
Impact of Amyloid-β on Platelet Mitochondrial Function and Platelet–Mediated Amyloid Aggregation in Alzheimer’s Disease

 ,
,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
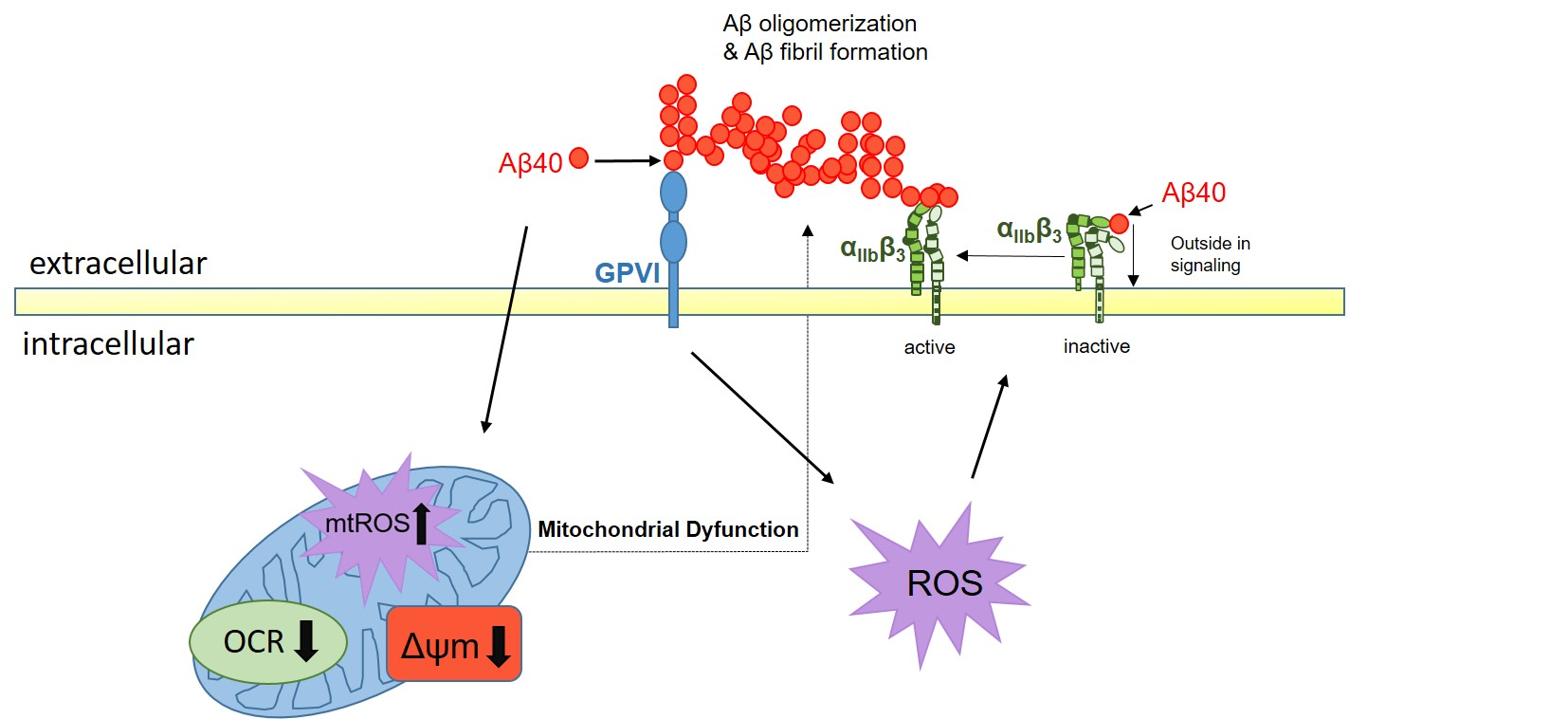
1. Introduction
2. Results
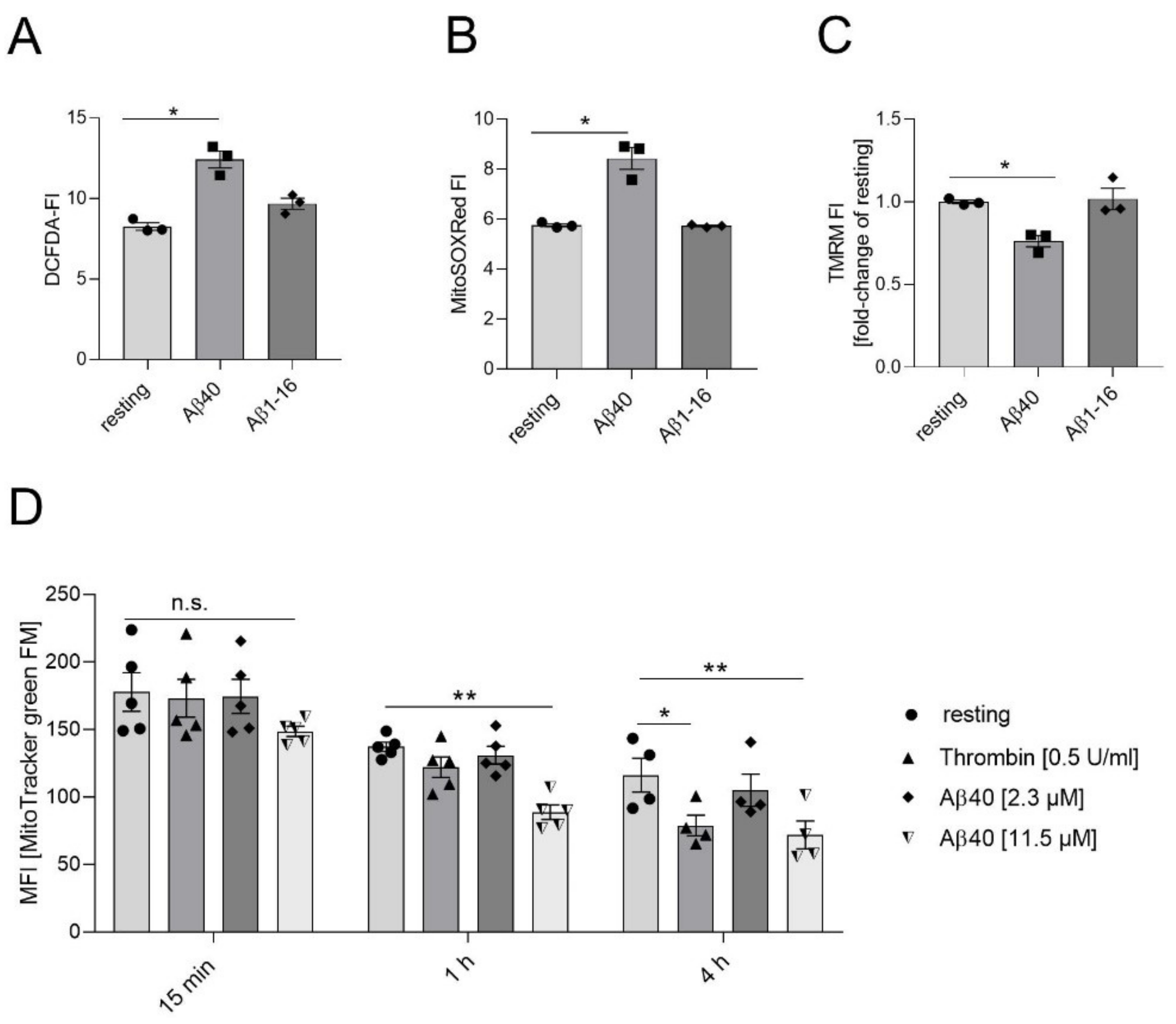
2.1. Effects of Aβ40 on Mitochondria in Platelets
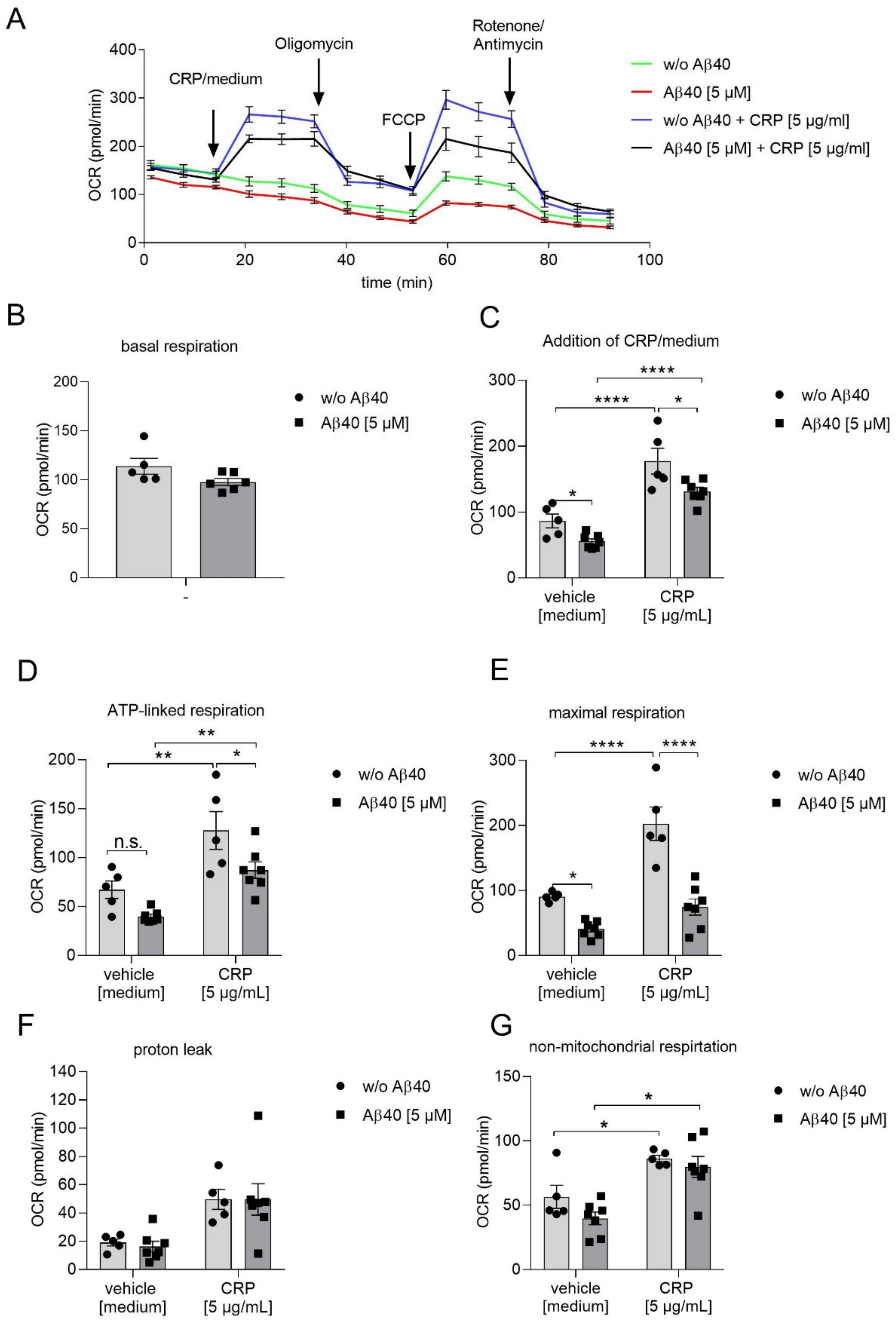
2.2. Reduced Mitochondrial Respiration in Platelets Following Aβ40 Treatment
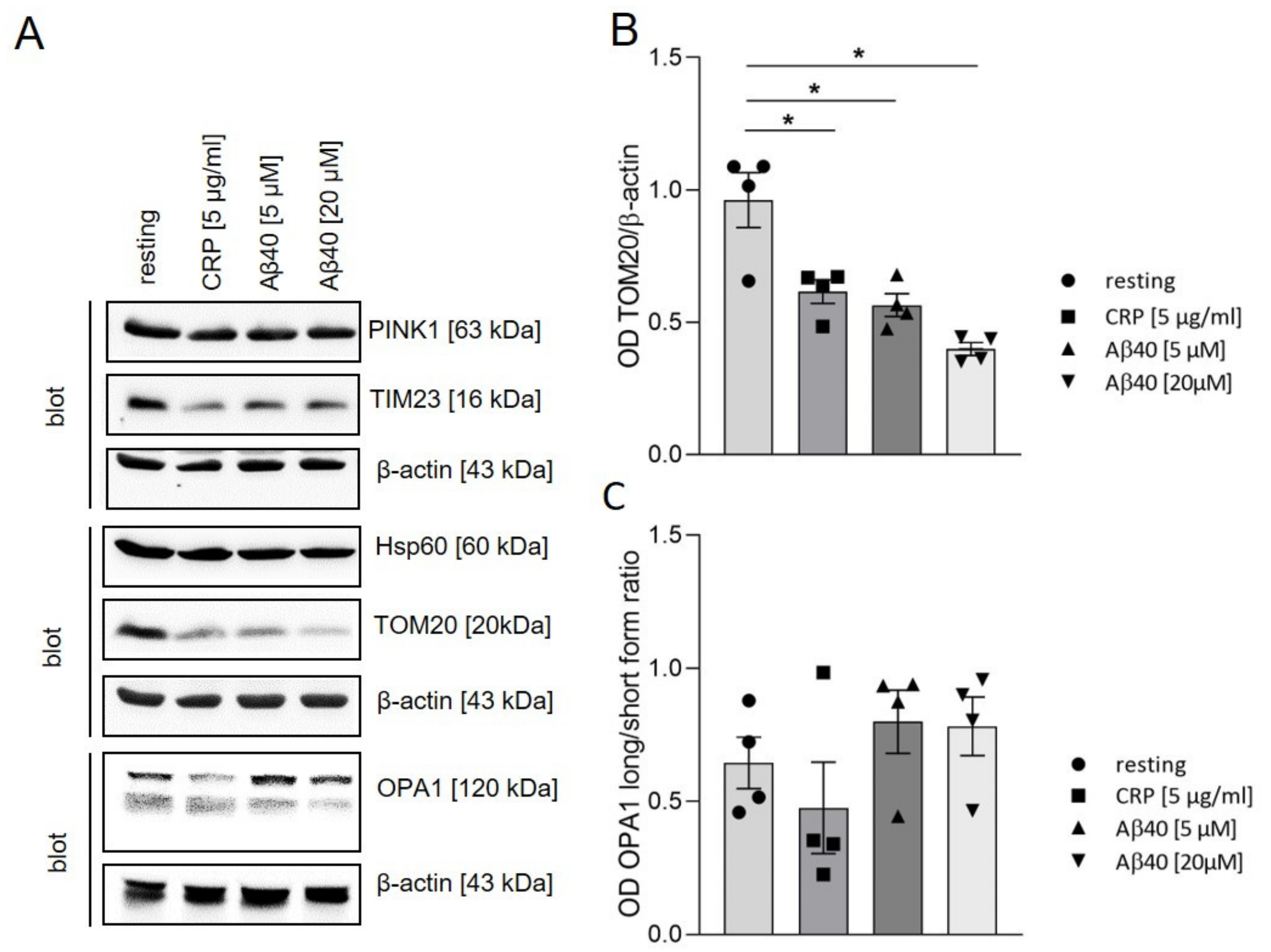
2.3. Impact of Extracellular Aβ40 on Mitochondrial Proteins
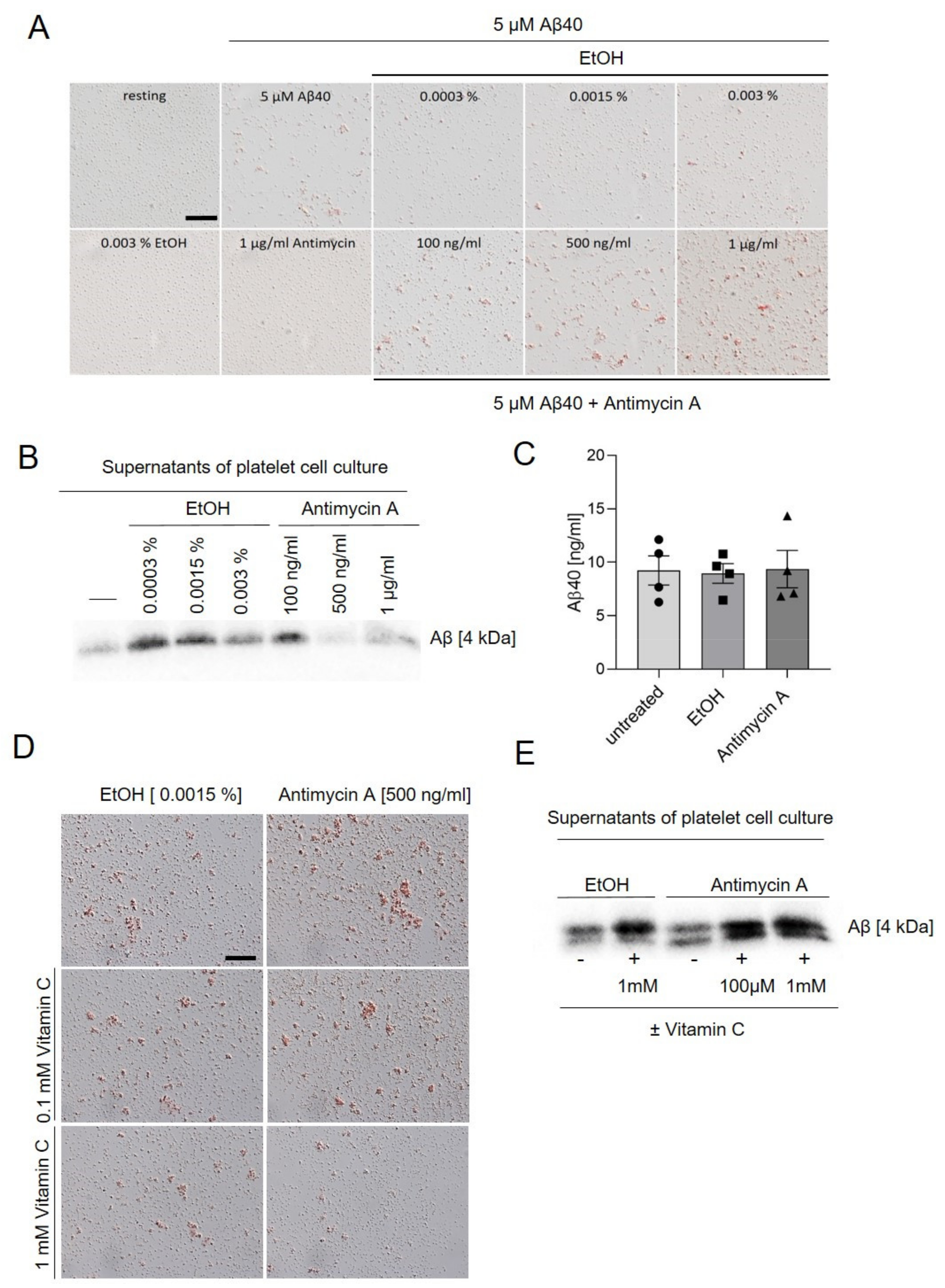
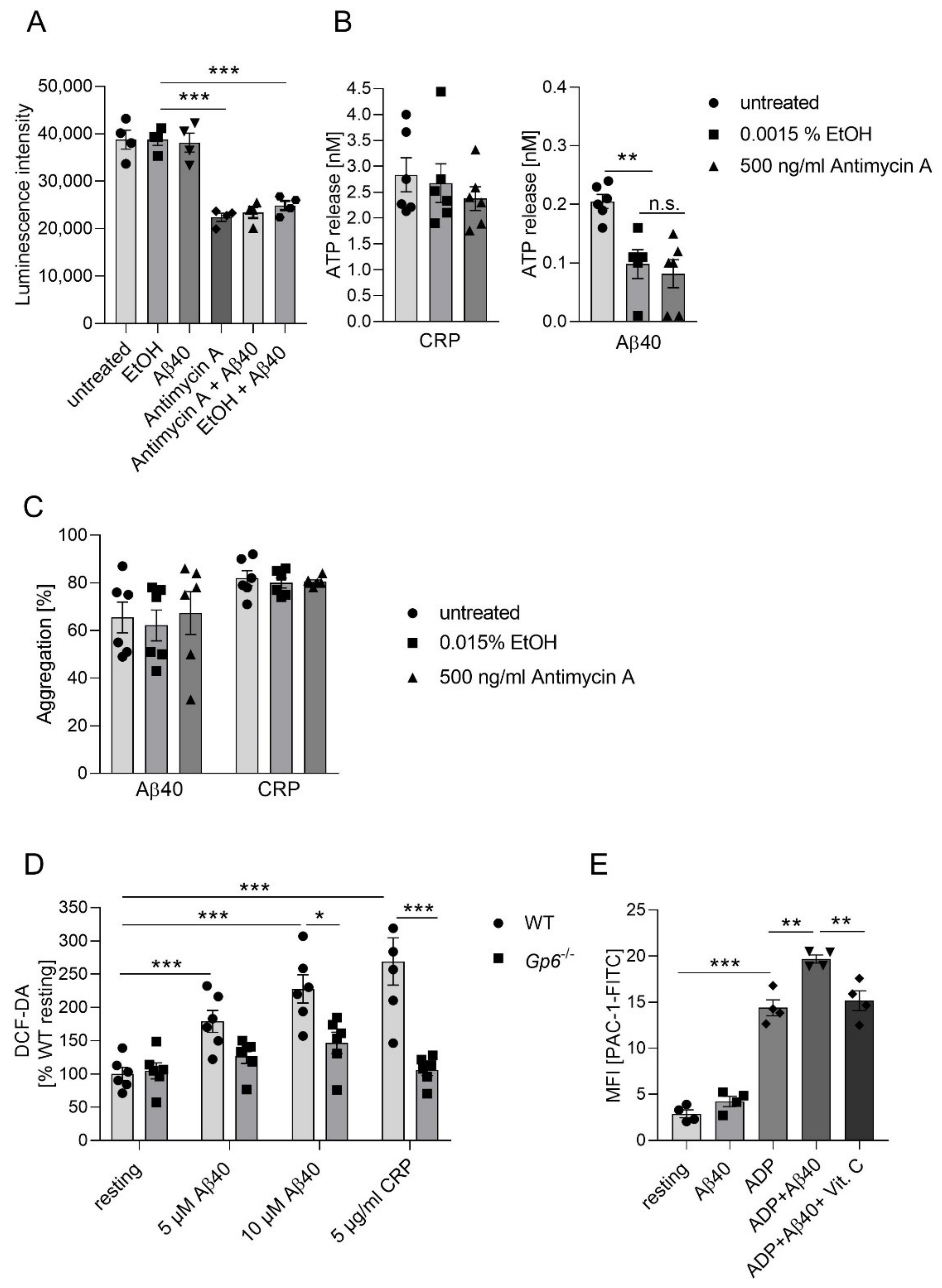
2.4. Inhibition of Complex III Leads to Enhanced Platelet Mediated Aβ Aggregate Formation In Vitro
2.5. Aβ Induced GPVI-Mediated ROS Production and Integrin αIIbβ3 Activation In Vitro
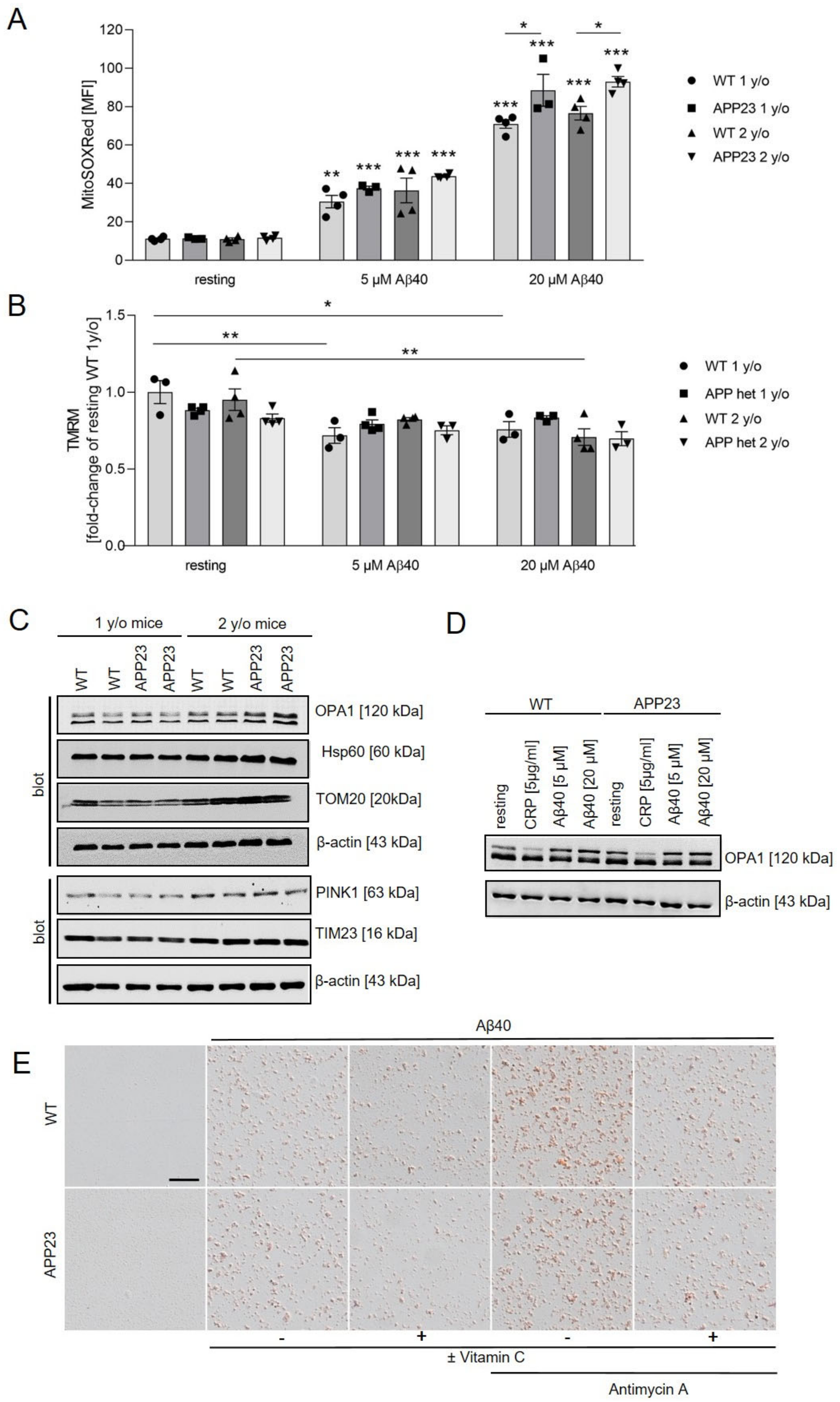
2.6. Mitochondrial ROS Production and Mitochondrial Membrane Potential in Platelets from Alzheimer’s Disease Transgenic Mice APP23
3. Discussion
4. Materials and Methods
4.1. Chemicals, Peptides and Antibodies
4.2. Animals
4.3. Murine Platelet Preparation
4.4. Human Platelet Preparation
4.5. Human and Murine Platelet Culture
4.6. Measurement of Intracellular ROS Level
4.7. Measurement of Mitochondrial Superoxide
4.8. Measurement of Mitochondrial Membrane Potential
4.9. Measurement of Mitochondria Release Using MitoTrackerTM Green FM
4.10. Cell Lysis and Immunoblotting
4.11. Platelet Aggregation
4.12. Measurement of Intracellular ATP Level and ATP Release
4.13. Aβ40 Quantification by Enzyme-Linked Immunosorbent Assay (ELISA)
4.14. Measurement of the Oxygen Consumption Rate
4.15. Flow Cytometry Measurement of PAC-1 Binding
