
Generation and Characterization of a New FRET-Based Ca2+ Sensor Targeted to the Nucleus

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
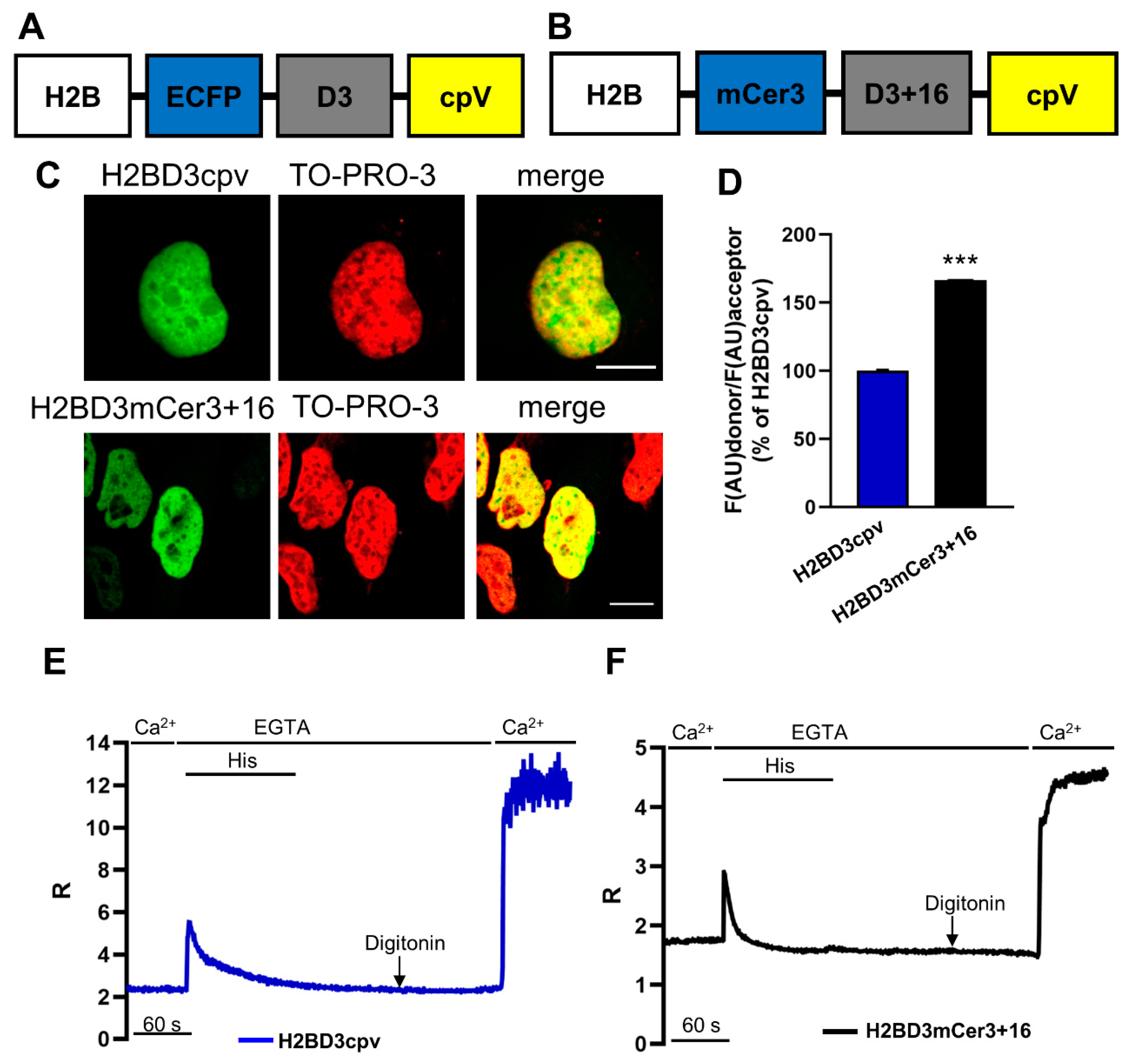
2.1. Generation of H2BD3mCerulean3+16
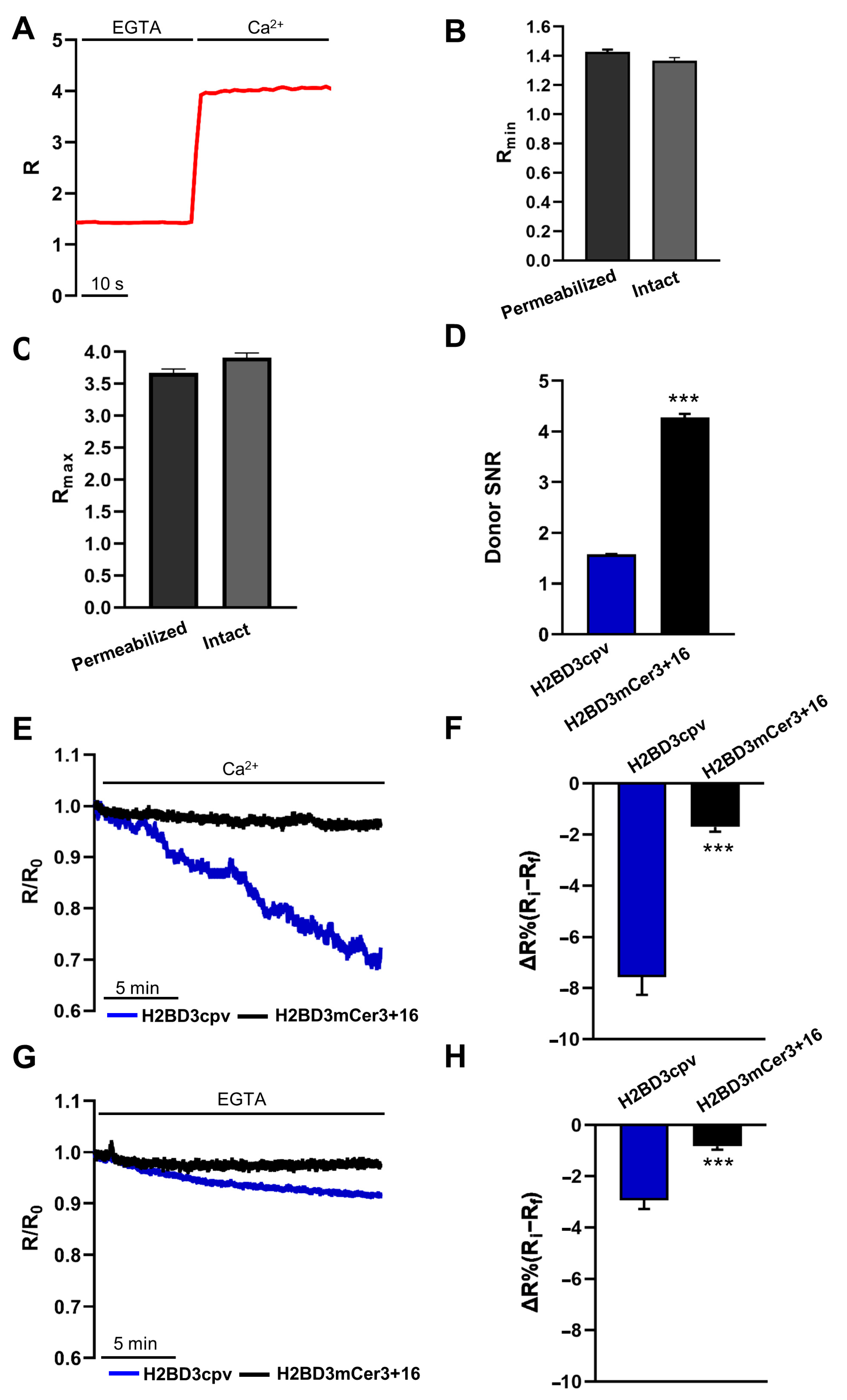
2.2. Biophysical Characterization of Nuclear Cameleons
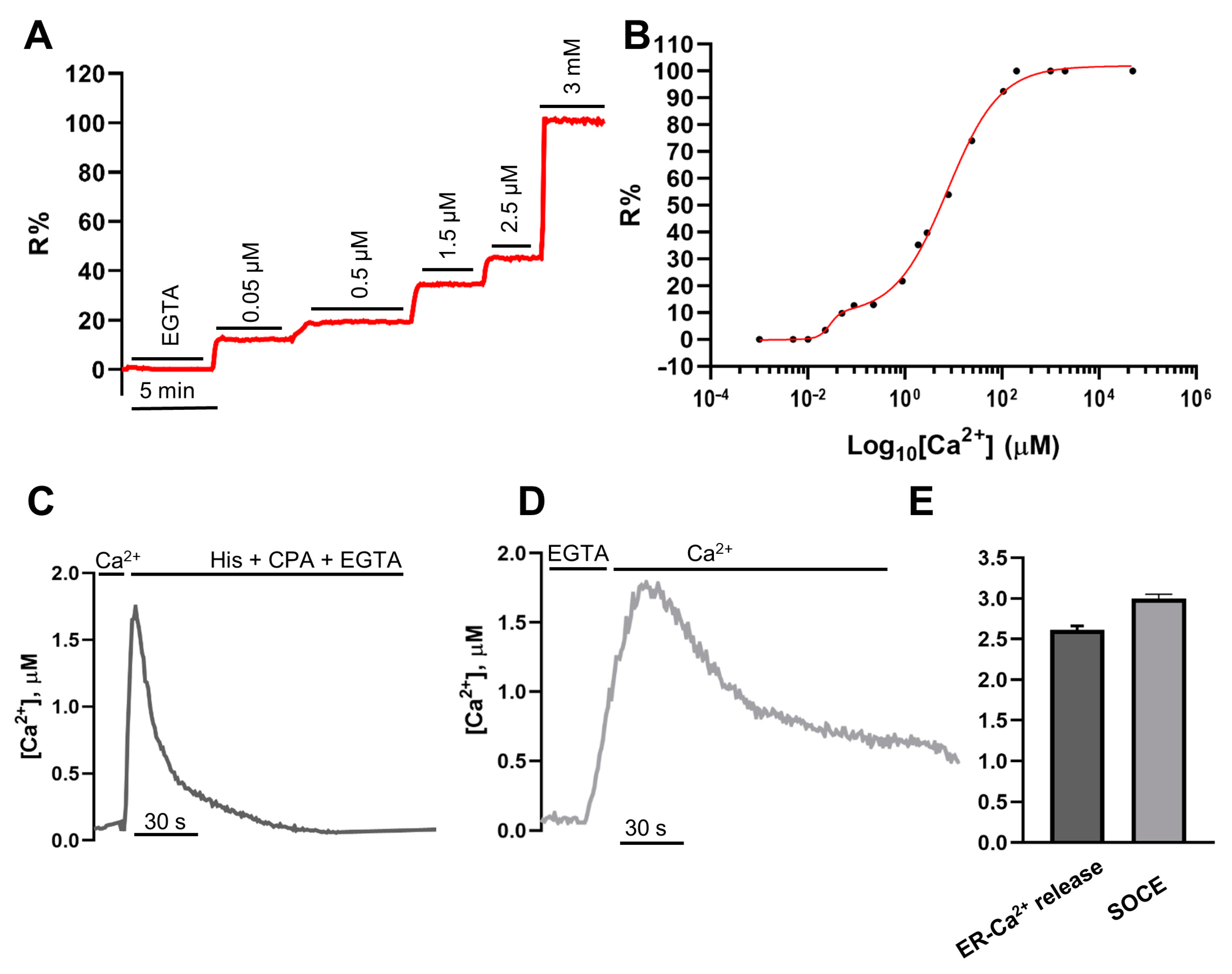
