
Impaired Mitophagy in Neurons and Glial Cells during Aging and Age-Related Disorders

 , , and
, , and
Abstract
:1. Introduction
2. Mitophagy in Neurons in Aging and Neurodegeneration
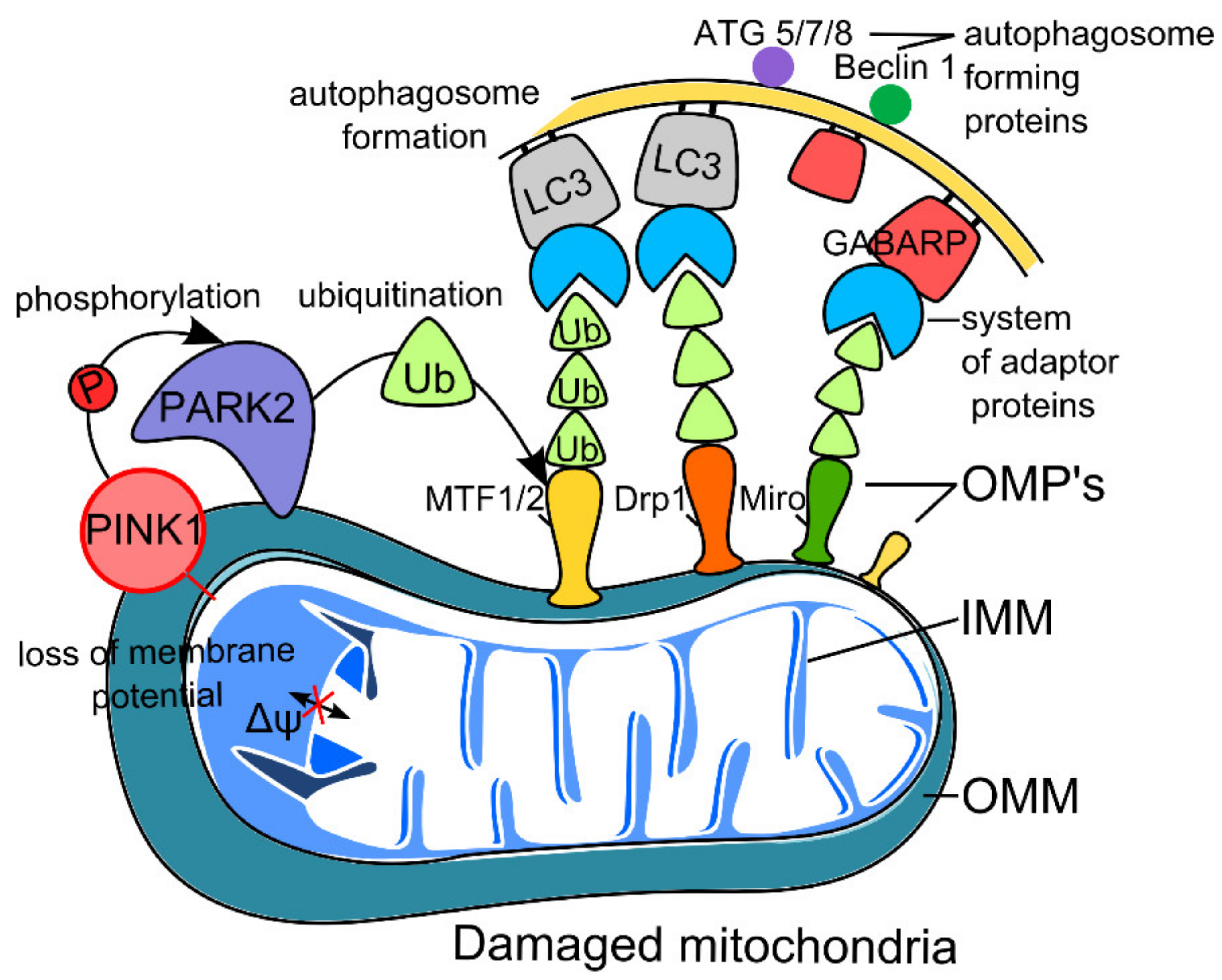
2.1. PINK1–Parkin Pathway of Neuron Mitophagy (Non-Receptor-Mediated Mitophagy)
2.1.1. Mitophagy and Aging
2.1.2. Mitophagy and Neurodegeneration
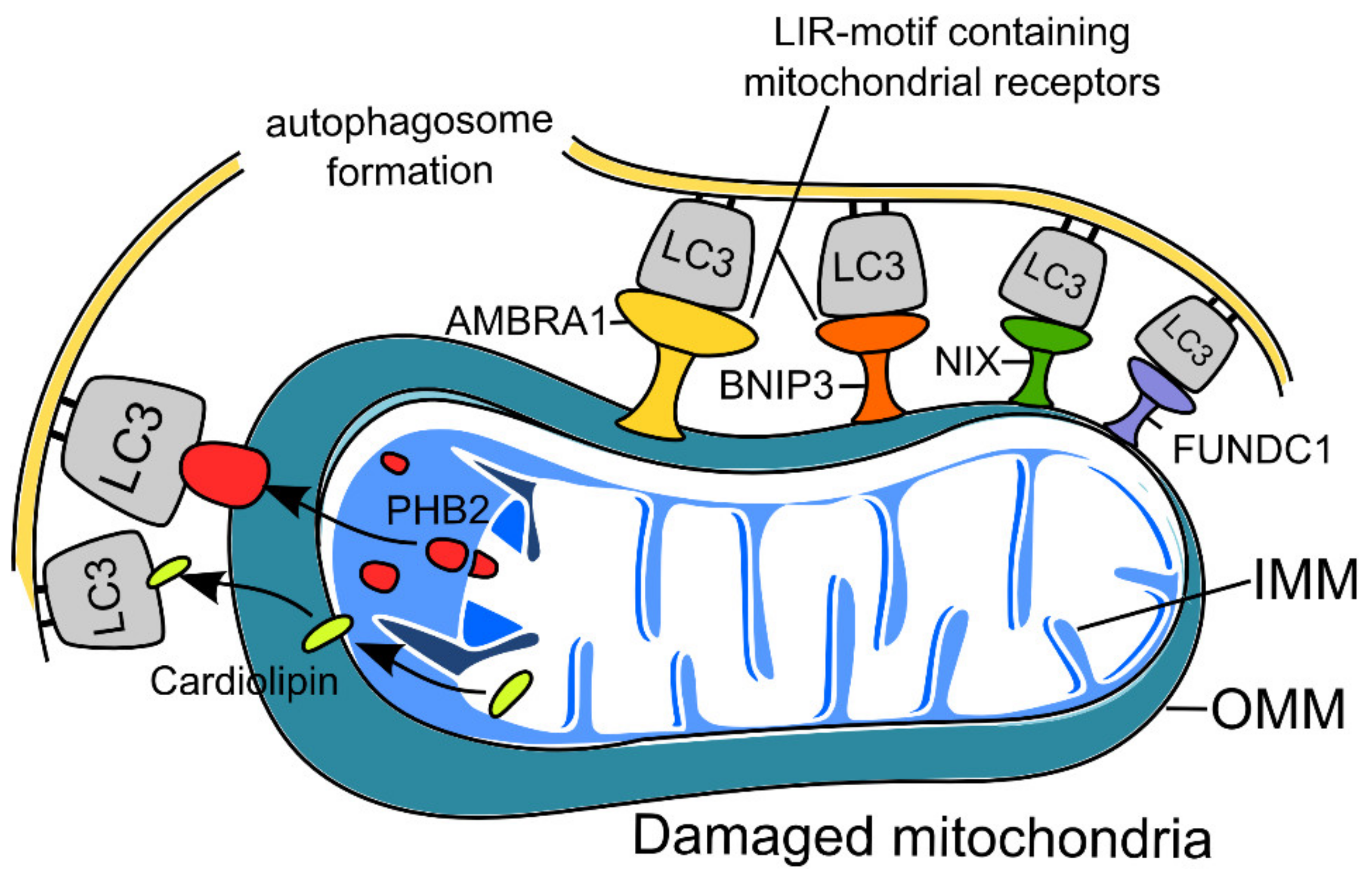
2.2. Receptor-Mediated Mitophagy in Neurons
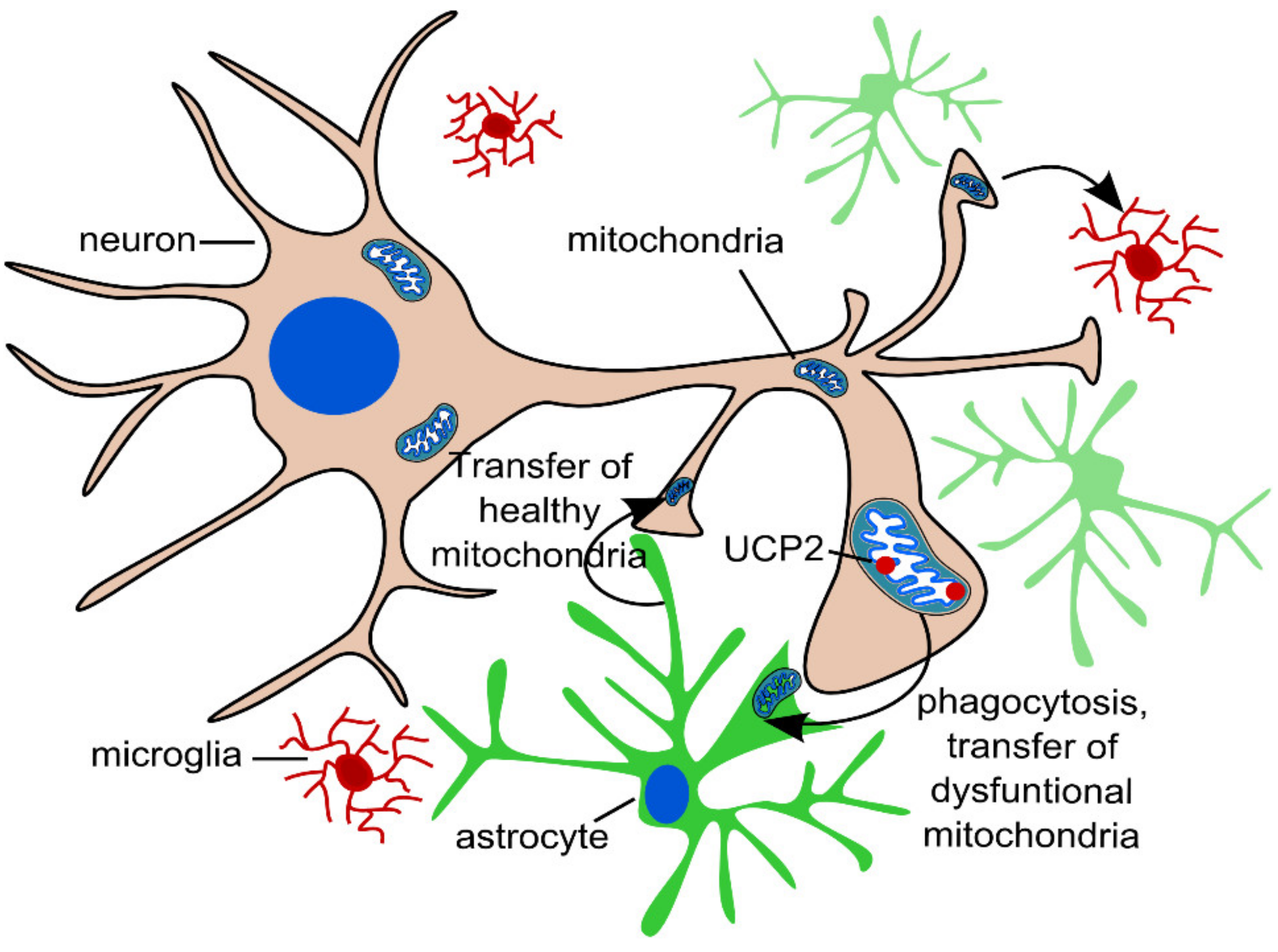
2.3. Transcellular Mitophagy of Neurons
3. Mitophagy in Glial Cells in Aging and Neurodegeneration
3.1. PINK1–Parkin Pathway of Astrocyte Mitophagy (Non-Receptor-Mediated Mitophagy)
3.2. Receptor-Mediated Mitophagy in Glial Cells
3.3. Autophagosome-Forming Proteins
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Vilhelm, A.B. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otın, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farr, J.N.; Almeida, M. The Spectrum of Fundamental Basic Science Discoveries Contributing to Organismal Aging. J. Bone Miner. Res. 2018, 33, 1568–1584. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Tian, F.; Wang, S.; Wang, F.; Xiong, L. Astrocyte Autophagy Flux Protects Neurons Against Oxygen-Glucose Deprivation and Ischemic/Reperfusion Injury. Rejuvenation Res. 2018, 21, 1999. [Google Scholar] [CrossRef] [PubMed]
- Bakula, D.; Scheibye-Knudsen, M. MitophAging: Mitophagy in Aging and Disease. Front Cell Dev. Biol. 2020, 8, 239. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.Y.; Liu, J.; Finkel, T. The role of mitochondria in aging. J. Clin. Investig. 2018, 128, 3662–3670. [Google Scholar] [CrossRef] [Green Version]
- Fivenson, E.M.; Lautrup, S.; Sun, N.; Scheibye-Knudsen, M.; Stevnsner, T.V.; Nilsen, H.; Bohr, V.A.; Fanga, F.E. Mitophagy in neurodegeneration and aging. Neurochem. Int. 2017, 109, 202–209. [Google Scholar] [CrossRef]
- Garza-Lombó, C.; Pappa, A.; Mihalis, I. Panayiotidis, Rodrigo Franco. Redox Homeostasis, Oxidative Stress and Mitophagy. Mitochondrion 2020, 51, 105–117. [Google Scholar] [CrossRef]
