
Immunomodulatory Properties of BRAF and MEK Inhibitors Used for Melanoma Therapy—Paradoxical ERK Activation and Beyond



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Constitutive MAPK Signaling Is a Major Cause of Melanoma Induction/Progression
2.1. BRAF Mutations Are a Dominant Driver of Melanoma
2.2. Dysregulated Signaling Pathways
2.3. Additional Melanoma-Promoting Mutations
3. Melanoma-Induced Immune Modulation
4. MAPK-Targeted Treatment of Metastatic Melanoma
4.1. Limitations of BRAFi Monotherapy
4.2. BRAFi/MEKi Combination Therapy
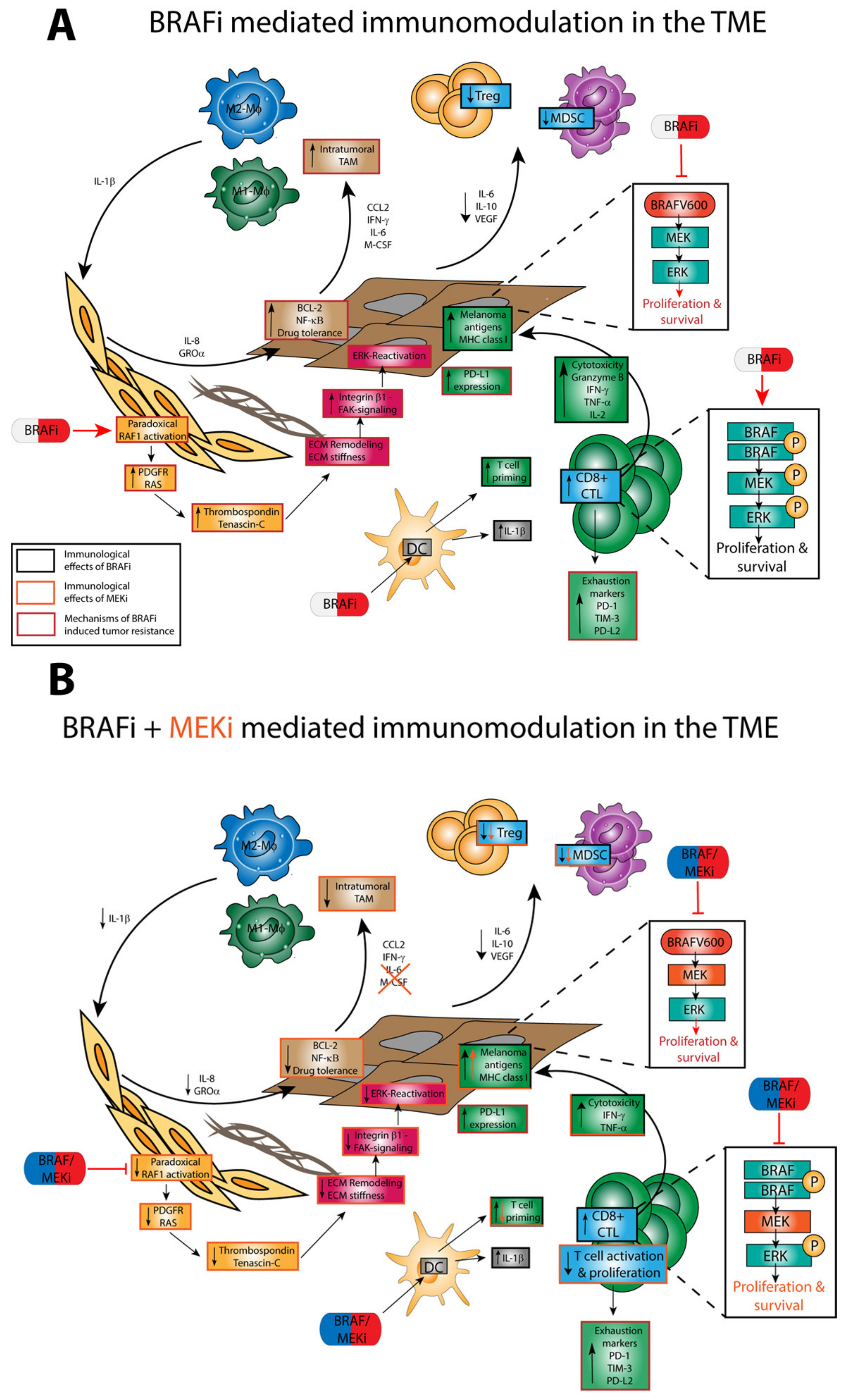
4.3. Off-Target Effects of BRAFi/MEKi on Immune Cells
4.3.1. Paradoxical ERK Activation
4.3.2. TME
4.3.3. Regulatory Immune Cells
4.3.4. DC
4.3.5. T Cells
5. Conclusions/Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| AE | Adverse event |
| APC | Antigen-presenting cell |
| BCL | B-Cell chronic lymphocytic leukemia/lymphoma |
| BRAF | v-Raf murine sarcoma viral oncogene homolog B |
| BRAFi | BRAF inhibitors (BRAFi) |
| CAF | Cancer-associated fibroblast |
| cDC | Conventional DC |
| CDK | Cyclin-dependent kinase |
| CDKN | Cyclin-dependent kinase inhibitor |
| CTLA-4 | Cytotoxic T-lymphocyte associated protein |
| DC | Dendritic cell |
| ERK | Extracellular signal-regulated kinase |
| GTP | Guanosine 5’-triphosphate |
| HGF | Hepatocyte growth factor |
| ICI | Immune checkpoint inhibitor |
| IFN | Interferon |
| IL | Interleukin |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| MDSC | Myeloid-derived suppressor cell |
| MEK | MAPK/ERK kinase |
| MEKi | MEK inhibitor |
| MHC | Major histocompatibility complex |
| MMP | Matrix metalloprotease |
| MO-DC | Monocyte-derived DC |
| NF1 | Neurofibroma 1 |
| NRAS | Neuroblastoma RAS viral oncogene homolog |
| PBMC | Peripheral blood mononuclear cells |
| PD-1 | Programmed cell death |
| PD-L1 | PD-1 ligand 1 |
| pDC | Plasmacytoid DC |
| PI3K | Phosphoinositide 3-kinases |
| PLCγ | Phospholipase Cγ |
| PFS | Progression-free survival |
| PTEN | Phosphatase and tensin homolog |
| RAF | Rat fibrosarcoma |
| RAS | Rat sarcoma |
| RTK | Receptor tyrosine kinase |
| siRNA | Silencer RNA |
| STAT | Signal transducer and activator of transcription |
| TAM | Tumor-associated macrophage |
| TERT | Telomerase reverse transcriptase |
| TIL | Tumor-infiltrating leukocyte |
| TNF | Tumor necrosis factor |
| TLRT | Toll-like receptor |
| TME | Tumor microenvironment |
| Treg | Regulatory T cell |
| VEGF | Vascular endothelial growth factor |
References
- Leiter, U.; Keim, U.; Garbe, C. Epidemiology of Skin Cancer: Update 2019. Adv. Exp. Med. Biol. 2020, 1268, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.-T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, C.; Livingstone, E.; Roesch, A.; Schilling, B.; Schadendorf, D. Immunotherapy in melanoma: Recent advances and future directions. Eur. J. Surg. Oncol. 2017, 43, 604–611. [Google Scholar] [CrossRef]
- Singh, S.; Numan, A.; Agrawal, N.; Tambuwala, M.M.; Singh, V.; Kesharwani, P. Role of immune checkpoint inhibitors in the revolutionization of advanced melanoma care. Int. Immunopharmacol. 2020, 83, 106417. [Google Scholar] [CrossRef]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef]
- Czarnecka, A.M.; Bartnik, E.; Fiedorowicz, M.; Rutkowski, P. Targeted Therapy in Melanoma and Mechanisms of Resistance. Int. J. Mol. Sci. 2020, 21, 4576. [Google Scholar] [CrossRef]
