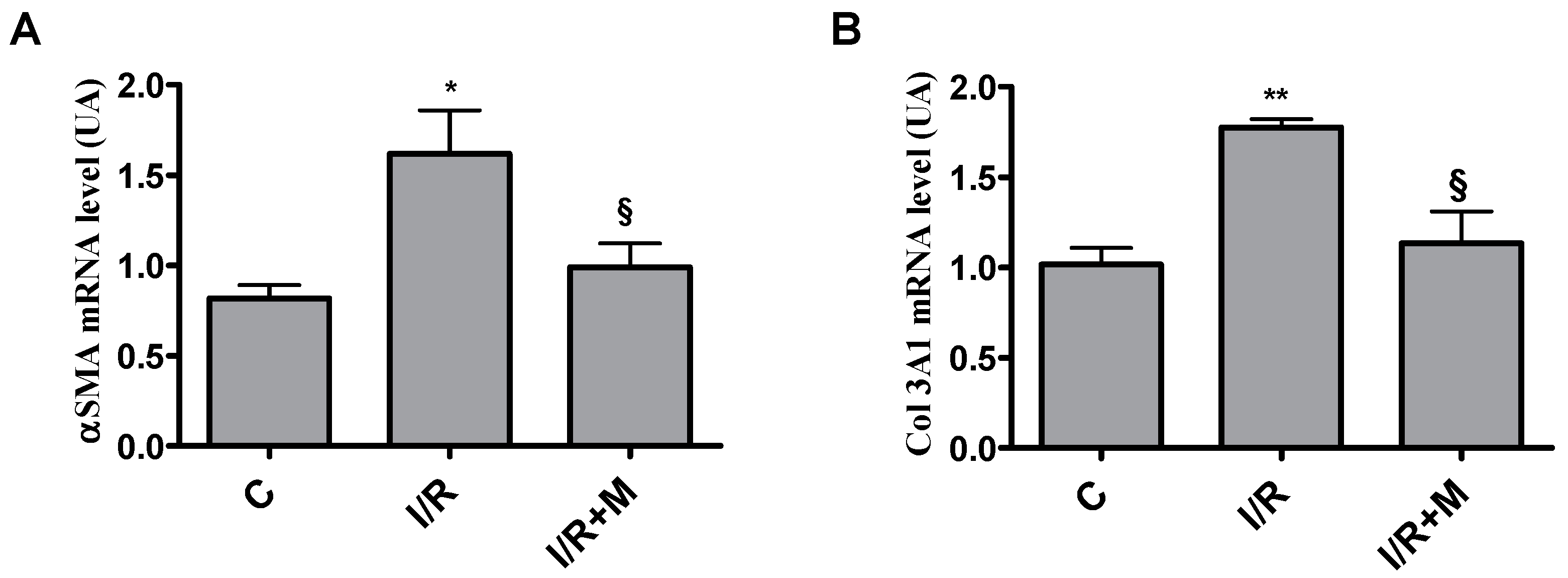
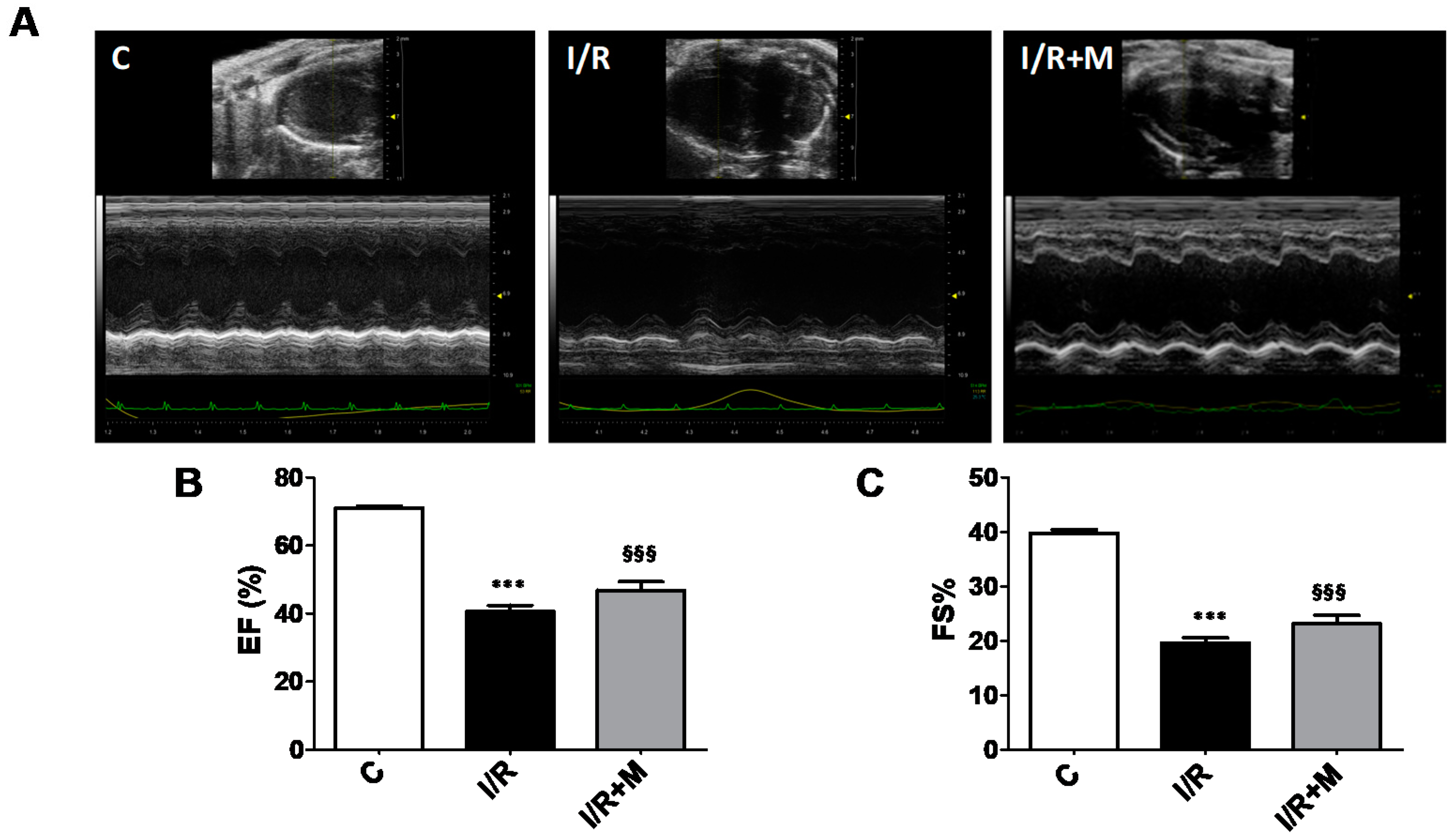
2.1. Metformin Treatment Decreases Fibrotic Remodeling in a Mouse Model of Cardiac I/R Injury
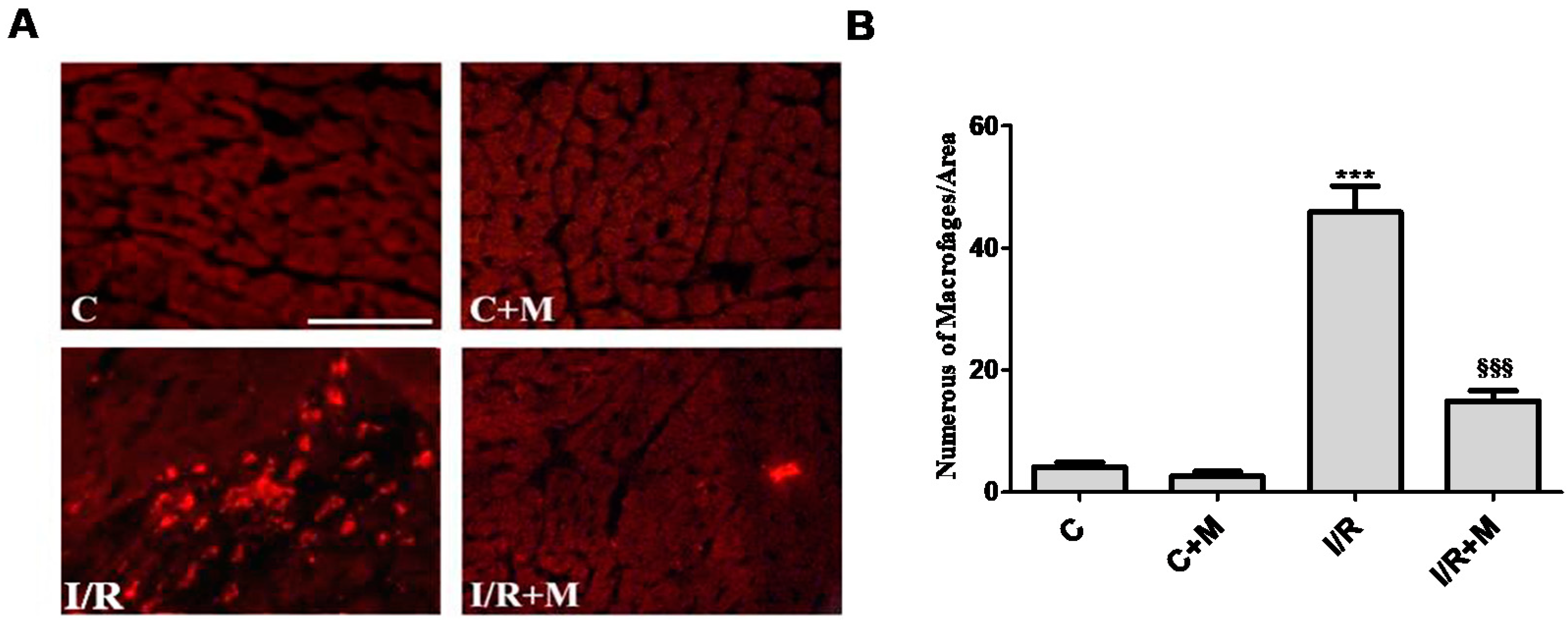
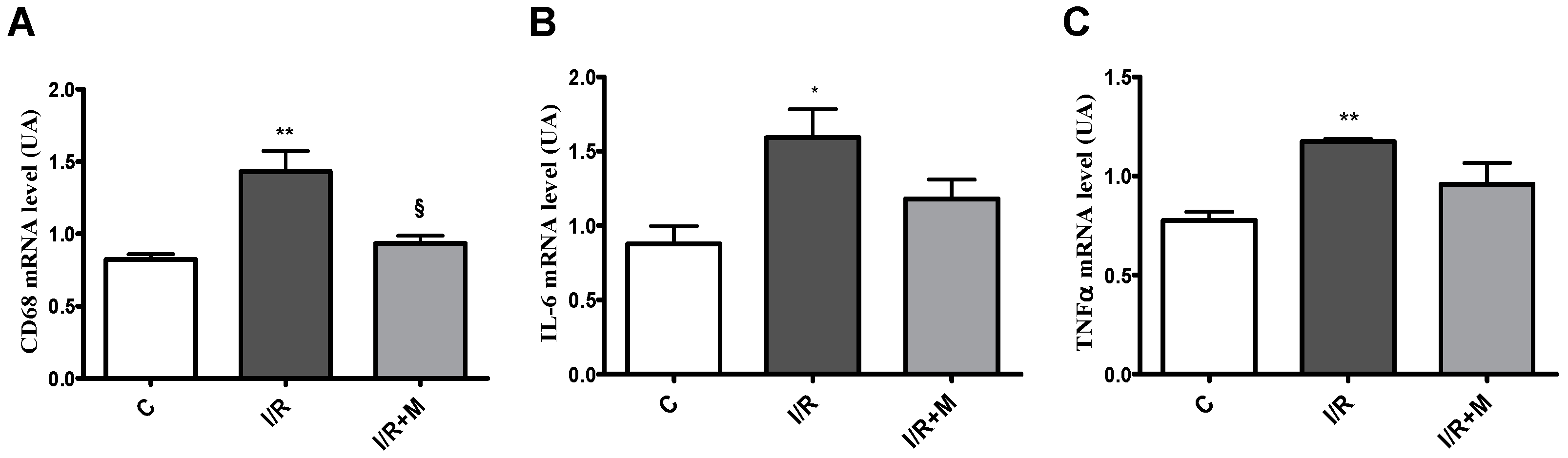
2.2. Metformin Exerts Anti-Inflammatory Activity in Cardiac Section
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Protocol
4.3. Echocardiography
Immunofluorescence and Histological Studies
4.4. Reagents and Antibodies
4.5. Quantitative RT-PCR Analysis
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Haffner, S.M.; Lehto, S.; Rönnemaa, T.; Pyörälä, K.; Laakso, M. Mortality from Coronary Heart Disease in Subjects with Type 2 Diabetes and in Nondiabetic Subjects with and without Prior Myocardial Infarction. N. Engl. J. Med. 1998, 339, 229–234. [Google Scholar] [CrossRef]
- Dei Cas, A.; Khan, S.S.; Butler, J.; Mentz, R.J.; Bonow, R.O.; Avogaro, A.; Tschoepe, D.; Doehner, W.; Greene, S.J.; Senni, M.; et al. Impact of diabetes on epidemiology, treatment, and outcomes of patients with heart failure. JACC Hear Fail. 2015, 3, 136–145. [Google Scholar] [CrossRef]
- Paulson, D.J. The diabetic heart is more sensitive to ischemic injury. Cardiovasc. Res. 1997, 34, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Ding, M.; Lei, J.; Han, H.; Li, W.; Qu, Y.; Fu, E.; Fu, F.; Wang, X. SIRT1 protects against myocardial ischemia–reperfusion injury via activating eNOS in diabetic rats. Cardiovasc. Diabetol. 2015, 14, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, A.; Bonney, M.; Bonney, S.; Weitzel, L.; Koeppen, M.; Eckle, T. Myocardial ischemia reperfusion injury—From basic science to clinical bedside. Semin. Cardiothorac. Vasc. Anesth. 2012, 16, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Raedschelders, K.; Ansley, D.M.; Chen, D.D. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacol. Ther. 2012, 33, 230–255. [Google Scholar] [CrossRef]