- Kondadi, A.K.; Anand, R.; Reichert, A.S. Functional Interplay between Cristae Biogenesis, Mitochondrial Dynamics and Mitochondrial DNA Integrity. Int. J. Mol. 2019, 20, 4311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maday, S.; Holzbaur, E.L. Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev. Cell 2014, 30, 71–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, C.S.; Holzbaur, E.L. Degradation of engulfed mitochondria is rate-limiting in Optineurin-mediated mitophagy in neurons. eLife Sci. 2019, 9, e50260. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Walker, J.E.; Youle, R. Mitochondrial Quality Control Mediated by PINK1 and Parkin: Links to Parkinsonism. Cold Spring Harb. Perspect. Biol. 2012, 4, a011338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, N.S.; Wilkins, H.M. Mitophagy and the Brain. Int. J. Mol. Sci. 2020, 21, 9661. [Google Scholar] [CrossRef] [PubMed]
- Tseng, A.H.; Shieh, S.S.; Wang, D.L. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 2013, 63, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schlehe, J.S.; LaVoie, M.J.; Schwarz, T.L. Mitophagy of Damaged Mitochondria Occurs Locally in Distal Neuronal Axons and Requires PINK1 and Parkin. J. Cell Biol. 2014, 206, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Zhang, M.; Jeong, Y.Y.; Margolis, D.J.; Cai, Q. The role of mitophagy in the regulation of mitochondrial energetic status in neurons. Autophagy 2021, 1–20. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Valla, J.; Berndt, J.D.; Gonzalez-Lima, F. Energy hypometabolism in posterior cingulate cortex of Alzheimer’s patients: Superficial laminar cytochrome oxidase associated with disease duration. J. Neurosci. 2001, 21, 4923–4930. [Google Scholar] [CrossRef] [PubMed]
- Cen, X.; Zhang, M.; Zhou, M.; Ye, L.; Xia, H. Mitophagy Regulates Neurodegenerative Diseases. Cells 2021, 10, 1876. [Google Scholar] [CrossRef]
