
Good Cholesterol Gone Bad? HDL and COVID-19

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Virus Infection and Cholesterol
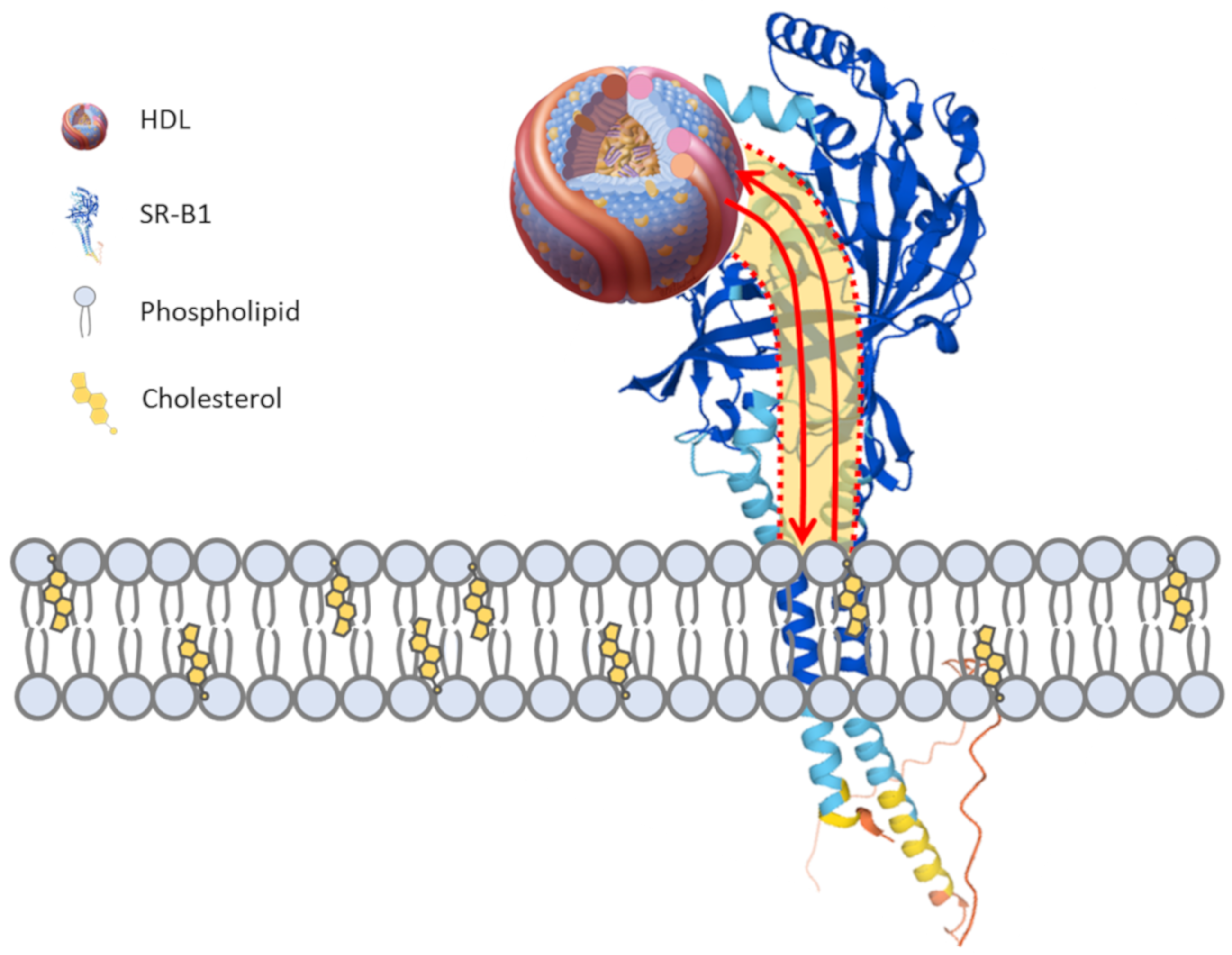
3. The HDL Receptor SR-B1
4. SR-B1 and Viral Infection
5. HDL Structural Complexity and Function
6. Anti-Oxidative and Anti-Inflammatory Properties of HDL
7. Effects of Chronic Disease on HDL Structure and Function and Implications for COVID-19
8. HDL Alterations in COVID-19
9. Spike Protein, HDL, and Endothelial Cell Function
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A.D.M.E.; et al. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J. Pathol. 2020, 251, 228–248. [Google Scholar] [CrossRef] [PubMed]
- Coronavirus Disease (COVID-19). Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 27 August 2021).
- Glebov, O.O. Understanding SARS-CoV-2 endocytosis for COVID-19 drug repurposing. FEBS J. 2020, 287, 3664–3671. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e5. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Herzog, P.; Pfefferle, S.; Steffen, I.; Muench, M.O.; Simmons, G.; Hofmann, H.; Kuri, T.; Weber, F.; et al. Differential Downregulation of ACE2 by the Spike Proteins of Severe Acute Respiratory Syndrome Coronavirus and Human Coronavirus NL63. J. Virol. 2010, 84, 1198–1205. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020, 12, 8. [Google Scholar] [CrossRef]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Sluimer, J.C.; Gasc, J.M.; Hamming, I.; Van Goor, H.; Michaud, A.; Van Den Akker, L.H.; Jütten, B.; Cleutjens, J.; Bijnens, A.P.J.J.; Corvol, P.; et al. Angiotensin-converting enzyme 2 (ACE2) expression and activity in human carotid atherosclerotic lesions. J. Pathol. 2008, 215, 273–279. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.Y.; Ma, Y.T.; Zhang, J.Y.; Xie, X. COVID-19 and the cardiovascular system. Nat. Rev. Cardiol. 2020, 17, 259–260. [Google Scholar] [CrossRef] [Green Version]
- Kochi, A.N.; Tagliari, A.P.; Forleo, G.B.; Fassini, G.M.; Tondo, C. Cardiac and arrhythmic complications in patients with COVID-19. J. Cardiovasc. Electrophysiol. 2020, 31, 1003–1008. [Google Scholar] [CrossRef] [Green Version]
- Nicin, L.; Abplanalp, W.T.; Mellentin, H.; Kattih, B.; Tombor, L.; John, D.; Schmitto, J.D.; Heineke, J.; Emrich, F.; Arsalan, M.; et al. Cell type-specific expression of the putative SARS-CoV-2 receptor ACE2 in human hearts. Eur. Heart J. 2020, 41, 1804–1806. [Google Scholar] [CrossRef] [Green Version]
- Thum, T. SARS-CoV-2 receptor ACE2 expression in the human heart: Cause of a post-pandemic wave of heart failure? Eur. Heart J. 2020, 41, 1807–1809. [Google Scholar] [CrossRef]
- Hu, X.; Chen, D.; Wu, L.; He, G.; Ye, W. Low Serum Cholesterol Level Among Patients with COVID-19 Infection in Wenzhou, China. SSRN Electron. J. 2020. [Google Scholar] [CrossRef]
- Wei, X.; Zeng, W.; Su, J.; Wan, H.; Yu, X.; Cao, X.; Tan, W.; Wang, H. Hypolipidemia is associated with the severity of COVID-19. J. Clin. Lipidol. 2020, 14, 297–304. [Google Scholar] [CrossRef]
- Tanaka, S.; De Tymowski, C.; Assadi, M.; Zappella, N.; Jean-Baptiste, S.; Robert, T.; Peoch, K.; Lortat-Jacob, B.; Fontaine, L.; Bouzid, D.; et al. Lipoprotein concentrations over time in the intensive care unit COVID-19 patients: Results from the ApoCOVID study. PLoS ONE 2020, 15, e0239573. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, Q.; Zhao, X.; Dong, H.; Wu, C.; Wu, F.; Yu, B.; Lv, J.; Zhang, S.; Wu, G.; et al. Low high-density lipoprotein level is correlated with the severity of COVID-19 patients: An observational study. Lipids Health Dis. 2020, 19. [Google Scholar] [CrossRef]
- Hu, X.; Chen, D.; Wu, L.; He, G.; Ye, W. Declined serum high density lipoprotein cholesterol is associated with the severity of COVID-19 infection. Clin. Chim. Acta 2020, 510, 105–110. [Google Scholar] [CrossRef]
- Wang, H.; Yuan, Z.; Pavel, M.A.; Hobson, R.; Hansen, S. The role of high cholesterol in age-related COVID19 lethality. bioRxiv Prepr. Serv. Biol. 2020. [Google Scholar] [CrossRef]
- Ding, X.; Zhang, J.; Liu, L.; Yuan, X.; Zang, X.; Lu, F.; He, P.; Wang, Q.; Zhang, X.; Xu, Y.; et al. High-density lipoprotein cholesterol as a factor affecting virus clearance in covid-19 patients. Respir. Med. 2020, 175, 106218. [Google Scholar] [CrossRef]
- Roccaforte, V.; Daves, M.; Lippi, G.; Spreafico, M.; Bonato, C. Altered lipid profile in patients with COVID-19 infection. J. Lab. Precis. Med. 2021, 6. [Google Scholar] [CrossRef]
- Caricchio, R.; Gallucci, M.; Dass, C.; Zhang, X.; Gallucci, S.; Fleece, D.; Bromberg, M.; Criner, G.J. Preliminary predictive criteria for COVID-19 cytokine storm. Ann. Rheum. Dis. 2021, 80, 88–95. [Google Scholar] [CrossRef]
- Wei, C.; Wan, L.; Yan, Q.; Wang, X.; Zhang, J.; Yang, X.; Zhang, Y.; Fan, C.; Li, D.; Deng, Y.; et al. HDL-scavenger receptor B type 1 facilitates SARS-CoV-2 entry. Nat. Metab. 2020, 2, 1391–1400. [Google Scholar] [CrossRef]
- Cho, K.-H.; Kim, J.-R.; Lee, I.-C.; Kwon, H.-J. Native High-Density Lipoproteins (HDL) with Higher Paraoxonase Exerts a Potent Antiviral Effect against SARS-CoV-2 (COVID-19), While Glycated HDL Lost the Antiviral Activity. Antioxidants 2021, 10, 209. [Google Scholar] [CrossRef] [PubMed]
- Henrich, S.E.; McMahon, K.M.; Palacio, N.; Bhalla, P.; Penaloza-MacMaster, P.; Thaxton, C.S. Targeting Scavenger Receptor Type B-1 (SR-B1) and Cholesterol Inhibits Entry of SARS-CoV-2 Pseudovirus in Cell Culture. bioRxiv 2020. [Google Scholar] [CrossRef]
- PrabhuDas, M.; Bowdish, D.; Drickamer, K.; Febbraio, M.; Herz, J.; Kobzik, L.; Krieger, M.; Loike, J.; Means, T.K.; Moestrup, S.K.; et al. Standardizing Scavenger Receptor Nomenclature. J. Immunol. 2014, 192, 1997–2006. [Google Scholar] [CrossRef] [Green Version]
- PrabhuDas, M.R.; Baldwin, C.L.; Bollyky, P.L.; Bowdish, D.M.E.; Drickamer, K.; Febbraio, M.; Herz, J.; Kobzik, L.; Krieger, M.; Loike, J.; et al. A Consensus Definitive Classification of Scavenger Receptors and Their Roles in Health and Disease. J. Immunol. 2017, 198, 3775–3789. [Google Scholar] [CrossRef] [Green Version]
- Heaton, N.S.; Randall, G. Multifaceted roles for lipids in viral infection. Trends Microbiol. 2011, 19, 368–375. [Google Scholar] [CrossRef]
- Mazzon, M.; Mercer, J. Lipid interactions during virus entry and infection. Cell. Microbiol. 2014, 16, 1493–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Li, Y.; Sadaoka, T.; Tang, H.; Yamamoto, T.; Yamanishi, K.; Mori, Y. Human herpesvirus 6 envelope cholesterol is required for virus entry. J. Gen. Virol. 2006, 87, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Osuna-Ramos, J.F.; Reyes-Ruiz, J.M.; Del Ángel, R.M. The Role of Host Cholesterol During Flavivirus Infection. Front. Cell. Infect. Microbiol. 2018, 8, 388. [Google Scholar] [CrossRef] [PubMed]
