
The Role of Neutrophilic Granulocytes in Philadelphia Chromosome Negative Myeloproliferative Neoplasms

 , and
, and
Abstract
:1. Introduction
2. Clinical Picture and Course of Disease
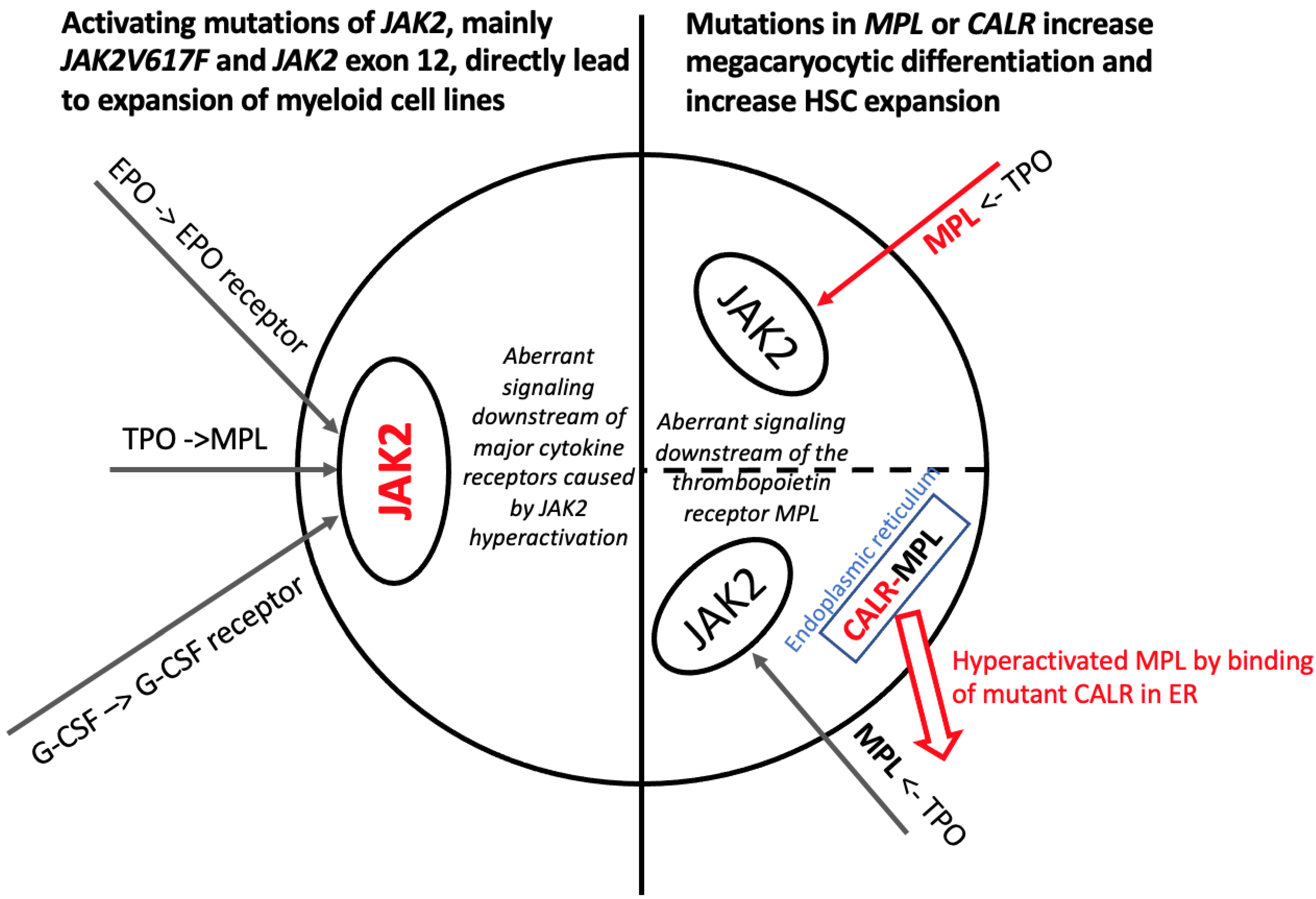
3. Molecular Biology of Ph-Negative Myeloproliferative Neoplasms and Predictive/Prognostic Implications
4. Inflammation and Neutrophilic Granulocytes in MPN
5. Principles of Clinical Management and Conclusions
5.1. Phlebotomy and Cytoreductive Therapy
5.2. Interferon
5.3. JAK Inhibitors
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AML | acute myleoid leukemia |
| CALR | calreticulin |
| CEL, NOS | chronic eosinophilic leukemia not otherwise specified |
| CHIP | clonal hematopoiesis of indetermined potential |
| CML | chronic myeloid leukemia |
| CNL | chronic neutrophilic leukemia |
| CRP | C-reactive protein |
| GvHD | Graft-versus-Host Disease |
| FGF | Fibroblast growth factor |
| HSC | hematopoietic stem cell |
| EPO | erythropoietin |
| ER | endoplasmic reticulum |
| ET | essential thrombocythemia |
| HGF | hepatocyte growth factor |
| JAK | Janus kinase |
| LDH | lactate dehydrogenase |
| MF | myelofibrosis |
| MPN | myeloproliferative neoplasms |
| MPL | myeloproliferative leukemia virus oncogene/protein |
| NG | neutrophilic granulocyte |
| NGAL | neutrophil gelatinase-associated lipocalin |
| PMF | primary myelofibrosis |
| PV | polycythemia vera |
| OS | overall survival |
| R-2b | ropeginterferon alfa-2b |
| TPO | thrombopoietin |
| TNF-alpha | Tumor necrosis factor-alpha |
| YKL-40 | Chitinase-3-like protein 1 |
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Barbui, T.; Thiele, J.; Gisslinger, H.; Kvasnicka, H.M.; Vannucchi, A.M.; Guglielmelli, P.; Orazi, A.; Tefferi, A. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: Document summary and in-depth discussion. Blood Cancer J. 2018, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Mead, A.J.; Mullally, A. Myeloproliferative neoplasm stem cells. Blood 2017, 129, 1607–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabbour, E.; Kantarjian, H. Chronic myeloid leukemia: 2020 update on diagnosis, therapy and monitoring. Am. J. Hematol. 2020, 95, 691–709. [Google Scholar] [CrossRef] [PubMed]
- Scherber, R.; Dueck, A.C.; Johansson, P.; Barbui, T.; Barosi, G.; Vannucchi, A.M.; Passamonti, F.; Andreasson, B.; Ferarri, M.L.; Rambaldi, A.; et al. The myeloproliferative neoplasm symptom assessment form (MPN-SAF): International prospective validation and reliability trial in 402 patients. Blood 2011, 118, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Siegel, F.P.; Tauscher, J.; Petrides, P.E. Aquagenic pruritus in polycythemia vera: Characteristics and influence on quality of life in 441 patients. Am. J. Hematol. 2013, 88, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Mann, N.; King, T.; Murphy, R. Review of primary and secondary erythromelalgia. Clin. Exp. Derm. 2019, 44, 477–482. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Knudson, R.A.; Ketterling, R.; Hanson, C.H.; Maffioli, M.; Caramazza, D.; Passamonti, F.; Pardanani, A. CALR vs. JAK2 vs. MPL-mutated or triple-negative myelofibrosis: Clinical, cytogenetic and molecular comparisons. Leukemia 2014, 28, 1472–1477. [Google Scholar] [CrossRef]
- Tefferi, A.; Barbui, T. Polycythemia vera and essential thrombocythemia: 2019 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2019, 94, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerquozzi, S.; Tefferi, A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: A literature review of incidence and risk factors. Blood Cancer J. 2015, 5, e366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunbar, A.J.; Rampal, R.K.; Levine, R. Leukemia secondary to myeloproliferative neoplasms. Blood 2020, 136, 61–70. [Google Scholar] [CrossRef]
- Finazzi, G. How to manage essential thrombocythemia. Leukemia 2012, 26, 875–882. [Google Scholar] [CrossRef] [Green Version]
- Carobbio, A.; Ferrari, A.; Masciulli, A.; Ghirardi, A.; Barosi, G.; Barbui, T. Leukocytosis and thrombosis in essential thrombocythemia and polycythemia vera: A systematic review and meta-analysis. Blood Adv. 2019, 3, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Verstovsek, S. Updates in the management of polycythemia vera and essential thrombocythemia. Adv. Hematol. 2019, 10, 2040620719870052. [Google Scholar] [CrossRef] [Green Version]
- Iurlo, A.; Cattaneo, D.; Gianelli, U. Blast transformation in myeloproliferative neoplasms: Risk factors, biological findings, and targeted therapeutic options. Int. J. Mol. Sci. 2019, 20, 1839. [Google Scholar] [CrossRef] [Green Version]