- Sanchez-Martin, P.; Komatsu, M. p62/SQSTM1—Steering the cell through health and disease. J. Cell Sci. 2018, 131, jcs222836–jcs222849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Vicente, M. Neuronal Mitophagy in Neurodegenerative Diseases. Front. Mol. Neurosci. 2017, 10, 64. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Dhawan, A.; Kadam, A.; Shinde, A. Autophagy and Mitochondria: Targets in Neurodegenerative Disorders. CNS Neurol. Disord.-Drug Targets 2018, 17, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L.F. Autophagosome dynamics in neurodegeneration at a glance. J Cell Sci. 2015, 128, 1259–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiesel, F.C.; Springer, W. Disease relevance of phosphorylated ubiquitin (p-S65-Ub). Autophagy 2015, 11, 2125–2126. [Google Scholar] [CrossRef] [Green Version]
- Barodia, S.K.; McMeekin, L.J.; Creed, R.B.; Quinones, E.K.; Cowell, R.M.; Goldberg, M.S. PINK1 phosphorylates ubiquitin predominantly in astrocytes. NPJ Park. Dis. 2019, 5, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takanashi, M.; Li, Y.; Hattori, N. Absence of Lewy pathology associated with PINK1 homozygous mutation. Neurology 2016, 86, 2212–2213. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmstrom, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Todd, A.M.; Staveley, B.E. Expression of Pink1 with α-synuclein in the dopaminergic neurons of Drosophila leads to increases in both lifespan and healthspan. Genet. Mol. Res. 2012, 116, 1497–1502. [Google Scholar] [CrossRef] [Green Version]
- Rana, A.; Rera, M.; Walker, D.W. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc. Natl. Acad. Sci. USA. 2013, 110, 8638–8643. [Google Scholar] [CrossRef] [Green Version]
- Rana, A.; Oliveira, M.P.; Khamoui, A.V.; Aparicio, R.; Rera, M.; Rossiter, H.B.; Walker, D.W. Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat. Commun. 2017, 8, 448. [Google Scholar] [CrossRef]
- Liang, W.; Moyzis, A.G.; Lampert, M.A.; Diao, R.Y.; Najor, R.H.; Gustafsson, A.B. Aging is associated with a decline in Atg9b-mediated autophagosome formation and appearance of enlarged mitochondria in the heart. Aging Cell. 2020, 19, e13187. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, D.J.; Blin, M.G.; Song, J.; Wood, S.C.; Goldstein, D.R. Aging Impairs Mitochondrial Function and Mitophagy and Elevates Interleukin 6 within the Cerebral Vasculature. J. Am. Heart Assoc. 