- Sousa, I.P.; Carvalho, C.A.M.; Gomes, A.M.O. Current Understanding of the Role of Cholesterol in the Life Cycle of Alphaviruses. Viruses 2020, 13, 35. [Google Scholar] [CrossRef] [PubMed]
- Iša, P.; Realpe, M.; Romero, P.; López, S.; Arias, C.F. Rotavirus RRV associates with lipid membrane microdomains during cell entry. Virology 2004, 322, 370–381. [Google Scholar] [CrossRef] [Green Version]
- Yi, L.; Fang, J.; Isik, N.; Chim, J.; Jin, T. HIV gp120-induced interaction between CD4 and CCR5 requires cholesterol-rich microenvironments revealed by live cell fluorescence resonance energy transfer imaging. J. Biol. Chem. 2006, 281, 35446–35453. [Google Scholar] [CrossRef] [Green Version]
- Li, G.M.; Li, Y.G.; Yamate, M.; Li, S.M.; Ikuta, K. Lipid rafts play an important role in the early stage of severe acute respiratory syndrome-coronavirus life cycle. Microbes Infect. 2007, 9, 96–102. [Google Scholar] [CrossRef]
- Ripa, I.; Andreu, S.; López-Guerrero, J.A.; Bello-Morales, R. Membrane Rafts: Portals for Viral Entry. Front. Microbiol. 2021, 12, 1274. [Google Scholar] [CrossRef]
- Liao, Z.; Cimakasky, L.M.; Hampton, R.; Nguyen, D.H.; Hildreth, J.E.K. Lipid Rafts and HIV Pathogenesis: Host Membrane Cholesterol Is Required for Infection by HIV Type 1; Mary Ann Liebert, Inc.: Larchmont, NY, USA, 2001; Volume 17. [Google Scholar]
- Dou, X.; Li, Y.; Han, J.; Zarlenga, D.S.; Zhu, W.; Ren, X.; Dong, N.; Li, X.; Li, G. Cholesterol of lipid rafts is a key determinant for entry and post-entry control of porcine rotavirus infection. BMC Vet. Res. 2018, 14, 45. [Google Scholar] [CrossRef] [Green Version]
- Glende, J.; Schwegmann-Wessels, C.; Al-Falah, M.; Pfefferle, S.; Qu, X.; Deng, H.; Drosten, C.; Naim, H.Y.; Herrler, G. Importance of cholesterol-rich membrane microdomains in the interaction of the S protein of SARS-coronavirus with the cellular receptor angiotensin-converting enzyme 2. Virology 2008, 381, 215–221. [Google Scholar] [CrossRef] [Green Version]
- Acton, S.; Rigotti, A.; Landschulz, K.T.; Xu, S.; Hobbs, H.H.; Krieger, M. Identification of Scavenger Receptor SR-BI as a High Density Lipoprotein Receptor. Science 1996, 271, 518–520. [Google Scholar] [CrossRef]
- Krieger, M. Scavenger receptor class B type I is a multiligand HDL receptor that influences diverse physiologic systems. J. Clin. Investig. 2001, 108, 793–797. [Google Scholar] [CrossRef]
- Huang, R.; Silva, R.A.G.D.; Jerome, W.G.; Kontush, A.; Chapman, M.J.; Curtiss, L.K.; Hodges, T.J.; Davidson, W.S. Apolipoprotein A-I structural organization in high-density lipoproteins isolated from human plasma. Nat. Struct. Mol. Biol. 2011, 18, 416–422. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Neculai, D.; Schwake, M.; Ravichandran, M.; Zunke, F.; Collins, R.F.; Peters, J.; Neculai, M.; Plumb, J.; Loppnau, P.; Pizarro, J.C.; et al. Structure of LIMP-2 provides functional insights with implications for SR-BI and CD36. Nature 2013, 504, 172–176. [Google Scholar] [CrossRef]
- Zannis, V.I.; Chroni, A.; Krieger, M. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J. Mol. Med. 2006, 84, 276–294. [Google Scholar] [CrossRef]
- Rothblat, G.H.; Phillips, M.C. High-density lipoprotein heterogeneity and function in reverse cholesterol transport. Curr. Opin. Lipidol. 2010, 21, 229–238. [Google Scholar] [CrossRef]
- Favari, E.; Chroni, A.; Tietge, U.J.F.; Zanotti, I.; Escolà-Gil, J.C.; Bernini, F. Cholesterol efflux and reverse cholesterol transport. Handb. Exp. Pharmacol. 2015, 224, 181–206. [Google Scholar] [CrossRef] [Green Version]
- Lewis, G.F.; Rader, D.J. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ. Res. 2005, 96, 1221–1232. [Google Scholar] [CrossRef] [Green Version]
- Trigatti, B.; Rayburn, H.; Viñals, M.; Braun, A.; Miettinen, H.; Penman, M.; Hertz, M.; Schrenzel, M.; Amigo, L.; Rigotti, A.; et al. Influence of the high density lipoprotein receptor SR-BI on reproductive and cardiovascular pathophysiology. Proc. Natl. Acad. Sci. USA 1999, 96, 9322–9327. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, H.E.; Rayburn, H.; Krieger, M. Abnormal lipoprotein metabolism and reversible female infertility in HDL receptor (SR-BI)-deficient mice. J. Clin. Investig. 2001, 108, 1717–1722. [Google Scholar] [CrossRef] [PubMed]
- Braun, A.; Zhang, S.; Miettinen, H.E.; Ebrahim, S.; Holm, T.M.; Vasile, E.; Post, M.J.; Yoerger, D.M.; Picard, M.H.; Krieger, J.L.; et al. Probucol prevents early coronary heart disease and death in the high-density lipoprotein receptor SR-BI/apolipoprotein E double knockout mouse. Proc. Natl. Acad. Sci. USA 2003, 100, 7283–7288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Eck, M.; Bos, I.S.T.; Hildebrand, R.B.; Van Rij, B.T.; Van Berkel, T.J.C. Dual role for scavenger receptor class B, type I on bone marrow-derived cells in atherosclerotic lesion development. Am. J. Pathol. 2004, 165, 785–794. [Google Scholar] [CrossRef] [Green Version]
- Rigotti, A.; Trigatti, B.; Babitt, J.; Penman, M.; Xu, S.; Krieger, M. Scavenger receptor BI-A cell surface receptor for high density lipoprotein. Curr. Opin. Lipidol. 1997, 8, 181–188. [Google Scholar] [CrossRef]
- Koenig, S.N.; Sucharski, H.C.; Jose, E.; Dudley, E.K.; Madiai, F.; Cavus, O.; Argall, A.D.; Williams, J.; Murphy, N.P.; Keith, C.B.; et al. Inherited Variants in SCARB1 Cause Severe Early-Onset Coronary Artery Disease. Circ. Res. 2021, 129, 296–307. [Google Scholar] [CrossRef]
- Zhang, S.; Picard, M.H.; Vasile, E.; Zhu, Y.; Raffai, R.L.; Weisgraber, K.H.; Krieger, M. Diet-induced occlusive coronary atherosclerosis, myocardial infarction, cardiac dysfunction, and premature death in scavenger receptor class B type I-deficient, hypomorphic apolipoprotein ER61 mice. Circulation 2005, 111, 3457–3464. [Google Scholar] [CrossRef] [Green Version]
- Fuller, M.; Dadoo, O.; Serkis, V.; Abutouk, D.; MacDonald, M.; Dhingani, N.; Macri, J.; Igdoura, S.A.; Trigatti, B.L. The effects of diet on occlusive coronary artery atherosclerosis and myocardial infarction in scavenger receptor class B, type 1/low-density lipoprotein receptor double knockout mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2394–2403. [Google Scholar] [CrossRef] [Green Version]
- Tong, Y.; Zhu, Y.; Xia, X.; Liu, Y.; Feng, Y.; Hua, X.; Chen, Z.; Ding, H.; Gao, L.; Wang, Y.; et al. Tupaia CD81, SR-BI, Claudin-1, and Occludin Support Hepatitis C Virus Infection. J. Virol. 2011, 85, 2793–2802. [Google Scholar] [CrossRef] [Green Version]