- Savoia, P.; Fava, P.; Casoni, F.; Cremona, O. Targeting the ERK Signaling Pathway in Melanoma. Int. J. Mol. Sci. 2019, 20, 1483. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proietti, I.; Skroza, N.; Michelini, S.; Mambrin, A.; Balduzzi, V.; Bernardini, N.; Marchesiello, A.; Tolino, E.; Volpe, S.; Maddalena, P.; et al. BRAF Inhibitors: Molecular Targeting and Immunomodulatory Actions. Cancers 2020, 12, 1823. [Google Scholar] [CrossRef] [PubMed]
- Aplin, A.E.; Kaplan, F.M.; Shao, Y. Mechanisms of resistance to RAF inhibitors in melanoma. J. Invest. Dermatol. 2011, 131, 1817–1820. [Google Scholar] [CrossRef] [Green Version]
- Ottaviano, M.; Giunta, E.F.; Tortora, M.; Curvietto, M.; Attademo, L.; Bosso, D.; Cardalesi, C.; Rosanova, M.; De Placido, P.; Pietroluongo, E.; et al. BRAF Gene and Melanoma: Back to the Future. Int. J. Mol. Sci. 2021, 22, 3474. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilmott, J.S.; Long, G.V.; Howle, J.R.; Haydu, L.E.; Sharma, R.N.; Thompson, J.F.; Kefford, R.F.; Hersey, P.; Scolyer, R.A. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin. Cancer Res. 2012, 18, 1386–1394. [Google Scholar] [CrossRef] [Green Version]
- Hajek, E.; Krebs, F.; Bent, R.; Haas, K.; Bast, A.; Steinmetz, I.; Tuettenberg, A.; Grabbe, S.; Bros, M. BRAF inhibitors stimulate inflammasome activation and interleukin 1 beta production in dendritic cells. Oncotarget 2018, 9, 28294–28308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karki, R.; Kanneganti, T.D. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat. Rev. Cancer 2019, 19, 197–214. [Google Scholar] [CrossRef]
- Bent, R.; Moll, L.; Grabbe, S.; Bros, M. Interleukin-1 Beta-A Friend or Foe in Malignancies? Int. J. Mol. Sci. 2018, 19, 2155. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Bhat, T.; Gutmann, D.H.; Johnson, K.J. Melanoma in individuals with neurofibromatosis type 1: A retrospective study. Dermatol. Online J. 2019, 25, 2. [Google Scholar]
- Delyon, J.; Lebbe, C.; Dumaz, N. Targeted therapies in melanoma beyond BRAF: Targeting NRAS-mutated and KIT-mutated melanoma. Curr. Opin. Oncol. 2020, 32, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef]
- Dong, J.; Phelps, R.G.; Qiao, R.; Yao, S.; Benard, O.; Ronai, Z.; Aaronson, S.A. BRAF oncogenic mutations correlate with progression rather than initiation of human melanoma. Cancer Res. 2003, 63, 3883–3885. [Google Scholar]
- Liang, W.S.; Hendricks, W.; Kiefer, J.; Schmidt, J.; Sekar, S.; Carpten, J.; Craig, D.W.; Adkins, J.; Cuyugan, L.; Manojlovic, Z.; et al. Integrated genomic analyses reveal frequent TERT aberrations in acral melanoma. Genome Res. 2017, 27, 524–532. [Google Scholar] [CrossRef] [Green Version]
- Hernando, B.; Swope, V.B.; Guard, S.; Starner, R.J.; Choi, K.; Anwar, A.; Cassidy, P.; Leachman, S.; Kadekaro, A.L.; Bennett, D.C.; et al. In vitro behavior and UV response of melanocytes derived from carriers of CDKN2A mutations and MC1R variants. Pigment Cell Melanoma Res. 2019, 32, 259–268. [Google Scholar] [CrossRef]
- Mandal, R.; Becker, S.; Strebhardt, K. Stamping out RAF and MEK1/2 to inhibit the ERK1/2 pathway: An emerging threat to anticancer therapy. Oncogene 2016, 35, 2547–2561. [Google Scholar] [CrossRef] [PubMed]