2020, 9, e017820. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Hayakawa, H.; Nihira, T.; Ren, Y.; Nakata, Y.; Nagai, M.; Hattori, N.; Miyake, K.; Takada, M.; Shimada, T.; et al. Parkin-Mediated Protection of Dopaminergic Neurons in a Chronic MPTP-Minipump Mouse Model of Parkinson Disease. Neuropathol. Exp. Neurol. 2011, 70, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Noda, S.; Sato, S.; Fukuda, T.; Tada, N.; Uchiyama, Y.; Tanaka, K.; Hattori, N. Loss of Parkin contributes to mitochondrial turnover and dopaminergic neuronal loss in aged mice. Neurobiol. Disease 2020, 136, 104717. [Google Scholar] [CrossRef] [PubMed]
- Wood-Kaczmar, A.; Gandhi, S.; Yao, Z.; Abramov, A.S.Y.; Miljan, E.A.; Keen, G.; Stanyer, L.; Hargreaves, L.; Klupsch, K.; Deas, E.; et al. PINK1 Is Necessary for Long Term Survival and Mitochondrial Function in Human Dopaminergic Neurons. PLoS ONE 2018, 3, e2455. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.; Zhang, Z.; Chang, R.; Cheng, H.; Mu, C.; Zhao, T.; Chen, L.; Zhang, C.; Luo, Q.; Lin, J.; et al. Dynamic PGAM5 multimers dephosphorylate BCL-xL or FUNDC1 to regulate mitochondrial and cellular fate. Cell Death Differ. 2020, 27, 1036–1051. [Google Scholar] [CrossRef]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener 2019, 14, 36. [Google Scholar] [CrossRef] [Green Version]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef] [Green Version]
- Martinez, J.H.; Alaimo, A.; Gorojod, R.M.; Porte Alcon, S.; Fuentes, F.; Coluccio Leskow, F.; Kotler, M.L. Drp-1 dependent mitochondrial fragmentation and protective autophagy in dopaminergic SH-SY5Y cells overexpressing alpha-synuclein. Mol. Cell Neurosci. 2018, 88, 107–117. [Google Scholar] [CrossRef]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Jeong, Y.Y. Mitophagy in Alzheimer’s Disease and Other Age-Related Neurodegenerative Diseases. Cells 2020, 9, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín-Maestro, P.; Sproul, A.; Martinez, H.; Paquet, D.; Gerges, M.; Noggle, S.; Starkov, A.A. Autophagy induction by bexarotene promotes mitophagy in Presenilin 1 familial Alzheimer’s disease iPSC-derived neural stem cells. Mol. Neurobiol. 2019, 56, 8220–8236. [Google Scholar] [CrossRef] [PubMed]