- Cohn, J.N.; Ferrari, R.; Sharpe, N. Cardiac remodeling—Concepts and clinical implications: A consensus paper from an international forum on cardiac remodeling. J. Am. Coll. Cardiol. 2000, 35, 569–582. [Google Scholar] [CrossRef] [Green Version]
- Lindman, B.R.; Arnold, S.V.; Madrazo, J.A.; Zajarias, A.; Johnson, S.N.; Pérez, J.E.; Mann, D. The Adverse Impact of Diabetes Mellitus on Left Ventricular Remodeling and Function in Patients with Severe Aortic Stenosis. Circ. Heart Fail. 2011, 4, 286–292. [Google Scholar] [CrossRef] [Green Version]
- Gjesdal, O.; Bluemke, D.; Lima, J.A. Cardiac remodeling at the population level—risk factors, screening, and outcomes. Nat. Rev. Cardiol. 2011, 8, 673–685. [Google Scholar] [CrossRef]
- Enomoto, S.; Yoshiyama, M.; Omura, T.; Matsumoto, R.; Kusuyama, T.; Kim, S.; Izumi, Y.; Akioka, K.; Iwao, H.; Takeuchi, K.; et al. Effects of eplerenone on transcriptional factors and mRNA expression related to cardiac remodelling after myocardial infarction. Heart 2005, 91, 1595–1600. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.M.; Lopez, E.F.; Lindsey, M.L. Macrophage roles following myocardial infarction. Int. J. Cardiol. 2008, 130, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Killingsworth, M.C.; Myasoedova, V.; Orekhov, A.N.; Bobryshev, Y.V. CD68/macrosialin: Not just a histochemical marker. Lab. Investig. 2016, 97, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Nian, M.; Lee, P.; Khaper, N.; Liu, P. Inflammatory Cytokines and Postmyocardial Infarction Remodeling. Circ. Res. 2004, 94, 1543–1553. [Google Scholar] [CrossRef]
- Eguchi, M.; Kim, Y.H.; Kang, K.W.; Shim, C.Y.; Jang, Y.; Dorval, T.; Kim, K.J.; Sweeney, G. Ischemia-Reperfusion Injury Leads to Distinct Temporal Cardiac Remodeling in Normal versus Diabetic Mice. PLoS ONE 2012, 7, e30450. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, M.S.; Kamalov, G.; Shahbaz, A.U.; Bhattacharya, S.K.; Ahokas, R.A.; Sun, Y.; Gerling, I.C.; Weber, K.T. Cellular and molecular pathways to myocardial necrosis and replacement fibrosis. Heart Fail. Rev. 2010, 16, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Steffens, S.; Montecucco, F.; Mach, F. The inflammatory response as a target to reduce myocardial ischaemia and reperfusion injury. Thromb. Haemost. 