
Metabolic Consequences of Gestational Cannabinoid Exposure



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Impact of Cannabinoids on the Placenta
2.1. The Endocannabinoid System in Placental Development and the Influence of Exogenous Cannabinoids
2.2. Prenatal Exogenous Cannabinoid Exposure on Placental Insufficiency and Birth Outcomes
2.3. Underlying Mechanisms: Cannabinoids and Placental Subcellular Stress
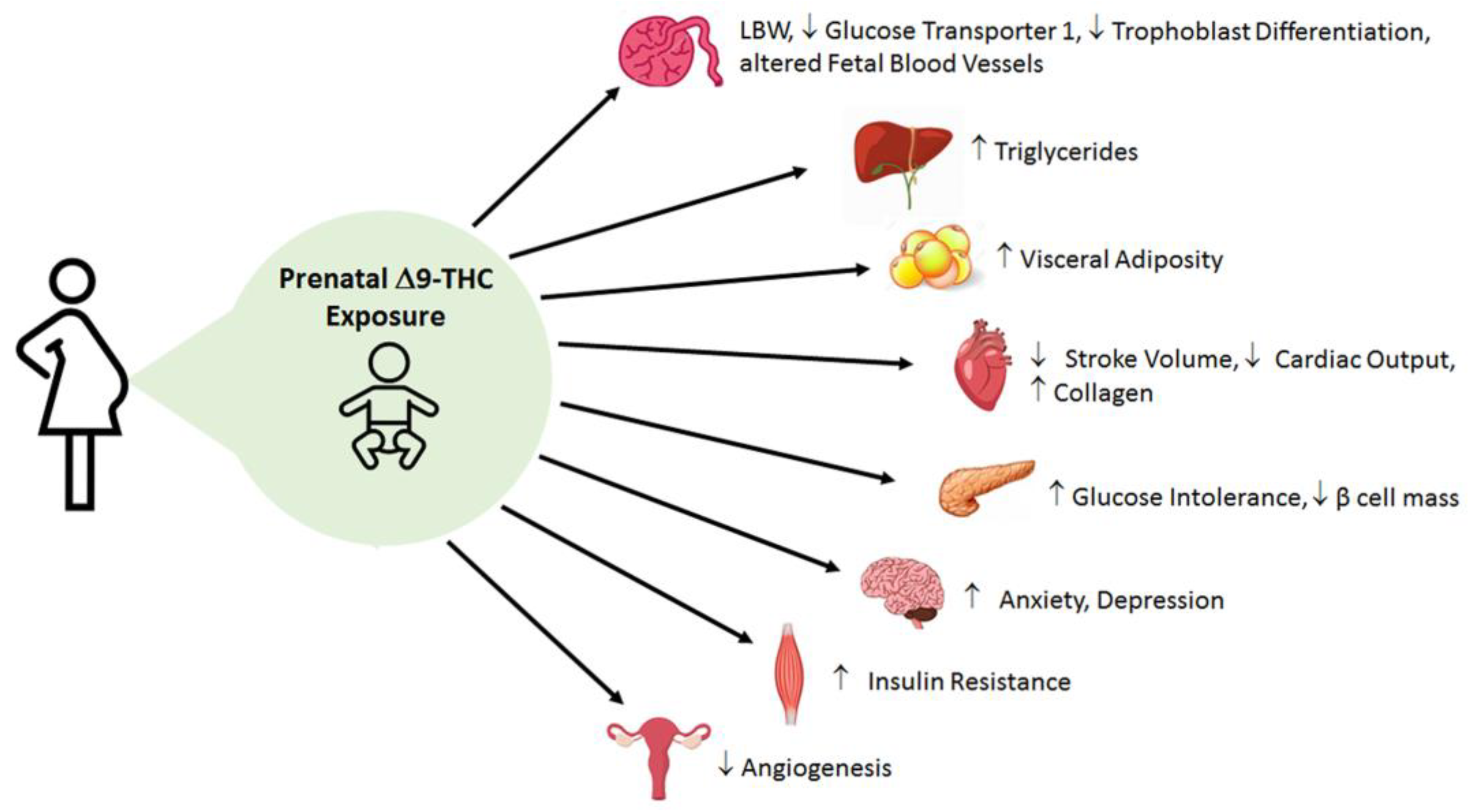
3. Cannabinoid-Induced FGR and Postnatal Hepatic Function and Lipid Metabolism
4. Cannabinoid-Induced FGR and Postnatal Glucose Homeostasis
5. Cannabinoid-Induced FGR and Postnatal Cardiovascular Function
6. Cannabinoid-Induced FGR and Postnatal Reproductive Function
7. Future Studies
7.1. Windows of Maternal Exposure
7.2. Intergenerational Effects of Maternal Cannabinoid Exposure
7.3. Paternal Cannabinoid Exposure
7.4. Other Receptors That Are Targeted by Cannabinoids
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Δ9-THC | Δ9-tetrahydrocannabinol |
| 2-AG | 2-arachidonoylglycerol |
| AEA | anandamide |
| BCRP | breast cancer resistant protein |
| CB1 | cannabinoid receptor type 1 |
| CB2 | cannabinoid receptor type 2 |
| CBD | cannabidiol |
| CBN | cannabinol |
| CNS | central nervous system |
| DOHaD | developmental origins of health and disease |
| eCB | endocannabinoid |
| EPCAM | epithelial cell adhesion molecule |
| ER | endoplasmic reticulum |
| FAAH | fatty acid amide hydrolase |
| FGR | fetal growth restriction |
| GR | glucocorticoid receptor |
| Glut1 | glucose transporter 1 |
| GPCR | g protein-coupled receptor |
| GPR | orphan G protein-coupled receptor |
| HUVEC | human umbilical vein endothelial cells |
| IUGR | intrauterine growth restriction |
| MAM | mitochondrial-associated ER membrane |
| mtCB1 | mitochondrial cannabinoid receptor type 1 |
| NAFLD | non-alcoholic liver disease |
| NAPE-PLD | N-acylphosphatidylethanolamine-specific phospholipase D |
| P-gp | P-glycoprotein |
| ROS | reactive oxygen species |
| T2D | type II diabetes |
| VEGFR-1 | vascular endothelial growth factor 1 |
References
- Degenhardt, L.; Ferrari, A.J.; Calabria, B.; Hall, W.D.; Norman, R.E.; McGrath, J.; Flaxman, A.D.; Engell, R.E.; Freedman, G.D.; Whiteford, H.A.; et al. The Global Epidemiology and Contribution of Cannabis Use and Dependence to the Global Burden of Disease: Results from the GBD 2010 Study. PLoS ONE 2013, 8, e76635. [Google Scholar] [CrossRef] [Green Version]
- Young-Wolff, K.C.; Tucker, L.-Y.; Alexeeff, S.; Armstrong, M.A.; Conway, A.; Weisner, C.; Goler, N. Trends in Self-Reported and Biochemically Tested Marijuana Use Among Pregnant Females in California from 2009–2016. JAMA 2017, 318, 2490–2491. [Google Scholar] [CrossRef]
- Mark, K.; Gryczynski, J.; Axenfeld, E.; Schwartz, R.P.; Terplan, M. Pregnant Women’s Current and Intended Cannabis Use in Relation to Their Views Toward Legalization and Knowledge of Potential Harm. J. Addict. Med. 2017, 11, 211–216. [Google Scholar] [CrossRef]
- Jarlenski, M.; Koma, J.W.; Zank, J.; Bodnar, L.M.; Bogen, D.L.; Chang, J.C. Trends in Perception of Risk of Regular Marijuana Use among US Pregnant and Nonpregnant Reproductive-Aged Women. Am. J. Obs. Gynecol. 2017, 217, 705–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westfall, R.E.; Janssen, P.A.; Lucas, P.; Capler, R. Survey of Medicinal Cannabis Use among Childbearing Women: Patterns of Its Use in Pregnancy and Retroactive Self-Assessment of Its Efficacy against “Morning Sickness”. Complement Ther. Clin. Pract. 2006, 12, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C.; Tarr, J.A.; Holland, C.L.; De Genna, N.M.; Richardson, G.A.; Rodriguez, K.L.; Sheeder, J.; Kraemer, K.L.; Day, N.L.; Rubio, D.; et al. Beliefs and Attitudes Regarding Prenatal Marijuana Use: Perspectives of Pregnant Women Who Report Use. Drug Alcohol Depend. 2019, 196, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Dickson, B.; Mansfield, C.; Guiahi, M.; Allshouse, A.A.; Borgelt, L.M.; Sheeder, J.; Silver, R.M.; Metz, T.D. Recommendations From Cannabis Dispensaries About First-Trimester Cannabis Use. Obs. Gynecol. 2018, 131, 1031–1038. [Google Scholar] [CrossRef]
- Bartlett, K.; Kaarid, K.; Gervais, N.; Vu, N.; Sharma, S.; Patel, T.; Shea, A.K. Pregnant Canadians’ Perceptions About the Transmission of Cannabis in Pregnancy and While Breastfeeding and the Impact of Information From Health Care Providers on Discontinuation of Use. J. Obs. Gynaecol. Can. 2020, 42, 1346–1350. [Google Scholar] [CrossRef]
- English, D.R.; Hulse, G.K.; Milne, E.; Holman, C.D.; Bower, C.I. Maternal Cannabis Use and Birth Weight: A Meta-Analysis. Addiction 1997, 92, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Gunn, J.K.L.; Rosales, C.B.; Center, K.E.; Nuñez, A.; Gibson, S.J.; Christ, C.; Ehiri, J.E. Prenatal Exposure to Cannabis and Maternal and Child Health Outcomes: A Systematic Review and Meta-Analysis. BMJ Open 2016, 6, e009986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conner, S.N.; Bedell, V.; Lipsey, K.; Macones, G.A.; Cahill, A.G.; Tuuli, M.G. Maternal Marijuana Use and Adverse Neonatal Outcomes: A Systematic Review and Meta-Analysis. Obs. Gynecol. 2016, 128, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.C.; Wainwright, H.; Molteno, C.D.; Georgieff, M.K.; Dodge, N.C.; Warton, F.; Meintjes, E.M.; Jacobson, J.L.; Jacobson, S.W. Alcohol, Methamphetamine, and Marijuana Exposure Have Distinct Effects on the Human Placenta. Alcohol. Clin. Exp. Res. 2016, 40, 753–764. [Google Scholar] [CrossRef]
- Scheyer, A.F.; Melis, M.; Trezza, V.; Manzoni, O.J.J. Consequences of Perinatal Cannabis Exposure. Trends Neurosci. 2019, 42, 871–884. [Google Scholar] [CrossRef]
- Nashed, M.G.; Hardy, D.B.; Laviolette, S.R. Prenatal Cannabinoid Exposure: Emerging Evidence of Physiological and Neuropsychiatric Abnormalities. Front. Psychiatry 2021, 11, 1577. [Google Scholar] [CrossRef]
- Cox, P.; Marton, T. Pathological Assessment of Intrauterine Growth Restriction. Best Pract. Res. Clin. Obstet. Gynaecol. 2009, 23, 751–764. [Google Scholar] [CrossRef]
- Gardosi, J. Intrauterine Growth Restriction: New Standards for Assessing Adverse Outcome. Best Pract. Res. Clin. Obstet. Gynaecol. 2009, 23, 741–749. [Google Scholar] [CrossRef]
- Barker, D.J.; Osmond, C.; Law, C.M. The Intrauterine and Early Postnatal Origins of Cardiovascular Disease and Chronic Bronchitis. J. Epidemiol. Commun. Health 1989, 43, 237–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, D.J. The Fetal and Infant Origins of Adult Disease. BMJ 1990, 301, 1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osmond, C.; Barker, D.J.; Winter, P.D.; Fall, C.H.; Simmonds, S.J. Early Growth and Death from Cardiovascular Disease in Women. BMJ 1993, 307, 1519–1524. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, J.; Forsén, T.; Tuomilehto, J.; Osmond, C.; Barker, D. Size at Birth, Childhood Growth and Obesity in Adult Life. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 735–740. [Google Scholar] [CrossRef] [Green Version]
