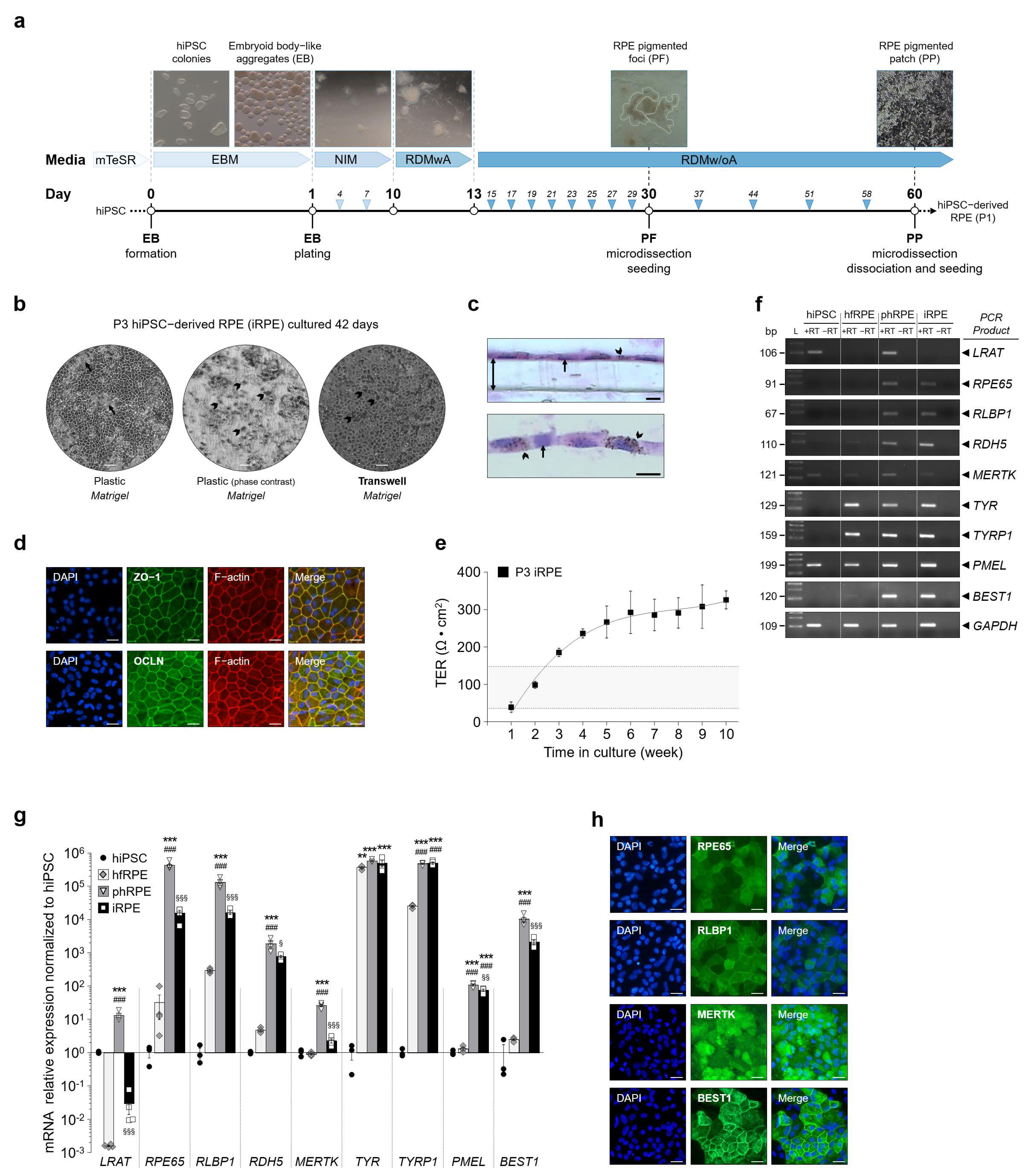
Figure 1.
Human induced pluripotent stem cell (hiPSC)-derived RPE cells (iRPE) express RPE characteristic marker genes and exhibit transepithelial resistance. (a) Protocol used to expand and differentiate hiPSC into human iRPE cells in 60 days. RPE pigmented foci are marked by white dashed lines. (b) Monolayer of pigmented iRPE cells at passage 3 (arrowhead). When grown on plastic, iRPE cells form dome-like cellular structures across cell culture (arrow). (c) Transversal sections of pigmented iRPE cells cultured on transwell (double arrow). Pigment granules (arrowhead). Scale bar: 20 µm. (d) Immunofluorescence of endogenous TJP1 (encoding ZO-1), OCLN and F-actin proteins in iRPE cells. Scale bar = 30 µm. (e) Weekly measurements of transepithelial resistance (TER) in iRPE cells cultured. TER is measured with an EVOM2 epithelial voltohmmeter and calculated as net Ω cm2. Gray zone between the two dashed lines corresponds to proposed functional TER values measured in native human adult RPE monolayers. Values are means ± SEM, n = 5–12 per time-point measurements. (f) Analysis by qualitative RT-PCR of the expression profile of specific RPE genes involved in the visual cycle (LRAT, RPE65, RLBP1 and RDH5), phagocytosis (MERTK), pigment synthesis (TYR, TYRP1 and PMEL) and ion channel transport (BEST1). The housekeeping gene GAPDH served as a control for RNA integrity. (g) Quantification by RT-qPCR of specific RPE marker mRNA transcript expression relative to GAPDH in hfRPE (n = 4), phRPE (n = 4) and iRPE (n = 4) normalized to undifferentiated hiPSC (n = 3). Data are represented in a Log10 scale. Values are means ± SEM. ** p < 0.01, *** p < 0.001, hfRPE, phRPE and iRPE versus hiPSC, ### p < 0.001, phRPE and iRPE versus hfRPE, § p < 0.05, §§ p < 0.01, §§§ p < 0.001, iRPE versus phRPE. (h) Representative analysis by immunofluorescence of endogenous RPE65, RLBP1, MERTK and BEST1 protein expression in iRPE cells. RPE65, RLBP1, MERTK and BEST1 staining is in green, and DAPI staining is in blue. Scale bar = 30 µm. mTeSR, feeder-free maintenance medium for human ES and iPS cells; EBM, embryoid-body-like aggregate media; NIM, neural induction media; RDMwA, RPE differentiation media with vitamin A; RDMw/oA, RPE differentiation media without vitamin A; P, passage; ZO-1, zonula occludens-1; OCLN, occluding; DAPI, 4′,6-diamidino-2-phenylindole; hfRPE, human fetal RPE; phRPE, post-mortem human RPE tissue; bp, base pair; L, ladder; +RT, presence of reverse transcriptase; −RT, absence of reverse transcriptase; LRAT, lecithin retinol acyltransferase; RPE65, retinoid isomerohydrolase; RLBP1, retinaldehyde-binding protein 1; RDH5, retinol dehydrogenase 5; MERTK, MER proto-oncogene tyrosine kinase; TYR, tyrosinase; TYRP1, tyrosinase-related protein 1; PMEL, premelanosome protein; BEST1, bestrophin 1; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Figure 1.
Human induced pluripotent stem cell (hiPSC)-derived RPE cells (iRPE) express RPE characteristic marker genes and exhibit transepithelial resistance. (a) Protocol used to expand and differentiate hiPSC into human iRPE cells in 60 days. RPE pigmented foci are marked by white dashed lines. (b) Monolayer of pigmented iRPE cells at passage 3 (arrowhead). When grown on plastic, iRPE cells form dome-like cellular structures across cell culture (arrow). (c) Transversal sections of pigmented iRPE cells cultured on transwell (double arrow). Pigment granules (arrowhead). Scale bar: 20 µm. (d) Immunofluorescence of endogenous TJP1 (encoding ZO-1), OCLN and F-actin proteins in iRPE cells. Scale bar = 30 µm. (e) Weekly measurements of transepithelial resistance (TER) in iRPE cells cultured. TER is measured with an EVOM2 epithelial voltohmmeter and calculated as net Ω cm2. Gray zone between the two dashed lines corresponds to proposed functional TER values measured in native human adult RPE monolayers. Values are means ± SEM, n = 5–12 per time-point measurements. (f) Analysis by qualitative RT-PCR of the expression profile of specific RPE genes involved in the visual cycle (LRAT, RPE65, RLBP1 and RDH5), phagocytosis (MERTK), pigment synthesis (TYR, TYRP1 and PMEL) and ion channel transport (BEST1). The housekeeping gene GAPDH served as a control for RNA integrity. (g) Quantification by RT-qPCR of specific RPE marker mRNA transcript expression relative to GAPDH in hfRPE (n = 4), phRPE (n = 4) and iRPE (n = 4) normalized to undifferentiated hiPSC (n = 3). Data are represented in a Log10 scale. Values are means ± SEM. ** p < 0.01, *** p < 0.001, hfRPE, phRPE and iRPE versus hiPSC, ### p < 0.001, phRPE and iRPE versus hfRPE, § p < 0.05, §§ p < 0.01, §§§ p < 0.001, iRPE versus phRPE. (h) Representative analysis by immunofluorescence of endogenous RPE65, RLBP1, MERTK and BEST1 protein expression in iRPE cells. RPE65, RLBP1, MERTK and BEST1 staining is in green, and DAPI staining is in blue. Scale bar = 30 µm. mTeSR, feeder-free maintenance medium for human ES and iPS cells; EBM, embryoid-body-like aggregate media; NIM, neural induction media; RDMwA, RPE differentiation media with vitamin A; RDMw/oA, RPE differentiation media without vitamin A; P, passage; ZO-1, zonula occludens-1; OCLN, occluding; DAPI, 4′,6-diamidino-2-phenylindole; hfRPE, human fetal RPE; phRPE, post-mortem human RPE tissue; bp, base pair; L, ladder; +RT, presence of reverse transcriptase; −RT, absence of reverse transcriptase; LRAT, lecithin retinol acyltransferase; RPE65, retinoid isomerohydrolase; RLBP1, retinaldehyde-binding protein 1; RDH5, retinol dehydrogenase 5; MERTK, MER proto-oncogene tyrosine kinase; TYR, tyrosinase; TYRP1, tyrosinase-related protein 1; PMEL, premelanosome protein; BEST1, bestrophin 1; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
![Ijms 22 09618 g001]()
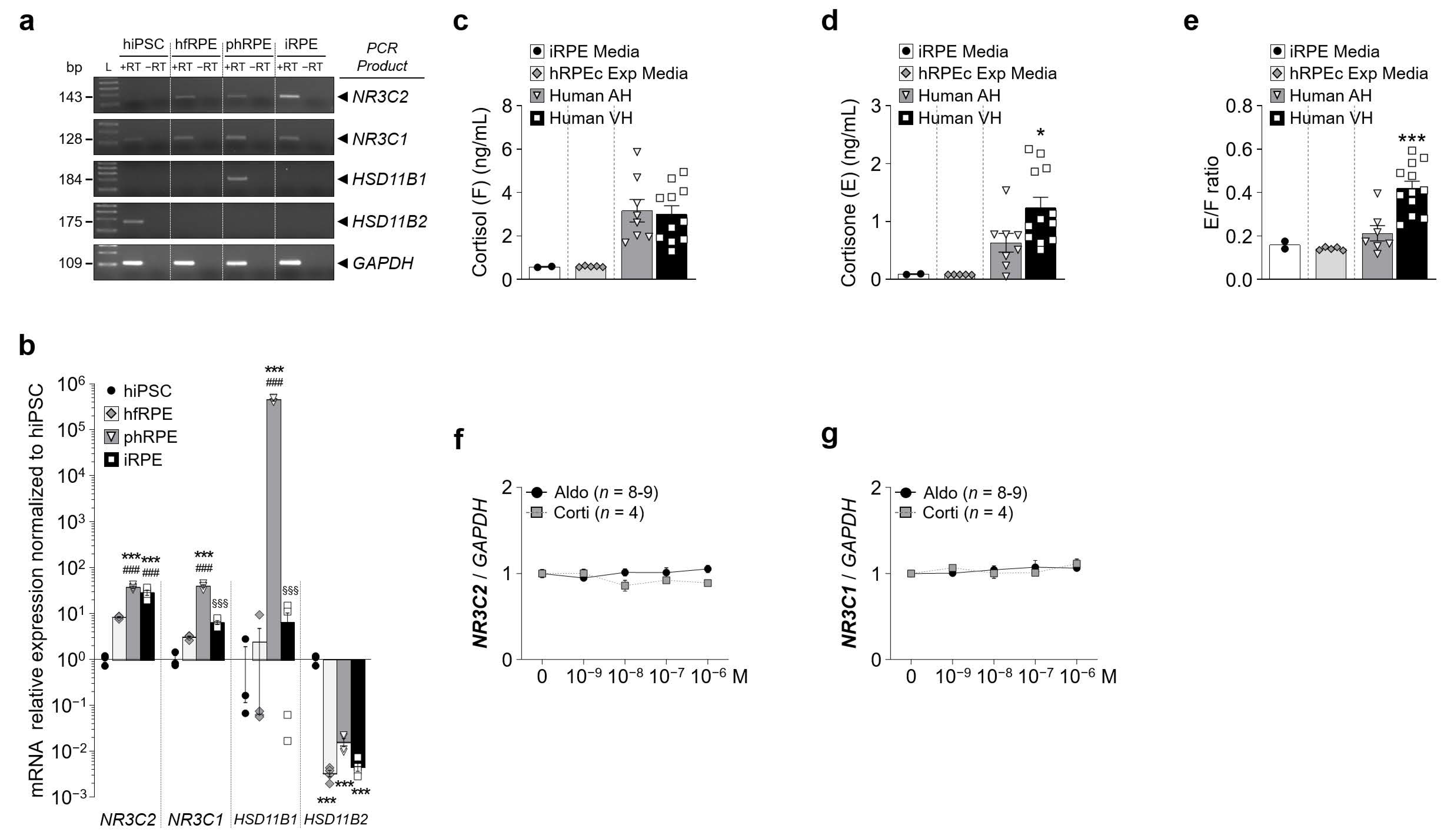
Figure 2.