- Coffey, E.E.; Beckel, J.M.; Laties, A.M.; Mitchell, C.H. Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer’s disease-linked presenilin 1 A246E mutation can be reversed with cAMP. Neuroscience 2014, 263, 111–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [Green Version]
- Lanzillotta, C.; Di Domenico, F.; Perluigi, M.; Butterfield, D.A. Targeting mitochondria in Alzheimer disease: Rationale and perspectives. CNS Drugs 2019, 33, 957–969. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of Aβ accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef]
- Ye, X.; Sun, X.; Starovoytov, V.; Cai, Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s Disease patient brains. Hum. Mol. Genet. 2015, 24, 2938–2951. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Plaza-Zabala, A.; Montpeyo, M.; Sebastian, D.; Vila, M.; Martinez-Vicente, M. Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy 2021, 17, 672–689. [Google Scholar] [CrossRef] [PubMed]
- Guedes-Dias, P.; de Proença, J.; Soares, T.R.; Leitão-Rocha, A.; Pinho, B.R.; Duchen, M.R.; Oliveira, J.M. HDAC6 inhibition induces mitochondrial fusion, autophagic flux and reduces diffuse mutant huntingtin in striatal neurons. Biochim. Biophys. Acta 2015, 1852, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Johansen, T.; Lamark, T. Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J. Mol. Biol. 2020, 432, 80–103. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Han, Z.; Feng, D.; Chen, Y.; Chen, L.; Wu, H.; Huang, L.; Zhou, C.; Cai, X.; Fu, C.; et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell. 2014, 54, 362–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Chiang, W.C.; Sumpter, R.; Mishra, P., Jr.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238.e210. [Google Scholar] [CrossRef] [Green Version]
- Bordi, M.; Berg, M.J.; Mohan, P.S.; Peterhoff, C.M.; Alldred, M.J.; Che, S.; Ginsberg, S.D.; Nixon, R.A. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 2016, 12, 2467–2483. [Google Scholar] [CrossRef] [PubMed]