- Hales, C.N.; Barker, D.J.P. The Thrifty Phenotype Hypothesis: Type 2 Diabetes. Br. Med. Bull. 2001, 60, 5–20. [Google Scholar] [CrossRef] [Green Version]
- Singhal, A. Long-Term Adverse Effects of Early Growth Acceleration or Catch-Up Growth. Ann. Nutr. Metab. 2017, 70, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Chandra, S.; Radwan, M.M.; Majumdar, C.G.; Church, J.C.; Freeman, T.P.; ElSohly, M.A. New Trends in Cannabis Potency in USA and Europe during the Last Decade (2008–2017). Eur. Arch. Psychiatry Clin. Neurosci. 2019, 269, 5–15. [Google Scholar] [CrossRef]
- Hutchings, D.E.; Martin, B.R.; Gamagaris, Z.; Miller, N.; Fico, T. Plasma Concentrations of Delta-9-Tetrahydrocannabinol in Dams and Fetuses Following Acute or Multiple Prenatal Dosing in Rats. Life Sci. 1989, 44, 697–701. [Google Scholar] [CrossRef]
- Bailey, J.R.; Cunny, H.C.; Paule, M.G.; Slikker, W. Fetal Disposition of Delta 9-Tetrahydrocannabinol (THC) during Late Pregnancy in the Rhesus Monkey. Toxicol. Appl. Pharm. 1987, 90, 315–321. [Google Scholar] [CrossRef]
- Corroon, J.; Phillips, J.A. A Cross-Sectional Study of Cannabidiol Users. Cannabis Cannabinoid Res. 2018, 3, 152–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battista, N.; Di Tommaso, M.; Bari, M.; Maccarrone, M. The Endocannabinoid System: An Overview. Front. Behav. Neurosci. 2012, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and Structure of a Brain Constituent That Binds to the Cannabinoid Receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef]
- Sugiura, T.; Kondo, S.; Sukagawa, A.; Nakane, S.; Shinoda, A.; Itoh, K.; Yamashita, A.; Waku, K. 2-Arachidonoylgylcerol: A Possible Endogenous Cannabinoid Receptor Ligand in Brain. Biochem. Biophys. Res. Commun. 1995, 215, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Reggio, P.H. Endocannabinoid Binding to the Cannabinoid Receptors: What Is Known and What Remains Unknown. Curr. Med. Chem. 2010, 17, 1468–1486. [Google Scholar] [CrossRef] [Green Version]
- Di Marzo, V.; De Petrocellis, L. Why Do Cannabinoid Receptors Have More than One Endogenous Ligand? Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 3216–3228. [Google Scholar] [CrossRef] [Green Version]
- Ibsen, M.S.; Connor, M.; Glass, M. Cannabinoid CB1 and CB2 Receptor Signaling and Bias. Cannabis Cannabinoid Res. 2017, 2, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Correa, F.; Wolfson, M.L.; Valchi, P.; Aisemberg, J.; Franchi, A.M. Endocannabinoid System and Pregnancy. Reproduction 2016, 152, R191–R200. [Google Scholar] [CrossRef] [PubMed]
- Buckley, N.E.; Hansson, S.; Harta, G.; Mezey, É. Expression of the CB1 and CB2 Receptor Messenger RNAs during Embryonic Development in the Rat. Neuroscience 1997, 82, 1131–1149. [Google Scholar] [CrossRef]
- Richardson, G.A.; Ryan, C.; Willford, J.; Day, N.L.; Goldschmidt, L. Prenatal Alcohol and Marijuana Exposure: Effects on Neuropsychological Outcomes at 10 Years. Neurotoxicol. Teratol. 2002, 24, 309–320. [Google Scholar] [CrossRef]
- Day, N.L.; Richardson, G.A.; Goldschmidt, L.; Robles, N.; Taylor, P.M.; Stoffer, D.S.; Cornelius, M.D.; Geva, D. Effect of Prenatal Marijuana Exposure on the Cognitive Development of Offspring at Age Three. Neurotoxicol. Teratol. 1994, 16, 169–175. [Google Scholar] [CrossRef]
- Leech, S.L.; Richardson, G.A.; Goldschmidt, L.; Day, N.L. Prenatal Substance Exposure: Effects on Attention and Impulsivity of 6-Year-Olds. Neurotoxicol. Teratol. 1999, 21, 109–118. [Google Scholar] [CrossRef]
- Smith, A.M.; Fried, P.A.; Hogan, M.J.; Cameron, I. Effects of Prenatal Marijuana on Visuospatial Working Memory: An FMRI Study in Young Adults. Neurotoxicol. Teratol. 2006, 28, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Campolongo, P.; Trezza, V.; Cassano, T.; Gaetani, S.; Morgese, M.G.; Ubaldi, M.; Soverchia, L.; Antonelli, T.; Ferraro, L.; Massi, M.; et al. Perinatal Exposure to Delta-9-Tetrahydrocannabinol Causes Enduring Cognitive Deficits Associated with Alteration of Cortical Gene Expression and Neurotransmission in Rats. Addict. Biol. 2007, 12, 485–495. [Google Scholar] [CrossRef]
- Silva, L.; Zhao, N.; Popp, S.; Dow-Edwards, D. Prenatal Tetrahydrocannabinol (THC) Alters Cognitive Function and Amphetamine Response from Weaning to Adulthood in the Rat. Neurotoxicol. Teratol. 2012, 34, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Beggiato, S.; Ieraci, A.; Tomasini, M.C.; Schwarcz, R.; Ferraro, L. Prenatal THC Exposure Raises Kynurenic Acid Levels in the Prefrontal Cortex of Adult Rats. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 100, 109883. [Google Scholar] [CrossRef]
- Ramírez-López, M.T.; Arco, R.; Decara, J.; Vázquez, M.; Noemí Blanco, R.; Alén, F.; Suárez, J.; Gómez de Heras, R.; Rodríguez de Fonseca, F. Exposure to a Highly Caloric Palatable Diet during the Perinatal Period Affects the Expression of the Endogenous Cannabinoid System in the Brain, Liver and Adipose Tissue of Adult Rat Offspring. PLoS ONE 2016, 11, e0165432. [Google Scholar] [CrossRef] [Green Version]
- Malenczyk, K.; Keimpema, E.; Piscitelli, F.; Calvigioni, D.; Björklund, P.; Mackie, K.; Di Marzo, V.; Hökfelt, T.G.M.; Dobrzyn, A.; Harkany, T. Fetal Endocannabinoids Orchestrate the Organization of Pancreatic Islet Microarchitecture. Proc. Natl. Acad. Sci. USA 2015, 112, E6185–E6194. [Google Scholar] [CrossRef] [Green Version]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.H.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid Receptors and Their Ligands: Beyond CB1 and CB2. Pharm. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Dey, S.K. Endocannabinoid Signaling in Female Reproduction. ACS Chem Neurosci 2012, 3, 349–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coskun, Z.M.; Bolkent, S. Evaluation of Δ(9)-Tetrahydrocannabinol Metabolites and Oxidative Stress in Type 2 Diabetic Rats. Iran. J. Basic Med. Sci 2016, 19, 154–158. [Google Scholar]
- Shrestha, N.; Cuffe, J.S.M.; Hutchinson, D.S.; Headrick, J.P.; Perkins, A.V.; McAinch, A.J.; Hryciw, D.H. Peripheral Modulation of the Endocannabinoid System in Metabolic Disease. Drug Discov. Today 2018, 23, 592–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Xie, H.; Yang, J.; Wang, H.; Bradshaw, H.B.; Dey, S.K. Endocannabinoid Signaling Directs Differentiation of Trophoblast Cell Lineages and Placentation. Proc. Natl. Acad. Sci. USA 2010, 107, 16887–16892. [Google Scholar] [CrossRef] [Green Version]