- Śmiech, M.; Leszczyński, P.; Kono, H.; Wardell, C.; Taniguchi, H. Emerging BRAF Mutations in Cancer Progression and Their Possible Effects on Transcriptional Networks. Genes 2020, 11, 1342. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; MacLennan, G.T.; Montironi, R. Molecular testing for BRAF mutations to inform melanoma treatment decisions: A move toward precision medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Couselo, E.; Adelantado, E.Z.; Ortiz, C.; García, J.S.; Perez-Garcia, J. NRAS-mutant melanoma: Current challenges and future prospect. Oncotargets Ther. 2017, 10, 3941–3947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sensi, M.; Nicolini, G.; Petti, C.; Bersani, I.; Lozupone, F.; Molla, A.; Vegetti, C.; Nonaka, D.; Mortarini, R.; Parmiani, G.; et al. Mutually exclusive NRASQ61R and BRAFV600E mutations at the single-cell level in the same human melanoma. Oncogene 2006, 25, 3357–3364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauthammer, M.; Kong, Y.; Bacchiocchi, A.; Evans, P.; Pornputtapong, N.; Wu, C.; McCusker, J.P.; Ma, S.; Cheng, E.; Straub, R.; et al. Exome sequencing identifies recurrent mutations in NF1 and RAS opathy genes in sun-exposed melanomas. Nat. Genet. 2015, 47, 996–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiuru, M.; Busam, K.J. The NF1 gene in tumor syndromes and melanoma. Lab. Investig. 2017, 97, 146–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nissan, M.H.; Pratilas, C.A.; Jones, A.M.; Ramirez, R.; Won, H.; Liu, C.; Tiwari, S.; Kong, L.; Hanrahan, A.J.; Yao, Z.; et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014, 74, 2340–2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef]
- Smith, S.L.; Pitt, A.R.; Spickett, C.M. Approaches to Investigating the Protein Interactome of PTEN. J. Proteome Res. 2021, 20, 60–77. [Google Scholar] [CrossRef] [PubMed]
- Aguissa-Touré, A.H.; Li, G. Genetic alterations of PTEN in human melanoma. Cell. Mol. Life Sci. 2012, 69, 1475–1491. [Google Scholar] [CrossRef]
- Hilke, F.J.; Sinnberg, T.; Gschwind, A.; Niessner, H.; Demidov, G.; Amaral, T.; Ossowski, S.; Bonzheim, I.; Röcken, M.; Riess, O.; et al. Distinct Mutation Patterns Reveal Melanoma Subtypes and Influence Immunotherapy Response in Advanced Melanoma Patients. Cancers 2020, 12, 2359. [Google Scholar] [CrossRef] [PubMed]
- Dankort, D.; Curley, D.P.; Cartlidge, R.A.; Nelson, B.; Karnezis, A.N.; Damsky, W.E., Jr.; You, M.J.; DePinho, R.A.; McMahon, M.; Bosenberg, M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat. Genet. 2009, 41, 544–552. [Google Scholar] [CrossRef] [Green Version]
- Bradley, S.D.; Chen, Z.; Melendez, B.; Talukder, A.; Khalili, J.S.; Rodriguez-Cruz, T.; Liu, S.; Whittington, M.; Deng, W.; Li, F.; et al. BRAFV600E Co-opts a Conserved MHC Class I Internalization Pathway to Diminish Antigen Presentation and CD8+ T-cell Recognition of Melanoma. Cancer Immunol. Res. 2015, 3, 602–609. [Google Scholar] [CrossRef] [Green Version]
- Shabaneh, T.B.; Molodtsov, A.K.; Steinberg, S.M.; Zhang, P.; Torres, G.M.; Mohamed, G.A.; Boni, A.; Curiel, T.J.; Angeles, C.V.; Turk, M.J. Oncogenic BRAF(V600E) Governs Regulatory T-cell Recruitment during Melanoma Tumorigenesis. Cancer Res. 2018, 78, 5038–5049. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.-C.; Meeth, K.M.; Tsui, Y.-C.; Srivastava, B.; Bosenberg, M.W.; Kaech, S.M. Immune-Based Antitumor Effects of BRAF Inhibitors Rely on Signaling by CD40L and IFNγ. Cancer Res. 2014, 74, 3205–3217. [Google Scholar] [CrossRef] [Green Version]