2009, 102, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction—from repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinde, A.V.; Frangogiannis, N.G. Fibroblasts in myocardial infarction: A role in inflammation and repair. J. Mol. Cell. Cardiol. 2014, 70, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Pchejetski, D.; Foussal, C.; Alfarano, C.; Lairez, O.; Calise, D.; Guilbeau-Frugier, C.; Schaak, S.; Seguelas, M.-H.; Wanecq, E.; Valet, P.; et al. Apelin prevents cardiac fibroblast activation and collagen production through inhibition of sphingosine kinase 1. Eur. Heart J. 2011, 33, 2360–2369. [Google Scholar] [CrossRef] [Green Version]
- Pandya, K.; Kim, H.S.; Smithies, O. Fibrosis, not cell size, delineates β-myosin heavy chain reexpression during cardiac hyperthrophy and normal aging in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 16864–16869. [Google Scholar] [CrossRef] [Green Version]
- merican Diabetes Association. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2020. Diabetes Care 2020, 43 (Suppl. S1), S98–S110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holman, R.R.; Paul, S.; Bethel, M.A.; Matthews, D.R.; Neil, H.A.W. 10-Year Follow-up of Intensive Glucose Control in Type 2 Diabetes. N. Engl. J. Med. 2008, 359, 1577–1589. [Google Scholar] [CrossRef] [Green Version]
- King, P.; Peacock, I.; Donnelly, R. The UK Prospective Diabetes Study (UKPDS): Clinical and therapeutic implications for type 2 diabetes. Br. J. Clin. Pharmacol. 1999, 48, 643–648. [Google Scholar] [CrossRef]
- Boyle, J.G.; Salt, I.P.; McKay, G.A. Metformin action on AMP-activated protein kinase: A translational research approach to understanding a potential new therapeutic target. Diabet Med. 2010, 27, 1097–1106. [Google Scholar] [CrossRef] [PubMed]