- Habayeb, O.M.H.; Taylor, A.H.; Evans, M.D.; Cooke, M.S.; Taylor, D.J.; Bell, S.C.; Konje, J.C. Plasma Levels of the Endocannabinoid Anandamide in Women—A Potential Role in Pregnancy Maintenance and Labor? J. Clin. Endocrinol. Metab. 2004, 89, 5482–5487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, B.M.; Correia-da-Silva, G.; Taylor, A.H.; Konje, J.C.; Bell, S.C.; Teixeira, N.A. Spatio-Temporal Expression Patterns of Anandamide-Binding Receptors in Rat Implantation Sites: Evidence for a Role of the Endocannabinoid System during the Period of Placental Development. Reprod. Biol. Endocrinol. 2009, 7, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, B.M.; Correia-da-Silva, G.; Taylor, A.H.; Lam, P.M.W.; Marczylo, T.H.; Konje, J.C.; Bell, S.C.; Teixeira, N.A. N-Acylethanolamine Levels and Expression of Their Metabolizing Enzymes during Pregnancy. Endocrinology 2010, 151, 3965–3974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, A.H.; Finney, M.; Lam, P.M.; Konje, J.C. Modulation of the Endocannabinoid System in Viable and Non-Viable First Trimester Pregnancies by Pregnancy-Related Hormones. Reprod. Biol. Endocrinol. 2011, 9, 152. [Google Scholar] [CrossRef] [Green Version]
- Neradugomma, N.K.; Drafton, K.; Mor, G.G.; Mao, Q. Marijuana-Derived Cannabinoids Inhibit Uterine Endometrial Stromal Cell Decidualization and Compromise Trophoblast-Endometrium Cross-Talk. Reprod. Toxicol. 2019, 87, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Deng, W.; Li, Y.; Tang, S.; Leishman, E.; Bradshaw, H.B.; Dey, S.K. Sustained Endocannabinoid Signaling Compromises Decidual Function and Promotes Inflammation-Induced Preterm Birth. J. Biol. Chem. 2016, 291, 8231–8240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, B.M.; Correia-da-Silva, G.; Almada, M.; Costa, M.A.; Teixeira, N.A. The Endocannabinoid System in the Postimplantation Period: A Role during Decidualization and Placentation. Int. J. Endocrinol. 2013, 2013, e510540. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, B.M.; Correia-da-Silva, G.; Teixeira, N.A. The Endocannabinoid Anandamide Induces Apoptosis of Rat Decidual Cells through a Mechanism Involving Ceramide Synthesis and P38 MAPK Activation. Apoptosis 2013, 18, 1526–1535. [Google Scholar] [CrossRef]
- Fonseca, B.M.; Battista, N.; Correia-da-Silva, G.; Rapino, C.; Maccarrone, M.; Teixeira, N.A. Activity of Anandamide (AEA) Metabolic Enzymes in Rat Placental Bed. Reprod. Toxicol. 2014, 49, 74–77. [Google Scholar] [CrossRef]
- Habayeb, O.M.H.; Taylor, A.H.; Bell, S.C.; Taylor, D.J.; Konje, J.C. Expression of the Endocannabinoid System in Human First Trimester Placenta and Its Role in Trophoblast Proliferation. Endocrinology 2008, 149, 5052–5060. [Google Scholar] [CrossRef] [Green Version]
- Costa, M.A.; Fonseca, B.M.; Teixeira, N.A.; Correia-da-Silva, G. The Endocannabinoid Anandamide Induces Apoptosis in Cytotrophoblast Cells: Involvement of Both Mitochondrial and Death Receptor Pathways. Placenta 2015, 36, 69–76. [Google Scholar] [CrossRef]
- Costa, M.A.; Fonseca, B.M.; Keating, E.; Teixeira, N.A.; Correia-da-Silva, G. 2-Arachidonoylglycerol Effects in Cytotrophoblasts: Metabolic Enzymes Expression and Apoptosis in BeWo Cells. Reproduction 2014, 147, 301–311. [Google Scholar] [CrossRef]
- Park, B.; Gibbons, H.M.; Mitchell, M.D.; Glassa, M. Identification of the CB1 Cannabinoid Receptor and Fatty Acid Amide Hydrolase (FAAH) in the Human Placenta. Placenta 2003, 24, 473–478. [Google Scholar] [CrossRef]
- Trabucco, E.; Acone, G.; Marenna, A.; Pierantoni, R.; Cacciola, G.; Chioccarelli, T.; Mackie, K.; Fasano, S.; Colacurci, N.; Meccariello, R.; et al. Endocannabinoid System in First Trimester Placenta: Low FAAH and High CB1 Expression Characterize Spontaneous Miscarriage. Placenta 2009, 30, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Meccariello, R.; Battista, N.; Bradshaw, H.B.; Wang, H. Endocannabinoids and Reproduction. Int. J. Endocrinol. 2014, 2014, e378069. [Google Scholar] [CrossRef] [PubMed]
- Maia, J.; Fonseca, B.M.; Teixeira, N.; Correia-da-Silva, G. The Fundamental Role of the Endocannabinoid System in Endometrium and Placenta: Implications in Pathophysiological Aspects of Uterine and Pregnancy Disorders. Hum. Reprod. Update 2020, 26, 586–602. [Google Scholar] [CrossRef] [PubMed]
- Maia, J.; Midão, L.; Cunha, S.C.; Almada, M.; Fonseca, B.M.; Braga, J.; Gonçalves, D.; Teixeira, N.; Correia-da-Silva, G. Effects of Cannabis Tetrahydrocannabinol on Endocannabinoid Homeostasis in Human Placenta. Arch. Toxicol. 2019, 93, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Costa, M.A.; Fonseca, B.M.; Mendes, A.; Braga, J.; Teixeira, N.A.; Correia da Silva, G. The Endocannabinoid Anandamide Affects the Synthesis of Human Syncytiotrophoblast-Related Proteins. Cell Tissue Res. 2015, 362, 441–446. [Google Scholar] [CrossRef]
- Chang, X.; Bian, Y.; He, Q.; Yao, J.; Zhu, J.; Wu, J.; Wang, K.; Duan, T. Suppression of STAT3 Signaling by Δ9-Tetrahydrocannabinol (THC) Induces Trophoblast Dysfunction. Cell. Physiol. Biochem. 2017, 42, 537–550. [Google Scholar] [CrossRef]
- Khare, M.; Taylor, A.H.; Konje, J.C.; Bell, S.C. Δ9-Tetrahydrocannabinol Inhibits Cytotrophoblast Cell Proliferation and Modulates Gene Transcription. Mol. Hum. Reprod. 2006, 12, 321–333. [Google Scholar] [CrossRef] [Green Version]
- Walker, O.S.; Ragos, R.; Gurm, H.; Lapierre, M.; May, L.L.; Raha, S. Delta-9-tetrahydrocannabinol Disrupts Mitochondrial Function and Attenuates Syncytialization in Human Placental BeWo Cells. Physiol. Rep. 2020, 8. [Google Scholar] [CrossRef]
- Almada, M.; Amaral, C.; Oliveira, A.; Fernandes, P.A.; Ramos, M.J.; Fonseca, B.M.; Correia-da-Silva, G.; Teixeira, N. Cannabidiol (CBD) but Not Tetrahydrocannabinol (THC) Dysregulate in Vitro Decidualization of Human Endometrial Stromal Cells by Disruption of Estrogen Signaling. Reprod. Toxicol. 2020, 93, 75–82. [Google Scholar] [CrossRef]
- Almada, M.; Amaral, C.; Diniz-Da-Costa, M.; Correia-Da-Silva, G.; Teixeira, N.A.; Fonseca, B.M. The Endocannabinoid Anandamide Impairs in Vitro Decidualization of Human Cells. Reproduction 2016, 152, 351–361. [Google Scholar] [CrossRef]
- Watanabe, K.; Kayano, Y.; Matsunaga, T.; Yamamoto, I.; Yoshimura, H. Inhibition of Anandamide Amidase Activity in Mouse Brain Microsomes by Cannabinoids. Biol. Pharm. Bull. 1996, 19, 1109–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Filion, K.B.; Abenhaim, H.A.; Eisenberg, M.J. Prevalence and Outcomes of Prenatal Recreational Cannabis Use in High-Income Countries: A Scoping Review. BJOG 2020, 127, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Metz, T.D.; Stickrath, E.H. Marijuana Use in Pregnancy and Lactation: A Review of the Evidence. Am. J. Obs. Gynecol. 2015, 213, 761–778. [Google Scholar] [CrossRef] [PubMed]