Mineralocorticoid and glucocorticoid receptors are expressed in iRPE cells. (a) Analysis by qualitative RT-PCR of the mineralocorticoid receptor (MR) encoded by NR3C2 and the glucocorticoid receptor (GR) encoded by NR3C1, HSD11B1 and HSD11B2 mRNA expression in iRPE cells. The housekeeping gene GAPDH served as a control for RNA integrity. (b) Quantification by RT-qPCR of expression of NR3C2, NR3C1, HSD11B1 and HSD11B2 relative to GAPDH in hfRPE (n = 4), phRPE (n = 4) and iRPE (n = 4) normalized to undifferentiated hiPSC (n = 3). Data are represented in a Log10 scale. Values are means ± SEM. *** p < 0.001, hfRPE, phRPE and iRPE versus hiPSC, ### p < 0.001, phRPE and iRPE versus hfRPE, §§§ p < 0.001, iRPE versus phRPE. (c–e) Cortisol (c) and cortisone (d) levels and cortisone/cortisol ratio (e) measured in iRPE apical cell culture media (n = 2), human RPE/choroid explant cell culture media (n = 5), human aqueous humor (AH) (n = 8) and human vitreous humor (VH) (n = 12). Values are means ± SEM. Unpaired t-test with Welch’s correction. ** p < 0.01, *** p < 0.001, Human VH versus Human AH. (f) and (g) Dose-dependency expression of NR3C2 (f) and NR3C1 (g) relative to GAPDH following aldosterone or cortisol treatment (10−9–10−6 M) in iRPE cells. Untreated cells’ mRNA expression was arbitrarily set to 1. Values are means ± SEM.
Figure 2.
Mineralocorticoid and glucocorticoid receptors are expressed in iRPE cells. (a) Analysis by qualitative RT-PCR of the mineralocorticoid receptor (MR) encoded by NR3C2 and the glucocorticoid receptor (GR) encoded by NR3C1, HSD11B1 and HSD11B2 mRNA expression in iRPE cells. The housekeeping gene GAPDH served as a control for RNA integrity. (b) Quantification by RT-qPCR of expression of NR3C2, NR3C1, HSD11B1 and HSD11B2 relative to GAPDH in hfRPE (n = 4), phRPE (n = 4) and iRPE (n = 4) normalized to undifferentiated hiPSC (n = 3). Data are represented in a Log10 scale. Values are means ± SEM. *** p < 0.001, hfRPE, phRPE and iRPE versus hiPSC, ### p < 0.001, phRPE and iRPE versus hfRPE, §§§ p < 0.001, iRPE versus phRPE. (c–e) Cortisol (c) and cortisone (d) levels and cortisone/cortisol ratio (e) measured in iRPE apical cell culture media (n = 2), human RPE/choroid explant cell culture media (n = 5), human aqueous humor (AH) (n = 8) and human vitreous humor (VH) (n = 12). Values are means ± SEM. Unpaired t-test with Welch’s correction. ** p < 0.01, *** p < 0.001, Human VH versus Human AH. (f) and (g) Dose-dependency expression of NR3C2 (f) and NR3C1 (g) relative to GAPDH following aldosterone or cortisol treatment (10−9–10−6 M) in iRPE cells. Untreated cells’ mRNA expression was arbitrarily set to 1. Values are means ± SEM.
![Ijms 22 09618 g002]()
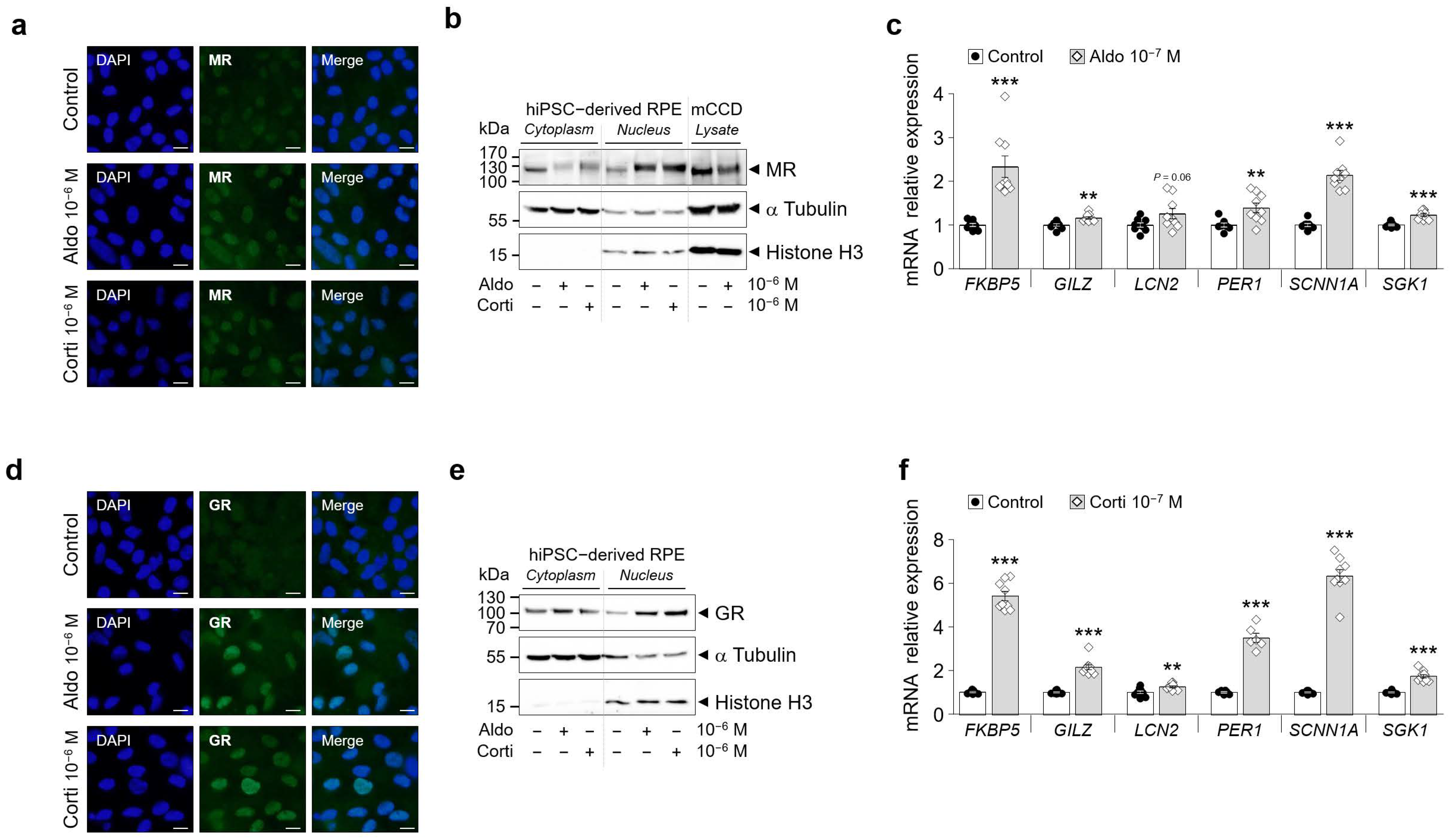
Figure 3.
GR and MR are functional in hiPSC-derived RPE cells (iRPR). (a) and (d) Representative analysis by immunofluorescence of endogenous MR (a) and GR (b) protein expression following aldosterone (aldo) or cortisol (corti) 1h treatment (10−6 M) in iRPE cells. MR and GR staining is in green, and DAPI staining is in blue. Scale bar: 5 µm. (b) and (e) Analysis by Western blot of endogenous MR (b) and GR (e) protein expression following aldosterone or cortisol 1h treatment (10−6 M) in iRPE cells cytoplasmic or nuclear fractions. As a positive control for MR protein expression, mouse cortical collecting duct cells (mCCD) with known functional MR expression were treated for 1 h with 50 nM aldosterone. α-Tubulin and Histone H3 were used as loading controls for the cytoplasmic and nuclear fractions, respectively. Arrows indicate the expected size of the 107 kDa MR, 94 kDa GR, 50 kDa α-Tubulin and 15 kDa Histone H3 proteins. (c) and (f) Analysis by RT-qPCR in iRPE cells following aldosterone (c) or cortisol (f) 24 h treatment (10−7 M) of the expression profile of genes (FKBP5, GILZ, LCN2, PER1, SCNN1A, SGK1) known to be up-regulated by corticosteroids. mRNA transcript expression was relative to the expression of the housekeeping gene GAPDH. n = 6–9 per experimental condition. Values are means ± SEM. ** p < 0.01, *** p < 0.001.
Figure 3.
GR and MR are functional in hiPSC-derived RPE cells (iRPR). (a) and (d) Representative analysis by immunofluorescence of endogenous MR (a) and GR (b) protein expression following aldosterone (aldo) or cortisol (corti) 1h treatment (10−6 M) in iRPE cells. MR and GR staining is in green, and DAPI staining is in blue. Scale bar: 5 µm. (b) and (e) Analysis by Western blot of endogenous MR (b) and GR (e) protein expression following aldosterone or cortisol 1h treatment (10−6 M) in iRPE cells cytoplasmic or nuclear fractions. As a positive control for MR protein expression, mouse cortical collecting duct cells (mCCD) with known functional MR expression were treated for 1 h with 50 nM aldosterone. α-Tubulin and Histone H3 were used as loading controls for the cytoplasmic and nuclear fractions, respectively. Arrows indicate the expected size of the 107 kDa MR, 94 kDa GR, 50 kDa α-Tubulin and 15 kDa Histone H3 proteins. (c) and (f) Analysis by RT-qPCR in iRPE cells following aldosterone (c) or cortisol (f) 24 h treatment (10−7 M) of the expression profile of genes (FKBP5, GILZ, LCN2, PER1, SCNN1A, SGK1) known to be up-regulated by corticosteroids. mRNA transcript expression was relative to the expression of the housekeeping gene GAPDH. n = 6–9 per experimental condition. Values are means ± SEM. ** p < 0.01, *** p < 0.001.