- Kirpichnikov, D.; Mcfarlane, S.I.; Sowers, J.R. Metformin: An Update. Ann. Intern. Med. 2002, 137, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Mann, D.L. Communication in the Heart: The Role of the Innate Immune System in Coordinating Cellular Responses to Ischemic Injury. J. Cardiovasc. Transl. Res. 2012, 5, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loi, H.; Boal, F.; Tronchere, H.; Cinato, M.; Kramar, S.; Oleshchuk, O.; Korda, M.; Kunduzova, O. Metformin protects the heart against hypertrophic and apoptotic remodeling after myocardial infarction. Front. Pharmacol. 2019, 10, 154. [Google Scholar] [CrossRef] [Green Version]
- Weber, K.T.; Sun, Y.; Bhattacharya, S.K.; Ahokas, R.A.; Gerling, I.C. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat. Rev. Cardiol. 2012, 10, 15–26. [Google Scholar] [CrossRef]
- Borne, S.W.M.V.D.; Diez, J.; Blankesteijn, W.M.; Verjans, J.; Hofstra, L.; Narula, J. Myocardial remodeling after infarction: The role of myofibroblasts. Nat. Rev. Cardiol. 2009, 7, 30–37. [Google Scholar] [CrossRef]
- Xiao, H.; Ma, X.; Feng, W.; Fu, Y.; Lu, Z.; Xu, M.; Shen, Q.; Zhu, Y.; Zhang, Y. Metformin attenuates cardiac fibrosis by inhibiting the TGF1-Smad3 signalling pathway. Cardiovasc. Res. 2010, 87, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yang, L.; Kang, L.; Li, J.; Yang, L.; Zhang, J.; Liu, J.; Zhu, M.; Zhang, Q.; Shen, Y.; et al. Metformin attenuates myocardial ischemia-reperfusion injury via up-regulation of antioxidant enzymes. PLoS ONE 2017, 12, e0182777. [Google Scholar] [CrossRef] [Green Version]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming Growth Factor (TGF)-β signaling in cardiac remodeling Introduction: The biology of TGF-β. J. Mol. Cell Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Gundewar, S.; Calvert, J.; Jha, S.; Toedt-Pingel, I.; Ji, S.Y.; Nunez, D.; Ramachandran, A.; Anaya-Cisneros, M.; Tian, R.; Lefer, D.J. Activation of AMP-Activated Protein Kinase by Metformin Improves Left Ventricular Function and Survival in Heart Failure. Circ. Res. 2009, 104, 403–411. [Google Scholar] [CrossRef] [Green Version]