- Bailey, B.A.; Wood, D.L.; Shah, D. Impact of Pregnancy Marijuana Use on Birth Outcomes: Results from Two Matched Population-Based Cohorts. J. Perinatol. 2020, 40, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Karanges, E.; Spiro, A.; Wong, A.; Spencer, J.; Huynh, T.; Gunasekaran, N.; Karl, T.; Long, L.E.; Huang, X.-F.; et al. Cannabidiol Potentiates Δ9-Tetrahydrocannabinol (THC) Behavioural Effects and Alters THC Pharmacokinetics during Acute and Chronic Treatment in Adolescent Rats. Psychopharmacology 2011, 218, 443–457. [Google Scholar] [CrossRef]
- Schwope, D.M.; Karschner, E.L.; Gorelick, D.A.; Huestis, M.A. Identification of Recent Cannabis Use: Whole-Blood and Plasma Free and Glucuronidated Cannabinoid Pharmacokinetics Following Controlled Smoked Cannabis Administration. Clin. Chem. 2011, 57, 1406–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falcon, M.; Pichini, S.; Joya, J.; Pujadas, M.; Sanchez, A.; Vall, O.; García Algar, O.; Luna, A.; de la Torre, R.; Rotolo, M.C.; et al. Maternal Hair Testing for the Assessment of Fetal Exposure to Drug of Abuse during Early Pregnancy: Comparison with Testing in Placental and Fetal Remains. Forensic Sci. Int. 2012, 218, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; de Castro, A.; Lendoiro, E.; Cruz-Landeira, A.; López-Rivadulla, M.; Concheiro, M. Detection of in Utero Cannabis Exposure by Umbilical Cord Analysis. Drug Test. Anal. 2018, 10, 636–643. [Google Scholar] [CrossRef]
- Ochiai, W.; Kitaoka, S.; Kawamura, T.; Hatogai, J.; Harada, S.; Iizuka, M.; Ariumi, M.; Takano, S.; Nagai, T.; Sasatsu, M.; et al. Maternal and Fetal Pharmacokinetic Analysis of Cannabidiol during Pregnancy in Mice. Drug Metab. Dispos. 2021, 49, 337–343. [Google Scholar] [CrossRef]
- Natale, B.V.; Gustin, K.N.; Lee, K.; Holloway, A.C.; Laviolette, S.R.; Natale, D.R.C.; Hardy, D.B. Δ9-Tetrahydrocannabinol Exposure during Rat Pregnancy Leads to Symmetrical Fetal Growth Restriction and Labyrinth-Specific Vascular Defects in the Placenta. Sci. Rep. 2020, 10, 544. [Google Scholar] [CrossRef]
- Chang, X.; Li, H.; Li, Y.; He, Q.; Yao, J.; Duan, T.; Wang, K. RhoA/MLC Signaling Pathway Is Involved in Δ9-Tetrahydrocannabinol-Impaired Placental Angiogenesis. Toxicol. Lett. 2018, 285, 148–155. [Google Scholar] [CrossRef]
- Tortoriello, G.; Morris, C.V.; Alpar, A.; Fuzik, J.; Shirran, S.L.; Calvigioni, D.; Keimpema, E.; Botting, C.H.; Reinecke, K.; Herdegen, T.; et al. Miswiring the Brain: Δ9-Tetrahydrocannabinol Disrupts Cortical Development by Inducing an SCG10/Stathmin-2 Degradation Pathway. EMBO J. 2014, 33, 668–685. [Google Scholar] [CrossRef]
- Brar, B.K.; Patil, P.S.; Jackson, D.N.; Gardner, M.O.; Alexander, J.M.; Doyle, N.M. Effect of Intrauterine Marijuana Exposure on Fetal Growth Patterns and Placental Vascular Resistance. J. Matern. Fetal Neonatal Med. 2021, 34, 3330–3334. [Google Scholar] [CrossRef]
- Benevenuto, S.G.; Domenico, M.D.; Martins, M.A.G.; Costa, N.S.; de Souza, A.R.L.; Costa, J.L.; Tavares, M.F.M.; Dolhnikoff, M.; Veras, M.M. Recreational Use of Marijuana during Pregnancy and Negative Gestational and Fetal Outcomes: An Experimental Study in Mice. Toxicology 2017, 376, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Hayward, C.E.; Lean, S.; Sibley, C.P.; Jones, R.L.; Wareing, M.; Greenwood, S.L.; Dilworth, M.R. Placental Adaptation: What Can We Learn from Birthweight:Placental Weight Ratio? Front. Physiol. 2016, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Solinas, M.; Massi, P.; Cantelmo, A.R.; Cattaneo, M.G.; Cammarota, R.; Bartolini, D.; Cinquina, V.; Valenti, M.; Vicentini, L.M.; Noonan, D.M.; et al. Cannabidiol Inhibits Angiogenesis by Multiple Mechanisms. Br. J. Pharm. 2012, 167, 1218–1231. [Google Scholar] [CrossRef] [Green Version]
- Feinshtein, V.; Erez, O.; Ben-Zvi, Z.; Eshkoli, T.; Sheizaf, B.; Sheiner, E.; Holcberg, G. Cannabidiol Enhances Xenobiotic Permeability through the Human Placental Barrier by Direct Inhibition of Breast Cancer Resistance Protein: An Ex Vivo Study. Am. J. Obstet. Gynecol. 2013, 209, 573.e1–573.e15. [Google Scholar] [CrossRef]
- Ni, Z.; Mao, Q. ATP-Binding Cassette Efflux Transporters in Human Placenta. Curr. Pharm. Biotechnol. 2011, 12, 674–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vähäkangas, K.; Myllynen, P. Drug Transporters in the Human Blood-Placental Barrier. Br. J. Pharmacol. 2009, 158, 665–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinshtein, V.; Erez, O.; Ben-Zvi, Z.; Erez, N.; Eshkoli, T.; Sheizaf, B.; Sheiner, E.; Huleihel, M.; Holcberg, G. Cannabidiol Changes P-Gp and BCRP Expression in Trophoblast Cell Lines. PeerJ 2013, 1, e153. [Google Scholar] [CrossRef] [Green Version]
- Lojpur, T.; Easton, Z.; Raez-Villanueva, S.; Laviolette, S.; Holloway, A.C.; Hardy, D.B. Δ9-Tetrahydrocannabinol Leads to Endoplasmic Reticulum Stress and Mitochondrial Dysfunction in Human BeWo Trophoblasts. Reprod. Toxicol. 2019, 87, 21–31. [Google Scholar] [CrossRef]
- Burton, G.J.; Yung, H.-W.; Cindrova-Davies, T.; Charnock-Jones, D.S. Placental Endoplasmic Reticulum Stress and Oxidative Stress in the Pathophysiology of Unexplained Intrauterine Growth Restriction and Early Onset Preeclampsia. Placenta 2009, 30, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, T.; Yoshimi, M.; Kadota, Y.; Inoue, M.; Sato, M.; Suzuki, S. Prolonged Endoplasmic Reticulum Stress Alters Placental Morphology and Causes Low Birth Weight. Toxicol. Appl. Pharm. 2014, 275, 134–144. [Google Scholar] [CrossRef]
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.-P. MAM: More than Just a Housekeeper. Trends Cell Biol. 2009, 19, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandò, C.; De Palma, C.; Stampalija, T.; Anelli, G.M.; Figus, M.; Novielli, C.; Parisi, F.; Clementi, E.; Ferrazzi, E.; Cetin, I. Placental Mitochondrial Content and Function in Intrauterine Growth Restriction and Preeclampsia. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E404–E413. [Google Scholar] [CrossRef]
- James, P.T.; Rigby, N.; Leach, R. International Obesity Task Force The Obesity Epidemic, Metabolic Syndrome and Future Prevention Strategies. Eur. J. Cardiovasc. Prev. Rehabil. 2004, 11, 3–8. [Google Scholar] [CrossRef]
- Suomela, E.; Oikonen, M.; Pitkänen, N.; Ahola-Olli, A.; Virtanen, J.; Parkkola, R.; Jokinen, E.; Laitinen, T.; Hutri-Kähönen, N.; Kähönen, M.; et al. Childhood Predictors of Adult Fatty Liver. The Cardiovascular Risk in Young Finns Study. J. Hepatol. 2016, 65, 784–790. [Google Scholar] [CrossRef] [Green Version]