![Ijms 22 09618 g003]()
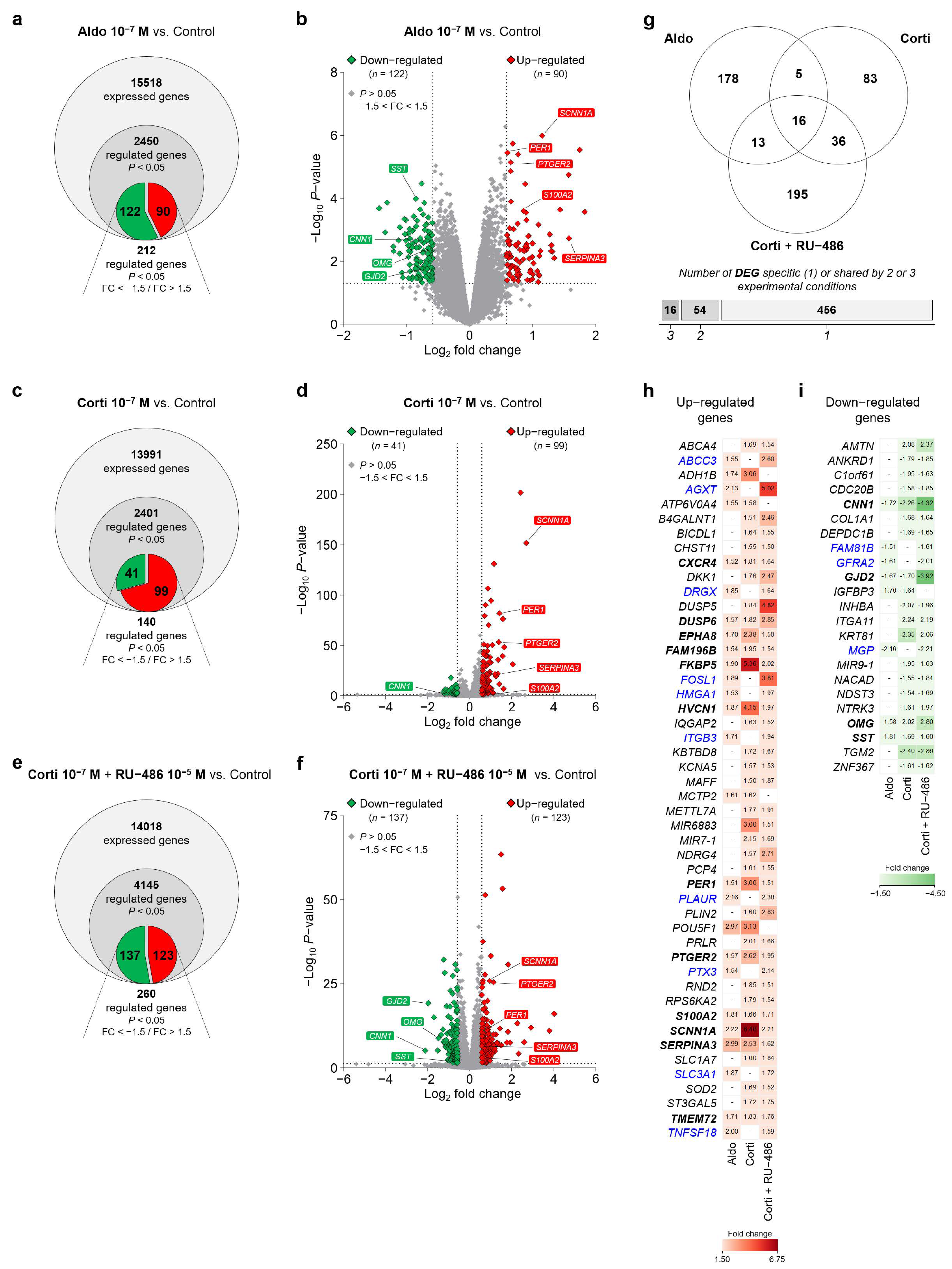
Figure 4.
RNA-Seq transcriptome analysis identified corticosteroid-regulated genes in iRPE cells. (a), (c) and (e) Venn diagram representing total expressed genes identified by the RNA-Seq transcriptome analysis, significantly regulated genes (p < 0.05) and significantly corticosteroid-regulated genes as predicted by the RNA-Seq data analysis with fold change (FC) values lower than −1.5 (down-regulated genes in green) and higher than 1.5 (up-regulated genes in red) (p < 0.05; FC < −1.5/FC > 1.5) in iRPE cells following aldosterone (aldo) 10−7 M (a), cortisol (corti) 10−7 M (c) or cortisol 10−7 M + RU-486 10−5 M (e) 24 h treatment. Corticosteroid-treated iRPE cells are compared to untreated cells. (b), (d) and (f) Volcano plots highlighting differentially expressed genes (DEGs) in aldosterone- (b), cortisol- (d) or cortisol + RU-486-treated (f) iRPE cells versus untreated cells. p-value (represented in a −Log10 scale) and fold-change (represented in a Log2 scale) cut-off is 0.05 (horizontal dotted line) and 1.5 (vertical dotted lines), respectively. Each up- and down-regulated expressed transcript is indicated in red and green diamonds, respectively. Representative corticosteroid-regulated DEGs of interest with comparable regulation in all three experimental treatments are shown in red (up-regulated) and green (down-regulated) rectangles. (g) Number of differentially expressed genes in corticosteroid-treated iRPE cells specific to one or shared by 2 or 3 of the following aldosterone (10−7 M), cortisol (10−7 M) and cortisol (10−7 M) + RU-486 (10−5 M) experimental treatments. (h) and (i) Lists of up-regulated (h) and down-regulated (i) genes identified by RNA-Seq data analysis in iRPE cells with a comparable differential expression in 2 or 3 experimental conditions. Fold-change values for each gene and specific to each experimental treatment are indicated in the tables. Genes regulated by corticosteroids in a similar manner in all three experimental treatments are in bold type. Genes regulated in a similar manner in the aldosterone 10−7 M and cortisol 10−7 M + RU-486 10−5 M experimental conditions in which aldosterone and cortisol agonists should mostly bind to and activate MR are in blue. SST, somatostatin; CNN1, calponin 1; OMG, oligodendrocyte myelin glycoprotein; GJD2, gap junction protein delta-2; SCNN1A, sodium channel epithelial 1 alpha subunit; PER1, period circadian clock 1; PTGER2, prostaglandin E receptor 2; S100A2, S100 calcium-binding protein A2; SERPINA3, serpin family A member 3.
Figure 4.
RNA-Seq transcriptome analysis identified corticosteroid-regulated genes in iRPE cells. (a), (c) and (e) Venn diagram representing total expressed genes identified by the RNA-Seq transcriptome analysis, significantly regulated genes (p < 0.05) and significantly corticosteroid-regulated genes as predicted by the RNA-Seq data analysis with fold change (FC) values lower than −1.5 (down-regulated genes in green) and higher than 1.5 (up-regulated genes in red) (p < 0.05; FC < −1.5/FC > 1.5) in iRPE cells following aldosterone (aldo) 10−7 M (a), cortisol (corti) 10−7 M (c) or cortisol 10−7 M + RU-486 10−5 M (e) 24 h treatment. Corticosteroid-treated iRPE cells are compared to untreated cells. (b), (d) and (f) Volcano plots highlighting differentially expressed genes (DEGs) in aldosterone- (b), cortisol- (d) or cortisol + RU-486-treated (f) iRPE cells versus untreated cells. p-value (represented in a −Log10 scale) and fold-change (represented in a Log2 scale) cut-off is 0.05 (horizontal dotted line) and 1.5 (vertical dotted lines), respectively. Each up- and down-regulated expressed transcript is indicated in red and green diamonds, respectively. Representative corticosteroid-regulated DEGs of interest with comparable regulation in all three experimental treatments are shown in red (up-regulated) and green (down-regulated) rectangles. (g) Number of differentially expressed genes in corticosteroid-treated iRPE cells specific to one or shared by 2 or 3 of the following aldosterone (10−7 M), cortisol (10−7 M) and cortisol (10−7 M) + RU-486 (10−5 M) experimental treatments. (h) and (i) Lists of up-regulated (h) and down-regulated (i) genes identified by RNA-Seq data analysis in iRPE cells with a comparable differential expression in 2 or 3 experimental conditions. Fold-change values for each gene and specific to each experimental treatment are indicated in the tables. Genes regulated by corticosteroids in a similar manner in all three experimental treatments are in bold type. Genes regulated in a similar manner in the aldosterone 10−7 M and cortisol 10−7 M + RU-486 10−5 M experimental conditions in which aldosterone and cortisol agonists should mostly bind to and activate MR are in blue. SST, somatostatin; CNN1, calponin 1; OMG, oligodendrocyte myelin glycoprotein; GJD2, gap junction protein delta-2; SCNN1A, sodium channel epithelial 1 alpha subunit; PER1, period circadian clock 1; PTGER2, prostaglandin E receptor 2; S100A2, S100 calcium-binding protein A2; SERPINA3, serpin family A member 3.
![Ijms 22 09618 g004]()
Figure 5.
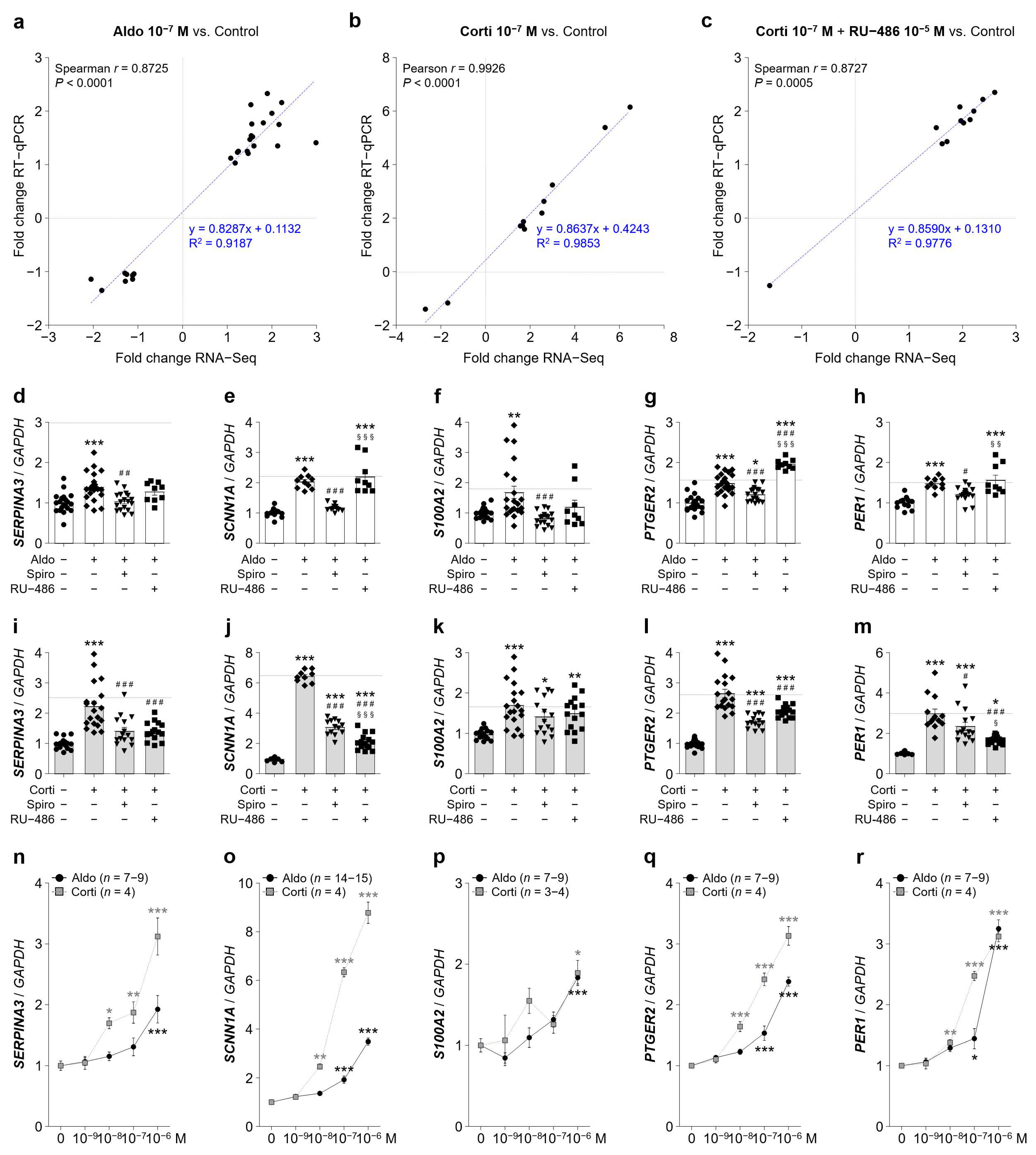
Aldosterone and cortisol up-regulated genes as predicted in RNA-Seq tested by RT-qPCR for validation. (a–c) Gene expression correlation analysis between RNA-Seq and RT-qPCR results generated in iRPE cells for the 3 experimental treatments aldosterone (aldo) 10−7 M (a), cortisol (corti) 10−7 M (b) and cortisol 10−7 M + RU-486 10−5 M (c). The Spearman’s rank correlation or Pearson’s correlation coefficient and the linear regression line (dashed blue line) are indicated. (d–h) Quantification by RT-qPCR of SERPINA3 (d), SCNN1A encoding α-ENaC (e), S100A2 (f), PTGER2 (g) and PER1 (h) mRNA expression relative to GAPDH in iRPE cells following 24 h treatment of aldosterone 10−7 M ± spironolactone (spiro) 10−5 M or RU-486 10−5 M. (i–m) Quantification by RT-qPCR of SERPINA3 (i), SCNN1A (j), S100A2 (k), PTGER2 (l) and PER1 (m) mRNA expression relative to GAPDH in iRPE cells following 24 h treatment of cortisol 10−7 M ± spironolactone 10−5 M or RU-486 10−5 M. Dashed lines represent relative gene expression as predicted by the RNA-Seq analysis. Values are means ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001, Aldo or Corti, Aldo or Corti + Spiro and Aldo or Corti + RU-486 versus untreated iRPE cells, # p < 0.05, ## p < 0.01, ### p < 0.001, Aldo or Corti + Spiro and Aldo or Corti + RU-486 versus Aldo or Corti, § p < 0.05, §§§ p < 0.001, Aldo or Corti + RU-486 versus Aldo or Corti + Spiro. (n–r) Dose-dependency expression of SERPINA3 (n), SCNN1A (o), S100A2 (p), PTGER2 (q) and PER1 (r) mRNA relative to GAPDH following 24 h treatment of aldosterone or cortisol treatment in iRPE cells (n = 9). Untreated cells’ mRNA expression was arbitrarily set to 1. Values are means ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001, relative values versus untreated condition. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; SERPINA3, serpin family A member 3; Spiro, spironolactone; SCNN1A, sodium channel epithelial 1 alpha subunit; S100A2, S100 calcium-binding protein A2; PTGER2, prostaglandin E receptor 2; PER1, period circadian clock 1.