2.3. In Situ Titration of H2BD3mCerulean3+16
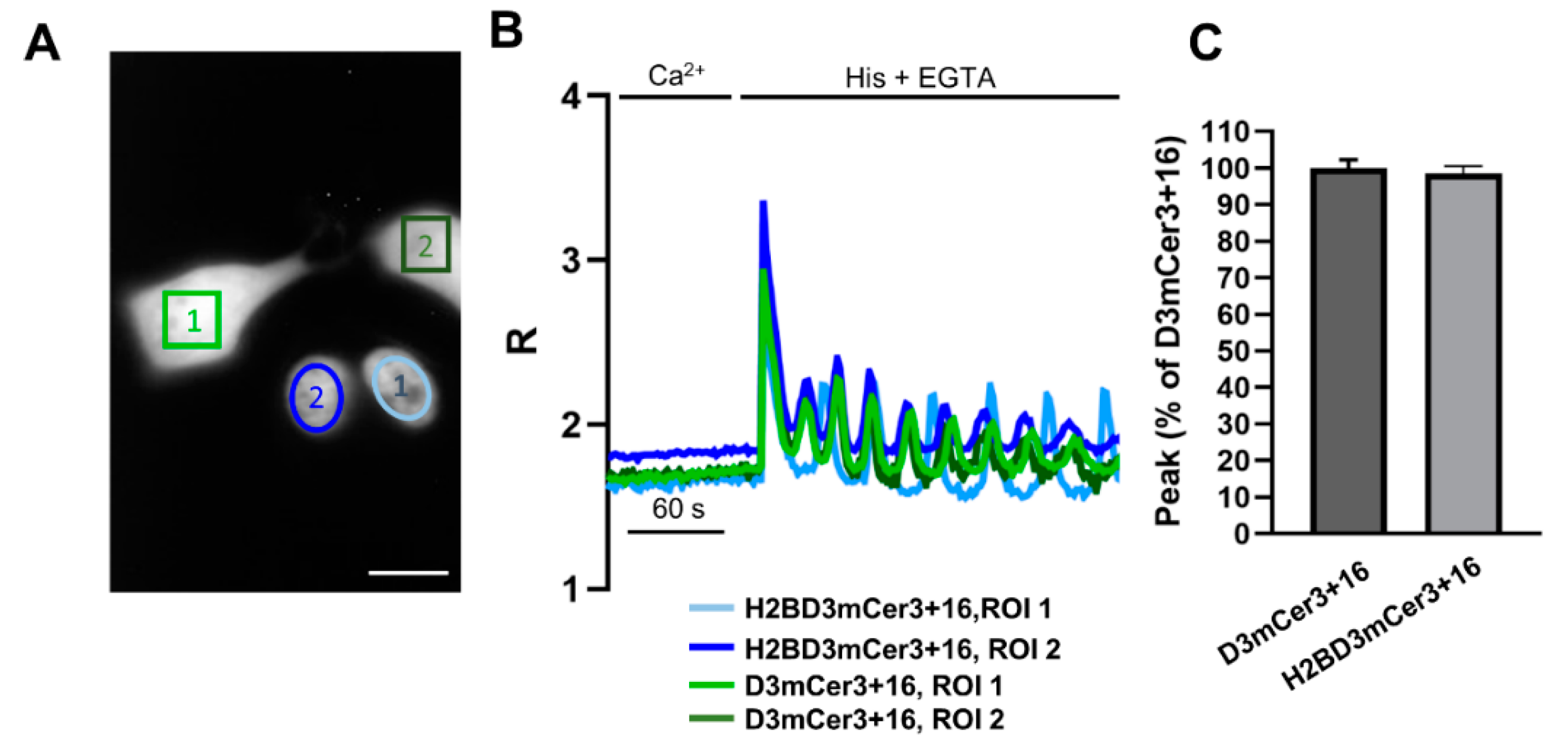
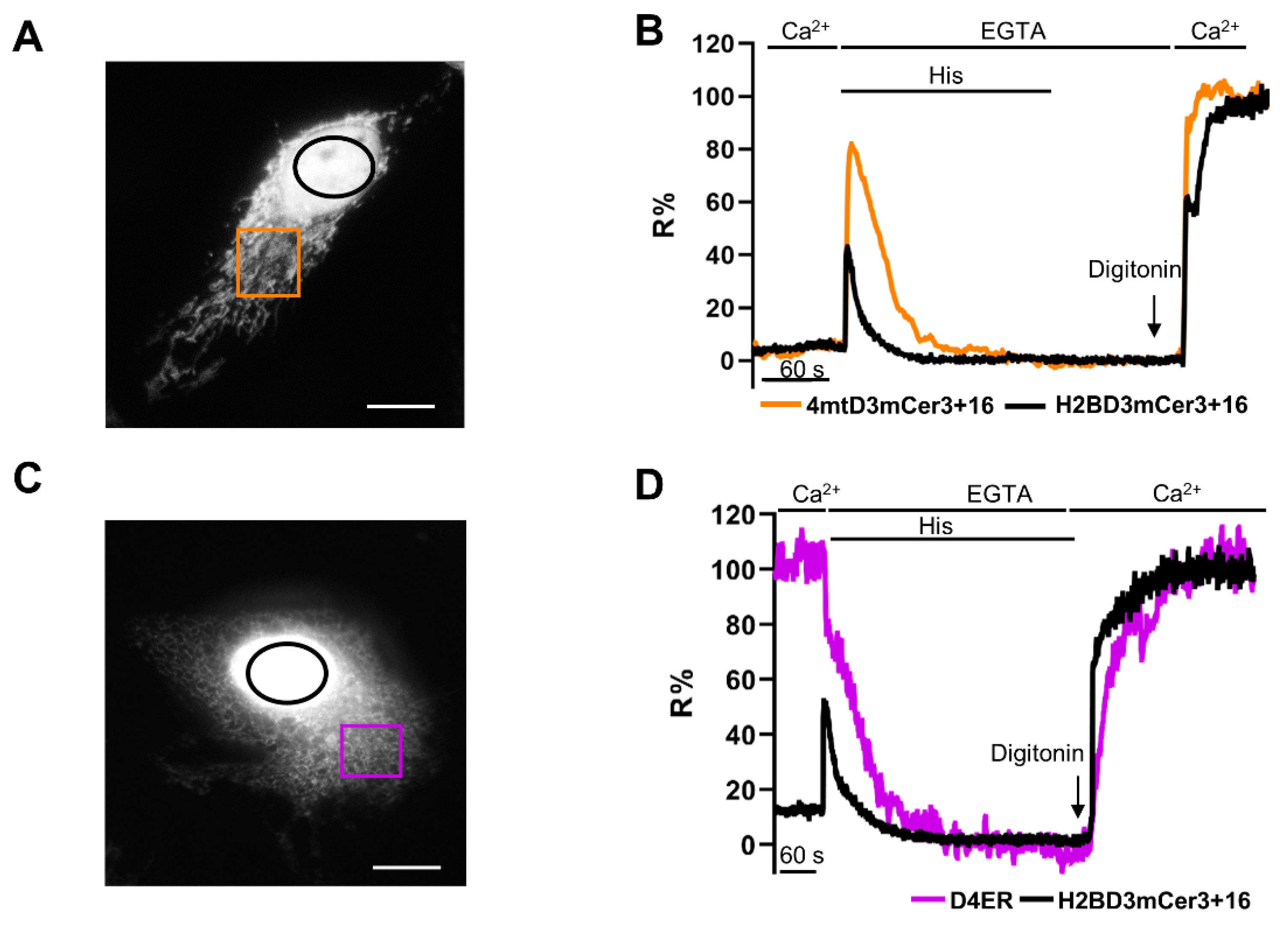
2.4. Simultaneous Monitoring of Nuclear and Cytosolic Ca2+ Signaling
2.5. Simultaneous Monitoring of Nuclear and Organelle Ca2+ Signaling in the Same Cell
3. Discussion
4. Materials and Methods
4.1. Constructs Generation
4.2. Cell Culture and Transfection
4.3. Confocal Analysis of mCerulean3 Fluorescence and Nuclear Localization
4.4. Fluorescence Microscope Settings for FRET Experiments
- (1)
- ER-Ca2+ release protocol: Classical experiments started in EC containing 1 mM CaCl2; after perfusion with 600 µM EGTA, cells were stimulated by perfusing with 100 µM histamine and 20 µM CPA, when indicated.
- (2)
- SOCE-activating protocol: Classical experiments started in EC containing 1 mM CaCl2; after 7 min of perfusion with EC containing 600 µM EGTA in the presence of 100 µM histamine and 20 µM CPA to empty the stores and maximize STIM1 oligomerization and translocation, cells were perfused with EC containing 2 mM CaCl2.
4.5. Buffer Titration with Spectrofluorometer
4.6. Materials
4.7. Data Analysis and Statistics
- -
- A normalized R/R0, where R0 is the R value at the beginning of the experiment (t0) and R is the R value at each time (t) of the experiment;
- -
- R% is calculated as R% = (R − Rmin)/(Rmax − Rmin) × 100;
- -
- [Ca2+] is calculated as described in [19].