- Burlone, M.E.; Budkowska, A. Hepatitis C virus cell entry: Role of lipoproteins and cellular receptors. J. Gen. Virol. 2009, 90, 1055–1070. [Google Scholar] [CrossRef]
- Li, Y.; Kakinami, C.; Li, Q.; Yang, B.; Li, H. Human Apolipoprotein A-I Is Associated with Dengue Virus and Enhances Virus Infection through SR-BI. PLoS ONE 2013, 8, e0070390. [Google Scholar] [CrossRef] [Green Version]
- Voisset, C.; Callens, N.; Blanchard, E.; Op De Beeck, A.; Dubuisson, J.; Vu-Dac, N. High density lipoproteins facilitate hepatitis C virus entry through the scavenger receptor class B type I. J. Biol. Chem. 2005, 280, 7793–7799. [Google Scholar] [CrossRef] [Green Version]
- Von Hahn, T.; Lindenbach, B.D.; Boullier, A.; Quehenberger, O.; Paulson, M.; Rice, C.M.; McKeating, J.A. Oxidized low-density lipoprotein inhibits hepatitis C virus cell entry in human hepatoma cells. Hepatology 2006, 43, 932–942. [Google Scholar] [CrossRef] [Green Version]
- Kocher, O.; Birrane, G.; Tsukamoto, K.; Fenske, S.; Yesilaltay, A.; Pal, R.; Daniels, K.; Ladias, J.A.A.; Krieger, M. In vitro and in vivo analysis of the binding of the C terminus of the HDL receptor scavenger receptor class B, type I (SR-BI), to the PDZ1 domain of its adaptor protein PDZK1. J. Biol. Chem. 2010, 285, 34999–35010. [Google Scholar] [CrossRef] [Green Version]
- Eyre, N.S.; Drummer, H.E.; Beard, M.R. The SR-BI Partner PDZK1 Facilitates Hepatitis C Virus Entry. PLoS Pathog. 2010, 6, e1001130. [Google Scholar] [CrossRef] [Green Version]
- Kocher, O.; Yesilaltay, A.; Cirovic, C.; Pal, R.; Rigotti, A.; Krieger, M. Targeted Disruption of the PDZK1 Gene in Mice Causes Tissue-specific Depletion of the High Density Lipoprotein Receptor Scavenger Receptor Class B Type I and Altered Lipoprotein Metabolism. J. Biol. Chem. 2003, 278, 52820–52825. [Google Scholar] [CrossRef] [Green Version]
- Dreux, M.; Pietschmann, T.; Granier, C.; Voisset, C.; Ricard-Blum, S.; Mangeot, P.E.; Keck, Z.; Foung, S.; Vu-Dac, N.; Dubuisson, J.; et al. High density lipoprotein inhibits hepatitis C virus-neutralizing antibodies by stimulating cell entry via activation of the scavenger receptor BI. J. Biol. Chem. 2006, 281, 18285–18295. [Google Scholar] [CrossRef] [Green Version]
- Voisset, C.; Op de Beeck, A.; Horellou, P.; Dreux, M.; Gustot, T.; Duverlie, G.; Cosset, F.L.; Vu-Dac, N.; Dubuisson, J. High-density lipoproteins reduce the neutralizing effect of hepatitis C virus (HCV)-infected patient antibodies by promoting HCV entry. J. Gen. Virol. 2006, 87, 2577–2581. [Google Scholar] [CrossRef]
- Eieenberg, S. High density lipoprotein metabolism. J. Lipids Res. 1984, 10, 1017–1058. [Google Scholar] [CrossRef]
- Lundberg, J.; Rudling, M.; Angelin, B. Interstitial fluid lipoproteins. Curr. Opin. Lipidol. 2013, 24, 327–331. [Google Scholar] [CrossRef]
- Cho, K.-H. Importance of Apolipoprotein A-I and A-II Composition in HDL and Its Potential for Studying COVID-19 and SARS-CoV-2. Medicines 2021, 8, 38. [Google Scholar] [CrossRef]
- Babitt, J.; Trigatti, B.; Rigotti, A.; Smart, E.J.; Anderson, R.G.W.; Xu, S.; Krieger, M. Murine SR-BI, a high density lipoprotein receptor that mediates selective lipid uptake, is N-glycosylated and fatty acylated and colocalizes with plasma membrane caveolae. J. Biol. Chem. 1997, 272, 13242–13249. [Google Scholar] [CrossRef] [Green Version]
- De La Llera-Moya, M.; Rothblat, G.H.; Connelly, M.A.; Kellner-Weibel, G.; Sakr, S.W.; Phillips, M.C.; Williams, D.L. Scavenger receptor BI (SR-BI) mediates free cholesterol flux independently of HDL tethering to the cell surface. J. Lipid Res. 1999, 40, 575–580. [Google Scholar] [CrossRef]
- Papale, G.A.; Nicholson, K.; Hanson, P.J.; Pavlovic, M.; Drover, V.A.; Sahoo, D. Extracellular hydrophobic regions in scavenger receptor BI play a key role in mediating HDL-cholesterol transport. Arch. Biochem. Biophys. 2010, 496, 132–139. [Google Scholar] [CrossRef] [Green Version]
- Papale, G.A.; Hanson, P.J.; Sahoo, D. Extracellular disulfide bonds support scavenger receptor-BI-mediated cholesterol transport. Biochemistry 2011, 50, 6245. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Jian, B.; Wang, N.; Sun, Y.; De La Llera Moya, M.; Phillips, M.C.; Rothblat, G.H.; Swaney, J.B.; Tall, A.R. Scavenger receptor BI promotes high density lipoprotein-mediated cellular cholesterol efflux. J. Biol. Chem. 1997, 272, 20982–20985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieland, T.J.F.; Penman, M.; Dori, L.; Krieger, M.; Kirchhausen, T. Discovery of chemical inhibitors of the selective transfer of lipids mediated by the HDL receptor SR-BI. Proc. Natl. Acad. Sci. USA 2002, 99, 15422–15427. [Google Scholar] [CrossRef] [Green Version]
- Bajimaya, S.; Frankl, T.; Hayashi, T.; Takimoto, T. Cholesterol is required for stability and infectivity of influenza A and respiratory syncytial viruses. Virology 2017, 510, 234–241. [Google Scholar] [CrossRef]
- Li, X.; Zhu, W.; Fan, M.; Zhang, J.; Peng, Y.; Huang, F.; Wang, N.; He, L.; Zhang, L.; Holmdahl, R.; et al. Dependence of SARS-CoV-2 infection on cholesterol-rich lipid raft and endosomal acidification. Comput. Struct. Biotechnol. J. 2021, 19, 1933–1943. [Google Scholar] [CrossRef]
- Kontush, A. HDL particle number and size as predictors of cardiovascular disease. Front. Pharmacol. 2015, 6, 218. [Google Scholar] [CrossRef] [Green Version]
- Laue, M.; Kauter, A.; Hoffmann, T.; Möller, L.; Michel, J.; Nitsche, A. Morphometry of SARS-CoV and SARS-CoV-2 particles in ultrathin plastic sections of infected Vero cell cultures. Sci. Rep. 2021, 11, 3515. [Google Scholar] [CrossRef]
- Zheng, Y.H.; Plemenitas, A.; Fielding, C.J.; Peterlin, B.M. Nef increases the synthesis of and transports cholesterol to lipid rafts and HIV-1 progeny virions. Proc. Natl. Acad. Sci. USA 2003, 100, 8460–8465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, D.H.; K Hildreth, J.E. Evidence for Budding of Human Immunodeficiency Virus Type 1 Selectively from Glycolipid-Enriched Membrane Lipid Rafts. Journal of Virology. 2000, 74, 3264–3272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, A.; Freed, E.O. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl. Acad. Sci. USA 2001, 98, 13925–13930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadras, M.A.; Greenberg, H.B. Rotavirus infectious particles use lipid rafts during replication for transport to the cell surface in vitro and in vivo. Virology 2003, 313, 308–321. [Google Scholar] [CrossRef] [Green Version]
- Guyader, M.; Kiyokawa, E.; Abrami, L.; Turelli, P.; Trono, D. Role for Human Immunodeficiency Virus Type 1 Membrane Cholesterol in Viral Internalization. J. Virol. 2002, 76, 10356–10364. [Google Scholar] [CrossRef] [Green Version]