Figure 5.
Aldosterone and cortisol up-regulated genes as predicted in RNA-Seq tested by RT-qPCR for validation. (a–c) Gene expression correlation analysis between RNA-Seq and RT-qPCR results generated in iRPE cells for the 3 experimental treatments aldosterone (aldo) 10−7 M (a), cortisol (corti) 10−7 M (b) and cortisol 10−7 M + RU-486 10−5 M (c). The Spearman’s rank correlation or Pearson’s correlation coefficient and the linear regression line (dashed blue line) are indicated. (d–h) Quantification by RT-qPCR of SERPINA3 (d), SCNN1A encoding α-ENaC (e), S100A2 (f), PTGER2 (g) and PER1 (h) mRNA expression relative to GAPDH in iRPE cells following 24 h treatment of aldosterone 10−7 M ± spironolactone (spiro) 10−5 M or RU-486 10−5 M. (i–m) Quantification by RT-qPCR of SERPINA3 (i), SCNN1A (j), S100A2 (k), PTGER2 (l) and PER1 (m) mRNA expression relative to GAPDH in iRPE cells following 24 h treatment of cortisol 10−7 M ± spironolactone 10−5 M or RU-486 10−5 M. Dashed lines represent relative gene expression as predicted by the RNA-Seq analysis. Values are means ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001, Aldo or Corti, Aldo or Corti + Spiro and Aldo or Corti + RU-486 versus untreated iRPE cells, # p < 0.05, ## p < 0.01, ### p < 0.001, Aldo or Corti + Spiro and Aldo or Corti + RU-486 versus Aldo or Corti, § p < 0.05, §§§ p < 0.001, Aldo or Corti + RU-486 versus Aldo or Corti + Spiro. (n–r) Dose-dependency expression of SERPINA3 (n), SCNN1A (o), S100A2 (p), PTGER2 (q) and PER1 (r) mRNA relative to GAPDH following 24 h treatment of aldosterone or cortisol treatment in iRPE cells (n = 9). Untreated cells’ mRNA expression was arbitrarily set to 1. Values are means ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001, relative values versus untreated condition. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; SERPINA3, serpin family A member 3; Spiro, spironolactone; SCNN1A, sodium channel epithelial 1 alpha subunit; S100A2, S100 calcium-binding protein A2; PTGER2, prostaglandin E receptor 2; PER1, period circadian clock 1.
![Ijms 22 09618 g005]()
Scheme 1.
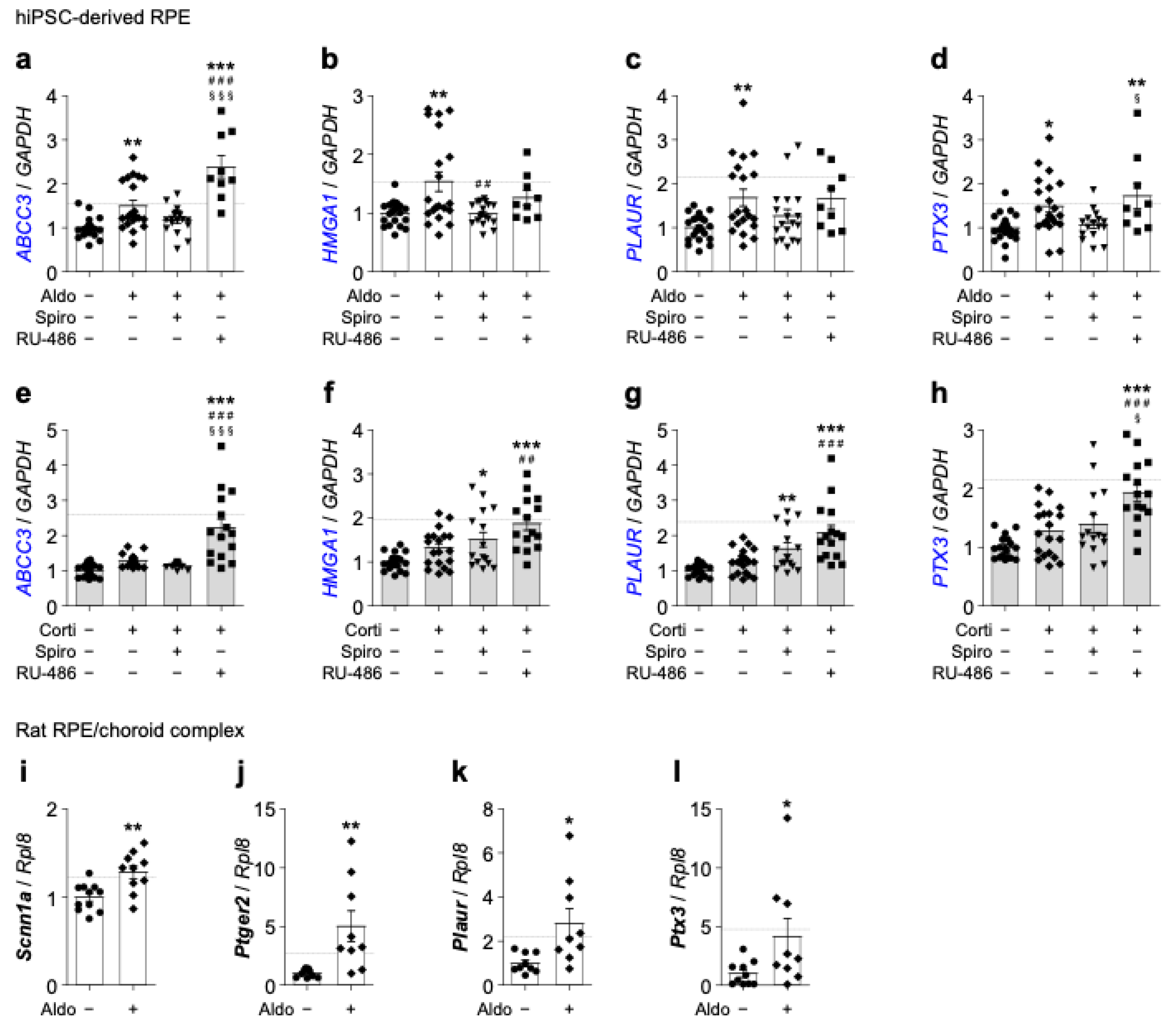
Additional corticosteroid up-regulated genes of interest as predicted in RNA-Seq tested by RT-qPCR for validation. (a–h) Quantification by RT-qPCR of ABCC3 (a) and (e), HMGA1 (b) and (f), PLAUR (c) and (g) and PTX3 (d) and (h) mRNA expression relative to GAPDH in iRPE cells following 24 h treatment of aldosterone (aldo) (10−7 M) (a–d) or cortisol (corti) (10−7 M) (e–h) ± spironolactone (spiro) (10−5 M) or RU-486 (10−5 M). Dashed lines represent relative gene expression as predicted by the RNA-Seq analysis for the experimental condition aldosterone (a–d) or cortisol + RU-486 (e–h). (i–l) Quantification by RT-qPCR of Scnn1a (i), Ptger2 (j), Plaur (k) and Ptx3 (l) expression relative to Rpl8 in Lewis rat RPE–choroid complexes isolated from intravitreally aldosterone-injected (10−7 M) and sham-injected eyes. Dashed lines represent relative gene expression as predicted by the RNA-Seq analysis for the experimental condition aldosterone. Values are means ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001, Aldo or Corti, Aldo or Corti + Spiro and Aldo or Corti + RU-486 versus untreated iRPE cells or rats RPE/choroid complexes, ## p < 0.01, ### p < 0.001, Aldo or Corti + Spiro and Aldo or Corti + RU-486 versus Aldo or Corti, § p < 0.05, §§§ p < 0.001, Aldo or Corti + RU-486 versus Aldo or Corti + Spiro. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; ABCC3, ATP-binding cassette subfamily C member 3; HMGA1, high-mobility group AT-hook 1; PLAUR and Plaur, urokinase plasminogen activator surface receptor; PTX3 and Ptx3, pentraxin 3; Scnn1a, sodium channel epithelial 1 alpha subunit; Ptger2, prostaglandin E receptor 2; Rpl8, ribosomal protein L8.
Scheme 1.
Additional corticosteroid up-regulated genes of interest as predicted in RNA-Seq tested by RT-qPCR for validation. (a–h) Quantification by RT-qPCR of ABCC3 (a) and (e), HMGA1 (b) and (f), PLAUR (c) and (g) and PTX3 (d) and (h) mRNA expression relative to GAPDH in iRPE cells following 24 h treatment of aldosterone (aldo) (10−7 M) (a–d) or cortisol (corti) (10−7 M) (e–h) ± spironolactone (spiro) (10−5 M) or RU-486 (10−5 M). Dashed lines represent relative gene expression as predicted by the RNA-Seq analysis for the experimental condition aldosterone (a–d) or cortisol + RU-486 (e–h). (i–l) Quantification by RT-qPCR of Scnn1a (i), Ptger2 (j), Plaur (k) and Ptx3 (l) expression relative to Rpl8 in Lewis rat RPE–choroid complexes isolated from intravitreally aldosterone-injected (10−7 M) and sham-injected eyes. Dashed lines represent relative gene expression as predicted by the RNA-Seq analysis for the experimental condition aldosterone. Values are means ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001, Aldo or Corti, Aldo or Corti + Spiro and Aldo or Corti + RU-486 versus untreated iRPE cells or rats RPE/choroid complexes, ## p < 0.01, ### p < 0.001, Aldo or Corti + Spiro and Aldo or Corti + RU-486 versus Aldo or Corti, § p < 0.05, §§§ p < 0.001, Aldo or Corti + RU-486 versus Aldo or Corti + Spiro. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; ABCC3, ATP-binding cassette subfamily C member 3; HMGA1, high-mobility group AT-hook 1; PLAUR and Plaur, urokinase plasminogen activator surface receptor; PTX3 and Ptx3, pentraxin 3; Scnn1a, sodium channel epithelial 1 alpha subunit; Ptger2, prostaglandin E receptor 2; Rpl8, ribosomal protein L8.
![Ijms 22 09618 sch001]()
Figure 6.