- Nesti, L.; Natali, A. Metformin effects on the heart and the cardiovascular system: A review of experimental and clinical data. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 657–669. [Google Scholar] [CrossRef]
- Moore-Morris, T.; Guimarães-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload–induced cardiac fibrosis. J. Clin. Investig. 2014, 124, 2921–2934. [Google Scholar] [CrossRef] [Green Version]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A Common Pathway to Organ Injury and Failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef]
- Kanisicak, O.; Khalil, H.; Ivey, M.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.; Lin, S.-C.J.; Aronow, B.J.; Tallquist, M.D.; et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 12260. [Google Scholar] [CrossRef] [Green Version]
- Hinz, B.; Lagares, D. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 2019, 16, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Mazer, C.D.; Yan, A.T.; Mason, T.; Garg, V.; Teoh, H.; Zuo, F.; Quan, A.; Farkouh, M.E.; Fitchett, D.H.; et al. Effect of Empagliflozin on Left Ventricular Mass in Patients with Type 2 Diabetes Mellitus and Coronary Artery Disease: The EMPA-HEART CardioLink-6 Randomized Clinical Trial. Circulation 2019, 140, 1693–1702. [Google Scholar] [CrossRef]
- Koska, J.; Sands, M.; Burciu, C.; Reaven, P. Cardiovascular effects of dipeptidyl peptidase-4 inhibitors in patients with type 2 diabetes. Diabetes Vasc. Dis. Res. 2015, 12, 154–163. [Google Scholar] [CrossRef]
- Martinelli, I.; Timotin, A.; Moreno-Corchado, P.; Marsal, D.; Kramar, S.; Loy, H.; Joffre, C.; Boal, F.; Tronchere, H.; Kunduzova, O. Galanin promotes autophagy and alleviates apoptosis in the hypertrophied heart through FoxO1 pathway. Redox Biol. 2021, 40, 101866. [Google Scholar] [CrossRef]
- Goto, P.L.; Cinato, M.; Merachli, F.; Vons, B.; Jimenez, T.; Marsal, D.; Todua, N.; Loi, H.; Santin, Y.; Cassel, S.; et al. In vitro and in vivo cardioprotective and metabolic efficacy of vitamin E TPGS/Apelin. J. Mol. Cell. Cardiol. 2019, 138, 165–174. [Google Scholar] [CrossRef]