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Puri, B.K. Calcium Signaling and Gene Expression. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2020; Volume 1131, pp. 537–545. [Google Scholar]
- Hoelz, A.; Debler, E.W.; Blobel, G. The structure of the nuclear pore complex. Annu. Rev. Biochem. 2011, 80, 613–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Secondo, A.; Petrozziello, T.; Tedeschi, V.; Boscia, F.; Pannaccione, A.; Molinaro, P.; Annunziato, L. Nuclear localization of NCX: Role in Ca2+ handling and pathophysiological implications. Cell Calcium 2020, 86, 102143. [Google Scholar] [CrossRef]
- Thomas, D.; Tovey, S.C.; Collins, T.J.; Bootman, M.D.; Berridge, M.J.; Lipp, P. A comparison of fluorescent Ca2+ indicator properties and their use in measuring elementary and global Ca2+ signals. Cell Calcium 2000, 28, 213–223. [Google Scholar] [CrossRef]
- Echevarria, W.; Leite, M.F.; Guerra, M.T.; Zipfel, W.R.; Nathanson, M.H. Regulation of calcium signals in the nucleus by a nucleoplasmic reticulum. Nat. Cell Biol. 2003, 5, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Brini, M.; Murgia, M.; Pasti, L.; Picard, D.; Pozzan, T.; Rizzuto, R. Nuclear Ca2+ concentration measured with specifically targeted recombinant aequorin. EMBO J. 1993, 12, 4813–4819. [Google Scholar] [CrossRef]
- Giacomello, M.; Drago, I.; Bortolozzi, M.; Scorzeto, M.; Gianelle, A.; Pizzo, P.; Pozzan, T. Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol. Cell 2010, 38, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Bengtson, C.P.; Freitag, H.E.; Weislogel, J.M.; Bading, H. Nuclear calcium sensors reveal that repetition of trains of synaptic stimuli boosts nuclear calcium signaling in CA1 pyramidal neurons. Biophys. J. 2010, 99, 4066–4077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonetti, M.; Hagenston, A.M.; Vardeh, D.; Freitag, H.E.; Mauceri, D.; Lu, J.; Satagopam, V.P.; Schneider, R.; Costigan, M.; Bading, H.; et al. Nuclear Calcium Signaling in Spinal Neurons Drives a Genomic Program Required for Persistent Inflammatory Pain. Neuron 2013, 77, 43–57. [Google Scholar] [CrossRef] [Green Version]
- Hayer, S.N.; Bading, H. Nuclear calcium signaling induces expression of the synaptic organizers Lrrtm1 and Lrrtm2. J. Biol. Chem. 2015, 290, 5523–5532. [Google Scholar] [CrossRef] [Green Version]
- Nakao, S.; Wakabayashi, S.; Nakamura, T.Y. Stimulus-dependent regulation of nuclear Ca2+ signaling in cardiomyocytes: A role of neuronal calcium sensor-1. PLoS ONE 2015, 10, e0125050. [Google Scholar] [CrossRef] [Green Version]
- Greotti, E.; Wong, A.; Pozzan, T.; Pendin, D.; Pizzo, P. Characterization of the ER-Targeted Low Affinity Ca(2+) Probe D4ER. Sensors 2016, 16, 1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Carrillo-Reid, L.; Bando, Y.; Peterka, D.S.; Yuste, R. Simultaneous two-photon imaging and two-photon optogenetics of cortical circuits in three dimensions. Elife 2018, 7, e32671. [Google Scholar] [CrossRef] [PubMed]
- Kelner, A.; Leitão, N.; Chabaud, M.; Charpentier, M.; de Carvalho-Niebel, F. Dual color sensors for simultaneous analysis of calcium signal dynamics in the nuclear and cytoplasmic compartments of plant cells. Front. Plant Sci. 2018, 9, 245. [Google Scholar] [CrossRef] [Green Version]
- Greotti, E.; Fortunati, I.; Pendin, D.; Ferrante, C.; Galla, L.; Zentilin, L.; Giacca, M.; Kaludercic, N.; Di Sante, M.; Mariotti, L.; et al. mCerulean3-Based Cameleon Sensor to Explore Mitochondrial Ca2+ Dynamics In Vivo. iScience 2019, 16, 340–355. [Google Scholar] [CrossRef] [Green Version]