- Nobili, V.; Marcellini, M.; Marchesini, G.; Vanni, E.; Manco, M.; Villani, A.; Bugianesi, E. Intrauterine Growth Retardation, Insulin Resistance, and Nonalcoholic Fatty Liver Disease in Children. Diabetes Care 2007, 30, 2638–2640. [Google Scholar] [CrossRef] [Green Version]
- Ravelli, G.P.; Stein, Z.A.; Susser, M.W. Obesity in Young Men after Famine Exposure in Utero and Early Infancy. N. Engl. J. Med. 1976, 295, 349–353. [Google Scholar] [CrossRef]
- Yang, Z.; Zhao, W.; Zhang, X.; Mu, R.; Zhai, Y.; Kong, L.; Chen, C. Impact of Famine during Pregnancy and Infancy on Health in Adulthood. Obes. Rev. 2008, 9 (Suppl. 1), 95–99. [Google Scholar] [CrossRef]
- Ma, N.; Nicholson, C.J.; Wong, M.; Holloway, A.C.; Hardy, D.B. Fetal and Neonatal Exposure to Nicotine Leads to Augmented Hepatic and Circulating Triglycerides in Adult Male Offspring Due to Increased Expression of Fatty Acid Synthase. Toxicol. Appl. Pharm. 2014, 275, 1–11. [Google Scholar] [CrossRef]
- Sohi, G.; Marchand, K.; Revesz, A.; Arany, E.; Hardy, D.B. Maternal Protein Restriction Elevates Cholesterol in Adult Rat Offspring Due to Repressive Changes in Histone Modifications at the Cholesterol 7alpha-Hydroxylase Promoter. Mol. Endocrinol. 2011, 25, 785–798. [Google Scholar] [CrossRef] [Green Version]
- Oke, S.L.; Lee, K.; Papp, R.; Laviolette, S.R.; Hardy, D.B. In Utero Exposure to Δ9-Tetrahydrocannabinol Leads to Postnatal Catch-Up Growth and Dysmetabolism in the Adult Rat Liver. Int. J. Mol. Sci. 2021, 22, 7502. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron Transfer between Cytochrome c and P66Shc Generates Reactive Oxygen Species That Trigger Mitochondrial Apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef]
- Berniakovich, I.; Trinei, M.; Stendardo, M.; Migliaccio, E.; Minucci, S.; Bernardi, P.; Pelicci, P.G.; Giorgio, M. P66Shc-Generated Oxidative Signal Promotes Fat Accumulation. J. Biol. Chem. 2008, 283, 34283–34293. [Google Scholar] [CrossRef] [Green Version]
- Oke, S.L.; Sohi, G.; Hardy, D.B. Perinatal Protein Restriction with Postnatal Catch-up Growth Leads to Elevated P66Shc and Mitochondrial Dysfunction in the Adult Rat Liver. Reproduction 2019, 159, 27–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Victora, C.G.; Barros, F.C.; Lima, R.C.; Behague, D.P.; Gonçalves, H.; Horta, B.L.; Gigante, D.P.; Vaughan, J.P. The Pelotas Birth Cohort Study, Rio Grande Do Sul, Brazil, 1982-2001. Cad. Saúde Pública 2003, 19, 1241–1256. [Google Scholar] [CrossRef] [Green Version]
- Perng, W.; Hajj, H.; Belfort, M.B.; Rifas-Shiman, S.L.; Kramer, M.S.; Gillman, M.W.; Oken, E. Birth Size, Early Life Weight Gain, and Midchildhood Cardiometabolic Health. J. Pediatrics 2016, 173, 122–130.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Blasco, D.; Busquets-Garcia, A.; Hebert-Chatelain, E.; Serrat, R.; Vicente-Gutierrez, C.; Ioannidou, C.; Gómez-Sotres, P.; Lopez-Fabuel, I.; Resch-Beusher, M.; Resel, E.; et al. Glucose Metabolism Links Astroglial Mitochondria to Cannabinoid Effects. Nature 2020, 583, 603–608. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, Y.; Zhao, H.; Zhou, J.; Feng, D.; Tang, F.; Li, Y.; Lv, L.; Chen, Z.; Ma, X.; et al. Inhibition of P66Shc Oxidative Signaling via CA-Induced Upregulation of MiR-203a-3p Alleviates Liver Fibrosis Progression. Mol. Ther. Nucleic Acids 2020, 21, 751–763. [Google Scholar] [CrossRef]
- Roderburg, C.; Urban, G.-W.; Bettermann, K.; Vucur, M.; Zimmermann, H.; Schmidt, S.; Janssen, J.; Koppe, C.; Knolle, P.; Castoldi, M.; et al. Micro-RNA Profiling Reveals a Role for MiR-29 in Human and Murine Liver Fibrosis. Hepatology 2011, 53, 209–218. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Wang, F.-S.; Yang, Y.-L.; Huang, Y.-H. MicroRNA-29a Suppresses CD36 to Ameliorate High Fat Diet-Induced Steatohepatitis and Liver Fibrosis in Mice. Cells 2019, 8, 1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.-L.; Kuo, H.-C.; Wang, F.-S.; Huang, Y.-H. MicroRNA-29a Disrupts DNMT3b to Ameliorate Diet-Induced Non-Alcoholic Steatohepatitis in Mice. Int. J. Mol. Sci. 2019, 20, 1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascio, S.; Zaret, K.S. Hepatocyte Differentiation Initiates during Endodermal-Mesenchymal Interactions Prior to Liver Formation. Development 1991, 113, 217–225. [Google Scholar] [CrossRef]
- Greengard, O.; Federman, M.; Knox, W.E. Cytomorphometry of developing rat liver and its application to enzymic differentiation. J. Cell Biol. 1972, 52, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Zelber-Sagi, S.; Azar, S.; Nemirovski, A.; Webb, M.; Halpern, Z.; Shibolet, O.; Tam, J. Serum Levels of Endocannabinoids Are Independently Associated with Nonalcoholic Fatty Liver Disease. Obesity 2017, 25, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.; Liu, J.; Mukhopadhyay, B.; Cinar, R.; Godlewski, G.; Kunos, G. Endocannabinoids in Liver Disease. Hepatology 2011, 53, 346–355. [Google Scholar] [CrossRef] [Green Version]
- Alswat, K.A. The Role of Endocannabinoids System in Fatty Liver Disease and Therapeutic Potentials. Saudi J. Gastroenterol. 2013, 19, 144–151. [Google Scholar] [CrossRef]
- Regnell, S.E. Cannabinoid 1 Receptor in Fatty Liver. Hepatol. Res. 2013, 43, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Osei-Hyiaman, D.; Liu, J.; Zhou, L.; Godlewski, G.; Harvey-White, J.; Jeong, W.; Bátkai, S.; Marsicano, G.; Lutz, B.; Buettner, C.; et al. Hepatic CB1 Receptor Is Required for Development of Diet-Induced Steatosis, Dyslipidemia, and Insulin and Leptin Resistance in Mice. J. Clin. Investig. 2008, 118, 3160–3169. [Google Scholar] [CrossRef] [PubMed]
- Osei-Hyiaman, D.; DePetrillo, M.; Pacher, P.; Liu, J.; Radaeva, S.; Bátkai, S.; Harvey-White, J.; Mackie, K.; Offertáler, L.; Wang, L.; et al. Endocannabinoid Activation at Hepatic CB1 Receptors Stimulates Fatty Acid Synthesis and Contributes to Diet-Induced Obesity. J. Clin. Investig. 2005, 115, 1298–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez-Sanchez, N.; Zamora-Valdes, D.; Pichardo-Bahena, R.; Barredo-Prieto, B.; Ponciano-Rodriguez, G.; Bermejo-Martínez, L.; Chavez-Tapia, N.C.; Baptista-González, H.A.; Uribe, M. Endocannabinoid Receptor CB2 in Nonalcoholic Fatty Liver Disease. Liver Int. 2007, 27, 215–219. [Google Scholar] [CrossRef]
- Ravinet Trillou, C.; Delgorge, C.; Menet, C.; Arnone, M.; Soubrié, P. CB1 Cannabinoid Receptor Knockout in Mice Leads to Leanness, Resistance to Diet-Induced Obesity and Enhanced Leptin Sensitivity. Int. J. Obes. Relat. Metab. Disord. 2004, 28, 640–648. [Google Scholar] [CrossRef] [Green Version]
- Gomes, T.M.; Dias da Silva, D.; Carmo, H.; Carvalho, F.; Silva, J.P. Epigenetics and the Endocannabinoid System Signaling: An Intricate Interplay Modulating Neurodevelopment. Pharmacol. Res. 2020, 162, 105237. [Google Scholar] [CrossRef]