- Choi, G.E.; Lee, H.J.; Chae, C.W.; Cho, J.H.; Jung, Y.H.; Kim, J.S.; Kim, S.Y.; Lim, J.R.; Han, H.J. BNIP3L/NIX-mediated mitophagy protects against glucocorticoid-induced synapse defects. Nat. Commun. 2021, 12, 487. [Google Scholar] [CrossRef]
- Cai, Y.; Yang, E.; Yao, X.; Zhang, X.; Wang, Q.; Wang, Y.; Liu, J.; Fan, W.; Kang, C.; Wu, J. FUNDC1-dependent mitophagy induced by tPA protects neurons against cerebral ischemia-reperfusion injury. Redox Biol. 2021, 38, 101792. [Google Scholar] [CrossRef]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimi, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 419–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, F.; Chen, D.; Si, J.; Hu, Q.; Qin, Z.; Fang, M.; Wang, G. The mitochondrial protein BNIP3L is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum. Mol. Genet. 2015, 24, 2528–2538. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Koentjoro, B.; Sue, C.M. Commentary: Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Front. Mol. Neurosci. 2017, 10, 297. [Google Scholar] [CrossRef] [PubMed]
- Koentjoro, B.; Park, J.S.; Sue, S.M. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci. Rep. 2017, 7, 44373. [Google Scholar] [CrossRef]
- Trecidi, K.D.; Braak, H. Review: Sporadic Parkinson’s disease: Development and distribution of a-synuclein pathology. Neuropathol. Appl. Neurobiol. 2016, 42, 33–50. [Google Scholar]
- Sepe, S.; Nardacci, R.; Fanelli, F.; Rosso, P.; Bernardi, C.; Cecconi, F.; Mastroberardino, P.G.; Piacentini, M.; Moreno, S. Expression of Ambra1 in mouse brain during physiological and Alzheimer type aging. Neurobiol. Aging 2014, 35, 96–108. [Google Scholar] [CrossRef]
- Davis, C.O.; Kim, K.-Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.A.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Zhang, Z.; Lu, J.; Pei, G. Mitochondria Are Dynamically Transferring Between Human Neural Cells and Alexander Disease-Associated GFAP Mutations Impair the Astrocytic Transfer. Front. Cell. Neurosci. 2019, 13, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motori, E.; Puyal, J.; Toni, N.; Ghanem, A.; Angeloni, C.; Malaguti, M.; Cantelli-Forti, G.; Berninger, B.; Conzelmann, K.