- Leslie, C.; Bowyer, S.E.; White, A.; Grieu-Iacopetta, F.; Trevenen, M.; Iacopetta, B.; Amanuel, B.; Millward, M. FOXP3+ T regulatory lymphocytes in primary melanoma are associated with BRAF mutation but not with response to BRAF inhibitor. Pathology 2015, 47, 557–563. [Google Scholar] [CrossRef]
- Kuske, M.; Westphal, D.; Wehner, R.; Schmitz, M.; Beissert, S.; Praetorius, C.; Meier, F. Immunomodulatory effects of BRAF and MEK inhibitors: Implications for Melanoma therapy. Pharmacol. Res. 2018, 136, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med. 2006, 203, 1651–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, P.A.; Henry, T.; Baranda, S.J.; Frleta, D.; Manches, O.; Bogunovic, D.; Bhardwaj, N. Inhibition of both BRAF and MEK in BRAF(V600E) mutant melanoma restores compromised dendritic cell (DC) function while having differential direct effects on DC properties. Cancer Immunol. Immunother. 2013, 62, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Jeon, S.; Kim, T.M.; Jung, C.K. Immune Gene Signature Delineates a Subclass of Papillary Thyroid Cancer with Unfavorable Clinical Outcomes. Cancers 2018, 10, 494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalili, J.S.; Liu, S.; Rodríguez-Cruz, T.G.; Whittington, M.; Wardell, S.; Liu, C.; Zhang, M.; Cooper, Z.A.; Frederick, D.T.; Li, Y.; et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin. Cancer Res. 2012, 18, 5329–5340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Whipple, C.A.; Boni, A.; Fisher, J.L.; Hampton, T.H.; Tsongalis, G.J.; Mellinger, D.L.; Yan, S.; Tafe, L.J.; Brinckerhoff, C.E.; Turk, M.J.; et al. The mitogen-activated protein kinase pathway plays a critical role in regulating immunological properties of BRAF mutant cutaneous melanoma cells. Melanoma Res. 2016, 26, 223–235. [Google Scholar] [CrossRef]
- Terai, M.; Eto, M.; Young, G.D.; Berd, D.; Mastrangelo, M.J.; Tamura, Y.; Harigaya, K.; Sato, T. Interleukin 6 mediates production of interleukin 10 in metastatic melanoma. Cancer Immunol. Immunother. 2012, 61, 145–155. [Google Scholar] [CrossRef]
- Caporali, S.; Alvino, E.; Lacal, P.M.; Levati, L.; Giurato, G.; Memoli, D.; Caprini, E.; Antonini Cappellini, G.C.; D’Atri, S. Targeting the PI3K/AKT/mTOR pathway overcomes the stimulating effect of dabrafenib on the invasive behavior of melanoma cells with acquired resistance to the BRAF inhibitor. Int. J. Oncol. 2016, 49, 1164–1174. [Google Scholar] [CrossRef]
- Dowling, J.; McGregor, S.P.; Williford, P. Update on Current Treatment Recommendations for Primary Cutaneous Melanoma. Dermatol. Clin. 2019, 37, 397–407. [Google Scholar] [CrossRef]
- Corrie, P.; Hategan, M.; Fife, K.; Parkinson, C. Management of melanoma. Br. Med. Bull. 2014, 111, 149–162. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, R.J. The role of targeted therapy for melanoma in the immunotherapy era. Semin. Cutan. Med. Surg. 2018, 37, 112–119. [Google Scholar] [CrossRef]
- Ny, L.; Hernberg, M.; Nyakas, M.; Koivunen, J.; Oddershede, L.; Yoon, M.; Wang, X.; Guyot, P.; Geisler, J. BRAF mutational status as a prognostic marker for survival in malignant melanoma: A systematic review and meta-analysis. Acta Oncol. 2020, 59, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Patel, H.; Yacoub, N.; Mishra, R.; White, A.; Long, Y.; Alanazi, S.; Garrett, J.T. Current Advances in the Treatment of BRAF-Mutant Melanoma. Cancers 2020, 12, 482. [Google Scholar] [CrossRef] [Green Version]