- De Long, N.E.; Barry, E.J.; Pinelli, C.; Wood, G.A.; Hardy, D.B.; Morrison, K.M.; Taylor, V.H.; Gerstein, H.C.; Holloway, A.C. Antenatal Exposure to the Selective Serotonin Reuptake Inhibitor Fluoxetine Leads to Postnatal Metabolic and Endocrine Changes Associated with Type 2 Diabetes in Wistar Rats. Toxicol. Appl. Pharm. 2015, 285, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Bruin, J.E.; Gerstein, H.C.; Morrison, K.M.; Holloway, A.C. Increased Pancreatic Beta-Cell Apoptosis Following Fetal and Neonatal Exposure to Nicotine Is Mediated via the Mitochondria. Toxicol. Sci. 2008, 103, 362–370. [Google Scholar] [CrossRef] [Green Version]
- Petrik, J.; Reusens, B.; Arany, E.; Remacle, C.; Coelho, C.; Hoet, J.J.; Hill, D.J. A Low Protein Diet Alters the Balance of Islet Cell Replication and Apoptosis in the Fetal and Neonatal Rat and Is Associated with a Reduced Pancreatic Expression of Insulin-like Growth Factor-II. Endocrinology 1999, 140, 4861–4873. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez-Silva, F.J.; Suárez, J.; Baixeras, E.; Cobo, N.; Bautista, D.; Cuesta-Muñoz, A.L.; Fuentes, E.; Juan-Pico, P.; Castro, M.J.; Milman, G.; et al. Presence of Functional Cannabinoid Receptors in Human Endocrine Pancreas. Diabetologia 2008, 51, 476–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starowicz, K.M.; Cristino, L.; Matias, I.; Capasso, R.; Racioppi, A.; Izzo, A.A.; Di Marzo, V. Endocannabinoid Dysregulation in the Pancreas and Adipose Tissue of Mice Fed with a High-Fat Diet. Obesity 2008, 16, 553–565. [Google Scholar] [CrossRef]
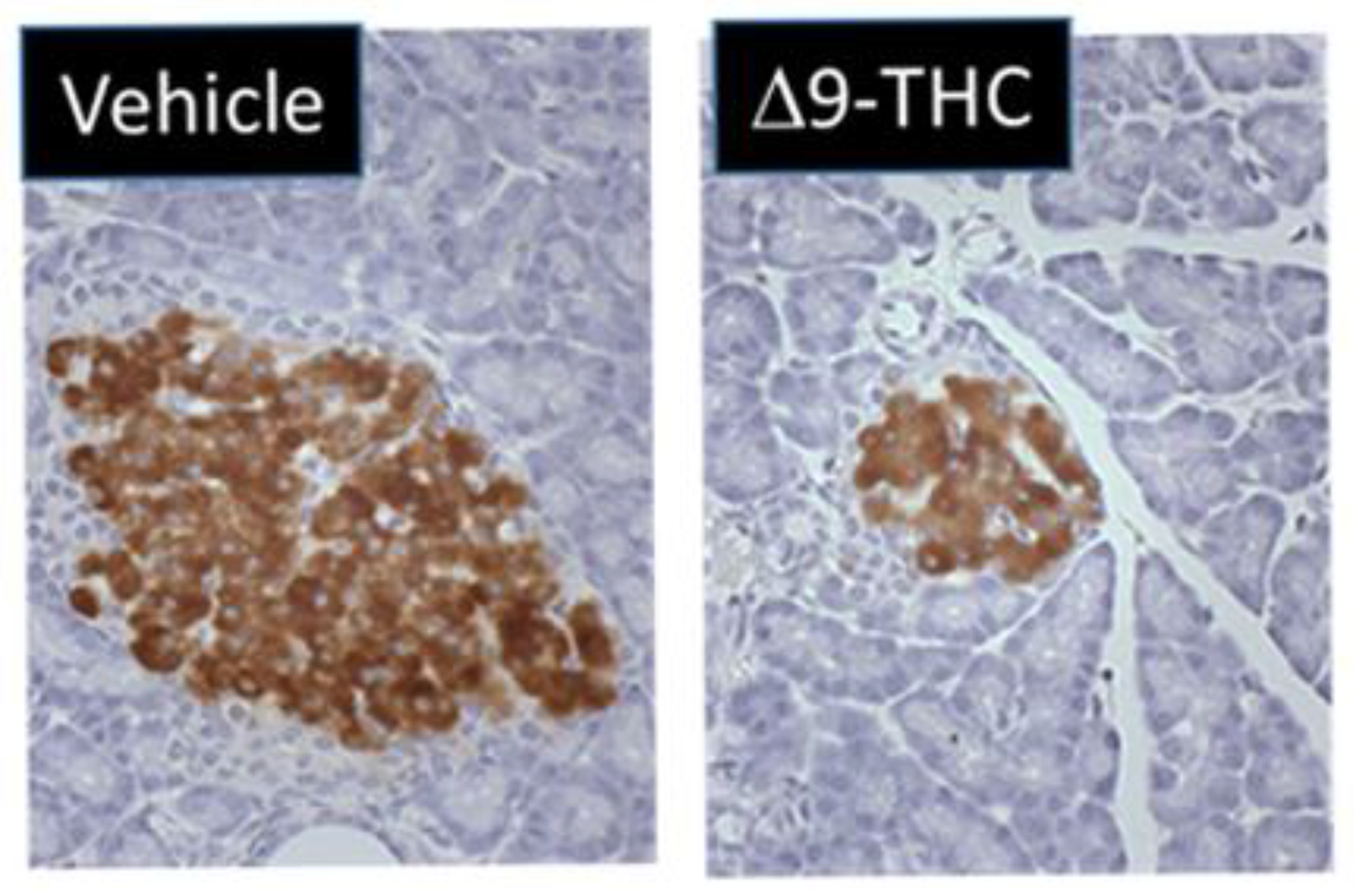
- Gillies, R.; Lee, K.; Vanin, S.; Laviolette, S.R.; Holloway, A.C.; Arany, E.; Hardy, D.B. Maternal Exposure to Δ9-Tetrahydrocannabinol Impairs Female Offspring Glucose Homeostasis and Endocrine Pancreatic Development in the Rat. Reprod. Toxicol. 2020, 94, 84–91. [Google Scholar] [CrossRef]
- Martínez-Peña, A.A.; Lee, K.; Petrik, J.J.; Hardy, D.B.; Holloway, A.C. Gestational Exposure to Δ9-THC Impacts Ovarian Follicular Dynamics and Angiogenesis in Adulthood in Wistar Rats. J. Dev. Orig. Health Dis. 2021, 1–5. [Google Scholar] [CrossRef]
- Mackenzie, R.W.; Elliott, B.T. Akt/PKB Activation and Insulin Signaling: A Novel Insulin Signaling Pathway in the Treatment of Type 2 Diabetes. Diabetes Metab. Syndr. Obes. 2014, 7, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Cohrs, C.M.; Stertmann, J.; Bozsak, R.; Speier, S. Human Beta Cell Mass and Function in Diabetes: Recent Advances in Knowledge and Technologies to Understand Disease Pathogenesis. Mol. Metab. 2017, 6, 943–957. [Google Scholar] [CrossRef]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-Cell Deficit and Increased Beta-Cell Apoptosis in Humans with Type 2 Diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Lee, K.J.; Kim, J.S.; Rho, J.G.; Shin, J.J.; Song, W.K.; Lee, E.K.; Egan, J.M.; Kim, W. Cannabinoids Regulate Bcl-2 and Cyclin D2 Expression in Pancreatic β Cells. PLoS ONE 2016, 11, e0150981. [Google Scholar] [CrossRef]
- Matias, I.; Gonthier, M.-P.; Orlando, P.; Martiadis, V.; De Petrocellis, L.; Cervino, C.; Petrosino, S.; Hoareau, L.; Festy, F.; Pasquali, R.; et al. Regulation, Function, and Dysregulation of Endocannabinoids in Models of Adipose and Beta-Pancreatic Cells and in Obesity and Hyperglycemia. J. Clin. Endocrinol. Metab. 2006, 91, 3171–3180. [Google Scholar] [CrossRef]
- Jourdan, T.; Godlewski, G.; Cinar, R.; Bertola, A.; Szanda, G.; Liu, J.; Tam, J.; Han, T.; Mukhopadhyay, B.; Skarulis, M.C.; et al. Activation of the Nlrp3 Inflammasome in Infiltrating Macrophages by Endocannabinoids Mediates Beta Cell Loss in Type 2 Diabetes. Nat. Med. 2013, 19, 1132–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenstock, J.; Hollander, P.; Chevalier, S.; Iranmanesh, A.; SERENADE Study Group. SERENADE: The Study Evaluating Rimonabant Efficacy in Drug-Naive Diabetic Patients: Effects of Monotherapy with Rimonabant, the First Selective CB1 Receptor Antagonist, on Glycemic Control, Body Weight, and Lipid Profile in Drug-Naive Type 2 Diabetes. Diabetes Care 2008, 31, 2169–2176. [Google Scholar] [CrossRef] [Green Version]
- Hollander, P.A.; Amod, A.; Litwak, L.E.; Chaudhari, U.; ARPEGGIO Study Group. Effect of Rimonabant on Glycemic Control in Insulin-Treated Type 2 Diabetes: The ARPEGGIO Trial. Diabetes Care 2010, 33, 605–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Williams, S.J.; O’Brien, D.; Davidge, S.T. Hypoxia or Nutrient Restriction during Pregnancy in Rats Leads to Progressive Cardiac Remodeling and Impairs Postischemic Recovery in Adult Male Offspring. FASEB J. 2006, 20, 1251–1253. [Google Scholar] [CrossRef]
- Yu, F.; Zheng, A.; Qian, J.; Li, Y.; Wu, L.; Yang, J.; Gao, X. Prenatal Nicotine Exposure Results in the Myocardial Fibrosis in the Adult Male Offspring Rats. Exp. Toxicol. Pathol. 2016, 68, 445–450. [Google Scholar] [CrossRef]
- Rueda-Clausen, C.F.; Morton, J.S.; Davidge, S.T. Effects of Hypoxia-Induced Intrauterine Growth Restriction on Cardiopulmonary Structure and Function during Adulthood. Cardiovasc. Res. 2009, 81, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Zimanyi, M.A.; Black, M.J. Effect of Maternal Protein Restriction in Rats on Cardiac Fibrosis and Capillarization in Adulthood. Pediatr. Res. 2006, 60, 83–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisano, J.K.; Kisiolek, J.N.; Smoak, P.; Phillips, K.T.; Stewart, L.K. Chronic Cannabis Use and Circulating Biomarkers of Neural Health, Stress, and Inflammation in Physically Active Individuals. Appl. Physiol. Nutr. Metab. 2020, 45, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Page, R.L.; Allen, L.A.; Kloner, R.A.; Carriker, C.R.; Martel, C.; Morris, A.A.; Piano, M.R.; Rana, J.S.; Saucedo, J.F.; American Heart Association Clinical Pharmacology Committee and Heart Failure and Transplantation Committee of the Council on Clinical Cardiology; et al. Medical Marijuana, Recreational Cannabis, and Cardiovascular Health: A Scientific Statement From the American Heart Association. Circulation 2020, 142, e131–e152. [Google Scholar] [CrossRef]
- Shi, Y.; Zhu, B.; Liang, D. The Associations between Prenatal Cannabis Use Disorder and Neonatal Outcomes. Addiction 2021. [Google Scholar] [CrossRef]
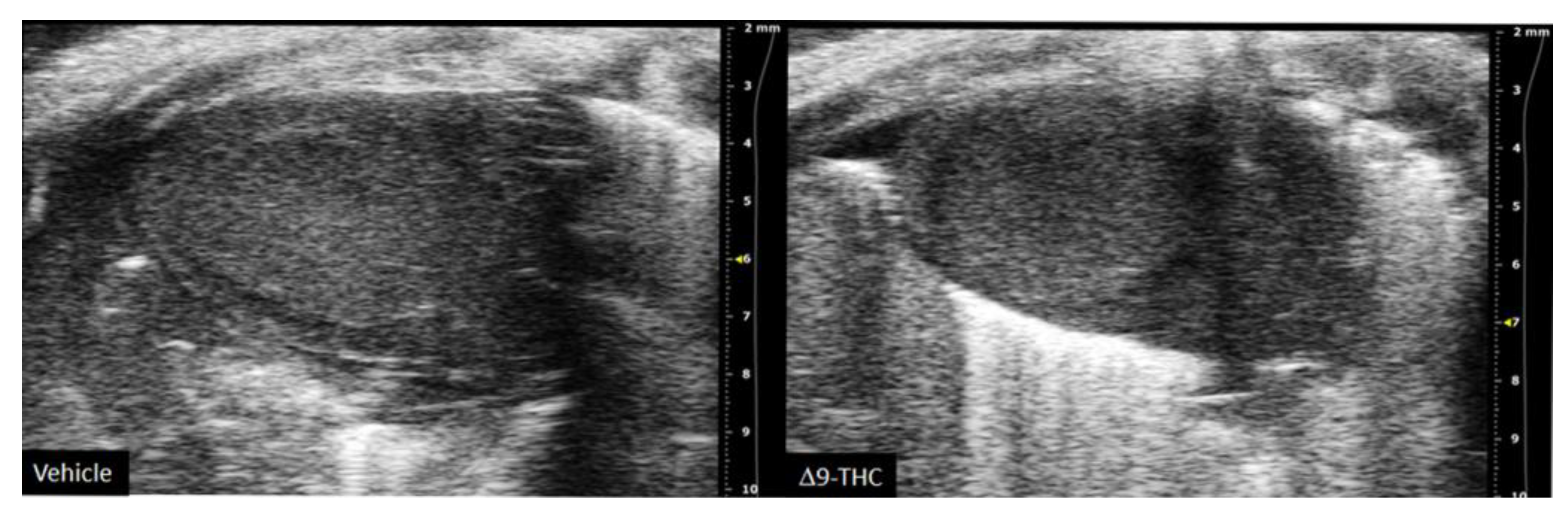
- Lee, K.; Laviolette, S.R.; Hardy, D.B. Exposure to Δ9-Tetrahydrocannabinol during Rat Pregnancy Leads to Impaired Cardiac Dysfunction in Postnatal Life. Pediatric Res. 2021, 1–8. [Google Scholar] [CrossRef]