- Domanska, M.K.; Dunning, R.A.; Dryden, K.A.; Zawada, K.E.; Yeager, M.; Kasson, P.M. Hemagglutinin Spatial Distribution Shifts in Response to Cholesterol in the Influenza Viral Envelope. Biophys. J. 2015, 109, 1917–1924. [Google Scholar] [CrossRef] [Green Version]
- Davidson, W.S.; Silva, R.A.G.D.; Chantepie, S.; Lagor, W.R.; Chapman, M.J.; Kontush, A. Proteomic analysis of defined hdl subpopulations reveals particle-specific protein clusters: Relevance to antioxidative function. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 870–876. [Google Scholar] [CrossRef]
- Havel, R.J.; Eder, H.A.; Bragdon, J.H. The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J. Clin. Investig. 1955, 34, 1345–1353. [Google Scholar] [CrossRef] [Green Version]
- Vaisar, T.; Pennathur, S.; Green, P.S.; Gharib, S.A.; Hoofnagle, A.N.; Cheung, M.C.; Byun, J.; Vuletic, S.; Kassim, S.; Singh, P.; et al. Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J. Clin. Investig. 2007, 117, 746–756. [Google Scholar] [CrossRef]
- Gordon, S.M.; Deng, J.; Tomann, A.B.; Shah, A.S.; Lu, L.J.; Davidson, W.S. Multi-dimensional co-separation analysis reveals protein-protein interactions defining plasma lipoprotein subspecies. Mol. Cell. Proteom. 2013, 12, 3123–3134. [Google Scholar] [CrossRef] [Green Version]
- Riwanto, M.; Rohrer, L.; Roschitzki, B.; Besler, C.; Mocharla, P.; Mueller, M.; Perisa, D.; Heinrich, K.; Altwegg, L.; Von Eckardstein, A.; et al. Altered activation of endothelial anti-and proapoptotic pathways by high-density lipoprotein from patients with coronary artery disease: Role of high-density lipoprotein-proteome remodeling. Circulation 2013, 127, 891–904. [Google Scholar] [CrossRef] [Green Version]
- Heinecke, J.W. HDL’s protein cargo: Friend or foe in cardioprotection? Circulation 2013, 127, 868–869. [Google Scholar] [CrossRef] [Green Version]
- HDL Proteome Watch Page. Available online: https://homepages.uc.edu/~davidswm/HDLproteome.html (accessed on 11 August 2021).
- Rye, K.A.; Barter, P.J. Thematic review series: High density lipoprotein structure, function, and metabolism cardioprotective functions of HDLs 1. J. Lipid Res. 2014, 55, 168–179. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, R.; Mu, H.; Wang, X.; Yao, Q.; Chen, C. Reverse cholesterol transport and cholesterol efflux in atherosclerosis. QJM-Mon. J. Assoc. Phys. 2005, 98, 845–856. [Google Scholar] [CrossRef]
- Kontush, A.; Lhomme, M.; Chapman, M.J. Thematic review series: High density lipoprotein structure, function, and metabolism: Unraveling the complexities of the HDL lipidome. J. Lipid Res. 2013, 54, 2950–2963. [Google Scholar] [CrossRef] [Green Version]
- Mardones, P.; Strobel, P.; Miranda, S.; Leighton, F.; Quiñones, V.; Amigo, L.; Rozowski, J.; Krieger, M.; Rigotti, A. α-tocopherol metabolism is abnormal in scavenger receptor class B type I (SR-BI)-deficient mice. J. Nutr. 2002, 132, 443–449. [Google Scholar] [CrossRef] [Green Version]
- Michell, D.L.; Vickers, K.C. Lipoprotein carriers of microRNAs. Biochim. Biophys. Acta 2016, 1861, 2069. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.S.; Tan, L.; Long, J.L.; Davidson, W.S. Proteomic diversity of high density lipoproteins: Our emerging understanding of its importance in lipid transport and beyond. J. Lipid Res. 2013, 54, 2575–2585. [Google Scholar] [CrossRef] [Green Version]
- Rader, D.J. Molecular regulation of HDL metabolism and function: Implications for novel therapies. J. Clin. Investig. 2006, 116, 3090–3100. [Google Scholar] [CrossRef] [Green Version]
- Brooks-Wilson, A.; Marcil, M.; Clee, S.M.; Zhang, L.H.; Roomp, K.; Van Dam, M.; Yu, L.; Brewer, C.; Collins, J.A.; Molhuizen, H.O.F.; et al. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat. Genet. 1999, 22, 336–345. [Google Scholar] [CrossRef]
- Bodzioch, M.; Orsó, E.; Klucken, J.; Langmann, T.; Böttcher, A.; Diederich, W.; Drobnik, W.; Barlage, S.; Büchler, C.; Porsch-Özcürümez, M.; et al. The gene encoding ATP-binding cassette transporter I is mutated in Tangier disease. Nat. Genet. 1999, 22, 347–351. [Google Scholar] [CrossRef]
- Zannis, V.I.; Fotakis, P.; Koukos, G.; Kardassis, D.; Ehnholm, C.; Jauhiainen, M.; Chroni, A. Hdl biogenesis, remodeling, and catabolism. In Proceedings of the Handbook of Experimental Pharmacology; Springer: New York, NY, USA, 2015; Volume 224, pp. 53–111. [Google Scholar]
- Ahsan, L.; Ossoli, A.F.; Freeman, L.; Vaisman, B.; Amar, M.J.; Shamburek, R.D.; Remaley, A.T. Role of Lecithin: Cholesterol Acyltransferase in HDL Metabolism and Atherosclerosis. In The HDL Handbook: Biological Functions and Clinical Implications, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; pp. 159–194. ISBN 9780124078673. [Google Scholar]
- Thacker, S.G.; Rousset, X.; Esmail, S.; Zarzour, A.; Jin, X.; Collins, H.L.; Sampson, M.; Stonik, J.; Demosky, S.; Malide, D.A.; et al. Increased plasma cholesterol esterification by LCAT reduces diet-induced atherosclerosis in SR-BI knockout mice. J. Lipid Res. 2015, 56, 1282–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desforges, J.F.; Gordon, D.J.; Rifkind, B.M. High-Density Lipoprotein—The Clinical Implications of Recent Studies. N. Engl. J. Med. 1989, 321, 1311–1316. [Google Scholar] [CrossRef]
- Glass, C.; Pittman, R.C.; Weinstein, D.B.; Steinberg, D. Dissociation of tissue uptake of cholesterol ester from that of apoprotein A-I of rat plasma high density lipoprotein: Selective delivery of cholesterol ester to liver, adrenal, and gonad. Proc. Natl. Acad. Sci. USA 1983, 80, 5435–5439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barter, P.J.; Nicholls, S.; Rye, K.A.; Anantharamaiah, G.M.; Navab, M.; Fogelman, A.M. Antiinflammatory properties of HDL. Circ. Res. 2004, 95, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Navab, M.; Hama, S.Y.; Cooke, C.J.; Anantharamaiah, G.M.; Chaddha, M.; Jin, L.; Subbanagounder, G.; Faull, K.F.; Reddy, S.T.; Miller, N.E.; et al. Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: Step 1. J. Lipid Res. 2000, 41, 1481–1494. [Google Scholar] [CrossRef]