- Su, F.; Viros, A.; Milagre, C.; Trunzer, K.; Bollag, G.; Spleiss, O.; Reis-Filho, J.S.; Kong, X.; Koya, R.C.; Flaherty, K.T.; et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N. Engl. J. Med. 2012, 366, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzano, J.L.; Layos, L.; Bugés, C.; de Los Llanos Gil, M.; Vila, L.; Martínez-Balibrea, E.; Martínez-Cardús, A. Resistant mechanisms to BRAF inhibitors in melanoma. Ann. Transl. Med. 2016, 4, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 2012, 22, 668–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emery, C.M.; Vijayendran, K.G.; Zipser, M.C.; Sawyer, A.M.; Niu, L.; Kim, J.J.; Hatton, C.; Chopra, R.; Oberholzer, P.A.; Karpova, M.B.; et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc. Natl. Acad. Sci. USA 2009, 106, 20411–20416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagle, N.; Van Allen, E.M.; Treacy, D.J.; Frederick, D.T.; Cooper, Z.A.; Taylor-Weiner, A.; Rosenberg, M.; Goetz, E.M.; Sullivan, R.J.; Farlow, D.N.; et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014, 4, 61–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dratkiewicz, E.; Simiczyjew, A.; Mazurkiewicz, J.; Ziętek, M.; Matkowski, R.; Nowak, D. Hypoxia and Extracellular Acidification as Drivers of Melanoma Progression and Drug Resistance. Cells 2021, 10, 862. [Google Scholar] [CrossRef]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [Green Version]
- Hirata, E.; Girotti, M.R.; Viros, A.; Hooper, S.; Spencer-Dene, B.; Matsuda, M.; Larkin, J.; Marais, R.; Sahai, E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin β1/FAK signaling. Cancer Cell 2015, 27, 574–588. [Google Scholar] [CrossRef] [Green Version]
- Young, H.L.; Rowling, E.J.; Bugatti, M.; Giurisato, E.; Luheshi, N.; Arozarena, I.; Acosta, J.C.; Kamarashev, J.; Frederick, D.T.; Cooper, Z.A.; et al. An adaptive signaling network in melanoma inflammatory niches confers tolerance to MAPK signaling inhibition. J. Exp. Med. 2017, 214, 1691–1710. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Xiao, M.; Ge, Y.; Krepler, C.; Belser, E.; Lopez-Coral, A.; Xu, X.; Zhang, G.; Azuma, R.; Liu, Q.; et al. BRAF Inhibition Stimulates Melanoma-Associated Macrophages to Drive Tumor Growth. Clin. Cancer Res. 2015, 21, 1652–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arozarena, I.; Wellbrock, C. Overcoming resistance to BRAF inhibitors. Ann. Transl. Med. 2017, 5, 387. [Google Scholar] [CrossRef] [Green Version]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Spain, L.; Julve, M.; Larkin, J. Combination dabrafenib and trametinib in the management of advanced melanoma with BRAFV600 mutations. Expert. Opin. Pharmacother. 2016, 17, 1031–1038. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Dummer, R.; Gogas, H.J.; Flaherty, K.T.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; et al. Update on tolerability and overall survival in COLUMBUS: Landmark analysis of a randomised phase 3 trial of encorafenib plus binimetinib vs vemurafenib or encorafenib in patients with BRAF V600-mutant melanoma. Eur. J. Cancer 2020, 126, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Moriceau, G.; Hugo, W.; Hong, A.; Shi, H.; Kong, X.; Yu, C.C.; Koya, R.C.; Samatar, A.A.; Khanlou, N.; Braun, J.; et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 2015, 27, 240–256. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Liu, S.; Zhang, G.; Bin, W.; Zhu, Y.; Frederick, D.T.; Hu, Y.; Zhong, W.; Randell, S.; Sadek, N.; et al. PAK signalling drives acquired drug resistance to MAPK inhibitors in BRAF-mutant melanomas. Nature 2017, 550, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Broman, K.K.; Dossett, L.A.; Sun, J.; Eroglu, Z.; Zager, J.S. Update on BRAF and MEK inhibition for treatment of melanoma in metastatic, unresectable, and adjuvant settings. Expert Opin. Drug Saf. 2019, 18, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Dumaz, N.; Lebbé, C. New perspectives on targeting RAF, MEK and ERK in melanoma. Curr. Opin. Oncol. 2021, 33, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Lavoie, H.; Sahmi, M.; David, M.; Hilt, C.; Hammell, A.; Therrien, M. RAF inhibitors promote RAS-RAF interaction by allosterically disrupting RAF autoinhibition. Nat. Commun. 2017, 8, 1211. [Google Scholar] [CrossRef] [Green Version]
- Karoulia, Z.; Wu, Y.; Ahmed, T.A.; Xin, Q.; Bollard, J.; Krepler, C.; Wu, X.; Zhang, C.; Bollag, G.; Herlyn, M.; et al. An Integrated Model of RAF Inhibitor Action Predicts Inhibitor Activity against Oncogenic BRAF Signaling. Cancer Cell 2016, 30, 485–498. [Google Scholar] [CrossRef] [Green Version]
- Karoulia, Z.; Gavathiotis, E.; Poulikakos, P.I. New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 2017, 17, 676–691. [Google Scholar] [CrossRef]
- Mukai, K.; Kamata, M.; Miyazaki, M.; Nagata, M.; Fukaya, S.; Hayashi, K.; Fukuyasu, A.; Ishikawa, T.; Ohnishi, T.; Tada, Y.; et al. Edoxaban prevented adverse effects including pyrexia and elevation of D-dimer caused by the combination of BRAF and MEK inhibitors in a patient with BRAF-mutant melanoma. J. Dermatol. 2021, 48, 707–709. [Google Scholar] [CrossRef]
- Callahan, M.K.; Masters, G.; Pratilas, C.A.; Ariyan, C.; Katz, J.; Kitano, S.; Russell, V.; Gordon, R.A.; Vyas, S.; Yuan, J.; et al. Paradoxical activation of T cells via augmented ERK signaling mediated by a RAF inhibitor. Cancer Immunol. Res. 2014, 2, 70–79. [Google Scholar] [CrossRef] [Green Version]
- Arthur, J.S.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [Green Version]
- Heidorn, S.J.; Milagre, C.; Whittaker, S.; Nourry, A.; Niculescu-Duvas, I.; Dhomen, N.; Hussain, J.; Reis-Filho, J.S.; Springer, C.J.; Pritchard, C.; et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010, 140, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Wang, Y.; Hong, Y.; Ye, X.; Shi, P.; Zhang, J.; Zhao, Q. Incidence and relative risk of cutaneous squamous cell carcinoma with single-agent BRAF inhibitor and dual BRAF/MEK inhibitors in cancer patients: A meta-analysis. Oncotarget 2017, 8, 83280–83291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Mayes, P.A.; Eastman, S.; Shi, H.; Yadavilli, S.; Zhang, T.; Yang, J.; Seestaller-Wehr, L.; Zhang, S.Y.; Hopson, C.; et al. The BRAF and MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination with Immunomodulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. 2015, 21, 1639–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frederick, D.T.; Piris, A.; Cogdill, A.P.; Cooper, Z.A.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin. Cancer Res. 2013, 19, 1225–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schilling, B.; Paschen, A. Immunological consequences of selective BRAF inhibitors in malignant melanoma: Neutralization of myeloid-derived suppressor cells. Oncoimmunology 2013, 2, e25218. [Google Scholar] [CrossRef] [Green Version]
- Hu-Lieskovan, S.; Mok, S.; Homet Moreno, B.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci. Transl. Med. 2015, 7, 279ra241. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.P.; Sanchez-Laorden, B.; O’Brien, K.; Brunton, H.; Ferguson, J.; Young, H.; Dhomen, N.; Flaherty, K.T.; Frederick, D.T.; Cooper, Z.A.; et al. The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage-derived TNFα. Cancer Discov. 2014, 4, 1214–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tel, J.; Koornstra, R.; de Haas, N.; van Deutekom, V.; Westdorp, H.; Boudewijns, S.; van Erp, N.; Di Blasio, S.; Gerritsen, W.; Figdor, C.G.; et al. Preclinical exploration of combining plasmacytoid and myeloid dendritic cell vaccination with BRAF inhibition. J. Transl. Med. 2016, 14, 88. [Google Scholar] [CrossRef] [Green Version]
- Vella, L.J.; Pasam, A.; Dimopoulos, N.; Andrews, M.; Knights, A.; Puaux, A.L.; Louahed, J.; Chen, W.; Woods, K.; Cebon, J.S. MEK inhibition, alone or in combination with BRAF inhibition, affects multiple functions of isolated normal human lymphocytes and dendritic cells. Cancer Immunol. Res. 2014, 2, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.B.; Henry, J.R.; Kaufman, M.D.; Lu, W.P.; Smith, B.D.; Vogeti, S.; Rutkoski, T.J.; Wise, S.; Chun, L.; Zhang, Y.; et al. Inhibition of RAF Isoforms and Active Dimers by LY3009120 Leads to Anti-tumor Activities in RAS or BRAF Mutant Cancers. Cancer Cell 2015, 28, 384–398. [Google Scholar] [CrossRef] [Green Version]
- Riegel, K.; Schlöder, J.; Sobczak, M.; Jonuleit, H.; Thiede, B.; Schild, H.; Rajalingam, K. RAF kinases are stabilized and required for dendritic cell differentiation and function. Cell Death Differ. 2020, 27, 1300–1315. [Google Scholar] [CrossRef] [PubMed]
- Riegel, K.; Rajalingam, K. The non-linearity of RAF-MEK signaling in dendritic cells. Cell Cycle 2020, 19, 2249–2259. [Google Scholar] [CrossRef] [PubMed]
- Schilling, B.; Sondermann, W.; Zhao, F.; Griewank, K.G.; Livingstone, E.; Sucker, A.; Zelba, H.; Weide, B.; Trefzer, U.; Wilhelm, T.; et al. Differential influence of vemurafenib and dabrafenib on patients’ lymphocytes despite similar clinical efficacy in melanoma. Ann. Oncol. 2014, 25, 747–753. [Google Scholar] [CrossRef]
- Ebert, P.J.R.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.; Deng, J.; Li, S.; Silk, T.; Dong, L.; Brea, E.J.; Houghton, S.; Redmond, D.; Zhong, H.; Boiarsky, J.; et al. Pulsatile MEK Inhibition Improves Anti-tumor Immunity and T Cell Function in Murine Kras Mutant Lung Cancer. Cell Rep. 2019, 27, 806–819.e5. [Google Scholar] [CrossRef] [Green Version]
- Mandalà, M.; De Logu, F.; Merelli, B.; Nassini, R.; Massi, D. Immunomodulating property of MAPK inhibitors: From translational knowledge to clinical implementation. Lab. Investig. 2017, 97, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Ascierto, P.A.; Dummer, R. Immunological effects of BRAF+MEK inhibition. OncoImmunology 2018, 7, e1468955. [Google Scholar] [CrossRef]
- Kakavand, H.; Wilmott, J.S.; Menzies, A.M.; Vilain, R.; Haydu, L.E.; Yearley, J.H.; Thompson, J.F.; Kefford, R.F.; Hersey, P.; Long, G.V.; et al. PD-L1 Expression and Tumor-Infiltrating Lymphocytes Define Different Subsets of MAPK Inhibitor-Treated Melanoma Patients. Clin. Cancer Res. 2015, 21, 3140–3148. [Google Scholar] [CrossRef] [Green Version]
- Wilmott, J.S.; Haydu, L.E.; Menzies, A.M.; Lum, T.; Hyman, J.; Thompson, J.F.; Hersey, P.; Kefford, R.F.; Scolyer, R.A.; Long, G.V. Dynamics of Chemokine, Cytokine, and Growth Factor Serum Levels in BRAF-Mutant Melanoma Patients during BRAF Inhibitor Treatment. J. Immunol. 2014, 192, 2505–2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Lawrence, D.; Atkinson, V.; Agarwal, S.; Miller, W.H.; Carlino, M.S.; Fisher, R.; Long, G.V.; Hodi, F.S.; Tsoi, J.; et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat. Med. 2019, 25, 936–940. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Lebbé, C.; Atkinson, V.; Mandalà, M.; Nathan, P.D.; Arance, A.; Richtig, E.; Yamazaki, N.; Robert, C.; Schadendorf, D.; et al. Combined PD-1, BRAF and MEK inhibition in advanced BRAF-mutant melanoma: Safety run-in and biomarker cohorts of COMBI-i. Nat. Med. 2020, 26, 1557–1563. [Google Scholar] [CrossRef]
- Ribas, A.; Algazi, A.; Ascierto, P.A.; Butler, M.O.; Chandra, S.; Gordon, M.; Hernandez-Aya, L.; Lawrence, D.; Lutzky, J.; Miller, W.H., Jr.; et al. PD-L1 blockade in combination with inhibition of MAPK oncogenic signaling in patients with advanced melanoma. Nat. Commun. 2020, 11, 6262. [Google Scholar] [CrossRef]
- Gogas, H.; Dréno, B.; Larkin, J.; Demidov, L.; Stroyakovskiy, D.; Eroglu, Z.; Francesco Ferrucci, P.; Pigozzo, J.; Rutkowski, P.; Mackiewicz, J.; et al. Cobimetinib plus atezolizumab in BRAF(V600) wild-type melanoma: Primary results from the randomized phase III IMspire170 study. Ann. Oncol. 2021, 32, 384–394. [Google Scholar] [CrossRef]
- Varayathu, H.; Sarathy, V.; Thomas, B.E.; Mufti, S.S.; Naik, R. Combination Strategies to Augment Immune Check Point Inhibitors Efficacy—Implications for Translational Research. Front. Oncol. 2021, 11, 559161. [Google Scholar] [CrossRef] [PubMed]
- Yeon, M.; Kim, Y.; Jung, H.S.; Jeoung, D. Histone Deacetylase Inhibitors to Overcome Resistance to Targeted and Immuno Therapy in Metastatic Melanoma. Front. Cell Dev. Biol. 2020, 8, 486. [Google Scholar] [CrossRef]
- Gibbons Johnson, R.M.; Dong, H. Functional Expression of Programmed Death-Ligand 1 (B7-H1) by Immune Cells and Tumor Cells. Front. Immunol. 2017, 8, 961. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, T.; Haist, M.; Kuske, M.; Grabbe, S.; Bros, M. Immunomodulatory Properties of BRAF and MEK Inhibitors Used for Melanoma Therapy—Paradoxical ERK Activation and Beyond. Int. J. Mol. Sci. 2021, 22, 9890. https://doi.org/10.3390/ijms22189890
Jung T, Haist M, Kuske M, Grabbe S, Bros M. Immunomodulatory Properties of BRAF and MEK Inhibitors Used for Melanoma Therapy—Paradoxical ERK Activation and Beyond. International Journal of Molecular Sciences. 2021; 22(18):9890. https://doi.org/10.3390/ijms22189890
Chicago/Turabian StyleJung, Thomas, Maximilian Haist, Michael Kuske, Stephan Grabbe, and Matthias Bros. 2021. "Immunomodulatory Properties of BRAF and MEK Inhibitors Used for Melanoma Therapy—Paradoxical ERK Activation and Beyond" International Journal of Molecular Sciences 22, no. 18: 9890. https://doi.org/10.3390/ijms22189890
APA StyleJung, T., Haist, M., Kuske, M., Grabbe, S., & Bros, M. (2021). Immunomodulatory Properties of BRAF and MEK Inhibitors Used for Melanoma Therapy—Paradoxical ERK Activation and Beyond. International Journal of Molecular Sciences, 22(18), 9890. https://doi.org/10.3390/ijms22189890