- Lu, Y.; Akinwumi, B.C.; Shao, Z.; Anderson, H.D. Ligand Activation of Cannabinoid Receptors Attenuates Hypertrophy of Neonatal Rat Cardiomyocytes. J. Cardiovasc. Pharm. 2014, 64, 420–430. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, P.; Bátkai, S.; Rajesh, M.; Czifra, N.; Harvey-White, J.; Haskó, G.; Zsengeller, Z.; Gerard, N.P.; Liaudet, L.; Kunos, G.; et al. Pharmacological Inhibition of CB1 Cannabinoid Receptor Protects against Doxorubicin-Induced Cardiotoxicity. J. Am. Coll. Cardiol. 2007, 50, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Athanasiou, A.; Clarke, A.B.; Turner, A.E.; Kumaran, N.M.; Vakilpour, S.; Smith, P.A.; Bagiokou, D.; Bradshaw, T.D.; Westwell, A.D.; Fang, L.; et al. Cannabinoid Receptor Agonists Are Mitochondrial Inhibitors: A Unified Hypothesis of How Cannabinoids Modulate Mitochondrial Function and Induce Cell Death. Biochem. Biophys. Res. Commun. 2007, 364, 131–137. [Google Scholar] [CrossRef]
- Hiley, C.R. Endocannabinoids and the Heart. J. Cardiovasc. Pharm. 2009, 53, 267–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfulaij, N.; Meiners, F.; Michalek, J.; Small-Howard, A.L.; Turner, H.C.; Stokes, A.J. Cannabinoids, the Heart of the Matter. J. Am. Heart Assoc. 2018, 7, e009099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulmer, M.L.; Thewke, D.P. The Endocannabinoid System and Heart Disease: The Role of Cannabinoid Receptor Type 2. Cardiovasc. Hematol. Disord. Drug Targets 2018, 18, 34–51. [Google Scholar] [CrossRef]
- Montecucco, F.; Di Marzo, V. At the Heart of the Matter: The Endocannabinoid System in Cardiovascular Function and Dysfunction. Trends Pharmacol. Sci. 2012, 33, 331–340. [Google Scholar] [CrossRef]
- Chan, K.A.; Bernal, A.B.; Vickers, M.H.; Gohir, W.; Petrik, J.J.; Sloboda, D.M. Early Life Exposure to Undernutrition Induces ER Stress, Apoptosis, and Reduced Vascularization in Ovaries of Adult Rat Offspring1. Biol. Reprod. 2015, 92. [Google Scholar] [CrossRef] [PubMed]
- Jazwiec, P.A.; Li, X.; Matushewski, B.; Richardson, B.S.; Sloboda, D.M. Fetal Growth Restriction Is Associated With Decreased Number of Ovarian Follicles and Impaired Follicle Growth in Young Adult Guinea Pig Offspring. Reprod. Sci. 2019, 26, 1557–1567. [Google Scholar] [CrossRef]
- Allais, A.; Albert, O.; Lefèvre, P.L.C.; Wade, M.G.; Hales, B.F.; Robaire, B. In Utero and Lactational Exposure to Flame Retardants Disrupts Rat Ovarian Follicular Development and Advances Puberty. Toxicol. Sci. 2020, 175, 197–209. [Google Scholar] [CrossRef]
- Castel, P.; Barbier, M.; Poumerol, E.; Mandon-Pépin, B.; Tassistro, V.; Lepidi, H.; Pelissier-Alicot, A.-L.; Manzoni, O.J.; Courbiere, B. Prenatal Cannabinoid Exposure Alters the Ovarian Reserve in Adult Offspring of Rats. Arch. Toxicol. 2020, 94, 4131–4141. [Google Scholar] [CrossRef] [PubMed]
- Dalterio, S.L.; de Rooij, D.G. Maternal Cannabinoid Exposure. Effects on Spermatogenesis in Male Offspring. Int. J. Androl. 1986, 9, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.K.; Itchon-Ramos, N.; Visco, Z.; Huang, Z.; Grenier, C.; Schrott, R.; Acharya, K.; Boudreau, M.-H.; Price, T.M.; Raburn, D.J.; et al. Cannabinoid Exposure and Altered DNA Methylation in Rat and Human Sperm. Epigenetics 2018, 13, 1208–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kung, J.W.C.; Currie, I.S.; Forbes, S.J.; Ross, J.A. Liver Development, Regeneration, and Carcinogenesis. J. Biomed. Biotechnol. 2010, 2010, 984248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenwood, M.R.; Hirsch, J. Postnatal Development of Adipocyte Cellularity in the Normal Rat. J. Lipid Res. 1974, 15, 474–483. [Google Scholar] [CrossRef]
- Dor, Y.; Brown, J.; Martinez, O.I.; Melton, D.A. Adult Pancreatic β-Cells Are Formed by Self-Duplication Rather than Stem-Cell Differentiation. Nature 2004, 429, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Wang, X.; Capasso, J.M.; Gerdes, A.M. Rapid Transition of Cardiac Myocytes from Hyperplasia to Hypertrophy during Postnatal Development. J. Mol. Cell Cardiol. 1996, 28, 1737–1746. [Google Scholar] [CrossRef]
- Painter, R.C.; Osmond, C.; Gluckman, P.; Hanson, M.; Phillips, D.I.W.; Roseboom, T.J. Transgenerational Effects of Prenatal Exposure to the Dutch Famine on Neonatal Adiposity and Health in Later Life. BJOG 2008, 115, 1243–1249. [Google Scholar] [CrossRef]
- Veenendaal, M.V.E.; Painter, R.C.; de Rooij, S.R.; Bossuyt, P.M.M.; van der Post, J.A.M.; Gluckman, P.D.; Hanson, M.A.; Roseboom, T.J. Transgenerational Effects of Prenatal Exposure to the 1944-45 Dutch Famine. BJOG 2013, 120, 548–553. [Google Scholar] [CrossRef]
- Zambrano, E.; Martínez-Samayoa, P.M.; Bautista, C.J.; Deás, M.; Guillén, L.; Rodríguez-González, G.L.; Guzmán, C.; Larrea, F.; Nathanielsz, P.W. Sex Differences in Transgenerational Alterations of Growth and Metabolism in Progeny (F2) of Female Offspring (F1) of Rats Fed a Low Protein Diet during Pregnancy and Lactation. J. Physiol. 2005, 566, 225–236. [Google Scholar] [CrossRef]
- Martínez, D.; Pentinat, T.; Ribó, S.; Daviaud, C.; Bloks, V.W.; Cebrià, J.; Villalmanzo, N.; Kalko, S.G.; Ramón-Krauel, M.; Díaz, R.; et al. In Utero Undernutrition in Male Mice Programs Liver Lipid Metabolism in the Second-Generation Offspring Involving Altered Lxra DNA Methylation. Cell Metab. 2014, 19, 941–951. [Google Scholar] [CrossRef] [Green Version]
- Thamotharan, M.; Garg, M.; Oak, S.; Rogers, L.M.; Pan, G.; Sangiorgi, F.; Lee, P.W.N.; Devaskar, S.U. Transgenerational Inheritance of the Insulin-Resistant Phenotype in Embryo-Transferred Intrauterine Growth-Restricted Adult Female Rat Offspring. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1270–E1279. [Google Scholar] [CrossRef] [Green Version]
- Gniuli, D.; Calcagno, A.; Caristo, M.E.; Mancuso, A.; Macchi, V.; Mingrone, G.; Vettor, R. Effects of High-Fat Diet Exposure during Fetal Life on Type 2 Diabetes Development in the Progeny. J. Lipid Res. 2008, 49, 1936–1945. [Google Scholar] [CrossRef] [Green Version]
- Dunn, G.A.; Bale, T.L. Maternal High-Fat Diet Effects on Third-Generation Female Body Size via the Paternal Lineage. Endocrinology 2011, 152, 2228–2236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jazwiec, P.A.; Patterson, V.S.; Ribeiro, T.A.; Yeo, E.; Kennedy, K.M.; Mathias, P.C.F.; Petrik, J.J.; Sloboda, D.M. Paternal Obesity Results in Placental Hypoxia and Sex-Specific Impairments in Placental Vascularization and Offspring Metabolic Function. bioRxiv 2021. [Google Scholar] [CrossRef]
- Sanchez-Garrido, M.A.; Ruiz-Pino, F.; Velasco, I.; Barroso, A.; Fernandois, D.; Heras, V.; Manfredi-Lozano, M.; Vazquez, M.J.; Castellano, J.M.; Roa, J.; et al. Intergenerational Influence of Paternal Obesity on Metabolic and Reproductive Health Parameters of the Offspring: Male-Preferential Impact and Involvement of Kiss1-Mediated Pathways. Endocrinology 2018, 159, 1005–1018. [Google Scholar] [CrossRef]
- Mao, Z.; Xia, W.; Chang, H.; Huo, W.; Li, Y.; Xu, S. Paternal BPA Exposure in Early Life Alters Igf2 Epigenetic Status in Sperm and Induces Pancreatic Impairment in Rat Offspring. Toxicol. Lett. 2015, 238, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Freitas, I.N.; Dos Reis Araujo, T.; Vettorazzi, J.F.; Magalhães, E.A.; Carneiro, E.M.; Bonfleur, M.L.; Ribeiro, R.A. Taurine Supplementation in High-Fat Diet Fed Male Mice Attenuates Endocrine Pancreatic Dysfunction in Their Male Offspring. Amino Acids 2019, 51, 727–738. [Google Scholar] [CrossRef]
- Anderson, R.A.; Furby, J.E.; Oswald, C.; Zaneveld, L.J. Tetratological Evaluation of Mouse Fetuses after Paternal Alcohol Ingestion. Neurobehav. Toxicol. Teratol. 1981, 3, 117–120. [Google Scholar]
- Cicero, T.J.; Adams, M.L.; O’Connor, L.; Nock, B.; Meyer, E.R.; Wozniak, D. Influence of Chronic Alcohol Administration on Representative Indices of Puberty and Sexual Maturation in Male Rats and the Development of Their Progeny. J. Pharm. Exp. Ther. 1990, 255, 707–715. [Google Scholar]
- Pembrey, M.E.; Bygren, L.O.; Kaati, G.; Edvinsson, S.; Northstone, K.; Sjöström, M.; Golding, J. Sex-Specific, Male-Line Transgenerational Responses in Humans. Eur. J. Hum. Genet. 2006, 14, 159–166. [Google Scholar] [CrossRef]
- Holloway, Z.R.; Hawkey, A.B.; Pippin, E.; White, H.; Wells, C.; Kenou, B.; Rezvani, A.H.; Murphy, S.K.; Levin, E.D. Paternal Factors in Neurodevelopmental Toxicology: THC Exposure of Male Rats Causes Long-Lasting Neurobehavioral Effects in Their Offspring. Neurotoxicology 2020, 78, 57–63. [Google Scholar] [CrossRef]
- Levin, E.D.; Hawkey, A.B.; Hall, B.J.; Cauley, M.; Slade, S.; Yazdani, E.; Kenou, B.; White, H.; Wells, C.; Rezvani, A.H.; et al. Paternal THC Exposure in Rats Causes Long-Lasting Neurobehavioral Effects in the Offspring. Neurotoxicol. Teratol. 2019, 74, 106806. [Google Scholar] [CrossRef]
- Slotkin, T.A.; Skavicus, S.; Levin, E.D.; Seidler, F.J. Paternal Δ9-Tetrahydrocannabinol Exposure Prior to Mating Elicits Deficits in Cholinergic Synaptic Function in the Offspring. Toxicol. Sci. 2020, 174, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Innocenzi, E.; De Domenico, E.; Ciccarone, F.; Zampieri, M.; Rossi, G.; Cicconi, R.; Bernardini, R.; Mattei, M.; Grimaldi, P. Paternal Activation of CB2 Cannabinoid Receptor Impairs Placental and Embryonic Growth via an Epigenetic Mechanism. Sci. Rep. 2019, 9, 17034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHugh, D.; Page, J.; Dunn, E.; Bradshaw, H.B. Δ9-Tetrahydrocannabinol and N-Arachidonyl Glycine Are Full Agonists at GPR18 Receptors and Induce Migration in Human Endometrial HEC-1B Cells. Br. J. Pharmacol. 2012, 165, 2414–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Z.-L.; Carroll, C.; Chen, R.; Alfonso, J.; Gutierrez, V.; He, H.; Lucman, A.; Xing, C.; Sebring, K.; Zhou, J.; et al. N-Oleoyldopamine Enhances Glucose Homeostasis through the Activation of GPR119. Mol. Endocrinol. 2010, 24, 161–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balenga, N.A.; Martínez-Pinilla, E.; Kargl, J.; Schröder, R.; Peinhaupt, M.; Platzer, W.; Bálint, Z.; Zamarbide, M.; Dopeso-Reyes, I.G.; Ricobaraza, A.; et al. Heteromerization of GPR55 and Cannabinoid CB2 Receptors Modulates Signalling. Br. J. Pharm. 2014, 171, 5387–5406. [Google Scholar] [CrossRef] [Green Version]
- Briand-Mésange, F.; Pons, V.; Allart, S.; Masquelier, J.; Chicanne, G.; Beton, N.; Payrastre, B.; Muccioli, G.G.; Ausseil, J.; Davignon, J.-L.; et al. Glycerophosphodiesterase 3 (GDE3) Is a Lysophosphatidylinositol-Specific Ectophospholipase C Acting as an Endocannabinoid Signaling Switch. J. Biol. Chem. 2020, 295, 15767–15781. [Google Scholar] [CrossRef]
- Tsutsumi, T.; Matsuda, R.; Morito, K.; Kawabata, K.; Yokota, M.; Nikawadori, M.; Inoue-Fujiwara, M.; Kawashima, S.; Hidaka, M.; Yamamoto, T.; et al. Identification of Human Glycerophosphodiesterase 3 as an Ecto Phospholipase C That Converts the G Protein-Coupled Receptor 55 Agonist Lysophosphatidylinositol to Bioactive Monoacylglycerols in Cultured Mammalian Cells. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2020, 1865, 158761. [Google Scholar] [CrossRef]
- Senn, L.; Cannazza, G.; Biagini, G. Receptors and Channels Possibly Mediating the Effects of Phytocannabinoids on Seizures and Epilepsy. Pharmaceuticals 2020, 13, 174. [Google Scholar] [CrossRef]
- Alhouayek, M.; Masquelier, J.; Muccioli, G.G. Lysophosphatidylinositols, from Cell Membrane Constituents to GPR55 Ligands. Trends Pharm. Sci. 2018, 39, 586–604. [Google Scholar] [CrossRef]
- Romero-Zerbo, S.Y.; Rafacho, A.; Díaz-Arteaga, A.; Suárez, J.; Quesada, I.; Imbernon, M.; Ross, R.A.; Dieguez, C.; de Fonseca, F.R.; Nogueiras, R.; et al. A Role for the Putative Cannabinoid Receptor GPR55 in the Islets of Langerhans. J. Endocrinol. 2011, 211, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vong, C.T.; Tseng, H.H.L.; Kwan, Y.W.; Lee, S.M.-Y.; Hoi, M.P.M. Novel Protective Effect of O-1602 and Abnormal Cannabidiol, GPR55 Agonists, on ER Stress-Induced Apoptosis in Pancreatic β-Cells. Biomed. Pharmacother. 2019, 111, 1176–1186. [Google Scholar] [CrossRef]
- Puhl, S.-L.; Hilby, M.; Kohlhaas, M.; Keidel, L.M.; Jansen, Y.; Hristov, M.; Schindler, J.; Maack, C.; Steffens, S. Haematopoietic and Cardiac GPR55 Synchronize Post-Myocardial Infarction Remodelling. Sci. Rep. 2021, 11, 14385. [Google Scholar] [CrossRef] [PubMed]
- ElSohly, M.A.; Mehmedic, Z.; Foster, S.; Gon, C.; Chandra, S.; Church, J.C. Changes in Cannabis Potency Over the Last 2 Decades (1995–2014): Analysis of Current Data in the United States. Biol. Psychiatry 2016, 79, 613–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, J.Y.; Farr, S.L.; Tong, V.T.; Creanga, A.A.; Callaghan, W.M. Prevalence and Patterns of Marijuana Use among Pregnant and Nonpregnant Women of Reproductive Age. Am. J. Obs. Gynecol. 2015, 213, 201.e1–201.e10. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.; Hardy, D.B. Metabolic Consequences of Gestational Cannabinoid Exposure. Int. J. Mol. Sci. 2021, 22, 9528. https://doi.org/10.3390/ijms22179528
Lee K, Hardy DB. Metabolic Consequences of Gestational Cannabinoid Exposure. International Journal of Molecular Sciences. 2021; 22(17):9528. https://doi.org/10.3390/ijms22179528
Chicago/Turabian StyleLee, Kendrick, and Daniel B. Hardy. 2021. "Metabolic Consequences of Gestational Cannabinoid Exposure" International Journal of Molecular Sciences 22, no. 17: 9528. https://doi.org/10.3390/ijms22179528
APA StyleLee, K., & Hardy, D. B. (2021). Metabolic Consequences of Gestational Cannabinoid Exposure. International Journal of Molecular Sciences, 22(17), 9528. https://doi.org/10.3390/ijms22179528