- Murphy, A.J.; Woollard, K.J. High-density lipoprotein: A potent inhibitor of inflammation: Frontiers in research review: Physiological and pathological functions of high-density lipoprotein. Clin. Exp. Pharmacol. Physiol. 2010, 37, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Urundhati, A.; Huang, Y.; Lupica, J.A.; Smith, J.D.; DiDonato, J.A.; Hazen, S.L. Modification of high density lipoprotein by myeloperoxidase generates a pro-inflammatory particle. J. Biol. Chem. 2009, 284, 30825–30835. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, L.; Qian, A.S.; Tahir, U.; Yu, P.; Trigatti, B.L. Sphingosine-1-phosphate receptor 1, expressed in myeloid cells, slows diet-induced atherosclerosis and protects against macrophage apoptosis in ldlr KO mice. Int. J. Mol. Sci. 2017, 18, 2721. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Qian, A.S.; Chathely, K.M.; Trigatti, B.L. PDZK1 in leukocytes protects against cellular apoptosis and necrotic core development in atherosclerotic plaques in high fat diet fed ldl receptor deficient mice. Atherosclerosis 2018, 276, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Durham, K.K.; Chathely, K.M.; Mak, K.C.; Momen, A.; Thomas, C.T.; Zhao, Y.-Y.; MacDonald, M.E.; Curtis, J.M.; Husain, M.; Trigatti, B.L. HDL protects against doxorubicin-induced cardiotoxicity in a scavenger receptor class B type 1-, PI3K-, and Akt-dependent manner. Am. J. Physiol. Circ. Physiol. 2018, 314, H31–H44. [Google Scholar] [CrossRef]
- Durham, K.K.; Kluck, G.; Mak, K.C.; Deng, Y.D.; Trigatti, B.L. Treatment with apolipoprotein A1 protects mice against doxorubicin-induced cardiotoxicity in a scavenger receptor class B, type I-dependent manner. Am. J. Physiol. Circ. Physiol. 2019, 316, H1447–H1457. [Google Scholar] [CrossRef]
- Ruiz, M.; Okada, H.; Dahlbäck, B. HDL-associated ApoM is anti-apoptotic by delivering sphingosine 1-phosphate to S1P1 & S1P3 receptors on vascular endothelium. Lipids Health Dis. 2017, 16. [Google Scholar] [CrossRef] [Green Version]
- Nofer, J.-R.; Levkau, B.; Wolinska, I.; Junker, R.; Fobker, M.; von Eckardstein, A.; Seedorf, U.; Assmann, G. Suppression of Endothelial Cell Apoptosis by High Density Lipoproteins (HDL) and HDL-associated Lysosphingolipids*. J. Biol. Chem. 2001, 276, 34480–34485. [Google Scholar] [CrossRef] [Green Version]
- Feuerborn, R.; Becker, S.; Potì, F.; Nagel, P.; Brodde, M.; Schmidt, H.; Christoffersen, C.; Ceglarek, U.; Burkhardt, R.; Nofer, J.R. High density lipoprotein (HDL)-associated sphingosine 1-phosphate (S1P) inhibits macrophage apoptosis by stimulating STAT3 activity and survivin expression. Atherosclerosis 2017, 257, 29–37. [Google Scholar] [CrossRef]
- Terasaka, N.; Yu, S.; Yvan-Charvet, L.; Wang, N.; Mzhavia, N.; Langlois, R.; Pagler, T.; Li, R.; Welch, C.L.; Goldberg, I.J.; et al. ABCG1 and HDL protect against endothelial dysfunction in mice fed a high-cholesterol diet. J. Clin. Investig. 2008, 118, 3701–3713. [Google Scholar] [CrossRef] [Green Version]
- Terasaka, N.; Wang, N.; Yvan-Charvet, L.; Tall, A.R. High-density lipoprotein protects macrophages from oxidized low-density lipoprotein-induced apoptosis by promoting efflux of 7-ketocholesterol via ABCG1. Proc. Natl. Acad. Sci. USA 2007, 104, 15093–15098. [Google Scholar] [CrossRef] [Green Version]
- Cui, D.; Thorp, E.; Li, Y.; Wang, N.; Yvan-Charvet, L.; Tall, A.R.; Tabas, I. Pivotal Advance: Macrophages become resistant to cholesterol-induced death after phagocytosis of apoptotic cells. J. Leukoc. Biol. 2007, 82, 1040–1050. [Google Scholar] [CrossRef] [Green Version]
- Talbot, C.P.J.; Plat, J.; Ritsch, A.; Mensink, R.P. Determinants of cholesterol efflux capacity in humans. Prog. Lipid Res. 2018, 69, 21–32. [Google Scholar] [CrossRef]
- Niisuke, K.; Kuklenyik, Z.; Horvath, K.V.; Gardner, M.S.; Toth, C.A.; Asztalos, B.F. Composition-function analysis of HDL subpopulations: Influence of lipid composition on particle functionality. J. Lipid Res. 2020, 61, 306–315. [Google Scholar] [CrossRef]
- Wang, N.; Silver, D.L.; Thiele, C.; Tall, A.R. ATP-binding Cassette Transporter A1 (ABCA1) Functions as a Cholesterol Efflux Regulatory Protein*. J. Biol. Chem. 2001, 276, 23742–23747. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, M.A.; Barrera, G.C.; Nakamura, K.; Baldán, Á.; Tarr, P.; Fishbein, M.C.; Frank, J.; Francone, O.L.; Edwards, P.A. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 2005, 1, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Owen, J.S.; Wilson, M.D.; Li, H.; Griffiths, G.L.; Thomas, M.J.; Hiltbold, E.M.; Fessler, M.B.; Parks, J.S. Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J. Lipid Res. 2010, 51, 3196. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Lee, J.Y.; Timmins, J.M.; Brown, J.M.; Boudyguina, E.; Mulya, A.; Gebre, A.K.; Willingham, M.C.; Hiltbold, E.M.; Mishra, N.; et al. Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. J. Biol. Chem. 2008, 283, 22930–22941. [Google Scholar] [CrossRef] [Green Version]
- Koseki, M.; Hirano, K.I.; Masuda, D.; Ikegami, C.; Tanaka, M.; Ota, A.; Sandoval, J.C.; Nakagawa-Toyama, Y.; Sato, S.B.; Kobayashi, T.; et al. Increased lipid rafts and accelerated lipopolysaccharide-induced tumor necrosis factor-α secretion in Abca1-deficient macrophages. J. Lipid Res. 2007, 48, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Baker, P.W.; Rye, K.A.; Gamble, J.R.; Vadas, M.A.; Barter, P.J. Ability of reconstituted high density lipoproteins to inhibit cytokine- induced expression of vascular cell adhesion molecule-1 in human umbilical vein endothelial cells. J. Lipid Res. 1999, 40, 345–353. [Google Scholar] [CrossRef]
- Recalde, D.; Ostos, M.A.; Badell, E.; Garcia-Otin, A.L.; Pidoux, J.; Castro, G.; Zakin, M.M.; Scott-Algara, D. Human Apolipoprotein A-IV Reduces Secretion of Proinflammatory Cytokines and Atherosclerotic Effects of a Chronic Infection Mimicked by Lipopolysaccharide. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 756–761. [Google Scholar] [CrossRef] [Green Version]
- Christoffersen, C.; Obinata, H.; Kumaraswamy, S.B.; Galvani, S.; Ahnström, J.; Sevvana, M.; Egerer-Sieber, C.; Muller, Y.A.; Hla, T.; Nielsen, L.B.; et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc. Natl. Acad. Sci. USA 2011, 108, 9613–9618. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Allegood, J.; Zhu, X.; Seo, J.; Gebre, A.K.; Boudyguina, E.; Cheng, D.; Chuang, C.C.; Shelness, G.S.; Spiegel, S.; et al. Uncleaved ApoM Signal Peptide Is Required for Formation of Large ApoM/Sphingosine 1-Phosphate (S1P)-enriched HDL Particles. J. Biol. Chem. 2015, 290, 7861–7870. [Google Scholar] [CrossRef] [Green Version]
- Hughes, J.E.; Srinivasan, S.; Lynch, K.R.; Proia, R.L.; Ferdek, P.; Hedrick, C.C. Sphingosine-1-phosphate induces an antiinflammatory phenotype in macrophages. Circ. Res. 2008, 102, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Al-Jarallah, A.; Chen, X.; González, L.; Trigatti, B.L. High Density Lipoprotein Stimulated Migration of Macrophages Depends on the Scavenger Receptor Class B, Type I, PDZK1 and Akt1 and Is Blocked by Sphingosine 1 Phosphate Receptor Antagonists. PLoS ONE 2014, 9, e106487. [Google Scholar] [CrossRef] [Green Version]