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H.; Tripathi, R.; Troung, Q.; Tirumala, K.; Reddy, T.P.; Anekonda, V.; Shirendeb, U.P.; Calkins, M.J.; Reddy, A.P.; Mao, P.; et al. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: Implications to mitochondria-targeted antioxidant therapeutics. Biochim. Biophys. Acta 2012, 1822, 639–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 18670–18675. [Google Scholar] [CrossRef] [Green Version]
- Cai, Q.; Tammineni, P. Alterations in Mitochondrial Quality Control in Alzheimer’s Disease. Front. Cell. Neurosci. 2016, 10, 24. [Google Scholar] [CrossRef] [Green Version]
- Shefa, U.; Jeong, N.Y.; Song, I.O.; Chung, H.J.; Kim, D.; Jung, J.; Huh, Y. Mitophagy links oxidative stress conditions and neurodegenerative diseases. Neural Regen. Res. 2019, 14, 749–756. [Google Scholar]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Yin, X.; Grady, M.C.; Mitchell, A.; Tonk, S.; Kuruva, C.S.; Bhatti, J.S.; Kandimalla, R.; Vijayan, M.; et al. Protective Effects of Indian Spice Curcumin Against Amyloid-β in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 61, 843–866. [Google Scholar] [CrossRef]
- Reddy, P.H.; Williams, J.; Smith, F.; Bhatti, J.S.; Kumar, S.; Vijayan, M.; Kandimalla, R.; Kuruva, C.S.; Wang, R.; Manczak, M.; et al. MicroRNAs, Aging, Cellular Senescence, and Alzheimer’s Disease. Prog. Mol. Biol. Transl. Sci. 2017, 146, 127–171. [Google Scholar]
- Reddy, P.H.; McWeeney, S.; Park, B.S.; Manczak, M.; Gutala, R.V.; Partovi, D.; Jung, Y.; Yau, V.; Searles, R.; Mori, M.; et al. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: Up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum. Mol. Genet. 2004, 13, 1225–1240. [Google Scholar] [CrossRef] [Green Version]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Z.H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.X.; Whyte, S.; Tanner, J.E.; Evin, G.; Beyreuther, K.; Masters, C.L. Secretion of Alzheimer’s disease Abeta amyloid peptide by activated human platelets. Lab. Investig. A J. Tech. Methods Pathol. 1998, 78, 461–469. [Google Scholar]