-K.; Götz, M.; et al. Inflammation-induced alteration of astrocyte mitochondrial dynamics requires autophagy for mitochondrial network maintenance. Cell Metab. 2013, 18, 844–859. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Li, X.C.; Wang, Z.H.; Luo, Y.; Zhang, X.; Liu, X.P.; Feng, Q.; Wang, Q.; Yue, Z.; Chen, Z.; et al. Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget 2016, 7, 17356–17368. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, J.C.; Jackson, J.G.; Robinson, M.B. Transient Oxygen/Glucose Deprivation Causes a Delayed Loss of Mitochondria and Increases Spontaneous Calcium Signaling in Astrocytic Processes. J. Neurosci. 2016, 36, 7109–7127. [Google Scholar] [CrossRef] [Green Version]
- Zehnder, T.; Petrelli, F.; Romanos, J.; Figueiredo, E.C.D.V.; Lewis, T.L., Jr.; Déglon, N.; Polleux, F.; Santello, M.; Bezzi, P. Mitochondrial biogenesis in developing astrocytes regulates astrocyte maturation and synapse formation. Cell Rep. 2021, 35, 108952. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, B.; Qiao, H.; Lv, X.; Liang, Q.; Shi, Z.; Xia, W.; Ji, F.; Jiao, J. Autophagy-related gene Atg5 is essential for astrocyte differentiation in the developing mouse cortex. EMBO Rep. 2014, 15, 1053–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanner, S.; Goldin, M.; Galron, R.; Ben Jacob, E.; Bonifazi, P.; Barzilai, A. Astrocytes restore connectivity and synchronization in dysfunctional cerebellar networks. Proc. Natl. Acad. Sci. USA 2018, 115, 8025–8030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, R.H.; Koppel, S.; Weidling, I.; Hayley, C.; Ji, Y.; Wilkins, H.M. Mitochondria, Cybrids, Aging, and Alzheimer’s Disease. Prog. Mol. Biol. Transl. Sci. 2017, 146, 259–302. [Google Scholar] [CrossRef] [Green Version]
- Morales, I.; Sanchez, A.; Puertas-Avendaño, R.; Rodriguez-Sabate, C.; Perez-Barreto, A.; Rodriguez, M. Neuroglial transmitophagy and Parkinson’s disease. Glia 2020, 68, 2277–2299. [Google Scholar] [CrossRef]