- Galvani, S.; Sanson, M.; Blaho, V.A.; Swendeman, S.L.; Conger, H.; Dahlbäck, B.; Kono, M.; Proia, R.L.; Smith, J.D.; Hla, T. HDL-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor S1P1 to limit vascular inflammation. Sci. Signal. 2015, 8, ra79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Ai, J.; Zheng, Z.; Howatt, D.A.; Daugherty, A.; Huang, B.; Li, X.A. High density lipoprotein protects against polymicrobe-induced sepsis in mice. J. Biol. Chem. 2013, 288, 17947–17953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajkrt, D.; Doran, J.E.; Koster, F.; Lerch, P.G.; Arnet, B.; Van Der Poll, T.; Ten Cate, J.W.; Van Deventer, S.J.H. Antiinflammatory effects of reconstituted high-density lipoprotein during human endotoxemia. J. Exp. Med. 1996, 184, 1601–1608. [Google Scholar] [CrossRef] [Green Version]
- Gupta, H.; Dai, L.; Datta, G.; Garber, D.W.; Grenett, H.; Li, Y.; Mishra, V.; Palgunachari, M.N.; Handattu, S.; Gianturco, S.H.; et al. Inhibition of lipopolysaccharide-induced inflammatory responses by an apolipoprotein AI mimetic peptide. Circ. Res. 2005, 97, 236–243. [Google Scholar] [CrossRef] [Green Version]
- Trinder, M.; Wang, Y.; Madsen, C.M.; Ponomarev, T.; Bohunek, L.; Daisely, B.A.; Kong, H.J.; Blauw, L.L.; Nordestgaard, B.G.; Tybjærg-Hansen, A.; et al. Inhibition of Cholesteryl Ester Transfer Protein Preserves High-Density Lipoprotein Cholesterol and Improves Survival in Sepsis. Circulation 2021, 143, 921–934. [Google Scholar] [CrossRef]
- Jomard, A.; Osto, E. High Density Lipoproteins: Metabolism, Function, and Therapeutic Potential. Front. Cardiovasc. Med. 2020, 7, 39. [Google Scholar] [CrossRef]
- Kontush, A.; Therond, P.; Zerrad, A.; Couturier, M.; Négre-Salvayre, A.; De Souza, J.A.; Chantepie, S.; Chapman, M.J. Preferential sphingosine-1-phosphate enrichment and sphingomyelin depletion are key features of small dense HDL3 particles: Relevance to antiapoptotic and antioxidative activities. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1843–1849. [Google Scholar] [CrossRef] [Green Version]
- Panzenböck, U.; Stocker, R. Formation of methionine sulfoxide-containing specific forms of oxidized high-density lipoproteins. Biochim. Biophys. Acta-Proteins Proteom. 2005, 1703, 171–181. [Google Scholar] [CrossRef]
- Elsøe, S.; Ahnström, J.; Christoffersen, C.; Hoofnagle, A.N.; Plomgaard, P.; Heinecke, J.W.; Binder, C.J.; Björkbacka, H.; Dahlbäck, B.; Nielsen, L.B. Apolipoprotein M binds oxidized phospholipids and increases the antioxidant effect of HDL. Atherosclerosis 2012, 221, 91–97. [Google Scholar] [CrossRef]
- Kontush, A.; Chapman, M.J. Functionally defective high-density lipoprotein: A new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Pharmacol. Rev. 2006, 58, 342–374. [Google Scholar] [CrossRef]
- Kontush, A.; Chapman, M.J. Antiatherogenic small, dense HDL-Guardian angel of the arterial wall? Nat. Clin. Pract. Cardiovasc. Med. 2006, 3, 144–153. [Google Scholar] [CrossRef]
- Farid, A.S.; Horii, Y. Modulation of paraoxonases during infectious diseases and its potential impact on atherosclerosis. Lipids Health Dis. 2012, 11, 92. [Google Scholar] [CrossRef] [Green Version]
- Manolescu, B.N.; Busu, C.; Badita, D.; Stanculescu, R.; Berteanu, M. Paraoxonase 1-an Update of the Antioxidant Properties of High- Density Lipoproteins. Maedica 2015, 10, 173–177. [Google Scholar]
- Ahmed, Z.; Ravandi, A.; Maguire, G.F.; Emili, A.; Draganov, D.; La Du, B.N.; Kuksis, A.; Connelly, P.W. Multiple substrates for paraoxonase-1 during oxidation of phosphatidylcholine by peroxynitrite. Biochem. Biophys. Res. Commun. 2002, 290, 391–396. [Google Scholar] [CrossRef]
- Shih, D.M.; Xia, Y.R.; Wang, X.P.; Miller, E.; Castellani, L.W.; Subbanagounder, G.; Cheroutre, H.; Faull, K.F.; Berliner, J.A.; Witztum, J.L.; et al. Combined serum paraoxonase knockout/apolipoprotein E knockout mice exhibit increased lipoprotein oxidation and atherosclerosis. J. Biol. Chem. 2000, 275, 17527–17535. [Google Scholar] [CrossRef] [Green Version]
- Ng, D.S.; Chu, T.; Esposito, B.; Hui, P.; Connelly, P.W.; Gross, P.L. Paraoxonase-1 deficiency in mice predisposes to vascular inflammation, oxidative stress, and thrombogenicity in the absence of hyperlipidemia. Cardiovasc. Pathol. 2008, 17, 226–232. [Google Scholar] [CrossRef]
- Huang, Y.; Wu, Z.; Riwanto, M.; Gao, S.; Levison, B.S.; Gu, X.; Fu, X.; Wagner, M.A.; Besler, C.; Gerstenecker, G.; et al. Myeloperoxidase, paraoxonase-1, and HDL form a functional ternary complex. J. Clin. Investig. 2013, 123, 3815–3828. [Google Scholar] [CrossRef] [Green Version]
- Goyal, J.; Wang, K.; Liu, M.; Subbaiah, P.V. Novel function of lecithin-cholesterol acyltransferase: Hydrolysis of oxidized polar phospholipids generated during lipoprotein oxidation. J. Biol. Chem. 1997, 272, 16231–16239. [Google Scholar] [CrossRef] [Green Version]
- McPherson, P.A.C.; Young, I.S.; McEneny, J. A dual role for lecithin:cholesterol acyltransferase (EC 2.3.1.43) in lipoprotein oxidation. Free Radic. Biol. Med. 2007, 43, 1484–1493. [Google Scholar] [CrossRef]
- Holleboom, A.G.; Daniil, G.; Fu, X.; Zhang, R.; Hovingh, G.K.; Schimmel, A.W.; Kastelein, J.J.P.; Stroes, E.S.G.; Witztum, J.L.; Hutten, B.A.; et al. Lipid oxidation in carriers of lecithin: Cholesterol acyltransferase gene mutations. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 3066–3075. [Google Scholar] [CrossRef] [Green Version]
- Zerrad-Saadi, A.; Therond, P.; Chantepie, S.; Couturier, M.; Rye, K.A.; Chapman, M.J.; Kontush, A. HDL3-mediated inactivation of LDL-associated phospholipid hydroperoxides is determined by the redox status of apolipoprotein A-I and HDL particle surface lipid rigidity: Relevance to inflammation and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 2169–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goulinet, S.; Chapman, M.J. Plasma LDL and HDL subspecies are heterogenous in particle content of tocopherols and oxygenated and hydrocarbon carotenoids: Relevance to oxidative resistance and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 786–796. [Google Scholar] [CrossRef] [PubMed]
- Alwaili, K.; Bailey, D.; Awan, Z.; Bailey, S.D.; Ruel, I.; Hafiane, A.; Krimbou, L.; Laboissiere, S.; Genest, J. The HDL proteome in acute coronary syndromes shifts to an inflammatory profile. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2012, 1821, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Clifton, P.M.; Mackinnon, A.M.; Barter, P.J. Effects of serum amyloid A protein (SAA) on composition, size, and density of high density lipoproteins in subjects with myocardial infarction. J. Lipid Res. 1985, 26, 1389–1398. [Google Scholar] [CrossRef]
- Han, C.Y.; Tang, C.; Guevara, M.E.; Wei, H.; Wietecha, T.; Shao, B.; Subramanian, S.; Omer, M.; Wang, S.; O’Brien, K.D.; et al. Serum amyloid A impairs the antiinflammatory properties of HDL. J. Clin. Investig. 2016, 126, 266–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadler, J.T.; Marsche, G. Obesity-Related Changes in High-Density Lipoprotein Metabolism and Function. Int. J. Mol. Sci. 2020, 21, 8985. [Google Scholar] [CrossRef] [PubMed]
- Azmi, S.; Ferdousi, M.; Liu, Y.; Adam, S.; Siahmansur, T.; Ponirakis, G.; Marshall, A.; Petropoulos, I.N.; Ho, J.H.; Syed, A.A.; et al. The role of abnormalities of lipoproteins and HDL functionality in small fibre dysfunction in people with severe obesity. Sci. Rep. 2021, 11, 12573. [Google Scholar] [CrossRef]
- Han, C.Y.; Chiba, T.; Campbell, J.S.; Fausto, N.; Chaisson, M.; Orasanu, G.; Plutzky, J.; Chait, A. Reciprocal and coordinate regulation of serum amyloid A versus apolipoprotein A-I and paraoxonase-1 by inflammation in murine hepatocytes. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1806–1813. [Google Scholar] [CrossRef] [Green Version]
- Camarota, L.M.; Woollett, L.A.; Howles, P.N. Reverse cholesterol transport is elevated in carboxyl ester lipase-knockout mice. FASEB J. 2011, 25, 1370–1377. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, R.A.K. Dysfunctional HDL in diabetes mellitus and its role in the pathogenesis of cardiovascular disease. Mol. Cell. Biochem. 2018, 440, 167–187. [Google Scholar] [CrossRef]
- Farbstein, D.; Levy, A.P. HDL dysfunction in diabetes: Causes and possible treatments. Expert Rev. Cardiovasc. Ther. 2012, 10, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Curtiss, L.K.; Witztum, J.L. Plasma apolipoproteins AI, AII, B, CI, and E are glucosylated in hyperglycemic diabetic subjects. Diabetes 1985, 34, 452–461. [Google Scholar] [CrossRef]
- Park, K.H.; Shin, D.G.; Kim, J.R.; Cho, K.H. Senescence-Related Truncation and Multimerization of Apolipoprotein A-I in High-Density Lipoprotein With an Elevated Level of Advanced Glycated End Products and Cholesteryl Ester Transfer Activity. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2010, 65A, 600–610. [Google Scholar] [CrossRef] [Green Version]
- Nobecourt, E.; Davies, M.J.; Brown, B.E.; Curtiss, L.K.; Bonnet, D.J.; Charlton, F.; Januszewski, A.S.; Jenkins, A.J.; Barter, P.J.; Rye, K.-A. The impact of glycation on apolipoprotein A-I structure and its ability to activate lecithin:cholesterol acyltransferase. Diabetologia 2007, 50, 643–653. [Google Scholar] [CrossRef] [Green Version]
- Kashyap, S.R.; Osme, A.; Ilchenko, S.; Golizeh, M.; Lee, K.; Wang, S.; Bena, J.; Previs, S.F.; Smith, J.D.; Kasumov, T. Glycation Reduces the Stability of ApoAI and Increases HDL Dysfunction in Diet-Controlled Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2018, 103, 388–396. [Google Scholar] [CrossRef] [Green Version]
- Pietzsch, J.; Julius, U.; Nitzsche, S.; Hanefeld, M. In vivo evidence for increased apolipoprotein A-I catabolism in subjects with impaired glucose tolerance. Diabetes 1998, 47, 1928–1934. [Google Scholar] [CrossRef]
- Liu, D.; Ji, L.; Zhang, D.; Tong, X.; Pan, B.; Liu, P.; Zhang, Y.; Huang, Y.; Su, J.; Willard, B.; et al. Nonenzymatic glycation of high-density lipoprotein impairs its anti-inflammatory effects in innate immunity. Diabetes. Metab. Res. Rev. 2012, 28, 186–195. [Google Scholar] [CrossRef]
- Kasumov, T.; Golizeh, M.; Kashyap, S. Glycation and Deamidation Result in HDL Dysfunction in Patients with Type 2 Diabetes. Diabetes 2018, 67, 330-OR. [Google Scholar] [CrossRef]
- Seres, I.; Paragh, G.; Deschene, E.; Fulop, T.; Khalil, A. Study of factors influencing the decreased HDL associated PON1 activity with aging. Exp. Gerontol. 2004, 39, 59–66. [Google Scholar] [CrossRef]
- Berrougui, H.; Isabelle, M.; Cloutier, M.; Grenier, G.; Khalil, A. Age-related impairment of HDL-mediated cholesterol efflux. J. Lipid Res. 2007, 48, 328–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tracy, R.P.; Psaty, B.M.; Macy, E.; Bovill, E.G.; Cushman, M.; Cornell, E.S.; Kuller, L.H. Lifetime smoking exposure affects the association of C-reactive protein with cardiovascular disease risk factors and subclinical disease in healthy elderly subjects. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2167–2176. [Google Scholar] [CrossRef] [PubMed]
- Bergt, C.; Oram, J.F.; Heinecke, J.W. Oxidized HDL: The paradox-idation of lipoproteins. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1488–1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhou, C.; Xu, H.; Brook, R.D.; Liu, S.; Yi, T.; Wang, Y.; Feng, B.; Zhao, M.; Wang, X.; et al. Ambient air pollution is associated with HDL (High-Density Lipoprotein) dysfunction in healthy adults. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 513–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, B.; Oda, M.N.; Oram, J.F.; Heinecke, J.W. Myeloperoxidase: An inflammatory enzyme for generating dysfunctional high density lipoprotein. Curr. Opin. Cardiol. 2006, 21, 322–328. [Google Scholar] [CrossRef]
- Park, K.-H.; Shin, D.-G.; Cho, K.-H. Dysfunctional Lipoproteins from Young Smokers Exacerbate Cellular Senescence and Atherogenesis with Smaller Particle Size and Severe Oxidation and Glycation. Toxicol. Sci. 2014, 140, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-M.; Lim, S.-M.; Yoo, J.-A.; Woo, M.-J.; Cho, K.-H. Consumption of high-dose vitamin C (1250 mg per day) enhances functional and structural properties of serum lipoprotein to improve anti-oxidant, anti-atherosclerotic, and anti-aging effects via regulation of anti-inflammatory microRNA. Food Funct. 2015, 6, 3604–3612. [Google Scholar] [CrossRef]
- Schofield, J.D.; Liu, Y.; Rao-Balakrishna, P.; Malik, R.A.; Soran, H. Diabetes Dyslipidemia. Diabetes Ther. 2016, 7, 203. [Google Scholar] [CrossRef] [Green Version]
- MR, T. Diabetic dyslipidaemia: From basic research to clinical practice. Diabetologia 2003, 46, 733–749. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yang, H.; Li, S.; Li, W.-D.; Wang, J.; Wang, Y. Association analysis framework of genetic and exposure risks for COVID-19 in middle-aged and elderly adults. Mech. Ageing Dev. 2021, 194, 111433. [Google Scholar] [CrossRef]
- Williamson, E.J.; Walker, A.J.; Bhaskaran, K.; Bacon, S.; Bates, C.; Morton, C.E.; Curtis, H.J.; Mehrkar, A.; Evans, D.; Inglesby, P.; et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020, 584, 430–436. [Google Scholar] [CrossRef]
- Ho, F.K.; Celis-Morales, C.A.; Gray, S.R.; Katikireddi, S.V.; Niedzwiedz, C.L.; Hastie, C.; Ferguson, L.D.; Berry, C.; Mackay, D.F.; Gill, J.M.R.; et al. Modifiable and non-modifiable risk factors for COVID-19, and comparison to risk factors for influenza and pneumonia: Results from a UK Biobank prospective cohort study. BMJ Open 2020, 10, e040402. [Google Scholar] [CrossRef]
- Aung, N.; Khanji, M.Y.; Munroe, P.B.; Petersen, S.E. Causal Inference for Genetic Obesity, Cardiometabolic Profile and COVID-19 Susceptibility: A Mendelian Randomization Study. Front. Genet. 2020, 11, 586308. [Google Scholar] [CrossRef]
- Scalsky, R.J.; Chen, Y.-J.; Desai, K.; O’Connell, J.R.; Perry, J.A.; Hong, C.C. Baseline cardiometabolic profiles and SARS-CoV-2 infection in the UK Biobank. PLoS ONE 2021, 16, e0248602. [Google Scholar] [CrossRef]
- Lassale, C.; Hamer, M.; Hernáez, Á.; Gale, C.R.; Batty, G.D. High density lipoprotein cholesterol and risk of subsequent COVID-19 hospitalisation: The UK Biobank study. medRxiv Prepr. Serv. Heal. Sci. 2021. [Google Scholar] [CrossRef]
- Begue, F.; Tanaka, S.; Mouktadi, Z.; Rondeau, P.; Veeren, B.; Diotel, N.; Tran-Dinh, A.; Robert, T.; Vélia, E.; Mavingui, P.; et al. Altered high-density lipoprotein composition and functions during severe COVID-19. Sci. Rep. 2021, 11, 2291. [Google Scholar] [CrossRef]
- Shen, B.; Yi, X.; Sun, Y.; Bi, X.; Du, J.; Zhang, C.; Quan, S.; Zhang, F.; Sun, R.; Qian, L.; et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell 2020, 182, 59. [Google Scholar] [CrossRef]
- Marfia, G.; Navone, S.; Guarnaccia, L.; Campanella, R.; Mondoni, M.; Locatelli, M.; Barassi, A.; Fontana, L.; Palumbo, F.; Garzia, E.; et al. Decreased serum level of sphingosine-1-phosphate: A novel predictor of clinical severity in COVID-19. EMBO Mol. Med. 2021, 13, e13424. [Google Scholar] [CrossRef]
- Li, H.; Xiang, X.; Ren, H.; Xu, L.; Zhao, L.; Chen, X.; Long, H.; Wang, Q.; Wu, Q. Serum Amyloid A is a biomarker of severe Coronavirus Disease and poor prognosis. J. Infect. 2020, 80, 646–655. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Magro, C.M.; Mulvey, J.; Kubiak, J.; Mikhail, S.; Suster, D.; Crowson, A.N.; Laurence, J.; Nuovo, G. Severe COVID-19: A multifaceted viral vasculopathy syndrome. Ann. Diagn. Pathol. 2021, 50, 151645. [Google Scholar] [CrossRef] [PubMed]
- Teuwen, L.-A.; Geldhof, V.; Pasut, A.; Carmeliet, P. COVID-19: The vasculature unleashed. Nat. Rev. Immunol. 2020, 20, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Hamming, I.; Timens, W.; Bulthuis, M.L.C.; Lely, A.T.; Navis, G.J.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Huertas, A.; Montani, D.; Savale, L.; Pichon, J.; Tu, L.; Parent, F.; Guignabert, C.; Humbert, M. Endothelial cell dysfunction: A major player in SARS-CoV-2 infection (COVID-19)? Eur. Respir. J. 2020, 56, 2001634. [Google Scholar] [CrossRef]
- Nuovo, G.J.; Magro, C.; Shaffer, T.; Awad, H.; Suster, D.; Mikhail, S.; He, B.; Michaille, J.-J.; Liechty, B.; Tili, E. Endothelial cell damage is the central part of COVID-19 and a mouse model induced by injection of the S1 subunit of the spike protein. Ann. Diagn. Pathol. 2021, 51, 151682. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, J.; Schiavon, C.R.; He, M.; Chen, L.; Shen, H.; Zhang, Y.; Yin, Q.; Cho, Y.; Andrade, L.; et al. SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE 2. Circ. Res. 2021, 128, 1323–1326. [Google Scholar] [CrossRef]
- Mandal, S.; Barnett, J.; Brill, S.E.; Brown, J.S.; Denneny, E.K.; Hare, S.S.; Heightman, M.; Hillman, T.E.; Jacob, J.; Jarvis, H.C.; et al. Long-COVID’: A cross-sectional study of persisting symptoms, biomarker and imaging abnormalities following hospitalisation for COVID-19. Thorax 2021, 76, 396–398. [Google Scholar] [CrossRef]
- Evans, N.; Nichols, J.; Pruitt, K.; Almodovar, S. “Lung Time No See”: SARS-Cov-2 Spike Protein Changes Genetic Expression in Human Primary Bronchial Epithelial Cells After Recovery. FASEB J. 2021, 35. [Google Scholar] [CrossRef]
- Al-Jarallah, A.; Trigatti, B.L. A role for the scavenger receptor, class B type I in high density lipoprotein dependent activation of cellular signaling pathways. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2010, 1801, 1239–1248. [Google Scholar] [CrossRef]
- Mineo, C.; Shaul, P.W. Novel biological functions of high-density lipoprotein cholesterol. Circ. Res. 2012, 111, 1079–1090. [Google Scholar] [CrossRef] [Green Version]
- Robert, J.; Osto, E.; von Eckardstein, A. The Endothelium Is Both a Target and a Barrier of HDL’s Protective Functions. Cells 2021, 10, 1041. [Google Scholar] [CrossRef]
- Correa, Y.; Waldie, S.; Thépaut, M.; Micciula, S.; Moulin, M.; Fieschi, F.; Pichler, H.; Trevor Forsyth, V.; Haertlein, M.; Cárdenas, M. SARS-CoV-2 spike protein removes lipids from model membranes and interferes with the capacity of high density lipoprotein to exchange lipids. J. Colloid Interface Sci. 2021, 602, 732–739. [Google Scholar] [CrossRef]
- Hardardóttir, I.; Moser, A.H.; Fuller, J.; Fielding, C.; Feingold, K.; Grünfeld, C. Endotoxin and cytokines decrease serum levels and extra hepatic protein and mRNA levels of cholesteryl ester transfer protein in syrian hamsters. J. Clin. Investig. 1996, 97, 2585–2592. [Google Scholar] [CrossRef]
- Wurfel, M.M.; Kunitake, S.T.; Lichenstein, H.; Kane, J.P.; Wright, S.D. Lipopolysaccharide (LPS)-binding protein is carried on lipoproteins and acts as a cofactor in the neutralization of LPS. J. Exp. Med. 1994, 180, 1025–1035. [Google Scholar] [CrossRef] [Green Version]
- Levels, J.H.M.; Marquart, J.A.; Abraham, P.R.; van den Ende, A.E.; Molhuizen, H.O.F.; van Deventer, S.J.H.; Meijers, J.C.M. Lipopolysaccharide Is Transferred from High-Density to Low-Density Lipoproteins by Lipopolysaccharide-Binding Protein and Phospholipid Transfer Protein. Infect. Immun. 2005, 73, 2321–2326. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kluck, G.E.G.; Yoo, J.-A.; Sakarya, E.H.; Trigatti, B.L. Good Cholesterol Gone Bad? HDL and COVID-19. Int. J. Mol. Sci. 2021, 22, 10182. https://doi.org/10.3390/ijms221910182
Kluck GEG, Yoo J-A, Sakarya EH, Trigatti BL. Good Cholesterol Gone Bad? HDL and COVID-19. International Journal of Molecular Sciences. 2021; 22(19):10182. https://doi.org/10.3390/ijms221910182
Chicago/Turabian StyleKluck, George E. G., Jeong-Ah Yoo, Emmanuel H. Sakarya, and Bernardo L. Trigatti. 2021. "Good Cholesterol Gone Bad? HDL and COVID-19" International Journal of Molecular Sciences 22, no. 19: 10182. https://doi.org/10.3390/ijms221910182
APA StyleKluck, G. E. G., Yoo, J. -A., Sakarya, E. H., & Trigatti, B. L. (2021). Good Cholesterol Gone Bad? HDL and COVID-19. International Journal of Molecular Sciences, 22(19), 10182. https://doi.org/10.3390/ijms221910182