- Chen, M.; Inestrosa, N.C.; Ross, G.S.; Fernandez, H.L. Platelets are the primary source of amyloid beta-peptide in human blood. Biochem. Biophys. Res. Commun. 1995, 213, 96–103. [Google Scholar] [CrossRef]
- Gowert, N.S.; Donner, L.; Chatterjee, M.; Eisele, Y.S.; Towhid, S.T.; Munzer, P.; Walker, B.; Ogorek, I.; Borst, O.; Grandoch, M.; et al. Blood platelets in the progression of Alzheimer’s disease. PLoS ONE 2014, 9, e90523. [Google Scholar] [CrossRef] [Green Version]
- Zainaghi, I.A.; Talib, L.L.; Diniz, B.S.; Gattaz, W.F.; Forlenza, O.V. Reduced platelet amyloid precursor protein ratio (APP ratio) predicts conversion from mild cognitive impairment to Alzheimer’s disease. J. Neural Transm. 2012, 119, 815–819. [Google Scholar] [CrossRef]
- Johnston, J.A.; Liu, W.W.; Coulson, D.T.; Todd, S.; Murphy, S.; Brennan, S.; Foy, C.J.; Craig, D.; Irvine, G.B.; Passmore, A.P. Platelet beta-secretase activity is increased in Alzheimer’s disease. Neurobiol. Aging 2008, 29, 661–668. [Google Scholar] [CrossRef]
- Stellos, K.; Panagiota, V.; Kogel, A.; Leyhe, T.; Gawaz, M.; Laske, C. Predictive value of platelet activation for the rate of cognitive decline in Alzheimer’s disease patients. J. Cereb. Blood Flow Metab. 2010, 30, 1817–1820. [Google Scholar] [CrossRef] [Green Version]
- Jarre, A.; Gowert, N.S.; Donner, L.; Munzer, P.; Klier, M.; Borst, O.; Schaller, M.; Lang, F.; Korth, C.; Elvers, M. Pre-activated blood platelets and a pro-thrombotic phenotype in APP23 mice modeling Alzheimer’s disease. Cell Signal. 2014, 26, 2040–2050. [Google Scholar] [CrossRef]
- Prodan, C.I.; Ross, E.D.; Vincent, A.S.; Dale, G.L. Rate of progression in Alzheimer’s disease correlates with coated-platelet levels—A longitudinal study. Transl. Res. J. Lab. Clin. Med. 2008, 152, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Donner, L.; Falker, K.; Gremer, L.; Klinker, S.; Pagani, G.; Ljungberg, L.U.; Lothmann, K.; Rizzi, F.; Schaller, M.; Gohlke, H.; et al. Platelets contribute to amyloid-beta aggregation in cerebral vessels through integrin alphaIIbbeta3-induced outside-in signaling and clusterin release. Sci. Signal. 2016, 9, ra52. [Google Scholar] [CrossRef]
- Donner, L.; Gremer, L.; Ziehm, T.; Gertzen, C.G.W.; Gohlke, H.; Willbold, D.; Elvers, M. Relevance of N-terminal residues for amyloid-beta binding to platelet integrin alphaIIbbeta3, integrin outside-in signaling and amyloid-beta fibril formation. Cell Signal. 2018, 50, 121–130. [Google Scholar] [CrossRef]
- Donner, L.; Toska, L.M.; Krüger, I.; Gröniger, S.; Barroso, R.; Burleigh, A.; Mezzano, D.; Pfeiler, S.; Kelm, M.; Gerdes, N.; et al. The collagen receptor glycoprotein VI promotes platelet-mediated aggregation of β-amyloid. Sci. Signal. 2020, 13, eaba9872. [Google Scholar] [CrossRef]
- Hayashi, T.; Tanaka, S.; Hori, Y.; Hirayama, F.; Sato, E.F.; Inoue, M. Role of mitochondria in the maintenance of platelet function during in vitro storage. Transfus. Med. 2011, 21, 166–174. [Google Scholar] [CrossRef]
- Melchinger, H.; Jain, K.; Tyagi, T.; Hwa, J. Role of Platelet Mitochondria: Life in a Nucleus-Free Zone. Front. Cardiovasc. Med. 2019, 6, 153. [Google Scholar] [CrossRef]
- Fišar, Z.; Hroudová, J.; Hansíková, H.; Spáčilová, J.; Lelková, P.; Wenchich, L.; Jirák, R.; Zvěřová, M.; Zeman, J.; Martásek, P.; et al. Mitochondrial Respiration in the Platelets of Patients with Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 930–941. [Google Scholar] [CrossRef]
- Fišar, Z.; Jirák, R.; Zvěřová, M.; Setnička, V.; Habartová, L.; Hroudová, J.; Vaníčková, Z.; Raboch, J. Plasma amyloid beta levels and platelet mitochondrial respiration in patients with Alzheimer’s disease. Clin. Biochem. 2019, 72, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.M.; Proença, M.T.; Santos, S.; Santana, I.; Oliveira, C.R. Cytochrome c oxidase is decreased in Alzheimer’s disease platelets. Neurobiol. Aging 2004, 25, 105–110. [Google Scholar] [CrossRef]
- Masselli, E.; Pozzi, G.; Vaccarezza, M.; Mirandola, P.; Galli, D.; Vitale, M.; Carubbi, C.; Gobbi, G. ROS in Platelet Biology: Functional Aspects and Methodological Insights. Int. J. Mol. Sci. 2020, 21, 4866. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, L.H.; Duchez, A.C.; Cloutier, N.; Soulet, D.; Martin, N.; Bollinger, J.; Pare, A.; Rousseau, M.; Naika, G.S.; Levesque, T.; et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 2014, 124, 2173–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leuner, K.; Schutt, T.; Kurz, C.; Eckert, S.H.; Schiller, C.; Occhipinti, A.; Mai, S.; Jendrach, M.; Eckert, G.P.; Kruse, S.E.; et al. Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid. Redox Signal. 2012, 16, 1421–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begonja, A.J.; Gambaryan, S.; Geiger, J.; Aktas, B.; Pozgajova, M.; Nieswandt, B.; Walter, U. Platelet NAD(P)H-oxidase-generated ROS production regulates alphaIIbbeta3-integrin activation independent of the NO/cGMP pathway. Blood 2005, 106, 2757–2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Dotto, V.; Fogazza, M.; Carelli, V.; Rugolo, M.; Zanna, C. Eight human OPA1 isoforms, long and short: What are they for? Biochim. Biophys. Acta Bioenerg. 2018, 1859, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Aibibula, M.; Naseem, K.M.; Sturmey, R.G. Glucose metabolism and metabolic flexibility in blood platelets. J. Thromb. Haemost. 2018, 16, 2300–2314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zharikov, S.; Shiva, S. Platelet mitochondrial function: From regulation of thrombosis to biomarker of disease. Biochem. Soc. Trans. 2013, 41, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Duan, Q.; Wang, T.; Ahmed, M.; Zhang, N.; Li, Y.; Li, L.; Yao, X. Mitochondrial Respiratory Chain Inhibitors Involved in ROS Production Induced by Acute High Concentrations of Iodide and the Effects of SOD as a Protective Factor. Oxidative Med. Cell. Longev. 2015, 2015, 217670. [Google Scholar] [CrossRef] [Green Version]
- Rusak, T.; Tomasiak, M.; Ciborowski, M. Peroxynitrite can affect platelet responses by inhibiting energy production. Acta Biochim. Pol. 2006, 53, 769–776. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.H.; Hyon, L.; Lee, K.M. The possible role of antioxidant vitamin C in Alzheimer’s disease treatment and prevention. Am. J. Alzheimer’s Dis. Other Dement. 2013, 28, 120–125. [Google Scholar] [CrossRef]
- Huang, J.; May, J.M. Ascorbic acid protects SH-SY5Y neuroblastoma cells from apoptosis and death induced by beta-amyloid. Brain Res. 2006, 1097, 52–58. [Google Scholar] [CrossRef]
- Engelhart, M.J.; Geerlings, M.I.; Ruitenberg, A.; van Swieten, J.C.; Hofman, A.; Witteman, J.C.; Breteler, M.M. Dietary intake of antioxidants and risk of Alzheimer disease. JAMA 2002, 287, 3223–3229. [Google Scholar] [CrossRef] [Green Version]
- Baloyannis, S.J. Mitochondrial alterations in Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, H.M.; Wegiel, J. Beta-amyloid formation by myocytes of leptomeningeal vessels. Acta Neuropathol. 1994, 87, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Calhoun, M.E.; Burgermeister, P.; Phinney, A.L.; Stalder, M.; Tolnay, M.; Wiederhold, K.H.; Abramowski, D.; Sturchler-Pierrat, C.; Sommer, B.; Staufenbiel, M.; et al. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc. Natl. Acad. Sci. USA 1999, 96, 14088–14093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzig, M.C.; Winkler, D.T.; Burgermeister, P.; Pfeifer, M.; Kohler, E.; Schmidt, S.D.; Danner, S.; Abramowski, D.; Stürchler-Pierrat, C.; Bürki, K.; et al. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat. Neurosci. 2004, 7, 954–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagishi, S.I.; Edelstein, D.; Du, X.L.; Kaneda, Y.; Guzmán, M.; Brownlee, M. Leptin induces mitochondrial superoxide production and monocyte chemoattractant protein-1 expression in aortic endothelial cells by increasing fatty acid oxidation via protein kinase A. J. Biol. Chem. 2001, 276, 25096–25100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardenes, N.; Corey, C.; Geary, L.; Jain, S.; Zharikov, S.; Barge, S.; Novelli, E.M.; Shiva, S. Platelet bioenergetic screen in sickle cell patients reveals mitochondrial complex V inhibition, which contributes to platelet activation. Blood 2014, 123, 2864–2872. [Google Scholar] [CrossRef] [Green Version]
- Hottz, E.D.; Oliveira, M.F.; Nunes, P.C.; Nogueira, R.M.; Valls-de-Souza, R.; Da Poian, A.T.; Weyrich, A.S.; Zimmerman, G.A.; Bozza, P.T.; Bozza, F.A. Dengue induces platelet activation, mitochondrial dysfunction and cell death through mechanisms that involve DC-SIGN and caspases. J. Thromb. Haemost. 2013, 11, 951–962. [Google Scholar] [CrossRef]
- Puskarich, M.A.; Cornelius, D.C.; Tharp, J.; Nandi, U.; Jones, A.E. Plasma syndecan-1 levels identify a cohort of patients with severe sepsis at high risk for intubation after large-volume intravenous fluid resuscitation. J. Crit. Care 2016, 36, 125–129. [Google Scholar] [CrossRef]
- Lust, W.D.; Taylor, C.; Pundik, S.; Selman, W.R.; Ratcheson, R.A. Ischemic cell death: Dynamics of delayed secondary energy failure during reperfusion following focal ischemia. Metab. Brain Dis. 2002, 17, 113–121. [Google Scholar] [CrossRef]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol. 2014, 2, 702–714. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donner, L.; Feige, T.; Freiburg, C.; Toska, L.M.; Reichert, A.S.; Chatterjee, M.; Elvers, M. Impact of Amyloid-β on Platelet Mitochondrial Function and Platelet–Mediated Amyloid Aggregation in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 9633. https://doi.org/10.3390/ijms22179633
Donner L, Feige T, Freiburg C, Toska LM, Reichert AS, Chatterjee M, Elvers M. Impact of Amyloid-β on Platelet Mitochondrial Function and Platelet–Mediated Amyloid Aggregation in Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(17):9633. https://doi.org/10.3390/ijms22179633
Chicago/Turabian StyleDonner, Lili, Tobias Feige, Carolin Freiburg, Laura Mara Toska, Andreas S. Reichert, Madhumita Chatterjee, and Margitta Elvers. 2021. "Impact of Amyloid-β on Platelet Mitochondrial Function and Platelet–Mediated Amyloid Aggregation in Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 17: 9633. https://doi.org/10.3390/ijms22179633
APA StyleDonner, L., Feige, T., Freiburg, C., Toska, L. M., Reichert, A. S., Chatterjee, M., & Elvers, M. (2021). Impact of Amyloid-β on Platelet Mitochondrial Function and Platelet–Mediated Amyloid Aggregation in Alzheimer’s Disease. International Journal of Molecular Sciences, 22(17), 9633. https://doi.org/10.3390/ijms22179633