- Sher, A.A.; Gao, A.; Coombs, K.M. Autophagy Modulators Profoundly Alter the Astrocyte Cellular Proteome. Cells 2020, 9, 805. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, С.; Lopez-Rodriguez, A.B. Astrocytes: Heterogeneous and Dynamic Phenotypes in Neurodegeneration and Innate Immunity. Neuroscientist 2018, 25, 455–474. [Google Scholar] [CrossRef] [Green Version]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edward, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [Green Version]
- Choi, I.; Choi, D.-J.; Yang, H.; Woo, J.H.; Chang, M.-Y.; Kim, J.Y.; Sun, W.; Park, S.-M.; Jou, I.; Lee, S.-H.; et al. PINK1 expression increases during brain development and stem cell differentiation, and affects the development of GFAP-positive astrocytes. Mol. Brain. 2016, 9, 5. [Google Scholar] [CrossRef] [Green Version]
- Ledesma, M.D.; Galvan, C.; Hellias, B.; Dotti, C.; Jensen, P.H. Astrocytic but not neuronal increased expression and redistribution of parkin during unfolded protein stress. J. Neurochem. 2002, 83, 1431–1440. [Google Scholar] [CrossRef] [Green Version]
- Solano, R.M.; Casarejos, M.J.; Menéndez-Cuervo, J.; Rodriguez-Navarro, J.A.; García de Yébenes, J.; Mena, M.A. Glial dysfunction in parkin null mice: Effects of aging. J. Neurosci. 2008, 28, 598–611. [Google Scholar] [CrossRef]
- Wall, C.E.; Rose, C.M.; Adrian, M.; Zeng, Y.J.; Kirkpatrick, D.S.; Bingol, B. PPEF2 Opposes PINK1-Mediated Mitochondrial Quality Control by Dephosphorylating Ubiquitin. Cell Rep. 2019, 29, 3280–3292.e7. [Google Scholar] [CrossRef] [Green Version]
- Tsefou, E.; Walker, A.S.; Clark, E.H.; Hicks, A.R.; Luft, C.; Takeda, K.; Watanabe, T.; Ramazio, B.; Staddon, J.M.; Briston, T.; et al. Investigation of USP30 inhibition to enhance Parkin-mediated mitophagy: Tools and approaches. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kulkarni, A.; Dong, A.; Kulkarni, V.V.; Chen, J.; Laxton, O.; Anand, A.; Maday, S. Differential regulation of autophagy during metabolic stress in astrocytes and neurons. Autophagy 2020, 16, 1651–1667. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Pasinelli, P.; Trotti, D. Role of mitochondria in mutant SOD1 linked amyotrophic lateral sclerosis. Biochim. Biophys. Acta–Mol. