
Cellular Models for Primary CoQ Deficiency Pathogenesis Study

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Coenzyme Q Biosynthesis Pathway Overview
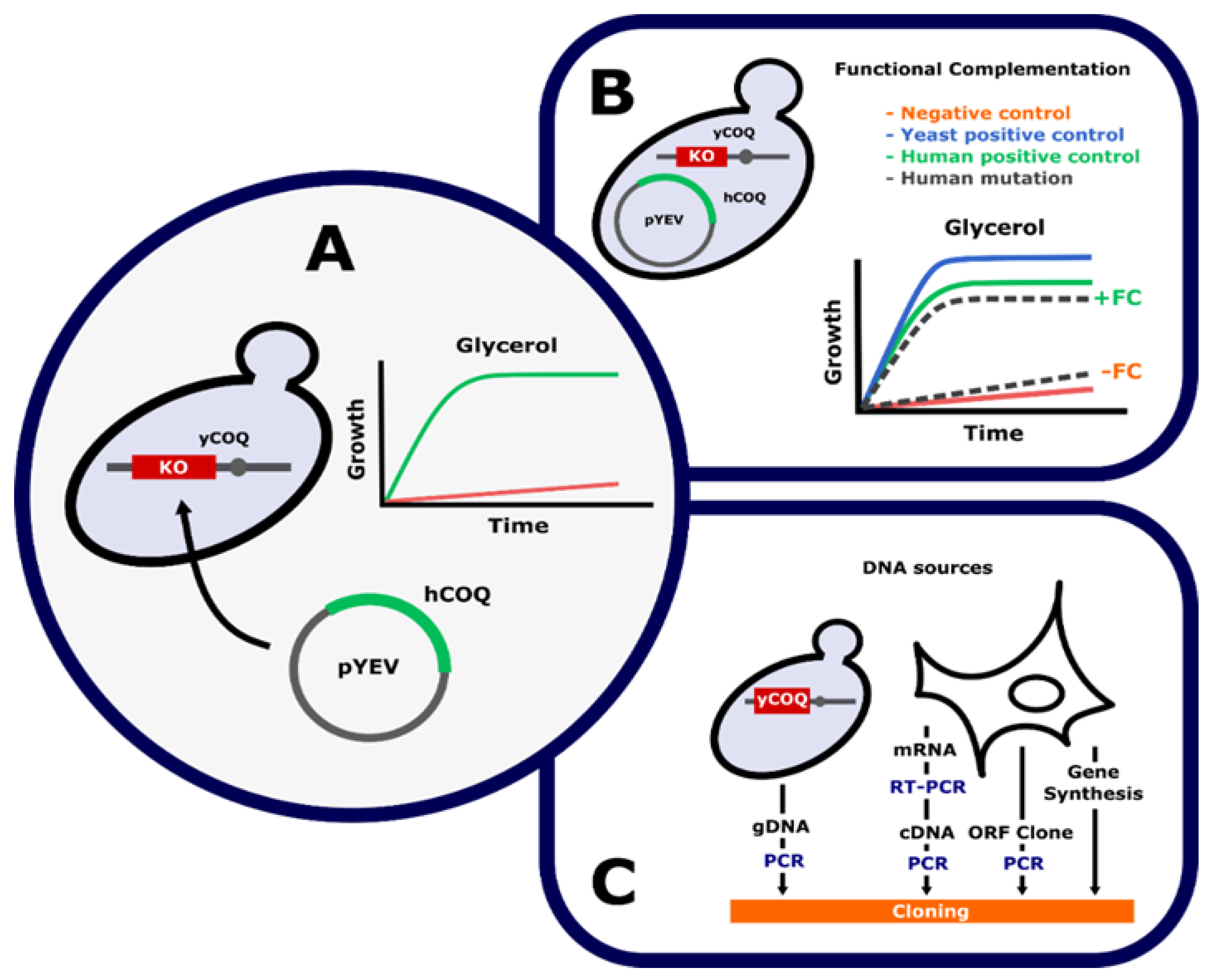
3. Functional Complementation of COQ Genes in Saccharomyces cerevisiae
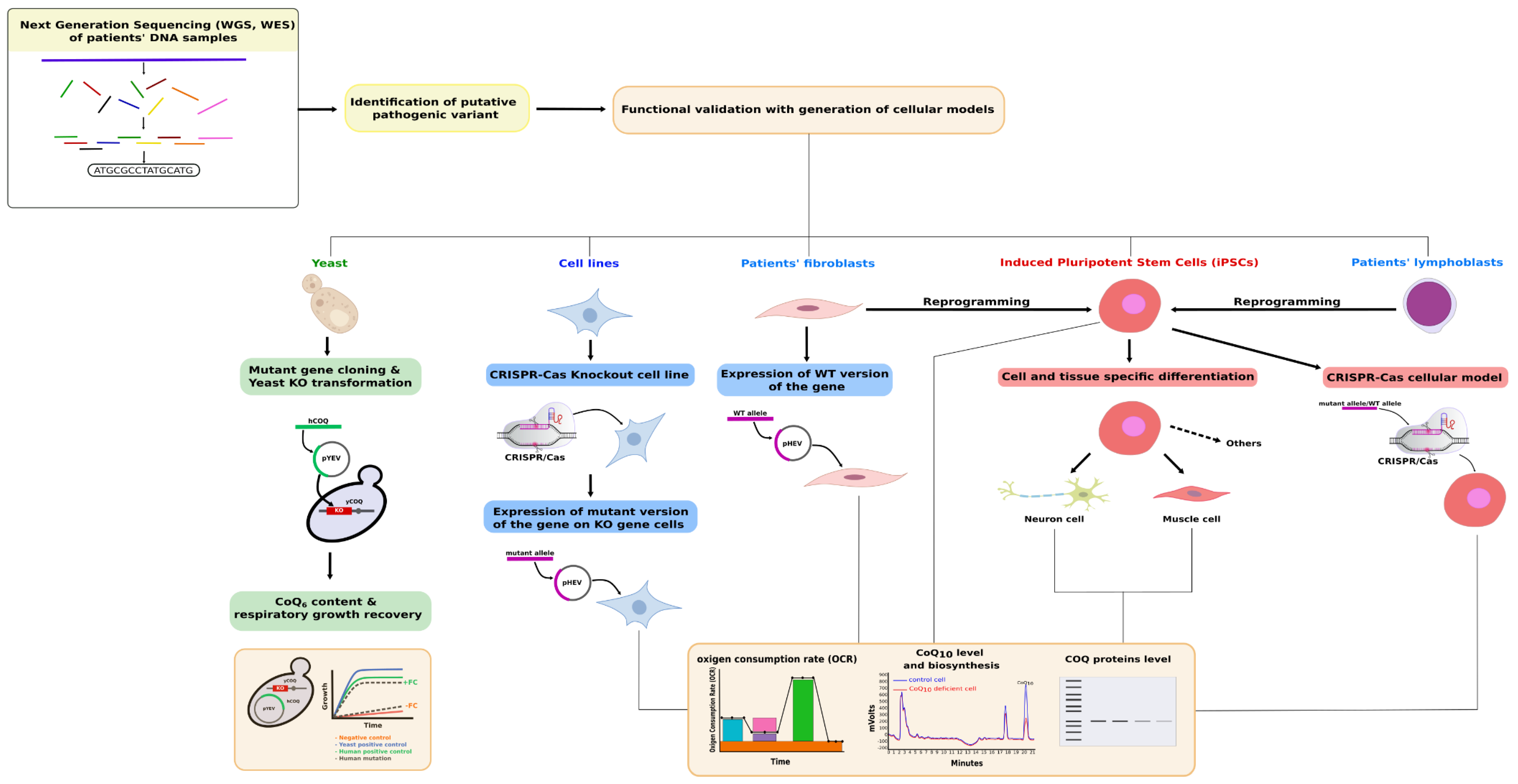
4. Mammalian Cell-Based Functional Tests for Primary CoQ Deficiencies Validation
4.1. Targeted Functional Tests
4.2. Rescue Experiments
4.3. Expression Studies in a Cell-Based Model System
4.4. Cellular Models Generated by Genome Editing Technologies
5. iPSCs as a Model for CoQ Deficiency Pathogenesis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Craven, L.; Alston, C.L.; Taylor, R.W.; Turnbull, D.M. Recent advances in mitochondrial disease. Annu. Rev. Genom. Hum. Genet. 2017, 18, 257–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiMauro, S.; Schon, E.A.; Carelli, V.; Hirano, M. The clinical maze of mitochondrial neurology. Nat. Rev. Neurol. 2013, 9, 429–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Herst, P.M.; Rowe, M.R.; Carson, G.M.; Berridge, M.V. Functional mitochondria in health and disease. Front. Endocrinol. 2017, 8, 296. [Google Scholar] [CrossRef] [Green Version]
- Calvo, G.T.B.; Haack, T.B.; Karall, D.; Ohtake, A.; Invernizzi, F.; Carrozzo, R.; Kremer, L.; Dusi, S.; Fauth, C.; Scholl-Bürgi, S.; et al. COQ4 mutations cause a broad spectrum of mitochondrial disorders associated with CoQ10 Deficiency. Am. J. Hum. Genet. 2015, 96, 309–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcazar-Fabra, M.; Navas, P.; Calvo, G.T.B. Coenzyme Q biosynthesis and its role in the respiratory chain structure. Biochim. Biophys. Acta BBA -Bioenergy 2016, 1857, 1073–1078. [Google Scholar] [CrossRef]
- Banerjee, R.; Purhonen, J.; Kallijärvi, J. The mitochondrial coenzyme Q junction and complex III: Biochemistry and pathophysiology. FEBS J. 2021. [Google Scholar] [CrossRef]
- Guarás, A.; Perales-Clemente, E.; Calvo, E.; Acin-Perez, R.; Loureiro, M.; Pujol, C.; Martínez-Carrascoso, I.; Nuñez, E.; Garcia-Marques, F.; Rodríguez-Hernández, Á.; et al. The CoQH2/CoQ ratio serves as a sensor of respiratory chain efficiency. Cell Rep. 2016, 15, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acín-Pérez, R.; Latorre-Pellicer, A.; Colás, C.; Balsa, E.; Perales-Clemente, E.; Quirós, P.M.; Calvo, E.; Rodríguez-Hernández, M.A.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef]
- Spinazzi, M.; Radaelli, E.; Horré, K.; Arranz, A.M.; Gounko, N.V.; Agostinis, P.; Maia, T.M.; Impens, F.; Morais, V.A.; Lopez-Lluch, G.; et al. PARL deficiency in mouse causes complex III defects, coenzyme Q depletion, and Leigh-like syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Alcazar-Fabra, M.; Trevisson, E.; Brea-Calvo, G. Clinical syndromes associated with coenzyme Q10 deficiency. Essays Biochem. 2018, 62, 377–398. [Google Scholar] [CrossRef]
- Salviati, L.; Trevisson, E.; Doimo, M.; Navas, P. Primary coenzyme Q10 deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2017. [Google Scholar]
- Hughes, B.G.; Harrison, P.M.; Hekimi, S. Estimating the occurrence of primary ubiquinone deficiency by analysis of large-scale sequencing data. Sci. Rep. 2017, 7, 17744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molyneux, S.L.; Florkowski, C.M.; Lever, M.; George, P.M. Biological variation of coenzyme Q10. Clin. Chem. 2005, 51, 455–457. [Google Scholar] [CrossRef] [Green Version]
- Duncan, A.J.; Heales, S.J.; Mills, K.; Eaton, S.; Land, J.M.; Hargreaves, I.P. Determination of coenzyme Q10 status in blood Mononuclear cells, skeletal muscle, and plasma by HPLC with Di-propoxy-coenzyme Q10 as an internal standard. Clin. Chem. 2005, 51, 2380–2382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hargreaves, I.P.; Duncan, A.J.; Heales, S.J.; Land, J.M. The effect of HMG-CoA reductase inhibitors on coenzyme Q10: Possible biochemical/clinical implications. Drug Saf. 2005, 28, 659–676. [Google Scholar] [CrossRef] [PubMed]
- del Pozo-Cruz, J.; Rodríguez-Bies, E.; Ballesteros-Simarro, M.; Navas-Enamorado, I.; Tung, B.T.; Navas, P.; López-Lluch, G. Physical activity affects plasma coenzyme Q10 levels differently in young and old humans. Biogerontology 2014, 15, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Bies, E.; Navas, P.; López-Lluch, G. Age-dependent effect of every-other-day feeding and aerobic exercise in ubiquinone levels and related antioxidant activities in mice muscle. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 33–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paredes-Fuentes, A.J.; Montero, R.; Codina, A.; Jou, C.; Fernández, G.; Maynou, J.; Santos-Ocaña, C.; Riera, J.; Navas, P.; Drobnic, F.; et al. Coenzyme Q10 treatment monitoring in different human biological samples. Antioxidants 2020, 9, 979. [Google Scholar] [CrossRef] [PubMed]
- Shults, C.W.; Haas, R.H.; Passov, D.; Beal, M.F. Coenzyme Q10 levels correlate with the activities of complexes I and II/III in mitochondria from parkinsonian and nonparkinsonian subjects. Ann. Neurol. 1997, 42, 261–264. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Tazir, M.; López, L.C.; Quinzii, C.M.; Assoum, M.; Drouot, N.; Busso, C.; Makri, S.; Ali-Pacha, L.; Benhassine, T.; et al. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am. J. Hum. Genet. 2008, 82, 661–672. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Aguilera, J.C.; Cortés, A.B.; Fernández-Ayala, D.J.M.; Navas, P. Biochemical assessment of coenzyme Q10 deficiency. J. Clin. Med. 2017, 6, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berardo, A.I.; Quinzii, C.M. Redefining infantile-onset multisystem phenotypes of coenzyme Q10-deficiency in the next-generation sequencing era. J. Transl. Genet. Genom. 2020, 4, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Pitceathly, R.; McFarland, R. Mitochondrial myopathies in adults and children. Curr. Opin. Neurol. 2014, 27, 576–582. [Google Scholar] [CrossRef]
- Suomalainen-Wartiovaara, A.; Battersby, B.J. Mitochondrial diseases: The contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 2018, 19, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef]
- Rodenburg, R.J. The functional genomics laboratory: Functional validation of genetic variants. J. Inherit. Metab. Dis. 2018, 41, 297–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, A.M.; Bradley, M.C.; del Rio, L.F.; Nag, A.; Tsui, H.S.; Clarke, C.F. Coenzyme Q10 deficiencies: Pathways in yeast and humans. Essays Biochem. 2018, 62, 361–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcázar-Fabra, M.; Rodríguez-Sánchez, F.; Trevisson, E.; Brea-Calvo, G. Primary coenzyme Q deficiencies: A literature review and online platform of clinical features to uncover genotype-phenotype correlations. Free Radic. Biol. Med. 2021, 167, 141–180. [Google Scholar] [CrossRef]
- Stefely, J.; Kwiecien, N.W.; Freiberger, E.C.; Richards, A.L.; Jochem, A.; Rush, M.J.P.; Ulbrich, A.; Robinson, K.P.; Hutchins, P.; Veling, M.T.; et al. Mitochondrial protein functions elucidated by multi-omic mass spectrometry profiling. Nat. Biotechnol. 2016, 34, 1191–1197. [Google Scholar] [CrossRef] [Green Version]
- Stefely, J.; Pagliarini, D.J. Biochemistry of Mitochondrial Coenzyme Q Biosynthesis. Trends Biochem. Sci. 2017, 42, 824–843. [Google Scholar] [CrossRef]
- Villalba, J.M.; Navas, P. Regulation of coenzyme Q biosynthesis pathway in eukaryotes. Free Radic. Biol. Med. 2021, 165, 312–323. [Google Scholar] [CrossRef]
- Lopez, M.J.A.; Trevisson, E.; Canton, M.; Vazquez-Fonseca, L.; Morbidoni, V.; Baschiera, E.; Frasson, C.; Pelosi, L.; Rascalou, B.; Desbats, M.A.; et al. Vanillic acid restores coenzyme q biosynthesis and ATP production in human cells lackingcoq6. Oxidative Med. Cell. Longev. 2019, 2019, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamukai, M. Biosynthesis of coenzyme Q in eukaryotes. Biosci. Biotechnol. Biochem. 2016, 80, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Ashby, M.N.; Edwards, P.A. Elucidation of the deficiency in two yeast coenzyme Q mutants. Characterization of the structural gene encoding hexaprenyl pyrophosphate synthetase. J. Biol. Chem. 1990, 265, 13157–13164. [Google Scholar] [CrossRef]
- Forsgren, M.; Attersand, A.; Lake, S.; Grünler, J.; Swiezewska, E.; Dallner, G.; Climent, I. Isolation and functional expression of human COQ2, a gene encoding a polyprenyl transferase involved in the synthesis of CoQ. Biochem. J. 2004, 382, 519–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonassen, T.; Clarke, C.F. Isolation and functional expression of human COQ3, a gene encoding a methyltransferase required for ubiquinone biosynthesis. J. Biol. Chem. 2000, 275, 12381–12387. [Google Scholar] [CrossRef] [Green Version]
- Herebian, D.; Seibt, A.; Smits, S.H.; Bünning, G.; Freyer, C.; Prokisch, H.; Karall, D.; Wredenberg, A.; Wedell, A.; López, L.C.; et al. Detection of 6-demethoxyubiquinone in CoQ 10 deficiency disorders: Insights into enzyme interactions and identification of potential therapeutics. Mol. Genet. Metab. 2017, 121, 216–223. [Google Scholar] [CrossRef]
- Nguyen, T.P.; Casarin, A.; Desbats, M.A.; Doimo, M.; Trevisson, E.; Santos-Ocaña, C.; Navas, P.; Clarke, C.F.; Salviati, L. Molecular characterization of the human COQ5 C-methyltransferase in coenzyme Q10 biosynthesis. Biochim. Biophys. Acta BBA -Mol. Cell Biol. Lipids 2014, 1841, 1628–1638. [Google Scholar] [CrossRef] [Green Version]
- Ozeir, M.; Mühlenhoff, U.; Webert, H.; Lill, R.; Fontecave, M.; Pierrel, F. Coenzyme Q biosynthesis: Coq6 is required for the C5-hydroxylation reaction and substrate analogs rescue Coq6 deficiency. Chem. Biol. 2011, 18, 1134–1142. [Google Scholar] [CrossRef]
- Tran, U.C.; Marbois, B.; Gin, P.; Gulmezian, M.; Jonassen, T.; Clarke, C.F. Complementation of saccharomyces cerevisiae coq7 mutants by mitochondrial targeting of the escherichia coli UbiF polypeptide: Two functions of yeast Coq7 polypeptide in coenzyme Q biosynthesis. J. Biol. Chem. 2006, 281, 16401–16409. [Google Scholar] [CrossRef] [Green Version]
- Stefely, J.; Reidenbach, A.G.; Ulbrich, A.; Oruganty, K.; Floyd, B.; Jochem, A.; Saunders, J.M.; Johnson, I.; Minogue, C.E.; Wrobel, R.L.; et al. Mitochondrial ADCK3 Employs an Atypical Protein Kinase-like Fold to Enable Coenzyme Q Biosynthesis. Mol. Cell 2015, 57, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widmeier, E.; Yu, S.; Nag, A.; Chung, Y.W.; Nakayama, M.; Fernández-Del-Río, L.; Hugo, H.; Schapiro, D.; Buerger, F.; Choi, W.-I.; et al. ADCK4 Deficiency destabilizes the coenzyme Q complex, which is rescued by 2,4-dihydroxybenzoic acid treatment. J. Am. Soc. Nephrol. 2020, 31, 1191–1211. [Google Scholar] [CrossRef] [PubMed]
- Lohman, D.C.; Aydin, D.; Von Bank, H.; Smith, R.W.; Linke, V.; Weisenhorn, E.; McDevitt, M.T.; Hutchins, P.; Wilkerson, E.M.; Wancewicz, B.; et al. An isoprene lipid-binding protein promotes eukaryotic coenzyme Q biosynthesis. Mol. Cell 2019, 73, 763–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vázquez-Fonseca, L.; Schäefer, J.; Navas-Enamorado, I.; Santos-Ocaña, C.; Hernández-Camacho, J.D.; Guerra, I.; Cascajo, M.V.; Sánchez-Cuesta, A.; Horvath, Z.; Siendones, E.; et al. ADCK2 Haploinsufficiency Reduces Mitochondrial Lipid Oxidation and Causes Myopathy Associated with CoQ Deficiency. J. Clin. Med. 2019, 8, 1374. [Google Scholar] [CrossRef] [Green Version]
- Caglayan, A.; Gumus, H.; Sandford, E.; Kubisiak, T.L.; Ma, Q.; Ozel, A.B.; Per, H.; Li, J.Z.; Shakkottai, V.G.; Burmeister, M. COQ4 Mutation leads to childhood-onset ataxia improved by CoQ10 administration. Cerebellum 2019, 18, 665–669. [Google Scholar] [CrossRef]
- Ashraf, S.; Gee, H.Y.; Woerner, S.; Xie, L.X.; Vega-Warner, V.; Lovric, S.; Fang, H.; Song, X.; Cattran, D.C.; Avila-Casado, C.; et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J. Clin. Investig. 2013, 123, 5179–5189. [Google Scholar] [CrossRef] [Green Version]
- Reidenbach, A.G.; Kemmerer, Z.A.; Aydin, D.; Jochem, A.; McDevitt, M.T.; Hutchins, P.D.; Stark, J.L.; Stefely, J.A.; Reddy, T.; Hebert, A.S.; et al. Conserved lipid and small-molecule modulation of COQ8 reveals regulation of the ancient kinase-like UbiB family. Cell Chem. Biol. 2018, 25, 154–165. [Google Scholar] [CrossRef] [Green Version]
- Tsui, H.S.; Pham, N.V.B.; Amer, B.R.; Bradley, M.C.; Gosschalk, J.E.; Gallagher-Jones, M.; Ibarra, H.; Clubb, R.T.; Blaby-Haas, C.E.; Clarke, C.F. Human COQ10A and COQ10B are distinct lipid-binding START domain proteins required for coenzyme Q function. J. Lipid Res. 2019, 60, 1293–1310. [Google Scholar] [CrossRef]
- Vasta, V.; Merritt, J.L., II; Saneto, R.P.; Hahn, S.H. Next-generation sequencing for mitochondrial diseases: A wide diagnostic spectrum. Pediatr. Int. 2012, 54, 585–601. [Google Scholar] [CrossRef]
- Mollet, J.; Giurgea, I.; Schlemmer, D.; Dallner, G.; Chretien, D.; Delahodde, A.; Bacq, D.; De Lonlay, P.; Munnich, A.; Rotig, A. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J. Clin. Investig. 2007, 117, 765–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, L.C.; Schuelke, M.; Quinzii, C.M.; Kanki, T.; Rodenburg, R.J.; Naini, A.; DiMauro, S.; Hirano, M. Leigh Syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) Mutations. Am. J. Hum. Genet. 2006, 79, 1125–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desbats, M.A.; Vetro, A.; Limongelli, I.; Lunardi, G.; Casarin, A.; Doimo, M.; Spinazzi, M.; Angelini, C.; Cenacchi, G.; Burlina, A.; et al. Primary coenzyme Q10 deficiency presenting as fatal neonatal multiorgan failure. Eur. J. Hum. Genet. 2015, 23, 1254–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desbats, M.A.; Morbidoni, V.; Silic-Benussi, M.; Doimo, M.; Ciminale, V.; Cassina, M.; Sacconi, S.; Hirano, M.; Basso, G.; Pierrel, F.; et al. TheCOQ2genotype predicts the severity of coenzyme Q10deficiency. Hum. Mol. Genet. 2016, 25, 4256–4265. [Google Scholar] [CrossRef] [Green Version]
- Jakobs, B.S.; van den Heuvel, L.P.; Smeets, R.J.; de Vries, M.C.; Hien, S.; Schaible, T.; Rodenburg, R.J. A novel mutation in COQ2 leading to fatal infantile multisystem disease. J. Neurol. Sci. 2013, 326, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Dinwiddie, D.L.; Smith, L.D.; Miller, N.A.; Atherton, A.M.; Farrow, E.G.; Strenk, M.E.; Soden, S.E.; Saunders, C.J.; Kingsmore, S. Diagnosis of mitochondrial disorders by concomitant next-generation sequencing of the exome and mitochondrial genome. Genomics 2013, 102, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Mollet, J.; Delahodde, A.; Serre, V.; Chretien, D.; Schlemmer, D.; Lombes, A.; Boddaert, N.; Desguerre, I.; De Lonlay, P.; De Baulny, H.O.; et al. CABC1 Gene Mutations Cause Ubiquinone Deficiency with Cerebellar Ataxia and Seizures. Am. J. Hum. Genet. 2008, 82, 623–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diomedi-Camassei, F.; Di Giandomenico, S.; Santorelli, F.M.; Caridi, G.; Piemonte, F.; Montini, G.; Ghiggeri, G.M.; Murer, L.; Barisoni, L.; Pastore, A.; et al. COQ2 nephropathy: A newly described inherited mitochondriopathy with primary renal involvement. J. Am. Soc. Nephrol. 2007, 18, 2773–2780. [Google Scholar] [CrossRef] [Green Version]
- Scalais, E.; Chafai, R.; Van Coster, R.; Bindl, L.; Nuttin, C.; Panagiotaraki, C.; Seneca, S.; Lissens, W.; Ribes, A.; Geers, C.; et al. Early myoclonic epilepsy, hypertrophic cardiomyopathy and subsequently a nephrotic syndrome in a patient with CoQ10 deficiency caused by mutations in para-hydroxybenzoate-polyprenyl transferase (COQ2). Eur. J. Paediatr. Neurol. 2013, 17, 625–630. [Google Scholar] [CrossRef]
- Quinzii, C.; Naini, A.; Salviati, L.; Trevisson, E.; Navas, P.; DiMauro, S.; Hirano, M. A mutation in Para-hydroxybenzoate-Polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am. J. Hum. Genet. 2006, 78, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Salviati, L.; Sacconi, S.; Murer, L.; Zacchello, G.; Franceschini, L.; Laverda, A.M.; Basso, G.; Quinzii, C.; Angelini, C.; Hirano, M.; et al. Infantile encephalomyopathy and nephropathy with CoQ10 deficiency: A CoQ10-responsive condition. Neurology 2005, 65, 606–608. [Google Scholar] [CrossRef]
- McCarthy, H.J.; Bierzynska, A.; Wherlock, M.; Ognjanovic, M.; Kerecuk, L.; Hegde, S.; Feather, S.; Gilbert, R.D.; Krischock, L.; Jones, C.; et al. Simultaneous sequencing of 24 genes associated with steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol. 2013, 8, 637–648. [Google Scholar] [CrossRef] [Green Version]
- Gigante, M.; Diella, S.; Santangelo, L.; Trevisson, E.; Acosta, M.; Amatruda, M.; Finzi, G.; Caridi, G.; Murer, L.; Accetturo, M.; et al. Further phenotypic heterogeneity of CoQ10 deficiency associated with steroid resistant nephrotic syndrome and novel COQ2 and COQ6 variants. Clin. Genet. 2017, 92, 224–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai-Okazaki, A.; Kishita, Y.; Kohda, M.; Mizuno, Y.; Fushimi, T.; Matsunaga, A.; Yatsuka, Y.; Hirata, T.; Harashima, H.; Takeda, A.; et al. Cardiomyopathy in children with mitochondrial disease: Prognosis and genetic background. Int. J. Cardiol. 2019, 279, 115–121. [Google Scholar] [CrossRef]
- Chung, W.K.; Martin, K.; Jalas, C.; Braddock, S.R.; Juusola, J.; Monaghan, K.G.; Warner, B.; Franks, S.; Yudkoff, M.; Lulis, L.; et al. Mutations inCOQ4, an essential component of coenzyme Q biosynthesis, cause lethal neonatal mitochondrial encephalomyopathy. J. Med. Genet. 2015, 52, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Sondheimer, N.; Hewson, S.; Cameron, J.M.; Somers, G.R.; Broadbent, J.D.; Ziosi, M.; Quinzii, C.M.; Naini, A.B. Novel recessive mutations in COQ4 cause severe infantile cardiomyopathy and encephalopathy associated with CoQ 10 deficiency. Mol. Genet. Metab. Rep. 2017, 12, 23–27. [Google Scholar] [CrossRef]
- Yu, M.H.-C.; Tsang, M.H.-Y.; Lai, S.; Ho, M.S.-P.; Tse, D.M.L.; Willis, B.; Kwong, A.K.-Y.; Chou, Y.-Y.; Lin, S.-P.; Quinzii, C.M.; et al. Primary coenzyme Q10 deficiency-7: Expanded phenotypic spectrum and a founder mutation in southern Chinese. NPJ Genom. Med. 2019, 4, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Zhou, Y.; Wang, Z.; Xia, Z.; Ren, J.; Guo, Q. Clinical phenotype, in silico and biomedical analyses, and intervention for an East Asian population-specific c.370G>A (p.G124S) COQ4 mutation in a Chinese family with CoQ10 deficiency-associated Leigh syndrome. J. Hum. Genet. 2019, 64, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Salviati, L.; Trevisson, E.; Rodriguez Hernandez, M.A.; Casarin, A.; Pertegato, V.; Doimo, M.; Cassina, M.; Agosto, C.; Desbats, M.A.; Sartori, G.; et al. Haploinsufficiency of COQ4causes coenzyme Q10deficiency. J. Med. Genet. 2012, 49, 187–191. [Google Scholar] [CrossRef] [Green Version]
- Ling, T.-K.; Law, C.-Y.; Yan, K.-W.; Fong, N.-C.; Wong, K.-C.; Lee, K.-L.; Chu, W.C.-W.; Brea-Calvo, G.; Lam, C.-W. Clinical whole-exome sequencing reveals a common pathogenic variant in patients with CoQ10 deficiency: An underdiagnosed cause of mitochondriopathy. Clin. Chim. Acta 2019, 497, 88–94. [Google Scholar] [CrossRef]
- Mero, S.; Salviati, L.; Leuzzi, V.; Rubegni, A.; Calderan, C.; Nardecchia, F.; Galatolo, D.; Desbats, M.A.; Naef, V.; Gemignani, F.; et al. New pathogenic variants in COQ4 cause ataxia and neurodevelopmental disorder without detectable CoQ10 deficiency in muscle or skin fibroblasts. J. Neurol. 2019, 497, 88–94. [Google Scholar] [CrossRef]
- Malicdan, M.C.V.; Vilboux, T.; Ben-Zeev, B.; Guo, J.; Eliyahu, A.; Pode-Shakked, B.; Dori, A.; Kakani, S.; Chandrasekharappa, S.C.; Ferreira, C.R.; et al. A novel inborn error of the coenzyme Q10 biosynthesis pathway: Cerebellar ataxia and static encephalomyopathy due to COQ5 C-methyltransferase deficiency. Hum. Mutat. 2018, 39, 69–79. [Google Scholar] [CrossRef]
- Doimo, M.; Trevisson, E.; Airik, R.; Bergdoll, M.; Santos-Ocaña, C.; Hildebrandt, F.; Navas, P.; Pierrel, F.; Salviati, L. Effect of vanillic acid on COQ6 mutants identified in patients with coenzyme Q10 deficiency. Biochim. Biophys. Acta BBA -Mol. Basis Dis. 2014, 1842, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heeringa, S.F.; Chernin, G.; Chaki, M.; Zhou, W.; Sloan, A.J.; Ji, Z.; Xie, L.X.; Salviati, L.; Hurd, T.W.; Vega-Warner, V.; et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J. Clin. Investig. 2011, 121, 2013–2024. [Google Scholar] [CrossRef] [Green Version]
- Koyun, M.; Çomak, E.; Akman, S. CoenzymeQ10 therapy in two sisters with CoQ6 mutations with long-term follow-up. Pediatr. Nephrol. 2018, 34, 737–738. [Google Scholar] [CrossRef]
- Park, E.; Ahn, Y.H.; Kang, H.G.; Yoo, K.H.; Won, N.H.; Lee, K.B.; Moon, K.C.; Seong, M.-W.; Gwon, T.R.; Park, S.S.; et al. COQ6 mutations in children with steroid-resistant focal segmental glomerulosclerosis and sensorineural hearing loss. Am. J. Kidney Dis. 2017, 70, 139–144. [Google Scholar] [CrossRef]
- Kwong, A.K.; Chiu, A.T.; Tsang, M.H.; Lun, K.; Rodenburg, R.J.T.; Smeitink, J.; Chung, B.H.; Fung, C. A fatal case of COQ7-associated primary coenzyme Q 10 deficiency. JIMD Rep. 2019, 47, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Freyer, C.; Stranneheim, H.; Naess, K.; Mourier, A.; Felser, A.; Maffezzini, C.; Lesko, N.; Bruhn, H.; Engvall, M.; Wibom, R.; et al. Rescue of primary ubiquinone deficiency due to a novel COQ7 defect using 2,4–dihydroxybensoic acid. J. Med Genet. 2015, 52, 779–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Smith, C.; Parboosingh, J.S.; Khan, A.; Innes, M.; Hekimi, S. Pathogenicity of two COQ7 mutations and responses to 2,4-dihydroxybenzoate bypass treatment. J. Cell. Mol. Med. 2017, 21, 2329–2343. [Google Scholar] [CrossRef]
- Gerards, M.; van den Bosch, B.; Calis, C.; Schoonderwoerd, K.; van Engelen, K.; Tijssen, M.; de Coo, R.; van der Kooi, A.; Smeets, H. Nonsense mutations in CABC1/ADCK3 cause progressive cerebellar ataxia and atrophy. Mitochondrion 2010, 10, 510–515. [Google Scholar] [CrossRef]
- Horvath, R.; Czermin, B.; Gulati, S.; Demuth, S.; Houge, G.; Pyle, A.; Dineiger, C.; Blakely, E.L.; Hassani, A.; Foley, C.; et al. Adult-onset cerebellar ataxia due to mutations inCABC1/ADCK3. J. Neurol. Neurosurg. Psychiatry 2011, 83, 174–178. [Google Scholar] [CrossRef]
- Mignot, C.; Apartis, E.; Durr, A.; Marques Lourenço, C.; Charles, P.; Devos, D.; Moreau, C.; de Lonlay, P.; Drouot, N.; Burglen, L.; et al. Phenotypic variability in ARCA2 and identification of a core ataxic phenotype with slow progression. Orphanet J. Rare Dis. 2013, 8, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anheim, M.; Fleury, M.; Monga, B.; Laugel, V.; Chaigne, D.; Rodier, G.; Ginglinger, E.; Boulay, C.; Courtois, S.; Drouot, N.; et al. Epidemiological, clinical, paraclinical and molecular study of a cohort of 102 patients affected with autosomal recessive progressive cerebellar ataxia from Alsace, Eastern France: Implications for clinical management. Neurogenetics 2009, 11, 1–12. [Google Scholar] [CrossRef]
- Jacobsen, J.C.; Whitford, W.; Swan, B.; Taylor, J.; Love, D.R.; Hill, R.; Molyneux, S.; George, P.M.; Mackay, R.; Robertson, S.P.; et al. Compound heterozygous inheritance of mutations in coenzyme Q8A results in autosomal recessive cerebellar ataxia and coenzyme q10 deficiency in a female Sib-Pair. JIMD Rep. 2017, 42, 31–36. [Google Scholar] [CrossRef]
- Hikmat, O.; Tzoulis, C.; Knappskog, P.M.; Johansson, S.; Boman, H.; Sztromwasser, P.; Lien, E.; Brodtkorb, E.; Ghezzi, D.; Bindoff, L.A. ADCK3 mutations with epilepsy, stroke-like episodes and ataxia: A POLG mimic? Eur. J. Neurol. 2016, 23, 1188–1194. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Ruiz-Lopez, M.; Slow, E.; Tarnopolsky, M.; Lang, A.E.; Munhoz, R.P. ADCK3-related Coenzyme Q10 Deficiency: A Potentially Treatable Genetic Disease. Mov. Disord. Clin. Pract. 2018, 5, 635–639. [Google Scholar] [CrossRef] [Green Version]
- Terracciano, A.; Renaldo, F.; Zanni, G.; D’Amico, A.; Pastore, A.; Barresi, S.; Valente, E.M.; Piemonte, F.; Tozzi, G.; Carrozzo, R.; et al. The use of muscle biopsy in the diagnosis of undefined ataxia with cerebellar atrophy in children. Eur. J. Paediatr. Neurol. 2012, 16, 248–256. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-T.; Hersheson, J.; Plagnol, V.; Fawcett, K.; Duberley, K.E.C.; Preza, E.; Hargreaves, I.P.; Chalasani, A.; Laurá, M.; Wood, N.; et al. Autosomal-recessive cerebellar ataxia caused by a novel ADCK3 mutation that elongates the protein: Clinical, genetic and biochemical characterisation. J. Neurol. Neurosurg. Psychiatry 2014, 85, 493–498. [Google Scholar] [CrossRef] [Green Version]
- Galosi, S.; Barca, E.; Carrozzo, R.; Schirinzi, T.; Quinzii, C.M.; Lieto, M.; Vasco, G.; Zanni, G.; Di Nottia, M.; Galatolo, D.; et al. Dystonia-Ataxia with early handwriting deterioration in COQ8A mutation carriers: A case series and literature review. Park. Relat. Disord. 2019, 68, 8–16. [Google Scholar] [CrossRef]
- Barca, E.; Musumeci, O.; Montagnese, F.; Marino, S.; Granata, F.; Nunnari, D.; Peverelli, L.; DiMauro, S.; Quinzii, C.; Toscano, A. Cerebellar ataxia and severe muscle CoQ10 deficiency in a patient with a novel mutation in ADCK3. Clin. Genet. 2016, 90, 156–160. [Google Scholar] [CrossRef] [Green Version]
- Atmaca, M.; Gulhan, B.; Korkmaz, E.; Inozu, M.; Soylemezoglu, O.; Candan, C.; Bayazıt, A.K.; Elmacı, A.M.; Parmaksiz, G.; Düzova, A.; et al. Follow-up results of patients with ADCK4 mutations and the efficacy of CoQ10 treatment. Pediatr. Nephrol. 2017, 32, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Vazquez Fonseca, L.; Doimo, M.; Calderan, C.; Desbats, M.A.; Acosta, M.J.; Cerqua, C.; Cassina, M.; Ashraf, S.; Hildebrandt, F.; Sartori, G.; et al. Mutations in COQ8B (ADCK4) found in patients with steroid-resistant nephrotic syndrome alter COQ8B function. Hum. Mutat. 2017, 39, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, E.; Lipska-Ziętkiewicz, B.S.; Boyer, O.; Gribouval, O.; Fourrage, C.; Tabatabaei, M.; Schnaidt, S.; Gucer, S.; Kaymaz, F.; Arici, M.; et al. ADCK4-Associated Glomerulopathy Causes Adolescence-Onset FSGS. J. Am. Soc. Nephrol. 2015, 27, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.C.; Ito, Y.; Ahmed, A.; Schwartzentruber, J.; Beaulieu, C.L.; Aberg, E.; Majewski, J.; Bulman, D.E.; Horsting-Wethly, K.; Koning, D.V.-D.; et al. A family segregating lethal neonatal coenzyme Q10 deficiency caused by mutations in COQ9. J. Inherit. Metab. Dis. 2018, 41, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Danhauser, K.; Herebian, D.; Haack, T.B.; Rodenburg, R.J.; Strom, T.M.; Meitinger, T.; Klee, D.; Mayatepek, E.; Prokisch, H.; Distelmaier, F. Fatal neonatal encephalopathy and lactic acidosis caused by a homozygous loss-of-function variant in COQ9. Eur. J. Hum. Genet. 2016, 24, 450–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, A.J.; Bitner-Glindzicz, M.; Meunier, B.; Costello, H.; Hargreaves, I.P.; Lopez, L.C.; Hirano, M.; Quinzii, C.M.; Sadowski, M.; Hardy, J.; et al. A Nonsense Mutation in COQ9 Causes Autosomal-Recessive Neonatal-Onset Primary Coenzyme Q10 Deficiency: A Potentially Treatable Form of Mitochondrial Disease. Am. J. Hum. Genet. 2009, 84, 558–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S.; Hargreaves, I.; Clayton, P.; Heales, S. Neonatal presentation of coenzyme Q10 deficiency. J. Pediatr. 2001, 139, 456–458. [Google Scholar] [CrossRef] [PubMed]
- Tzagoloff, A.; Dieckmann, C.L. PET genes of Saccharomyces cerevisiae. Microbiol. Rev. 1990, 54, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Tran, U.C.; Clarke, C.F. Endogenous synthesis of coenzyme Q in eukaryotes. Mitochondrion 2007, 7, S62–S71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saiki, R.; Nagata, A.; Kainou, T.; Matsuda, H.; Kawamukai, M. Characterization of solanesyl and decaprenyl diphosphate synthases in mice and humans. FEBS J. 2005, 272, 5606–5622. [Google Scholar] [CrossRef]
- Leonard, C.J.; Aravind, L.; Koonin, E.V. Novel Families of Putative Protein Kinases in Bacteria and Archaea: Evolution of the “Eukaryotic” Protein Kinase Superfamily. Genome Res. 1998, 8, 1038–1047. [Google Scholar] [CrossRef] [Green Version]
- He, C.H.; Xie, L.X.; Allan, C.M.; Tran, U.C.; Clarke, C.F. Coenzyme Q supplementation or over-expression of the yeast Coq8 putative kinase stabilizes multi-subunit Coq polypeptide complexes in yeast coq null mutants. Biochim. Biophys. Acta BBA -Mol. Cell Biol. Lipids 2014, 1841, 630–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.X.; Hsieh, E.J.; Watanabe, S.; Allan, C.M.; Chen, J.Y.; Tran, U.C.; Clarke, C.F. Expression of the human atypical kinase ADCK3 rescues coenzyme Q biosynthesis and phosphorylation of Coq polypeptides in yeast coq8 mutants. Biochim. Biophys. Acta BBA -Mol. Cell Biol. Lipids 2011, 1811, 348–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Martín, J.M.; Salviati, L.; Trevisson, E.; Montini, G.; DiMauro, S.; Quinzii, C.; Hirano, M.; Rodriguez-Hernandez, A.; Cordero, M.D.; Sánchez-Alcázar, J.A.; et al. Missense mutation of the COQ2 gene causes defects of bioenergetics and de novo pyrimidine synthesis. Hum. Mol. Genet. 2007, 16, 1091–1097. [Google Scholar] [CrossRef]
- Hsu, A.Y.; Poon, W.; Shepherd, J.A.; Myles, D.C.; Clarke, C.F. Complementation of coq3 Mutant Yeast by Mitochondrial Targeting of the Escherichia coli UbiG Polypeptide: Evidence That UbiG Catalyzes Both O-Methylation Steps in Ubiquinone Biosynthesis. Biochemistry 1996, 35, 9797–9806. [Google Scholar] [CrossRef]
- Marbois, B.N.; Clarke, C.F. The COQ7 gene encodes a protein in saccharomyces cerevisiae necessary for ubiquinone biosynthesis. J. Biol. Chem. 1996, 271, 2995–3004. [Google Scholar] [CrossRef] [Green Version]
- Arokium, H.; Ouerfelli, H.; Velours, G.; Camougrand, N.; Vallette, F.; Manon, S. Substitutions of potentially phosphorylatable serine residues of bax reveal how they may regulate its interaction with mitochondria. J. Biol. Chem. 2007, 282, 35104–35112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padilla, S.; Jonassen, T.; Jiménez-Hidalgo, M.A.; Fernández-Ayala, D.J.M.; López-Lluch, G.; Marbois, B.; Navas, P.; Clarke, C.F.; Santos-Ocaña, C. Demethoxy-Q, An Intermediate of coenzyme Q biosynthesis, fails to support respiration in saccharomyces cerevisiae and lacks antioxidant activity. J. Biol. Chem. 2004, 279, 25995–26004. [Google Scholar] [CrossRef] [Green Version]
- Villalba, J.; Palmgren, M.; Berberián, G.; Ferguson, C.; Serrano, R. Functional expression of plant plasma membrane H (+)-ATPase in yeast endoplasmic reticulum. J. Biol. Chem. 1992, 267, 12341–12349. [Google Scholar] [CrossRef]
- Westhoff, C.M.; Siegel, D.L.; Burd, C.; Foskett, J.K. Mechanism of genetic complementation of ammonium transport in yeast by human erythrocyte Rh-associated glycoprotein. J. Biol. Chem. 2004, 279, 17443–17448. [Google Scholar] [CrossRef] [Green Version]
- Yubero, D.; Montero, R.; Santos-Ocaña, C.; Salviati, L.; Navas, P.; Artuch, R. Molecular diagnosis of coenzyme Q10 deficiency: An update. Expert Rev. Mol. Diagn. 2018, 18, 491–498. [Google Scholar] [CrossRef]
- Stefely, J.; Licitra, F.; Laredj, L.; Reidenbach, A.G.; Kemmerer, Z.A.; Grangeray, A.; Jaeg-Ehret, T.; Minogue, C.E.; Ulbrich, A.; Hutchins, P.; et al. Cerebellar ataxia and coenzyme q deficiency through loss of unorthodox kinase activity. Mol. Cell 2016, 63, 608–620. [Google Scholar] [CrossRef] [Green Version]
- Liparulo, I.; Bergamini, C.; Bortolus, M.; Calonghi, N.; Gasparre, G.; Kurelac, I.; Masin, L.; Rizzardi, N.; Rugolo, M.; Wang, W.; et al. Coenzyme Q biosynthesis inhibition induces HIF-1α stabilization and metabolic switch toward glycolysis. FEBS J. 2021, 288, 1956–1974. [Google Scholar] [CrossRef] [PubMed]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA targeting with CRISPR–Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, M.J. The pursuit of precision mitochondrial medicine: Harnessing preclinical cellular and animal models to optimize mitochondrial disease therapeutic discovery. J. Inherit. Metab. Dis. 2021, 44, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Hermle, T.; Braun, D.A.; Helmstädter, M.; Huber, T.B.; Hildebrandt, F. Modeling Monogenic Human Nephrotic Syndrome in the Drosophila Garland Cell Nephrocyte. J. Am. Soc. Nephrol. 2016, 28, 1521–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Henderson, J.M.; Coote, K.; Cheng, Y.; Valley, H.C.; Zhang, X.-O.; Wang, Q.; Rhym, L.H.; Cao, Y.; Newby, G.A.; et al. Chemical modifications of adenine base editor mRNA and guide RNA expand its application scope. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Liu, G.; David, B.T.; Trawczynski, M.; Fessler, R.G. Advances in pluripotent stem cells: History, mechanisms, technologies, and applications. Stem Cell Rev. Rep. 2020, 16, 3–32. [Google Scholar] [CrossRef] [Green Version]
- McKnight, C.; Low, Y.; Elliott, D.; Thorburn, D.; Frazier, A. Modelling mitochondrial disease in human pluripotent stem cells: What have we learned? Int. J. Mol. Sci. 2021, 22, 7730. [Google Scholar] [CrossRef]
- Hsu, Y.-C.; Chen, C.-T.; Wei, Y.-H. Mitochondrial resetting and metabolic reprogramming in induced pluripotent stem cells and mitochondrial disease modeling. Biochim. Biophys. Acta BBA -Gen. Subj. 2016, 1860, 686–693. [Google Scholar] [CrossRef]
- Hatakeyama, H.; Goto, Y.-I. Concise Review: Heteroplasmic Mitochondrial DNA Mutations and Mitochondrial Diseases: Toward iPSC-Based Disease Modeling, Drug Discovery, and Regenerative Therapeutics. Stem Cells 2016, 34, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.-Y.; Zhuang, Q.-Q.; Qiu, Y.; Zhu, X.-F.; Yan, Q.-F. Cell models and drug discovery for mitochondrial diseases. J. Zhejiang Univ. Sci. B 2019, 20, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, I.; Heaton, R.A.; Mantle, D. Disorders of Human Coenzyme Q10 Metabolism: An Overview. Int. J. Mol. Sci. 2020, 21, 6695. [Google Scholar] [CrossRef]
- Galera-Monge, T.; Zurita-Díaz, F.; Canals, I.; Hansen, M.G.; Rufián-Vázquez, L.; Ehinger, J.K.; Elmér, E.; Martin, M.A.; Garesse, R.; Ahlenius, H.; et al. Mitochondrial Dysfunction and Calcium Dysregulation in Leigh Syndrome Induced Pluripotent Stem Cell Derived Neurons. Int. J. Mol. Sci. 2020, 21, 3191. [Google Scholar] [CrossRef] [PubMed]
- Soldner, F.; Jaenisch, R. Stem Cells, Genome Editing, and the Path to Translational Medicine. Cell 2018, 175, 615–632. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Hu, D.; Shang, Y.; Qi, X. Using induced pluripotent stem cell neuronal models to study neurodegenerative diseases. Biochim. Biophys. Acta BBA -Mol. Basis Dis. 2020, 1866, 165431. [Google Scholar] [CrossRef] [PubMed]
- Romero-Moya, D.; Castaño, J.; Santos-Ocaña, C.; Navas, P.; Menendez, P. Generation, genome edition and characterization of iPSC lines from a patient with coenzyme Q 10 deficiency harboring a heterozygous mutation in COQ4 gene. Stem Cell Res. 2017, 24, 144–147. [Google Scholar] [CrossRef]
- Romero-Moya, D.; Santos-Ocaña, C.; Castaño, J.; Garrabou, G.; Rodríguez-Gómez, J.A.; Ruiz-Bonilla, V.; Bueno, C.; Gonzalez, P.; Giorgetti, A.; Perdiguero, E.; et al. Genetic Rescue of Mitochondrial and Skeletal Muscle Impairment in an Induced Pluripotent Stem Cells Model of Coenzyme Q10 Deficiency. Stem Cells 2017, 35, 1687–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Camacho, J.D.; Bernier, M.; López-Lluch, G.; Navas, P. Coenzyme Q10 supplementation in aging and disease. Front. Physiol. 2018, 9, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-Y.; Ting, H.-C.; Liu, C.-A.; Su, H.-L.; Chiou, T.-W.; Lin, S.-Z.; Harn, H.-J.; Ho, T.-J. Induced pluripotent stem cell (iPSC)-based neurodegenerative disease models for phenotype recapitulation and drug screening. Molecules 2020, 25, 2000. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, F.K.; Okamoto, S.; Mitsui, J.; Sone, T.; Ishikawa, M.; Yamamoto, Y.; Kanegae, Y.; Nakatake, Y.; Imaizumi, K.; Ishiura, H.; et al. The pathogenesis linked to coenzyme Q10 insufficiency in iPSC-derived neurons from patients with multiple-system atrophy. Sci. Rep. 2018, 8, 14215. [Google Scholar] [CrossRef]
- Bonaventura, G.; Iemmolo, R.; Attaguile, G.; La Cognata, V.; Pistone, B.; Raudino, G.; D’Agata, V.; Cantarella, G.; Barcellona, M.; Cavallaro, S. iPSCs: A preclinical drug research tool for neurological disorders. Int. J. Mol. Sci. 2021, 22, 4596. [Google Scholar] [CrossRef]
- Cao, N.; Huang, Y.; Zheng, J.; Spencer, C.I.; Zhang, Y.; Fu, J.-D.; Nie, B.; Xie, M.; Zhang, M.; Wang, H.; et al. Conversion of human fibroblasts into functional cardiomyocytes by small molecules. Science 2016, 352, 1216–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grath, A.; Dai, G. Direct cell reprogramming for tissue engineering and regenerative medicine. J. Biol. Eng. 2019, 13, 14. [Google Scholar] [CrossRef]
- Ruggieri, M.; Riboldi, G.; Brajkovic, S.; Bucchia, M.; Bresolin, N.; Comi, G.; Corti, S. Induced neural stem cells: Methods of reprogramming and potential therapeutic applications. Prog. Neurobiol. 2014, 114, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Aqeilan, R.I. Engineering organoids: A promising platform to understand biology and treat diseases. Cell Death Differ. 2021, 28, 1–4. [Google Scholar] [CrossRef]
- Duong, A.; Evstratova, A.; Sivitilli, A.; Hernandez, J.J.; Gosio, J.; Wahedi, A.; Sondheimer, N.; Wrana, J.L.; Beaulieu, J.-M.; Attisano, L.; et al. Characterization of mitochondrial health from human peripheral blood mononuclear cells to cerebral organoids derived from induced pluripotent stem cells. Sci. Rep. 2021, 11, 4523. [Google Scholar] [CrossRef] [PubMed]
- Liput, M.; Magliaro, C.; Kuczynska, Z.; Zayat, V.; Ahluwalia, A.; Buzanska, L. Tools and approaches for analyzing the role of mitochondria in health, development and disease using human cerebral organoids. Dev. Neurobiol. 2021, 81, 591–607. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene | Enzyme Activity | Refs. |
|---|---|---|
| COQ1/PDSS1/PDSS2 | Isoprene units condensation | [35] |
| COQ2 | polyprenyl transferase | [36] |
| COQ3 | C5, C6 O-methylase | [37] |
| COQ4 | Unknown/stabilization of CoQ synthome * | [38] |
| COQ5 | C2 methylase | [39] |
| COQ6 | C5 hydroxylase | [40] |
| COQ7 | C6 hydroxylase | [41] |
| COQ8A/ADCK3 | Atypical protein kinase/ATPase * | [42] |
| COQ8B/ADCK4 | Atypical protein kinase/stability of CoQ synthome * | [43] |
| COQ9 | Lipid-binding protein/binds to COQ7 | [44] |
| ADCK2 | Atypical protein kinase/lipid transport into mitochondria * | [45] |
| Gene | Pathogenic Variants | Functional Validation | Refs. |
|---|---|---|---|
| PDSS1 | c.661_662insT; c.1108A > C | CoQ levels | [50] |
| c.924T > G; homozygous | Segregation, Biochemical study of CoQ synthesis rate, Yeast | [51] | |
| PDSS2 | c.964C > T; c.1145C > T | Segregation, Biochemical study of CoQ synthesis rate | [52] |
| COQ2 | c.395T > G; homozygous | Segregation, Biochemically, Yeast | [53] |
| c.755C > T; homozygous | Segregation, Yeast, CoQ levels | [54,55] | |
| c.287G > A; c.1009C > T; heteroplasmic mutation in MT-ND1 (3754C > A) which may contribute to the disease | Yeast | [54,56] | |
| c.1047delT; homozygous | Segregation, Yeast, biochemically | [57] | |
| c.287G > A; homozygous | Segregation, Yeast, CoQ levels | [54,58] | |
| c.287G > A; homozygous | Segregation, Biochemical characterisation (incorporation of labelled precursors) | [59] | |
| c.740A > G; homozygous | Biochemically, Yeast | [54,58,60,61] | |
| c.440G > A; c.533A > G | Segregation, Yeast, CoQ levels | [54,58] | |
| c.533A > G; c.551delT | Segregation, Yeast | [54,62] | |
| c.1019G > C; homozygous | Segregation, Biochemically, Yeast | [63] | |
| COQ4 | c.155T > C; c.521_523delCCA | Segregation, COQ4 protein levels, Yeast | [5] |
| c.433C > G; homozygous | |||
| c.421C > T; c.718C > T | Segregation, Yeast | [5,64] | |
| c.245T > A; c.473G > A | Segregation, CoQ levels | [65] | |
| c.23_33delTCCTCCGTCGG; c.311G > T and c.356C > T | Segregation, RNA/protein levels of coq4 | [66] | |
| c.370G > A; c.402 + 1G > C | CoQ levels | [67] | |
| c.370G > A; homozygous | Segregation, CoQ levels | [68] | |
| 3.9 Mb deletion of chromosome 9q34.13, including COQ4 gene | COQ4 protein levels, CoQ biosynthetic rate, Yeast | [69] | |
| c.370G > A; homozygous | CoQ levels | [67] | |
| c.370G > A; c.371G > T | Segregation, CoQ levels | [67,70] | |
| c.550T > C; c.402 + 1G > C | CoQ levels | [67] | |
| c.190C > T; homozygous | Segregation, COQ4 protein levels, Yeast | [5] | |
| c.577C > T; c.718C > T | Q levels, CII + III enzymatic activity, Oxygen consumption, Yeast | [71] | |
| c.284G > A; c.305G > A | |||
| COQ5 | 9590 pb tandem duplication of the last 4 exons of COQ5 after 1Kb of 3’UTR (modifies the 3’UTR) (base pair positions on Chr 12: 120,940,150-120,949,950/hg19); homozygous | Segregation, COQ5 mRNA and protein levels in fibroblasts | [72] |
| COQ6 | c.763G > A; homozygous | Segregation, Yeast | [73,74] |
| c.782C > T; homozygous | Segregation, Yeast | [63] | |
| c.1058C > A; homozygous | Segregation, Yeast | [73,74,75] | |
| c.189_191delGAA; c.782C > T | not validated, yeast | [63,76] | |
| c.1341G > A; c.1383delG | Segregation, Yeast | [73,74] | |
| c.484C > T; heterozygous | Yeast | [74] | |
| c.564G > A; heterozygous | Yeast | ||
| c.1235A > G; heterozygous | Yeast | [73] | |
| COQ7 | c.599_600delinsTAATGCATC; c.319C > T | Segregation, CoQ levels | [77] |
| c.422T > A: homozygous | Segregation, CoQ analogue bypass the reaction, Transient COQ7 expression in patient fibroblasts, heterologous expression in mouse cells | [78,79] | |
| c.332T > C (and c.308C > T); homozygous | Segregation, Heterologous expression in mouse cells, Does not respond to analogue treatment | [79] | |
| COQ8A | c.1651G > A; homocygous | Yeast, CoQ levels | [57] |
| c.1042C > T; c.1136T > A | Segregation, nonsense-mediated mRNA decay (NMD) | [80] | |
| c.1286A > G; heterozygous | Segregation, but lack of 2nd mutation, CoQ levels | [81] | |
| c.811C > T; homozygous | CoQ levels | [82] | |
| c.993C > T: c.1645G > A | Yeast, CoQ levels | [21,82,83] | |
| c.637C > T; c.815G > T | Yeast, CoQ levels | [57,82] | |
| c.830T > C; c.1506 + 1G > A | Segregation, CoQ levels | [84] | |
| c.1042C > T; homozygous | Segregation, nonsense-mediated mRNA decay (NMD) | [80] | |
| c.815G > A; c.1813dupG | Yeast, CoQ levels | [57,82] | |
| c.895C > T; c.1732T > G | CoQ levels | [85] | |
| c.500_521del22insTTG; homozygous | CoQ biosynthetic rate | [21] | |
| c.1541A > G; c.1750_1752delACC | Yeast, CoQ biosynthetic rate | [21] | |
| c.913G > T; homozygous | CoQ levels | [86] | |
| c.1042C > T; homozygous | Segregation, CoQ levels | [87] | |
| c.895C > T; homozygous | CoQ levels | [85] | |
| c.1398 + 2T > C; homozygous | Segregation, CoQ levels | [21] | |
| c.1844dupG; homozygous | Segregation, CoQ levels | [88] | |
| c.1750_1752delACC; c.1532C > T | CoQ levels | [86] | |
| c.901C > T; c.1399-3_1408del | Segregation, CoQ levels | [86] | |
| 27.6 kb deletion of 1q42.3; homozygous | Low levels of COQ8A mRNA, CoQ levels | [89] | |
| c.1511_1512delCT; homozygous | WB of the protein, CoQ levels | [90] | |
| c.911C > T; homozygous | Segregation, CoQ levels | [81] | |
| COQ8B | c.1199dupA; homozygous | Segregation, Yeast, CoQ levels | [47,91,92,93] |
| c.857A > G; c.1447G > T | Segregation, Yeast | [47,92] | |
| c.1339dupG; homozygous | Yeast | [91,92,93] | |
| c.532C > T; homozygous | Segregation, Yeast, CoQ levels | [47,92,93] | |
| c.645delT; c.1430G > A | Segregation, Yeast | [47,92] | |
| c.645delT; homozygous | Yeast | [92,93] | |
| c.857A > G; c.1447G > T | Segregation, Yeast | [47,92] | |
| c.1356_1362delGGGCCCT; homozygous | Segregation, CoQ levels | [47] | |
| c.958C > T; homozygous | Segregation, Yeast | [47,92] | |
| COQ9 | c.521 + 2T > C; c.711 + 3G > C | Segregation, no detectable COQ9 protein levels in patient fibroblasts | [94] |
| c.521 + 1delG; homozygous | Segregation, COQ9 protein levels in patient fibroblasts, heterologous expression of COQ9 in patient fibroblasts restore the phenotype | [95] | |
| c.730C > T; homozygous | Biochemically (CoQ biosynthesis rate), Yeast | [96,97] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos-Ocaña, C.; Cascajo, M.V.; Alcázar-Fabra, M.; Staiano, C.; López-Lluch, G.; Brea-Calvo, G.; Navas, P. Cellular Models for Primary CoQ Deficiency Pathogenesis Study. Int. J. Mol. Sci. 2021, 22, 10211. https://doi.org/10.3390/ijms221910211
Santos-Ocaña C, Cascajo MV, Alcázar-Fabra M, Staiano C, López-Lluch G, Brea-Calvo G, Navas P. Cellular Models for Primary CoQ Deficiency Pathogenesis Study. International Journal of Molecular Sciences. 2021; 22(19):10211. https://doi.org/10.3390/ijms221910211
Chicago/Turabian StyleSantos-Ocaña, Carlos, María V. Cascajo, María Alcázar-Fabra, Carmine Staiano, Guillermo López-Lluch, Gloria Brea-Calvo, and Plácido Navas. 2021. "Cellular Models for Primary CoQ Deficiency Pathogenesis Study" International Journal of Molecular Sciences 22, no. 19: 10211. https://doi.org/10.3390/ijms221910211
APA StyleSantos-Ocaña, C., Cascajo, M. V., Alcázar-Fabra, M., Staiano, C., López-Lluch, G., Brea-Calvo, G., & Navas, P. (2021). Cellular Models for Primary CoQ Deficiency Pathogenesis Study. International Journal of Molecular Sciences, 22(19), 10211. https://doi.org/10.3390/ijms221910211