- Mullally, A.; Poveromo, L.; Schneider, R.K.; Al-Shahrour, F.; Lane, S.W.; Ebert, B.L. Distinct roles for long-term hematopoietic stem cells and erythroid precursor cells in a murine model of Jak2V617F-mediated polycythemia vera. Blood 2012, 120, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Sangkhae, V.; Etheridge, S.L.; Kaushansky, K.; Hitchcock, I.S. The thrombopoietin receptor, MPL, is critical for development of a JAK2V617F-induced myeloproliferative neoplasm. Blood 2014, 124, 3956–3963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieber, M.; Crispino, J.D.; Stein, B. Myelofibrosis in 2019: Moving beyond JAK2 inhibition. Blood Cancer J. 2019, 9, 74. [Google Scholar] [CrossRef] [PubMed]
- Chou, F.S.; Mulloy, J.C. The thrombopoietin/MPL pathway in hematopoiesis and leukemogenesis. J. Cell. Biochem. 2011, 112, 1491–1498. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couédic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Pardanani, A. Myeloproliferative neoplasms: A contemporary review. JAMA Oncol. 2015, 1, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A. Primary myelofibrosis: 2019 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2018, 93, 1551–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef] [Green Version]
- Edahiro, Y.; Araki, M.; Komatsu, N. Mechanism underlying the development of myeloproliferative neoplasms through mutant calreticulin. Cancer Sci. 2020, 111, 2682–2688. [Google Scholar] [CrossRef]
- Perner, F.; Perner, C.; Ernst, T.; Heidel, F.H. Roles of JAK2 in aging, inflammation, hematopoiesis and malignant transformation. Cells 2019, 8, 854. [Google Scholar] [CrossRef] [Green Version]
- McKerrell, T.; Park, N.; Chi, J.; Collord, G.; Moreno, T.; Ponstingl, H.; Dias, J.; Gerasimou, P.; Melanthiou, K.; Prokopiou, C.; et al. V617F hematopoietic clones are present several years prior to MPN diagnosis and follow different expansion kinetics. Blood Adv. 2017, 1, 968–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, R.; Grosel, J. Acute portal vein thrombosis in a 59-year-old male with JAK2 V617F mutation. Radiol. Case Rep. 2018, 13, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Colaizzo, D.; Amitrano, L.; Tiscia, G.L.; Scenna, G.; Grandone, E.; Guardascione, M.A.; Brancaccio, V.; Margaglione, M. The JAK2 V617F mutation frequently occurs in patients with portal and mesenteric venous thrombosis. J. Thromb. Haemost. 2007, 5, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, P.; Takizawa, H.; Kubovcakova, L.; Guo, G.; Hao-Shen, H.; Dirnhofer, S.; Orkin, S.H.; Manz, M.G.; Skoda, R.C. Myeloproliferative neoplasms can be initiated from a single hematopoietic stem cell expressing JAK2-V617F. J. Exp. Med. 2014, 211, 2213–2230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delhommeau, F.; Dupont, S.; Tonetti, C.; Massé, A.; Godin, I.; Le Couedic, J.P.; Debili, N.; Saulnier, P.; Casadevall, N.; Vainchenker, W.; et al. Evidence that the JAK2 G1849T (V617F) mutation occurs in a lymphomyeloid progenitor in polycythemia vera and idiopathic myelofibrosis. Blood 2007, 109, 71–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mylonas, E.; Yoshida, K.; Frick, M.; Hoyer, K.; Christen, F.; Kaeda, J.; Obenaus, M.; Noerenberg, D.; Hennch, C.; Chan, W.; et al. Single-cell analysis based dissection of clonality in myelofibrosis. Nat. Commun. 2020, 11, 73. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Kent, D.G.; Chen, E.; Green, A.R. Mouse models of myeloproliferative neoplasms: JAK of all grades. Dis. Model. Mech. 2011, 4, 311–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleary, C.; Kralovics, R. Molecular basis and clonal evolution of myeloproliferative neoplasms. Clin. Chem. Lab. Med. 2013, 51, 1889–1896. [Google Scholar] [CrossRef] [Green Version]
- Prick, J.; de Haan, G.; Green, A.R.; Kent, D.G. Clonal heterogeneity as a driver of disease variability in the evolution of myeloproliferative neoplasms. Exp. Hematol. 2014, 42, 841–851. [Google Scholar] [CrossRef]
- Viny, A.D.; Levine, R.L. Genetics of myeloproliferative neoplasms. Cancer J. 2014, 20, 61–65. [Google Scholar] [CrossRef] [Green Version]
- Them, N.C.; Kralovics, R. Genetic basis of MPN: Beyond JAK2-V617F. Curr. Hematol. Malig. Rep. 2013, 8, 299–306. [Google Scholar] [CrossRef]