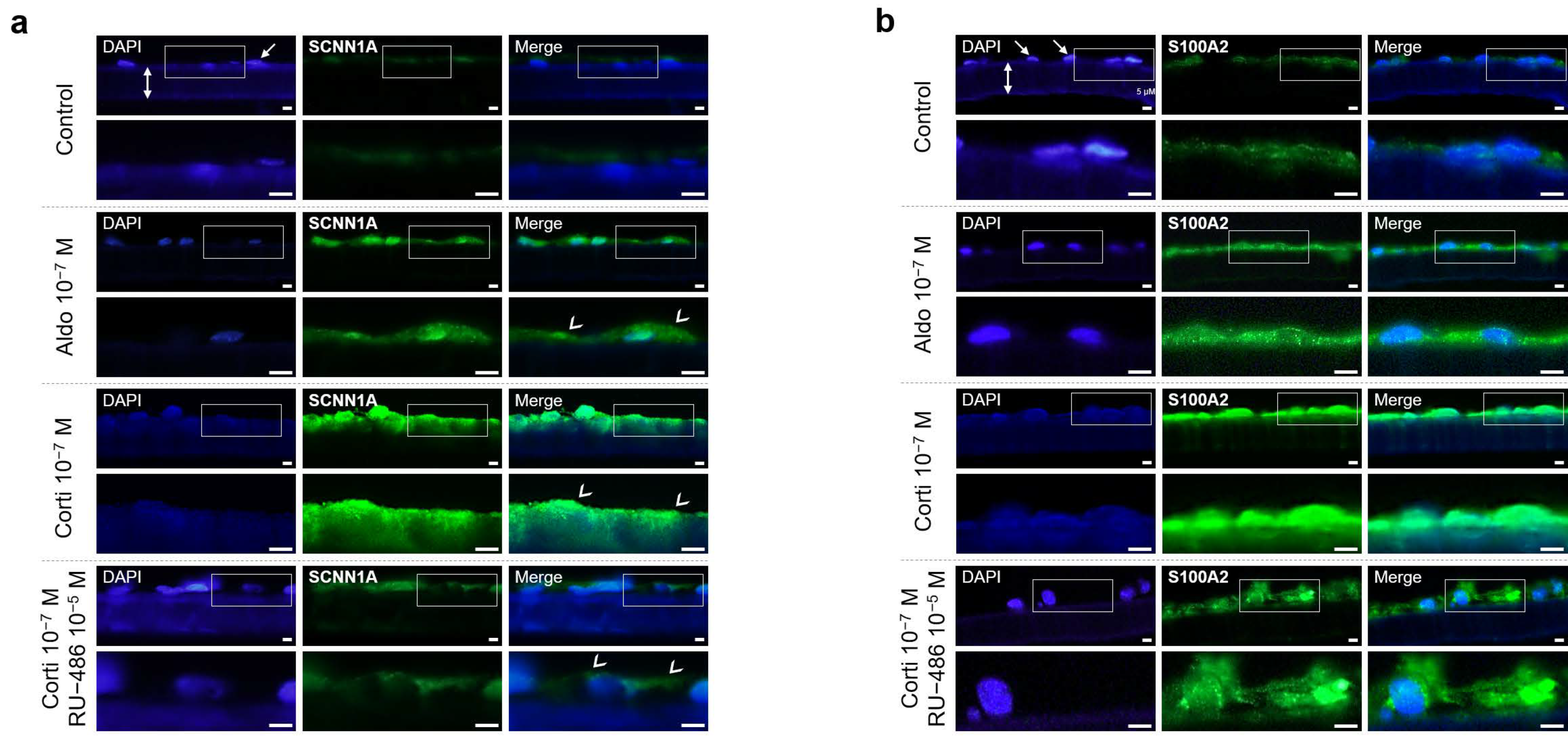
SCNN1A and S100A2 immunohistochemistry in iRPE cells. (a) and (b) Representative analysis by immunofluorescence of endogenous SCNN1A (a) and S100A2 (b) protein expression in iRPE cells cultured on transwell filters (double white arrows) following 24 h treatment of aldosterone (aldo) 10−7 M, cortisol (corti) 10−7 M or cortisol (10−7 M) ± RU-486 (10−5 M). SCNN1A and S100A2 staining is in green, and DAPI staining for cell nuclei (white arrows) is in blue, respectively. White arrowheads indicate apical staining (a). Scale bar: 30 µm. SCNN1A, sodium channel epithelial 1 α subunit; S100A2, S100 calcium-binding protein A2.
Figure 6.
SCNN1A and S100A2 immunohistochemistry in iRPE cells. (a) and (b) Representative analysis by immunofluorescence of endogenous SCNN1A (a) and S100A2 (b) protein expression in iRPE cells cultured on transwell filters (double white arrows) following 24 h treatment of aldosterone (aldo) 10−7 M, cortisol (corti) 10−7 M or cortisol (10−7 M) ± RU-486 (10−5 M). SCNN1A and S100A2 staining is in green, and DAPI staining for cell nuclei (white arrows) is in blue, respectively. White arrowheads indicate apical staining (a). Scale bar: 30 µm. SCNN1A, sodium channel epithelial 1 α subunit; S100A2, S100 calcium-binding protein A2.
Figure 7.
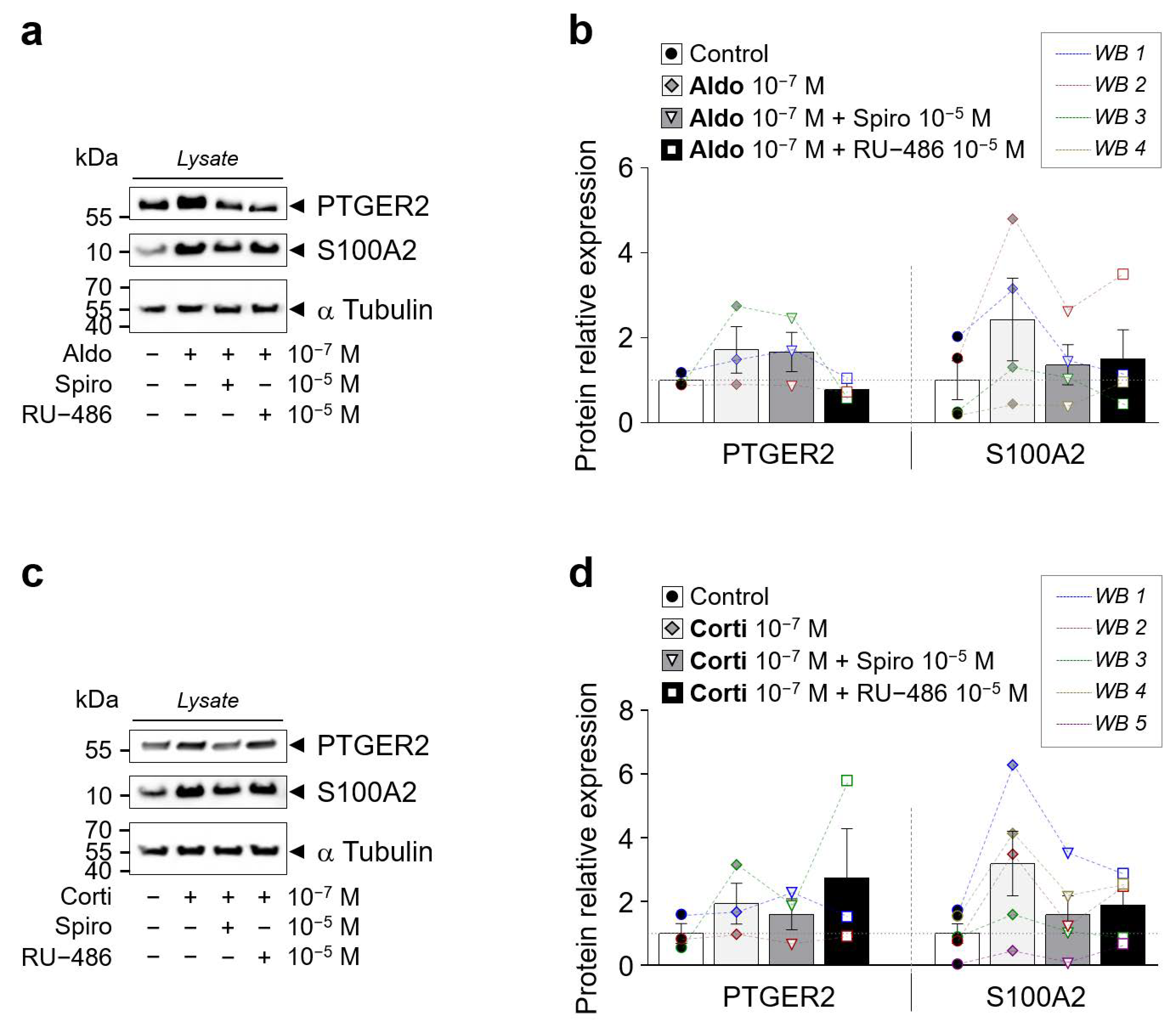
Western blot of PTGER2 and S100A2 in iRPE cells. (a) and (b) Western blot of PTGER2 and S100A2 protein expression in iRPE cells following 24 h treatment of aldosterone (aldo) 10−7 M (a) or cortisol (corti) 10−7 M (b) ± spironolactone (spiro) 10−5 M or RU-486 10−5 M. α-Tubulin is used as loading control. Arrows indicate the expected size of the 55 kDa PTGER2, 15 kDa S100A2 and 50 kDa α-Tubulin proteins. (c,d) Bar graphs represent densitometric quantification of PTGER2 (n = 3) and S100A2 (n = 4–5) protein expression relative to α-Tubulin levels in protein lysate (panel a and b). Values are means ± SEM. S100A2, S100 calcium-binding protein A2; PTGER2, prostaglandin E receptor 2.
Figure 7.
Western blot of PTGER2 and S100A2 in iRPE cells. (a) and (b) Western blot of PTGER2 and S100A2 protein expression in iRPE cells following 24 h treatment of aldosterone (aldo) 10−7 M (a) or cortisol (corti) 10−7 M (b) ± spironolactone (spiro) 10−5 M or RU-486 10−5 M. α-Tubulin is used as loading control. Arrows indicate the expected size of the 55 kDa PTGER2, 15 kDa S100A2 and 50 kDa α-Tubulin proteins. (c,d) Bar graphs represent densitometric quantification of PTGER2 (n = 3) and S100A2 (n = 4–5) protein expression relative to α-Tubulin levels in protein lysate (panel a and b). Values are means ± SEM. S100A2, S100 calcium-binding protein A2; PTGER2, prostaglandin E receptor 2.
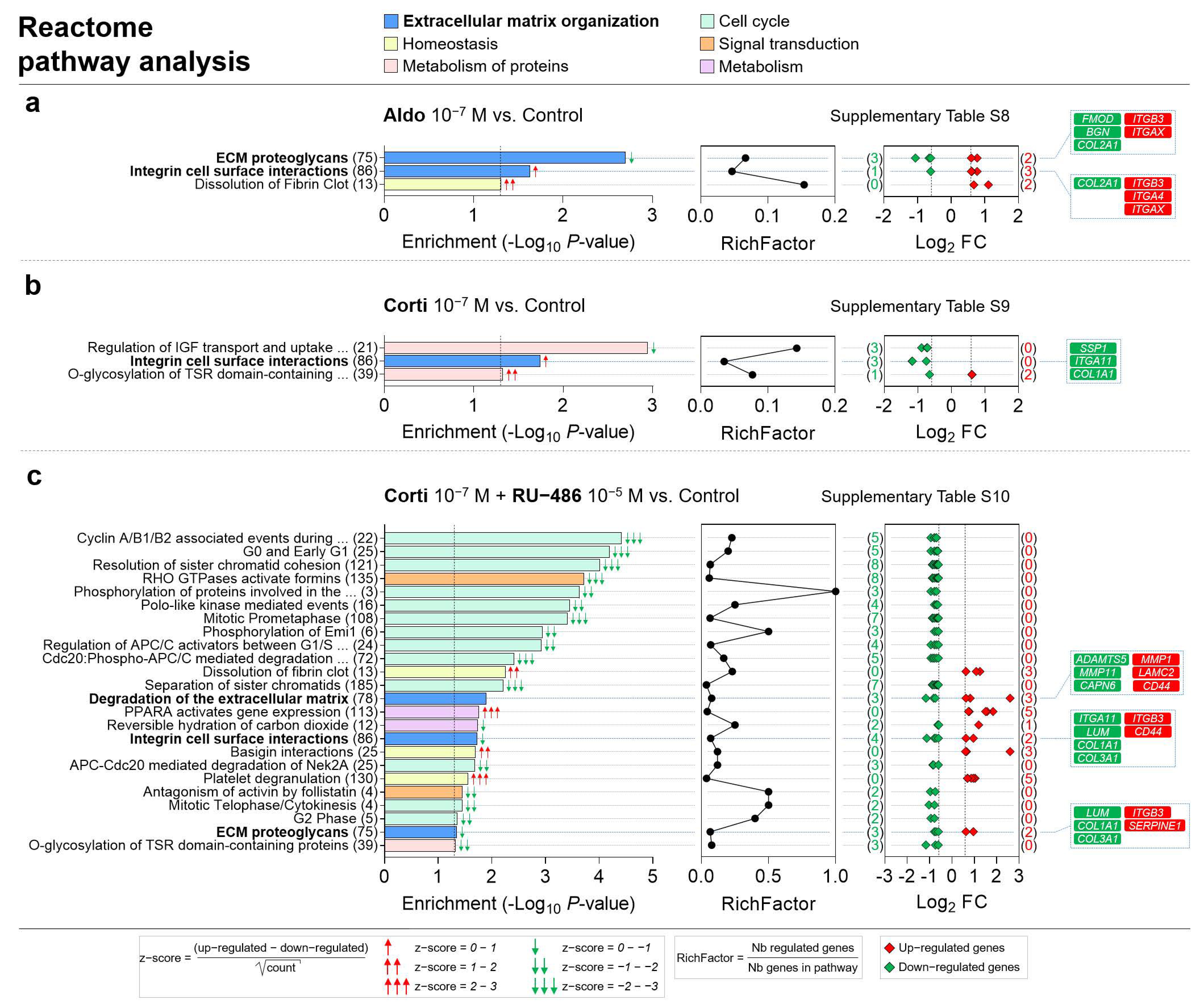
Figure 8.
ECM organization, homeostasis, metabolism of proteins, cell cycle, signal transduction and metabolism are the most represented Reactome pathway categories enriched in corticosteroid-treated iRPE cells. (a–c) Specific corticosteroid-regulated enriched Reactome pathways as predicted by bioinformatic enrichment analysis with their respective RichFactor value and the number of differentially expressed genes (DEGs) involved in the pathway, in iRPE cells treated for 24 h with aldosterone (aldo) 10−7 M (a), cortisol (corti) 10−7 M (b) or cortisol 10−7 M + RU-486 10−5 M (c) and compared to untreated cells. Reactome pathways are considered enriched if p value is <0.05 and the number of regulated genes in the pathways is ≥2. p-values, and fold-change (FC) values are represented in a −Log10 and a Log2 scale, respectively. Most represented enriched Reactome pathways and pathways of interest are highlighted in bold. Based on z-score values, green and red arrows in the left panel indicate down- and up-regulated pathways, respectively. Numbers between parentheses placed after enriched Reactome pathway names in the left panels represent the total number of genes implicated in the respective pathway. Numbers between parentheses in the right panels indicate the number of down- (in green) and up-regulated (in red) genes in their corresponding pathway. Corticosteroid-regulated genes in Reactome pathways of interest are listed in the right panel (Corti 10−7 M + RU-486 10−5 M vs. Control).
Figure 8.
ECM organization, homeostasis, metabolism of proteins, cell cycle, signal transduction and metabolism are the most represented Reactome pathway categories enriched in corticosteroid-treated iRPE cells. (a–c) Specific corticosteroid-regulated enriched Reactome pathways as predicted by bioinformatic enrichment analysis with their respective RichFactor value and the number of differentially expressed genes (DEGs) involved in the pathway, in iRPE cells treated for 24 h with aldosterone (aldo) 10−7 M (a), cortisol (corti) 10−7 M (b) or cortisol 10−7 M + RU-486 10−5 M (c) and compared to untreated cells. Reactome pathways are considered enriched if p value is <0.05 and the number of regulated genes in the pathways is ≥2. p-values, and fold-change (FC) values are represented in a −Log10 and a Log2 scale, respectively. Most represented enriched Reactome pathways and pathways of interest are highlighted in bold. Based on z-score values, green and red arrows in the left panel indicate down- and up-regulated pathways, respectively. Numbers between parentheses placed after enriched Reactome pathway names in the left panels represent the total number of genes implicated in the respective pathway. Numbers between parentheses in the right panels indicate the number of down- (in green) and up-regulated (in red) genes in their corresponding pathway. Corticosteroid-regulated genes in Reactome pathways of interest are listed in the right panel (Corti 10−7 M + RU-486 10−5 M vs. Control).
![Ijms 22 09618 g008]()
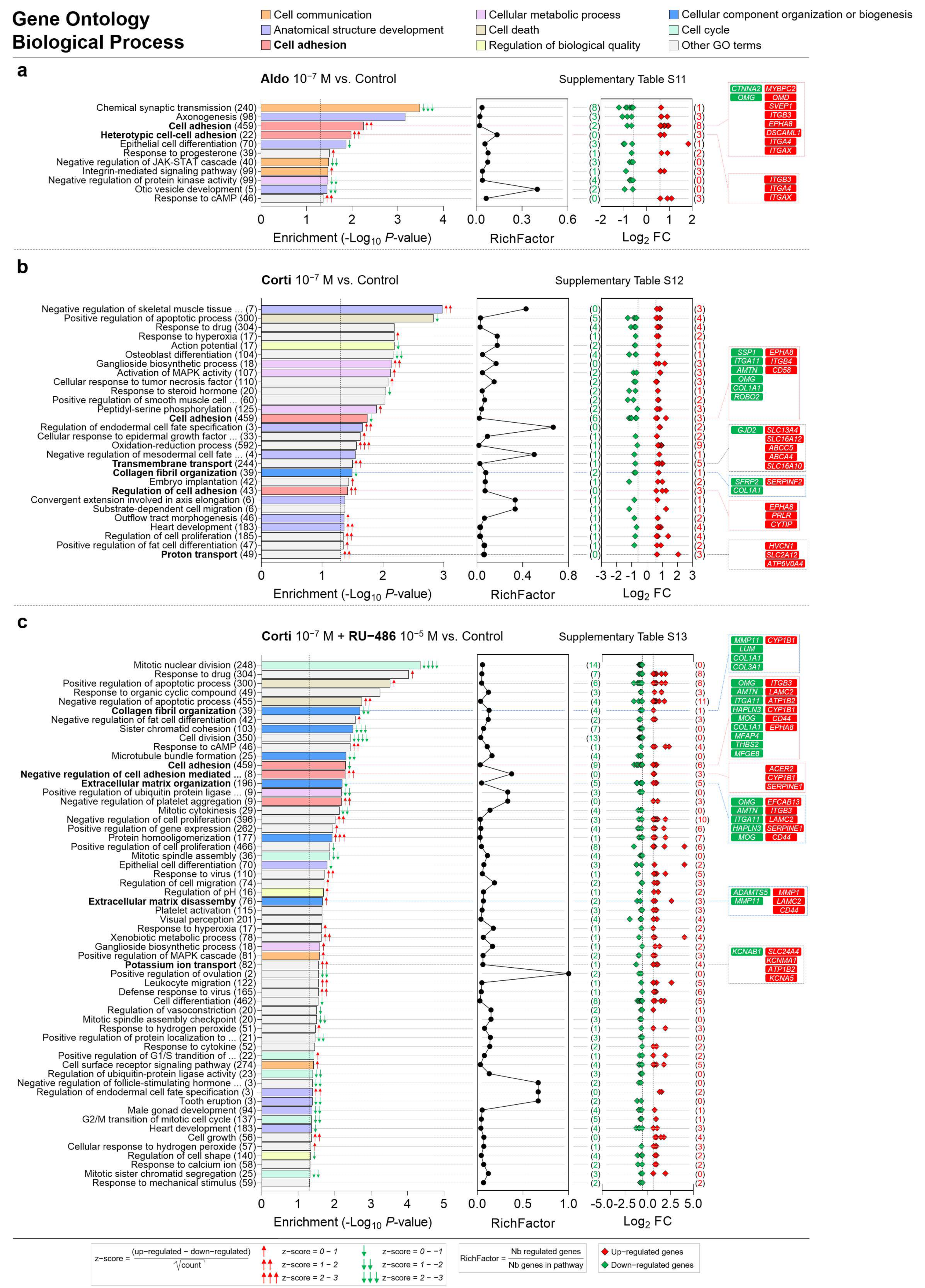
Figure 9.
Cell communication, anatomical structure development, cell adhesion, cellular metabolic process, cell death, regulation of biological quality, cellular component organization or biogenesis and cell cycle are the most represented biological process GO categories enriched in corticosteroid-treated iRPE cells. (a–c) Specific corticosteroid-regulated enriched biological process gene ontology (GO) terms as predicted by bioinformatic enrichment analysis with their respective RichFactor value and the number of differentially expressed genes (DEGs) involved in the process, in iRPE cells treated for 24 h with aldosterone (aldo) 10−7 M (a), cortisol (corti) 10−7 M (b) or cortisol 10−7 M + RU-486 10−5 M (c) and compared to untreated cells. Biological process GO terms are considered enriched if p value is <0.05 and the number of regulated genes in the pathways is ≥2. p-values and fold-change (FC) values are represented in a −Log10 and a Log2 scale, respectively. Most represented enriched biological process GO terms and GO terms of interest are highlighted in bold. Based on z-score values, green and red arrows in the left panel indicate down- and up-regulated GO terms, respectively. Numbers between parentheses placed after enriched biological process GO term names in the left panels represent the total number of genes implicated in the respective GO term. Numbers between parentheses in the right panels indicate the number of down- (in green) and up-regulated (in red) genes in their corresponding GO term. Corticosteroid-regulated genes in biological process GO terms of interest are listed in the right panel.
Figure 9.
Cell communication, anatomical structure development, cell adhesion, cellular metabolic process, cell death, regulation of biological quality, cellular component organization or biogenesis and cell cycle are the most represented biological process GO categories enriched in corticosteroid-treated iRPE cells. (a–c) Specific corticosteroid-regulated enriched biological process gene ontology (GO) terms as predicted by bioinformatic enrichment analysis with their respective RichFactor value and the number of differentially expressed genes (DEGs) involved in the process, in iRPE cells treated for 24 h with aldosterone (aldo) 10−7 M (a), cortisol (corti) 10−7 M (b) or cortisol 10−7 M + RU-486 10−5 M (c) and compared to untreated cells. Biological process GO terms are considered enriched if p value is <0.05 and the number of regulated genes in the pathways is ≥2. p-values and fold-change (FC) values are represented in a −Log10 and a Log2 scale, respectively. Most represented enriched biological process GO terms and GO terms of interest are highlighted in bold. Based on z-score values, green and red arrows in the left panel indicate down- and up-regulated GO terms, respectively. Numbers between parentheses placed after enriched biological process GO term names in the left panels represent the total number of genes implicated in the respective GO term. Numbers between parentheses in the right panels indicate the number of down- (in green) and up-regulated (in red) genes in their corresponding GO term. Corticosteroid-regulated genes in biological process GO terms of interest are listed in the right panel.
![Ijms 22 09618 g009]()
Scheme 2.
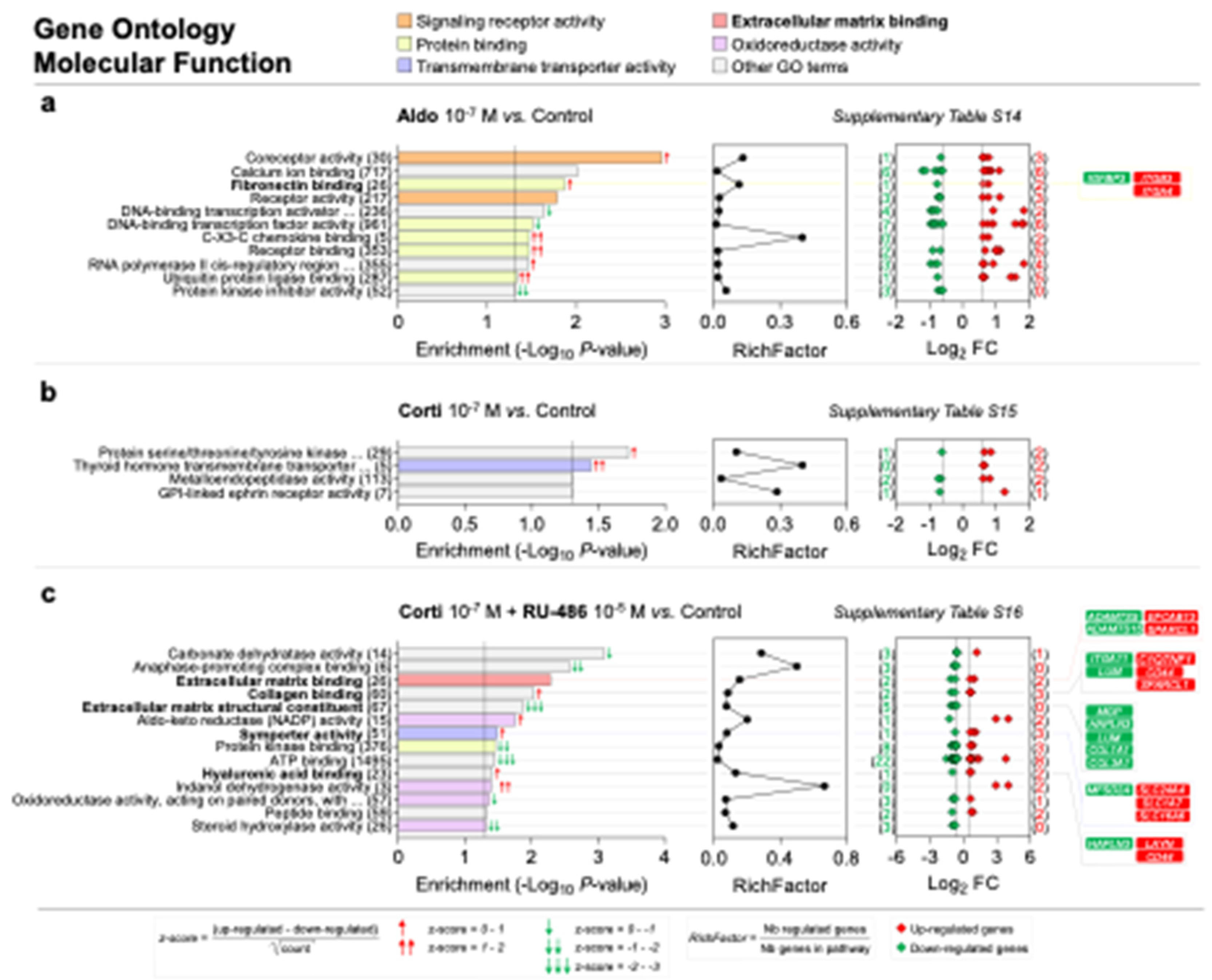
Signaling receptor activity, protein binding, transmembrane transporter activity, ECM binding and oxidoreductase activity are the most represented molecular function GO categories enriched in corticosteroid-treated iRPE cells. (a–c) Specific corticosteroid-regulated enriched molecular function gene ontology (GO) terms as predicted by bioinformatic enrichment analysis with their respective RichFactor value and the number of differentially expressed genes (DEGs) involved in the function, in iRPE cells treated for 24 h with aldosterone (aldo) 10−7 M (a), cortisol (corti) 10−7 M (b) or cortisol 10−7 M + RU-486 10−5 M (c) and compared to untreated cells. Molecular function GO terms are considered enriched if p value is <0.05 and the number of regulated genes in the pathways is ≥2. p-values and fold-change (FC) values are represented in a −Log10 and a Log2 scale, respectively. Most represented enriched molecular function GO terms are regrouped into five categories: signaling receptor activity, protein binding, transmembrane transporter activity, extracellular matrix binding and oxidoreductase activity; GO terms of interest are highlighted in bold. Based on z-score values, green and red arrows in the left panel indicate down- and up-regulated GO terms, respectively. Numbers between parentheses placed after enriched molecular function GO term names in the left panels represent the total number of genes implicated in the respective GO term. Numbers between parentheses in the right panels indicate the number of down- (in green) and up-regulated (in red) genes in their corresponding GO term. Corticosteroid-regulated genes in molecular function GO terms of interest are listed in the right panel.
Scheme 2.
Signaling receptor activity, protein binding, transmembrane transporter activity, ECM binding and oxidoreductase activity are the most represented molecular function GO categories enriched in corticosteroid-treated iRPE cells. (a–c) Specific corticosteroid-regulated enriched molecular function gene ontology (GO) terms as predicted by bioinformatic enrichment analysis with their respective RichFactor value and the number of differentially expressed genes (DEGs) involved in the function, in iRPE cells treated for 24 h with aldosterone (aldo) 10−7 M (a), cortisol (corti) 10−7 M (b) or cortisol 10−7 M + RU-486 10−5 M (c) and compared to untreated cells. Molecular function GO terms are considered enriched if p value is <0.05 and the number of regulated genes in the pathways is ≥2. p-values and fold-change (FC) values are represented in a −Log10 and a Log2 scale, respectively. Most represented enriched molecular function GO terms are regrouped into five categories: signaling receptor activity, protein binding, transmembrane transporter activity, extracellular matrix binding and oxidoreductase activity; GO terms of interest are highlighted in bold. Based on z-score values, green and red arrows in the left panel indicate down- and up-regulated GO terms, respectively. Numbers between parentheses placed after enriched molecular function GO term names in the left panels represent the total number of genes implicated in the respective GO term. Numbers between parentheses in the right panels indicate the number of down- (in green) and up-regulated (in red) genes in their corresponding GO term. Corticosteroid-regulated genes in molecular function GO terms of interest are listed in the right panel.
![Ijms 22 09618 sch002]()
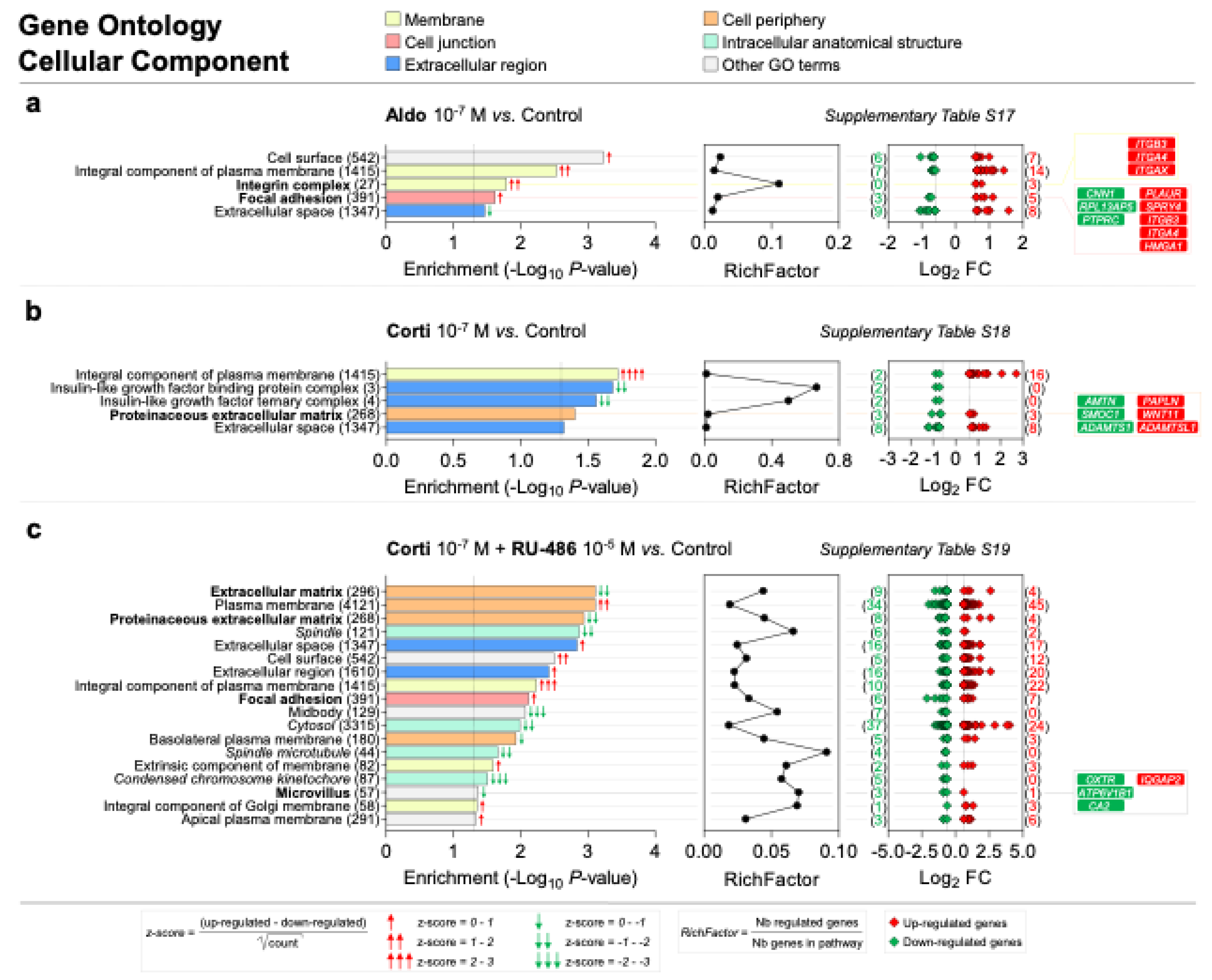
Scheme 3.
Membrane, cell junction, extracellular region, cell periphery and intracellular anatomical structure are the most represented cellular component GO categories enriched in corticosteroid-treated iRPE cells. (a–c) Specific corticosteroid-regulated enriched cellular component gene ontology (GO) terms as predicted by bioinformatic enrichment analysis with their respective RichFactor value and the number of differentially expressed genes (DEGs) involved in the component, in iRPE cells treated for 24 h with aldosterone (aldo) 10−7 M (a), cortisol (corti) 10−7 M (b) or cortisol 10−7 M + RU-486 10−5 M (c) and compared to untreated cells. Cellular component GO terms are considered enriched if p value is <0.05 and the number of regulated genes in the pathways is ≥2. p-values and fold-change (FC) values are represented in a −Log10 and a Log2 scale, respectively. Most represented enriched cellular component GO terms are regrouped into five categories: membrane, cell junction, extracellular region, cell periphery and intracellular anatomical structure; GO terms of interest are highlighted in bold. Based on z-score values, green and red arrows in the left panel indicate down- and up-regulated GO terms, respectively. Numbers between parentheses placed after enriched cellular component GO term names in the left panels represent the total number of genes implicated in the respective GO term. Numbers between parentheses in the right panels indicate the number of down- (in green) and up-regulated (in red) genes in their corresponding GO term. Corticosteroid-regulated genes in cellular component GO terms of interest are listed in the right panel.
Scheme 3.
Membrane, cell junction, extracellular region, cell periphery and intracellular anatomical structure are the most represented cellular component GO categories enriched in corticosteroid-treated iRPE cells. (a–c) Specific corticosteroid-regulated enriched cellular component gene ontology (GO) terms as predicted by bioinformatic enrichment analysis with their respective RichFactor value and the number of differentially expressed genes (DEGs) involved in the component, in iRPE cells treated for 24 h with aldosterone (aldo) 10−7 M (a), cortisol (corti) 10−7 M (b) or cortisol 10−7 M + RU-486 10−5 M (c) and compared to untreated cells. Cellular component GO terms are considered enriched if p value is <0.05 and the number of regulated genes in the pathways is ≥2. p-values and fold-change (FC) values are represented in a −Log10 and a Log2 scale, respectively. Most represented enriched cellular component GO terms are regrouped into five categories: membrane, cell junction, extracellular region, cell periphery and intracellular anatomical structure; GO terms of interest are highlighted in bold. Based on z-score values, green and red arrows in the left panel indicate down- and up-regulated GO terms, respectively. Numbers between parentheses placed after enriched cellular component GO term names in the left panels represent the total number of genes implicated in the respective GO term. Numbers between parentheses in the right panels indicate the number of down- (in green) and up-regulated (in red) genes in their corresponding GO term. Corticosteroid-regulated genes in cellular component GO terms of interest are listed in the right panel.
![Ijms 22 09618 sch003]()
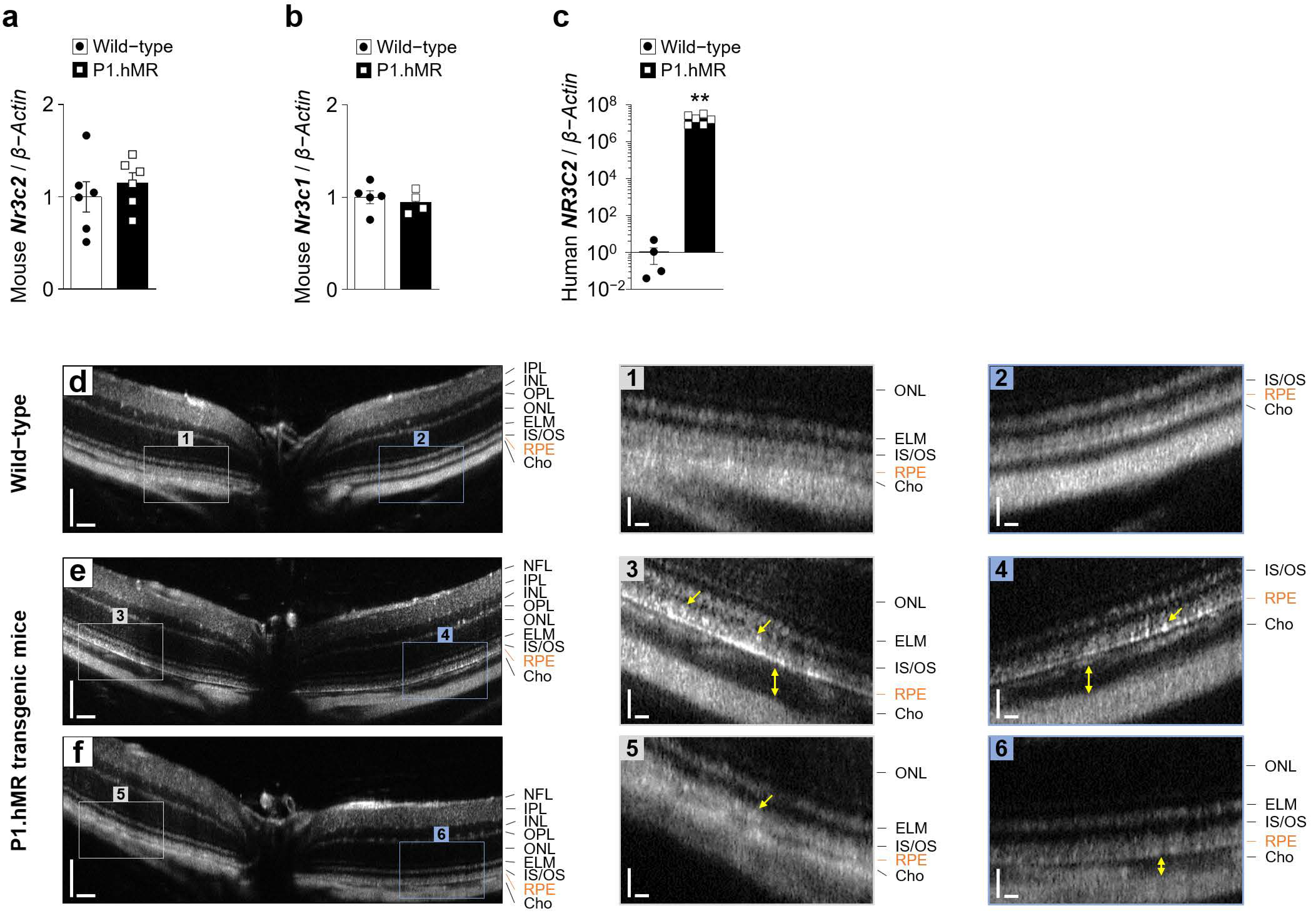
Figure 10.
Clinical phenotype of P1.hMR transgenic mice. (a–c) Quantification by RT-qPCR of the mouse Nr3c2 encoding MR (a), the mouse Nr3c1 encoding GR (b) and the human Nr3c2 (c) mRNA to β-Actin in RPE/choroid complexes (n = 4–6 eyes per condition) from wild-type animals and mice overexpressing a human MR P1.hMR transgene. Values are means ± SEM. Unpaired t-test with Welch’s correction. ** p < 0.01. (d–f) Representative in vivo MICRON III image-guided optical coherence tomography images of B-scans passing through the optic nerve of retina from wild-type (d) or transgenic P1.hMR (e,f) mice showing increased choroidal thickness (yellow double arrows) and irregular and hyper-reflective signal at the level of the RPE (yellow arrows). Scale bar: 100 µm (d–f) and 25 µm (insets 1–6). NFL, nerve fiber layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer, ONL, outer nuclear layer; ELM, external limiting membrane; IS, inner segment; OS, outer segment; RPE, retinal pigment epithelium; Cho, choroid.
Figure 10.
Clinical phenotype of P1.hMR transgenic mice. (a–c) Quantification by RT-qPCR of the mouse Nr3c2 encoding MR (a), the mouse Nr3c1 encoding GR (b) and the human Nr3c2 (c) mRNA to β-Actin in RPE/choroid complexes (n = 4–6 eyes per condition) from wild-type animals and mice overexpressing a human MR P1.hMR transgene. Values are means ± SEM. Unpaired t-test with Welch’s correction. ** p < 0.01. (d–f) Representative in vivo MICRON III image-guided optical coherence tomography images of B-scans passing through the optic nerve of retina from wild-type (d) or transgenic P1.hMR (e,f) mice showing increased choroidal thickness (yellow double arrows) and irregular and hyper-reflective signal at the level of the RPE (yellow arrows). Scale bar: 100 µm (d–f) and 25 µm (insets 1–6). NFL, nerve fiber layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer, ONL, outer nuclear layer; ELM, external limiting membrane; IS, inner segment; OS, outer segment; RPE, retinal pigment epithelium; Cho, choroid.
![Ijms 22 09618 g010]()
Figure 11.
Histologic phenotype of P1.hMR mice. (a–e) Representative histologic sections of the retina from wild-type and P1.hMR transgenic mice at nine months of age. Sections are stained with toluidine blue. Black triangles delineate the limit between the choroid and the sclera. Inset (1) showed normal mouse outer retina. Insets (2–5) show histological regions where choroidal vessels are dilated (blue double arrows), RPE cells are swollen (black asterisks) with accumulation of vacuoles (black arrowheads), and the outer retina is disorganized by fluid accumulation (black arrows) and elongation of the inner and outer segments of photoreceptors (black double arrows). Scale bar: 25 µm (a–e) and 10 µm (1–5). (f–h) Choroidal surface area (f), photoreceptor segments surface area (g) and RPE thickness (h) were quantified on retina sections from wild-type and P1.hMR transgenic mice (n = 8 eyes). Values are means ± SEM. Unpaired t-test with Welch’s correction. * p < 0.05, ** p < 0.01. P1, proximal functional alternative promoter 1 upstream of the first two untranslated exons 1α and 1β of the hMR gene; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer, ONL, outer nuclear layer; IS, inner segment; OS, outer segment; RPE, retinal pigment epithelium; Cho, choroid; Scl, sclera; Cc, choriocapillaris; CV, choroidal vessel; PRS, photoreceptor segments.
Figure 11.
Histologic phenotype of P1.hMR mice. (a–e) Representative histologic sections of the retina from wild-type and P1.hMR transgenic mice at nine months of age. Sections are stained with toluidine blue. Black triangles delineate the limit between the choroid and the sclera. Inset (1) showed normal mouse outer retina. Insets (2–5) show histological regions where choroidal vessels are dilated (blue double arrows), RPE cells are swollen (black asterisks) with accumulation of vacuoles (black arrowheads), and the outer retina is disorganized by fluid accumulation (black arrows) and elongation of the inner and outer segments of photoreceptors (black double arrows). Scale bar: 25 µm (a–e) and 10 µm (1–5). (f–h) Choroidal surface area (f), photoreceptor segments surface area (g) and RPE thickness (h) were quantified on retina sections from wild-type and P1.hMR transgenic mice (n = 8 eyes). Values are means ± SEM. Unpaired t-test with Welch’s correction. * p < 0.05, ** p < 0.01. P1, proximal functional alternative promoter 1 upstream of the first two untranslated exons 1α and 1β of the hMR gene; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer, ONL, outer nuclear layer; IS, inner segment; OS, outer segment; RPE, retinal pigment epithelium; Cho, choroid; Scl, sclera; Cc, choriocapillaris; CV, choroidal vessel; PRS, photoreceptor segments.
![Ijms 22 09618 g011]()
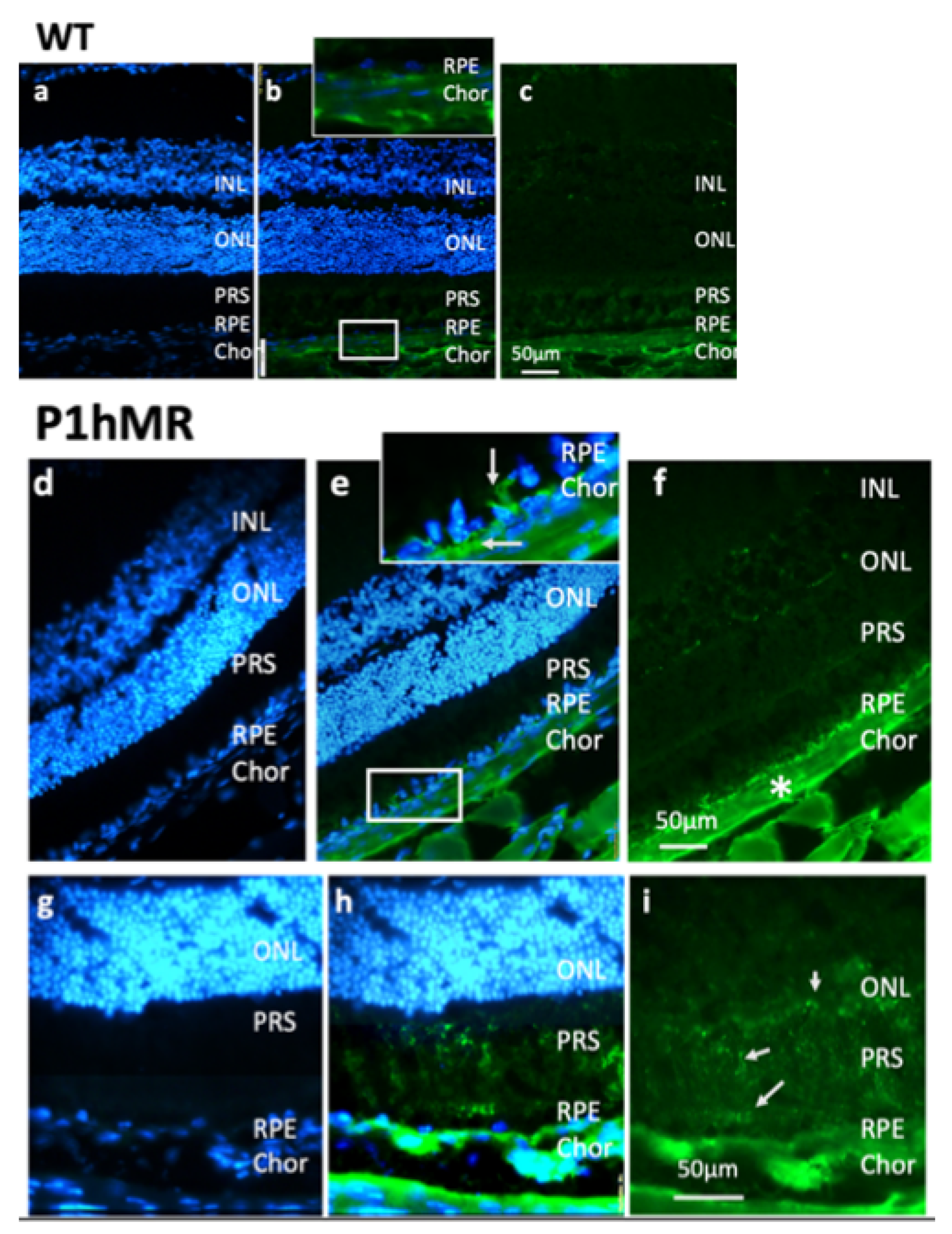
Scheme 4.
Albumin accumulates within RPE cells and outer retina in P1.hMR transgenic mice. (a–i) Albumin immunostaining (in green) on retinal cryosections from wild-type (WT, a–c) and P1.hMR transgenic (d–i) mice. Cell nuclei are stained with DAPI (in blue). White arrows in (e, inset) and the white asterisk in (f) indicate accumulation of albumin in P1.hMR mice within RPE cells and in the choroidal stroma, respectively. Focal albumin staining is observed within the outer retina and particularly at the level of photoreceptor segments (g–i, arrows). INL, inner nuclear layer; ONL, outer nuclear layer; PRS, photoreceptor inner and outer segments; RPE, retinal pigment epithelium; Chor, choroid.
Scheme 4.
Albumin accumulates within RPE cells and outer retina in P1.hMR transgenic mice. (a–i) Albumin immunostaining (in green) on retinal cryosections from wild-type (WT, a–c) and P1.hMR transgenic (d–i) mice. Cell nuclei are stained with DAPI (in blue). White arrows in (e, inset) and the white asterisk in (f) indicate accumulation of albumin in P1.hMR mice within RPE cells and in the choroidal stroma, respectively. Focal albumin staining is observed within the outer retina and particularly at the level of photoreceptor segments (g–i, arrows). INL, inner nuclear layer; ONL, outer nuclear layer; PRS, photoreceptor inner and outer segments; RPE, retinal pigment epithelium; Chor, choroid.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}