- Palmer, A.E.; Giacomello, M.; Kortemme, T.; Hires, S.A.; Lev-Ram, V.; Baker, D.; Tsien, R.Y. Ca2+ Indicators Based on Computationally Redesigned Calmodulin-Peptide Pairs. Chem. Biol. 2006, 13, 521–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markwardt, M.L.; Kremers, G.J.; Kraft, C.A.; Ray, K.; Cranfill, P.J.C.; Wilson, K.A.; Day, R.N.; Wachter, R.M.; Davidson, M.W.; Rizzo, M.A. An improved cerulean fluorescent protein with enhanced brightness and reduced reversible photoswitching. PLoS ONE 2011, 6, e17896. [Google Scholar] [CrossRef]
- Palmer, A.E.; Tsien, R.Y. Measuring calcium signaling using genetically targetable fluorescent indicators. Nat. Protoc. 2006, 1, 1057–1065. [Google Scholar] [CrossRef]
- Putney, J.W. Forms and functions of store-operated calcium entry mediators, STIM and Orai. Adv. Biol. Regul. 2018, 68, 88–96. [Google Scholar] [CrossRef]
- Thestrup, T.; Litzlbauer, J.; Bartholomaus, I.; Mues, M.; Russo, L.; Dana, H.; Kovalchuk, Y.; Liang, Y.; Kalamakis, G.; Laukat, Y.; et al. Optimized ratiometric calcium sensors for functional in vivo imaging of neurons and T lymphocytes. Nat. Methods 2014, 11, 175–182. [Google Scholar] [CrossRef]
- Miyawaki, A.; Llopis, J.; Heim, R.; McCaffery, J.M.; Adams, J.A.; Ikura, M.; Tsien, R.Y. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 1997, 388, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Manjarres, I.M.; Chamero, P.; Domingo, B.; Molina, F.; Llopis, J.; Alonso, M.T.; Garcia-Sancho, J. Red and green aequorins for simultaneous monitoring of Ca2+ signals from two different organelles. Pflügers Arch. 2008, 455, 961–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drago, I.; De Stefani, D.; Rizzuto, R.; Pozzan, T. Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 12986–12991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lia, A.; Henriques, V.J.; Zonta, M.; Chiavegato, A.; Carmignoto, G.; Gómez-Gonzalo, M.; Losi, G. Calcium Signals in Astrocyte Microdomains, a Decade of Great Advances. Front. Cell. Neurosci. 2021, 15, 177. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H2BD3mCerulean3+16 | DR | Kd (µM) | N |
|---|---|---|---|
| Kd1 | 2.95 ± 0.06 | 0.03 ± 0.01 | 4.1 ± 4.6 |
| Kd2 | 7.57 ± 0.81 | 0.8 ± 0.06 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galla, L.; Vajente, N.; Pendin, D.; Pizzo, P.; Pozzan, T.; Greotti, E. Generation and Characterization of a New FRET-Based Ca2+ Sensor Targeted to the Nucleus. Int. J. Mol. Sci. 2021, 22, 9945. https://doi.org/10.3390/ijms22189945
Galla L, Vajente N, Pendin D, Pizzo P, Pozzan T, Greotti E. Generation and Characterization of a New FRET-Based Ca2+ Sensor Targeted to the Nucleus. International Journal of Molecular Sciences. 2021; 22(18):9945. https://doi.org/10.3390/ijms22189945
Chicago/Turabian StyleGalla, Luisa, Nicola Vajente, Diana Pendin, Paola Pizzo, Tullio Pozzan, and Elisa Greotti. 2021. "Generation and Characterization of a New FRET-Based Ca2+ Sensor Targeted to the Nucleus" International Journal of Molecular Sciences 22, no. 18: 9945. https://doi.org/10.3390/ijms22189945
APA StyleGalla, L., Vajente, N., Pendin, D., Pizzo, P., Pozzan, T., & Greotti, E. (2021). Generation and Characterization of a New FRET-Based Ca2+ Sensor Targeted to the Nucleus. International Journal of Molecular Sciences, 22(18), 9945. https://doi.org/10.3390/ijms22189945