- Emde, B.; Heinen, A.; Gödecke, A.; Bottermann, K. Wheat germ agglutinin staining as a suitable method for detection and quantification of fibrosis in cardiac tissue after myocardial infarction. Eur. J. Histochem. 2014, 58, 2448. [Google Scholar] [CrossRef] [Green Version]
- Alfarano, C.; Foussal, C.; Lairez, O.; Calise, D.; Attané, C.; Anesia, R.; Daviaud, D.; Wanecq, E.; Parini, A.; Valet, P.; et al. Transition from metabolic adaptation to maladaptation of the heart in obesity: Role of apelin. Int. J. Obes. 2014, 39, 312–320. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loi, H.; Kramar, S.; Laborde, C.; Marsal, D.; Pizzinat, N.; Cussac, D.; Roncalli, J.; Boal, F.; Tronchere, H.; Oleshchuk, O.; et al. Metformin Attenuates Postinfarction Myocardial Fibrosis and Inflammation in Mice. Int. J. Mol. Sci. 2021, 22, 9393. https://doi.org/10.3390/ijms22179393
Loi H, Kramar S, Laborde C, Marsal D, Pizzinat N, Cussac D, Roncalli J, Boal F, Tronchere H, Oleshchuk O, et al. Metformin Attenuates Postinfarction Myocardial Fibrosis and Inflammation in Mice. International Journal of Molecular Sciences. 2021; 22(17):9393. https://doi.org/10.3390/ijms22179393
Chicago/Turabian StyleLoi, Halyna, Solomiia Kramar, Charlotte Laborde, Dimitri Marsal, Nathalie Pizzinat, Daniel Cussac, Jerome Roncalli, Frederic Boal, Helene Tronchere, Oleksandra Oleshchuk, and et al. 2021. "Metformin Attenuates Postinfarction Myocardial Fibrosis and Inflammation in Mice" International Journal of Molecular Sciences 22, no. 17: 9393. https://doi.org/10.3390/ijms22179393
APA StyleLoi, H., Kramar, S., Laborde, C., Marsal, D., Pizzinat, N., Cussac, D., Roncalli, J., Boal, F., Tronchere, H., Oleshchuk, O., Korda, M., & Kunduzova, O. (2021). Metformin Attenuates Postinfarction Myocardial Fibrosis and Inflammation in Mice. International Journal of Molecular Sciences, 22(17), 9393. https://doi.org/10.3390/ijms22179393