- Nangalia, J.; Green, A.R. Myeloproliferative neoplasms: From origins to outcomes. Blood 2017, 130, 2475–2483. [Google Scholar] [CrossRef]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of mutation order on myeloproliferative neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Larsen, T.S.; Pallisgaard, N.; Møller, M.B.; Hasselbalch, H.C. The JAK2 V617F allele burden in essential thrombocythemia, polycythemia vera and primary myelofibrosis--impact on disease phenotype. Eur. J. Haematol. 2007, 79, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, F.; Rumi, E.; Pietra, D.; Della Porta, M.G.; Boveri, E.; Pascutto, C.; Vanelli, L.; Arcaini, L.; Burcheri, S.; Malcovati, L.; et al. Relation between JAK2 (V617F) mutation status, granulocyte activation, and constitutive mobilization of CD34+ cells into peripheral blood in myeloproliferative disorders. Blood 2006, 107, 3676–3682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumi, E.; Pietra, D.; Ferretti, V.; Klampfl, T.; Harutyunyan, A.S.; Milosevic, J.D.; Them, N.C.; Berg, T.; Elena, C.; Casetti, I.C.; et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014, 123, 1544–1551. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A. Primary myelofibrosis: 2021 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2021, 96, 145–162. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C.; Bjørn, M.E. MPNs as inflammatory diseases: The evidence, consequences, and perspectives. Mediat. Inflamm. 2015, 2015, 102476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasselbalch, H.C. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk. Res. 2013, 37, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: Is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood 2012, 119, 3219–3225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lussana, F.; Rambaldi, A. Inflammation and myeloproliferative neoplasms. J. Autoimmun. 2017, 85, 58–63. [Google Scholar] [CrossRef]
- Koschmieder, S.; Mughal, T.I.; Hasselbalch, H.C.; Barosi, G.; Valent, P.; Kiladjian, J.J.; Jeryczynski, G.; Gisslinger, H.; Jutzi, J.S.; Pahl, H.L.; et al. Myeloproliferative neoplasms and inflammation: Whether to target the malignant clone or the inflammatory process or both. Leukemia 2016, 30, 1018–1024. [Google Scholar] [CrossRef]
- Longhitano, L.; Li Volti, G.; Giallongo, C.; Spampinato, M.; Barbagallo, I.; Di Rosa, M.; Romano, A.; Avola, R.; Tibullo, D.; Palumbo, G.A. The role of inflammation and inflammasome in myeloproliferative disease. J. Clin. Med. 2020, 9, 2334. [Google Scholar] [CrossRef] [PubMed]
- Fleischman, A.G.; Aichberger, K.J.; Luty, S.B.; Bumm, T.G.; Petersen, C.L.; Doratotaj, S.; Vasudevan, K.B.; LaTocha, D.H.; Yang, F.; Press, R.D.; et al. TNFα facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood 2011, 118, 6392–6398. [Google Scholar] [CrossRef]
- Mager, L.F.; Riether, C.; Schürch, C.M.; Banz, Y.; Wasmer, M.H.; Stuber, R.; Theocharides, A.P.; Li, X.; Xia, Y.; Saito, H.; et al. IL-33 signaling contributes to the pathogenesis of myeloproliferative neoplasms. J. Clin. Investig. 2015, 125, 2579–2591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boissinot, M.; Cleyrat, C.; Vilaine, M.; Jacques, Y.; Corre, I.; Hermouet, S. Anti-inflammatory cytokines hepatocyte growth factor and interleukin-11 are over-expressed in Polycythemia vera and contribute to the growth of clonal erythroblasts independently of JAK2V617F. Oncogene 2011, 30, 990–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manshouri, T.; Estrov, Z.; Quintás-Cardama, A.; Burger, J.; Zhang, Y.; Livun, A.; Knez, L.; Harris, D.; Creighton, C.J.; Kantarjian, H.M.; et al. Bone marrow stroma-secreted cytokines protect JAK2(V617F)-mutated cells from the effects of a JAK2 inhibitor. Cancer Res. 2011, 71, 3831–3840. [Google Scholar] [CrossRef] [Green Version]
- Goerttler, P.S.; Kreutz, C.; Donauer, J.; Faller, D.; Maiwald, T.; März, E.; Rumberger, B.; Sparna, T.; Schmitt-Gräff, A.; Wilpert, J.; et al. Gene expression profiling in polycythaemia vera: Overexpression of transcription factor NF-E2. Br. J. Haematol. 2005, 129, 138–150. [Google Scholar] [CrossRef]
- Wang, W.; Schwemmers, S.; Hexner, E.O.; Pahl, H.L. AML1 is overexpressed in patients with myeloproliferative neoplasms and mediates JAK2V617F-independent overexpression of NF-E2. Blood 2010, 116, 254–266. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. A role of NF-E2 in chronic inflammation and clonal evolution in essential thrombocythemia, polycythemia vera and myelofibrosis? Leuk. Res. 2014, 38, 263–266. [Google Scholar] [CrossRef]
- Sollazzo, D.; Forte, D.; Polverelli, N.; Romano, M.; Perricone, M.; Rossi, L.; Ottaviani, E.; Luatti, S.; Martinelli, G.; Vianelli, N.; et al. Crucial factors of the inflammatory microenvironment (IL-1β/TNF-α/TIMP-1) promote the maintenance of the malignant hemopoietic clone of myelofibrosis: An in vitro study. Oncotarget 2016, 7, 43974–43988. [Google Scholar] [CrossRef] [Green Version]
- Arranz, L.; Sánchez-Aguilera, A.; Martín-Pérez, D.; Isern, J.; Langa, X.; Tzankov, A.; Lundberg, P.; Muntión, S.; Tzeng, Y.S.; Lai, D.M.; et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 2014, 512, 78–81. [Google Scholar] [CrossRef]
- Wehrle, J.; Seeger, T.S.; Schwemmers, S.; Pfeifer, D.; Bulashevska, A.; Pahl, H.L. Transcription factor nuclear factor erythroid-2 mediates expression of the cytokine interleukin 8, a known predictor of inferior outcome in patients with myeloproliferative neoplasms. Haematologica 2013, 98, 1073–1080. [Google Scholar] [CrossRef] [Green Version]
- Hermouet, S.; Godard, A.; Pineau, D.; Corre, I.; Raher, S.; Lippert, E.; Jacques, Y. Abnormal production of interleukin (IL)-11 and IL-8 in polycythaemia vera. Cytokine 2002, 20, 178–183. [Google Scholar] [CrossRef]
- Emadi, S.; Clay, D.; Desterke, C.; Guerton, B.; Maquarre, E.; Charpentier, A.; Jasmin, C.; Le Bousse-Kerdilès, M.C. French INSERM Research Network on MMM. IL-8 and its CXCR1 and CXCR2 receptors participate in the control of megakaryocytic proliferation, differentiation, and ploidy in myeloid metaplasia with myelofibrosis. Blood 2005, 105, 464–473. [Google Scholar] [CrossRef] [Green Version]
- Laterveer, L.; Lindley, I.J.; Hamilton, M.S.; Willemze, R.; Fibbe, W.E. Interleukin-8 induces rapid mobilization of hematopoietic stem cells with radioprotective capacity and long-term myelolymphoid repopulating ability. Blood 1995, 85, 2269–2275. [Google Scholar] [CrossRef] [Green Version]
- Corre-Buscail, I.; Pineau, D.; Boissinot, M.; Hermouet, S. Erythropoietin-independent erythroid colony formation by bone marrow progenitors exposed to interleukin-11 and interleukin-8. Exp. Hematol. 2005, 33, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 2015, 5, 316–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Vaidya, R.; Caramazza, D.; Finke, C.; Lasho, T.; Pardanani, A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: A comprehensive cytokine profiling study. J. Clin. Oncol. 2011, 29, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Barosi, G.; Massa, M.; Campanelli, R.; Fois, G.; Catarsi, P.; Viarengo, G.; Villani, L.; Poletto, V.; Bosoni, T.; Magrini, U.; et al. Primary myelofibrosis: Older age and high JAK2V617F allele burden are associated with elevated plasma high-sensitivity C-reactive protein levels and a phenotype of progressive disease. Leuk. Res. 2017, 60, 18–23. [Google Scholar] [CrossRef]
- Barbui, T.; Carobbio, A.; Finazzi, G.; Guglielmelli, P.; Salmoiraghi, S.; Rosti, V.; Rambaldi, A.; Vannucchi, A.M.; Barosi, G. Elevated C-reactive protein is associated with shortened leukemia-free survival in patients with myelofibrosis. Leukemia 2013, 27, 2084–2086. [Google Scholar] [CrossRef]
- Barbui, T.; Carobbio, A.; Finazzi, G.; Vannucchi, A.M.; Barosi, G.; Antonioli, E.; Guglielmelli, P.; Pancrazzi, A.; Salmoiraghi, S.; Zilio, P.; et al. Inflammation and thrombosis in essential thrombocythemia and polycythemia vera: Different role of C-reactive protein and pentraxin 3. Haematologica 2011, 96, 315–318. [Google Scholar] [CrossRef]
- Bjørn, M.E.; Andersen, C.L.; Jensen, M.K.; Hasselbalch, H.C. Circulating YKL-40 in myelofibrosis a potential novel biomarker of disease activity and the inflammatory state. Eur. J. Haematol. 2014, 93, 224–228. [Google Scholar] [CrossRef]
- Andersen, C.L.; Bjørn, M.E.; McMullin, M.F.; Harrison, C.; Samuelsson, J.; Ejerblad, E.; Zweegman, S.; Fernandes, S.; Bareford, D.; Knapper, S.; et al. Circulating YKL-40 in patients with essential thrombocythemia and polycythemia vera treated with the novel histone deacetylase inhibitor vorinostat. Leuk. Res. 2014, 38, 816–821. [Google Scholar] [CrossRef]
- Allegra, A.; Alonci, A.; Bellomo, G.; Campo, S.; Cannavò, A.; Penna, G.; Russo, S.; Centorrino, R.; Gerace, D.; Petrungaro, A.; et al. Increased serum levels of neutrophil gelatinase-associated lipocalin in patients with essential thrombocythemia and polycythemia vera. Leuk. Lymphoma 2011, 52, 101–107. [Google Scholar] [CrossRef]
- Bauvois, B.; Susin, S.A. Revisiting neutrophil gelatinase-associated lipocalin (NGAL) in cancer: Saint or sinner? Cancers 2018, 10, 336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Xia, L.; Liu, Y.C.; Hochman, T.; Bizzari, L.; Aruch, D.; Lew, J.; Weinberg, R.; Goldberg, J.D.; Hoffman, R. Lipocalin produced by myelofibrosis cells affects the fate of both hematopoietic and marrow microenvironmental cells. Blood 2015, 126, 972–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagoya, Y.; Yoshimi, A.; Tsuruta-Kishino, T.; Arai, S.; Satoh, T.; Akira, S.; Kurokawa, M. JAK2V617F+ myeloproliferative neoplasm clones evoke paracrine DNA damage to adjacent normal cells through secretion of lipocalin-2. Blood 2014, 124, 2996–3006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonicelli, G.; Abdulkarim, K.; Mounier, M.; Johansson, P.; Rossi, C.; Jooste, V.; Andreasson, B.; Maynadié, M.; Girodon, F. Leucocytosis and thrombosis at diagnosis are associated with poor survival in polycythaemia vera: A population-based study of 327 patients. Br. J. Haematol. 2013, 160, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Rumi, E.; Finazzi, G.; Gisslinger, H.; Vannucchi, A.M.; Rodeghiero, F.; Randi, M.L.; Vaidya, R.; Cazzola, M.; Rambaldi, A.; et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: An international study. Leukemia 2013, 27, 1874–1881. [Google Scholar] [CrossRef] [Green Version]
- Boiocchi, L.; Gianelli, U.; Iurlo, A.; Fend, F.; Bonzheim, I.; Cattaneo, D.; Knowles, D.M.; Orazi, A. Neutrophilic leukocytosis in advanced stage polycythemia vera: Hematopathologic features and prognostic implications. Mod. Pathol. 2015, 28, 1448–1457. [Google Scholar] [CrossRef]
- Falanga, A.; Marchetti, M.; Barbui, T.; Smith, C.W. Pathogenesis of thrombosis in essential thrombocythemia and polycythemia vera: The role of neutrophils. Semin. Hematol. 2005, 42, 239–247. [Google Scholar] [CrossRef]
- Burgaleta, C.; González, N.; César, J. Increased CD11/CD18 expression and altered metabolic activity on polymorphonuclear leukocytes from patients with polycythemia vera and essential thrombocythemia. Acta Haematol. 2002, 108, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, W.; Fidler, T.; Wang, Y.; Tang, Y.; Woods, B.; Welch, C.; Cai, B.; Silvestre-Roig, C.; Ai, D.; et al. Macrophage inflammation, erythrophagocytosis, and accelerated atherosclerosis in Jak2. Circ. Res. 2018, 123, e35–e47. [Google Scholar] [CrossRef] [PubMed]
- Carulli, G.; Minnucci, S.; Azzara, A.; Gianfaldoni, M.L.; Angiolini, C.; Sagripanti, A.; Ferretti, A.; Ambrogi, F. Neutrophil functions in essential thrombocythemia. Hematol. Pathol. 1995, 9, 37–47. [Google Scholar] [CrossRef]
- Gupta, N.; Edelmann, B.; Schnoeder, T.M.; Saalfeld, F.C.; Wolleschak, D.; Kliche, S.; Schraven, B.; Heidel, F.H.; Fischer, T. JAK2-V617F activates β1-integrin-mediated adhesion of granulocytes to vascular cell adhesion molecule 1. Leukemia 2017, 31, 1223–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edelmann, B.; Gupta, N.; Schnoeder, T.M.; Oelschlegel, A.M.; Shahzad, K.; Goldschmidt, J.; Philipsen, L.; Weinert, S.; Ghosh, A.; Saalfeld, F.C.; et al. JAK2-V617F promotes venous thrombosis through β1/β2 integrin activation. J. Clin. Investig. 2018, 128, 4359–4371. [Google Scholar] [CrossRef]
- Arellano-Rodrigo, E.; Alvarez-Larrán, A.; Reverter, J.C.; Villamor, N.; Colomer, D.; Cervantes, F. Increased platelet and leukocyte activation as contributing mechanisms for thrombosis in essential thrombocythemia and correlation with the JAK2 mutational status. Haematologica 2006, 91, 169–175. [Google Scholar]
- Marin Oyarzún, C.P.; Heller, P.G. Platelets as mediators of thromboinflammation in chronic myeloproliferative neoplasms. Front. Immunol. 2019, 10, 1373. [Google Scholar] [CrossRef] [Green Version]
- Campbell, P.J.; MacLean, C.; Beer, P.A.; Buck, G.; Wheatley, K.; Kiladjian, J.J.; Forsyth, C.; Harrison, C.N.; Green, A.R. Correlation of blood counts with vascular complications in essential thrombocythemia: Analysis of the prospective PT1 cohort. Blood 2012, 120, 1409–1411. [Google Scholar] [CrossRef] [Green Version]
- Falanga, A.; Marchetti, M.; Vignoli, A.; Balducci, D.; Barbui, T. Leukocyte-platelet interaction in patients with essential thrombocythemia and polycythemia vera. Exp. Hematol. 2005, 33, 523–530. [Google Scholar] [CrossRef]
- Alvarez-Larrán, A.; Arellano-Rodrigo, E.; Reverter, J.C.; Domingo, A.; Villamor, N.; Colomer, D.; Cervantes, F. Increased platelet, leukocyte, and coagulation activation in primary myelofibrosis. Ann. Hematol. 2008, 87, 269–276. [Google Scholar] [CrossRef]
- Pircher, J.; Engelmann, B.; Massberg, S.; Schulz, C. Platelet-neutrophil crosstalk in atherothrombosis. Thromb. Haemost. 2019, 119, 1274–1282. [Google Scholar] [CrossRef]
- Schmitt, A.; Jouault, H.; Guichard, J.; Wendling, F.; Drouin, A.; Cramer, E.M. Pathologic interaction between megakaryocytes and polymorphonuclear leukocytes in myelofibrosis. Blood 2000, 96, 1342–1347. [Google Scholar] [CrossRef]
- Cunin, P.; Nigrovic, P.A. Megakaryocyte emperipolesis: A new frontier in cell-in-cell interaction. Platelets 2020, 31, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Centurione, L.; Di Baldassarre, A.; Zingariello, M.; Bosco, D.; Gatta, V.; Rana, R.A.; Langella, V.; Di Virgilio, A.; Vannucchi, A.M.; Migliaccio, A.R. Increased and pathologic emperipolesis of neutrophils within megakaryocytes associated with marrow fibrosis in GATA-1(low) mice. Blood 2004, 104, 3573–3580. [Google Scholar] [CrossRef] [Green Version]
- Cashell, A.W.; Buss, D.H. The frequency and significance of megakaryocytic emperipolesis in myeloproliferative and reactive states. Ann. Hematol. 1992, 64, 273–276. [Google Scholar] [CrossRef]
- Cunin, P.; Bouslama, R.; Machlus, K.R.; Martínez-Bonet, M.; Lee, P.Y.; Wactor, A.; Nelson-Maney, N.; Morris, A.; Guo, L.; Weyrich, A.; et al. Megakaryocyte emperipolesis mediates membrane transfer from intracytoplasmic neutrophils to platelets. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Wolach, O.; Sellar, R.S.; Martinod, K.; Cherpokova, D.; McConkey, M.; Chappell, R.J.; Silver, A.J.; Adams, D.; Castellano, C.A.; Schneider, R.K.; et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Laridan, E.; Martinod, K.; De Meyer, S.F. Neutrophil extracellular traps in arterial and venous thrombosis. Semin. Thromb. Hemost. 2019, 45, 86–93. [Google Scholar] [CrossRef]
- Ferrer-Marín, F.; Cuenca-Zamora, E.J.; Guijarro-Carrillo, P.J.; Teruel-Montoya, R. Emerging role of neutrophils in the thrombosis of chronic myeloproliferative neoplasms. Int. J. Mol. Sci. 2021, 22, 1143. [Google Scholar] [CrossRef]
- Marin Oyarzún, C.P.; Carestia, A.; Lev, P.R.; Glembotsky, A.C.; Castro Ríos, M.A.; Moiraghi, B.; Molinas, F.C.; Marta, R.F.; Schattner, M.; Heller, P.G. Neutrophil extracellular trap formation and circulating nucleosomes in patients with chronic myeloproliferative neoplasms. Sci. Rep. 2016, 6, 38738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guy, A.; Favre, S.; Labrouche-Colomer, S.; Deloison, L.; Gourdou-Latyszenok, V.; Renault, M.A.; Riviere, E.; James, C. High circulating levels of MPO-DNA are associated with thrombosis in patients with MPN. Leukemia 2019, 33, 2544–2548. [Google Scholar] [CrossRef] [PubMed]
- Slezak, S.; Jin, P.; Caruccio, L.; Ren, J.; Bennett, M.; Zia, N.; Adams, S.; Wang, E.; Ascensao, J.; Schechter, G.; et al. Gene and microRNA analysis of neutrophils from patients with polycythemia vera and essential thrombocytosis: Down-regulation of micro RNA-1 and -133a. J. Transl. Med. 2009, 7, 39. [Google Scholar] [CrossRef] [Green Version]
- Landolfi, R.; Marchioli, R.; Kutti, J.; Gisslinger, H.; Tognoni, G.; Patrono, C.; Barbui, T.; European Collaboration on Low-Dose Aspirin in Polycythemia Vera Investigators. Efficacy and safety of low-dose aspirin in polycythemia vera. N. Engl. J. Med. 2004, 350, 114–124. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Larrán, A.; Cervantes, F.; Pereira, A.; Arellano-Rodrigo, E.; Pérez-Andreu, V.; Hernández-Boluda, J.C.; Ayats, R.; Salvador, C.; Muntañola, A.; Bellosillo, B.; et al. Observation versus antiplatelet therapy as primary prophylaxis for thrombosis in low-risk essential thrombocythemia. Blood 2010, 116, 1205–1210. [Google Scholar] [CrossRef] [Green Version]
- Marchioli, R.; Finazzi, G.; Specchia, G.; Cacciola, R.; Cavazzina, R.; Cilloni, D.; De Stefano, V.; Elli, E.; Iurlo, A.; Latagliata, R.; et al. Cardiovascular events and intensity of treatment in polycythemia vera. N. Engl. J. Med. 2013, 368, 22–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Trillos, A.; Gaya, A.; Maffioli, M.; Arellano-Rodrigo, E.; Calvo, X.; Díaz-Beyá, M.; Cervantes, F. Efficacy and tolerability of hydroxyurea in the treatment of the hyperproliferative manifestations of myelofibrosis: Results in 40 patients. Ann. Hematol. 2010, 89, 1233–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, C.N.; Campbell, P.J.; Buck, G.; Wheatley, K.; East, C.L.; Bareford, D.; Wilkins, B.S.; van der Walt, J.D.; Reilly, J.T.; Grigg, A.P.; et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N. Engl. J. Med. 2005, 353, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Gisslinger, H.; Gotic, M.; Holowiecki, J.; Penka, M.; Thiele, J.; Kvasnicka, H.M.; Kralovics, R.; Petrides, P.E.; Group, A.S. Anagrelide compared with hydroxyurea in WHO-classified essential thrombocythemia: The ANAHYDRET Study, a randomized controlled trial. Blood 2013, 121, 1720–1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiladjian, J.J.; Cassinat, B.; Chevret, S.; Turlure, P.; Cambier, N.; Roussel, M.; Bellucci, S.; Grandchamp, B.; Chomienne, C.; Fenaux, P. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood 2008, 112, 3065–3072. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Kantarjian, H.; Manshouri, T.; Luthra, R.; Estrov, Z.; Pierce, S.; Richie, M.A.; Borthakur, G.; Konopleva, M.; Cortes, J.; et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J. Clin. Oncol. 2009, 27, 5418–5424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- How, J.; Hobbs, G. Use of interferon alfa in the treatment of myeloproliferative neoplasms: Perspectives and review of the literature. Cancers 2020, 12, 1954. [Google Scholar] [CrossRef]
- Wagner, S.M.; Melchardt, T.; Greil, R. Ropeginterferon alfa-2b for the treatment of patients with polycythemia vera. Drugs Today 2020, 56, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Gisslinger, H.; Klade, C.; Georgiev, P.; Krochmalczyk, D.; Gercheva-Kyuchukova, L.; Egyed, M.; Rossiev, V.; Dulicek, P.; Illes, A.; Pylypenko, H.; et al. Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): A randomised, non-inferiority, phase 3 trial and its extension study. Lancet Haematol. 2020, 7, e196–e208. [Google Scholar] [CrossRef]
- Barbui, T.; Vannucchi, A.M.; De Stefano, V.; Masciulli, A.; Carobbio, A.; Ferrari, A.; Ghirardi, A.; Rossi, E.; Ciceri, F.; Bonifacio, M.; et al. Ropeginterferon alfa-2b versus phlebotomy in low-risk patients with polycythaemia vera (Low-PV study): A multicentre, randomised phase 2 trial. Lancet Haematol. 2021, 8, e175–e184. [Google Scholar] [CrossRef]
- Bose, P.; Verstovsek, S. JAK2 inhibitors for myeloproliferative neoplasms: What is next? Blood 2017, 130, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef] [Green Version]
- Vannucchi, A.M.; Kiladjian, J.J.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Mesa, R.; et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N. Engl. J. Med. 2015, 372, 426–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passamonti, F.; Griesshammer, M.; Palandri, F.; Egyed, M.; Benevolo, G.; Devos, T.; Callum, J.; Vannucchi, A.M.; Sivgin, S.; Bensasson, C.; et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): A randomised, open-label, phase 3b study. Lancet Oncol. 2017, 18, 88–99. [Google Scholar] [CrossRef]
- Cervantes, F.; Vannucchi, A.M.; Kiladjian, J.J.; Al-Ali, H.K.; Sirulnik, A.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.S.; Passamonti, F.; et al. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood 2013, 122, 4047–4053. [Google Scholar] [CrossRef] [Green Version]
- Harrison, C.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Harrison, C.N.; Schaap, N.; Vannucchi, A.M.; Kiladjian, J.J.; Tiu, R.V.; Zachee, P.; Jourdan, E.; Winton, E.; Silver, R.T.; Schouten, H.C.; et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): A single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. 2017, 4, e317–e324. [Google Scholar] [CrossRef]
- Harrison, C.N.; Schaap, N.; Vannucchi, A.M.; Kiladjian, J.J.; Jourdan, E.; Silver, R.T.; Schouten, H.C.; Passamonti, F.; Zweegman, S.; Talpaz, M.; et al. Fedratinib in patients with myelofibrosis previously treated with ruxolitinib: An updated analysis of the JAKARTA2 study using stringent criteria for ruxolitinib failure. Am. J. Hematol. 2020, 95, 594–603. [Google Scholar] [CrossRef]
- Zhou, T.; Georgeon, S.; Moser, R.; Moore, D.J.; Caflisch, A.; Hantschel, O. Specificity and mechanism-of-action of the JAK2 tyrosine kinase inhibitors ruxolitinib and SAR302503 (TG101348). Leukemia 2014, 28, 404–407. [Google Scholar] [CrossRef]
- Mesa, R.A.; Vannucchi, A.M.; Mead, A.; Egyed, M.; Szoke, A.; Suvorov, A.; Jakucs, J.; Perkins, A.; Prasad, R.; Mayer, J.; et al. Pacritinib versus best available therapy for the treatment of myelofibrosis irrespective of baseline cytopenias (PERSIST-1): An international, randomised, phase 3 trial. Lancet Haematol. 2017, 4, e225–e236. [Google Scholar] [CrossRef]
- Tremblay, D.; Mesa, R.; Scott, B.; Buckley, S.; Roman-Torres, K.; Verstovsek, S.; Mascarenhas, J. Pacritinib demonstrates spleen volume reduction in patients with myelofibrosis independent of JAK2V617F allele burden. Blood Adv. 2020, 4, 5929–5935. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Hoffman, R.; Talpaz, M.; Gerds, A.T.; Stein, B.; Gupta, V.; Szoke, A.; Drummond, M.; Pristupa, A.; Granston, T.; et al. Pacritinib vs. best available therapy, including ruxolitinib, in patients with myelofibrosis: A randomized clinical trial. JAMA Oncol. 2018, 4, 652–659. [Google Scholar] [CrossRef]
- Mesa, R.A.; Kiladjian, J.J.; Catalano, J.V.; Devos, T.; Egyed, M.; Hellmann, A.; McLornan, D.; Shimoda, K.; Winton, E.F.; Deng, W.; et al. SIMPLIFY-1: A phase III randomized trial of momelotinib versus ruxolitinib in janus kinase inhibitor-naïve patients with myelofibrosis. J. Clin. Oncol. 2017, 35, 3844–3850. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.N.; Vannucchi, A.M.; Platzbecker, U.; Cervantes, F.; Gupta, V.; Lavie, D.; Passamonti, F.; Winton, E.F.; Dong, H.; Kawashima, J.; et al. Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): A randomised, open-label, phase 3 trial. Lancet Haematol. 2018, 5, e73–e81. [Google Scholar] [CrossRef]
- Greenfield, G.; McPherson, S.; Mills, K.; McMullin, M.F. The ruxolitinib effect: Understanding how molecular pathogenesis and epigenetic dysregulation impact therapeutic efficacy in myeloproliferative neoplasms. J. Transl. Med. 2018, 16, 360. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Wu, L.; Wang, X.; Jin, Z.; Gao, Z.; Wang, Z. Ruxolitinib for refractory/relapsed hemophagocytic lymphohistiocytosis. Haematologica 2020, 105, e210–e212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeiser, R.; von Bubnoff, N.; Butler, J.; Mohty, M.; Niederwieser, D.; Or, R.; Szer, J.; Wagner, E.M.; Zuckerman, T.; Mahuzier, B.; et al. Ruxolitinib for glucocorticoid-refractory acute graft-versus-host disease. N. Engl. J. Med. 2020, 382, 1800–1810. [Google Scholar] [CrossRef] [PubMed]
- Jagasia, M.; Perales, M.A.; Schroeder, M.A.; Ali, H.; Shah, N.N.; Chen, Y.B.; Fazal, S.; Dawkins, F.W.; Arbushites, M.C.; Tian, C.; et al. Ruxolitinib for the treatment of steroid-refractory acute GVHD (REACH1): A multicenter, open-label phase 2 trial. Blood 2020, 135, 1739–1749. [Google Scholar] [CrossRef]


{kind=link}
| Inflammatory Cell/Organelle/Protein | Role |
|---|---|
| Bone marrow stromal cells [52] | Expansion of JAK2V617F mutant HSC |
| Inflammasome [53] | Inflammation aiding disease progression |
| TNF-alpha [54] | Expansion of JAK2V617F mutant HSC and suppression of non-mutant HSC |
| IL-33 [55] | Prolonged survival of JAK2V617F-positive cell lines |
| IL-11, HGF [56] | Necessary for expansion of erythroid precursors harboring JAK2V617F |
| IL-6, CXCL10, FGF [57] | Protection of malignant clones against JAK2 inhibition |
| NF-E2 [60,63] | Leuko- and thrombocytosis, increases IL-8 |
| IL-8 [65,66,67] 0.75 | Alteration of megakaryopoiesis, neutrophilia, erythrocytosis |
| CRP [70,71,72] | Correlation with JAK2V617F allele burden in PV, ET, PMFPrognostic marker in PMF |
| IL-8, IL-2R, IL-12, IL-15 [69] | Prognostic markers in PMF |
| YKL-40 [48,73,74] | Correlated with disease progression, transition to post-PV MF, neutrophilia, thrombocytosis, CRP, LDH, JAK2V617 allele burden |
| Neutrophil gelatinase-associated lipocalin (NGAL) [75,76,77] | Correlation with neutrophilia in PV, ET, MF |
| Major Mechanism | Area of Involvement |
|---|---|
| Increased endothelial adhesion and accelerated atherosclerosis [82,86] | Arterial thrombosis |
| Increased NET formation [99] | Thrombosis |
| Aggregation of thromboyctes with neutrophils [82] | Thrombosis |
| Emperipolesis [94,96] | Bone marrow fibrosis, thrombocytosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiem, D.; Wagner, S.; Magnes, T.; Egle, A.; Greil, R.; Melchardt, T. The Role of Neutrophilic Granulocytes in Philadelphia Chromosome Negative Myeloproliferative Neoplasms. Int. J. Mol. Sci. 2021, 22, 9555. https://doi.org/10.3390/ijms22179555
Kiem D, Wagner S, Magnes T, Egle A, Greil R, Melchardt T. The Role of Neutrophilic Granulocytes in Philadelphia Chromosome Negative Myeloproliferative Neoplasms. International Journal of Molecular Sciences. 2021; 22(17):9555. https://doi.org/10.3390/ijms22179555
Chicago/Turabian StyleKiem, Dominik, Sandro Wagner, Teresa Magnes, Alexander Egle, Richard Greil, and Thomas Melchardt. 2021. "The Role of Neutrophilic Granulocytes in Philadelphia Chromosome Negative Myeloproliferative Neoplasms" International Journal of Molecular Sciences 22, no. 17: 9555. https://doi.org/10.3390/ijms22179555
APA StyleKiem, D., Wagner, S., Magnes, T., Egle, A., Greil, R., & Melchardt, T. (2021). The Role of Neutrophilic Granulocytes in Philadelphia Chromosome Negative Myeloproliferative Neoplasms. International Journal of Molecular Sciences, 22(17), 9555. https://doi.org/10.3390/ijms22179555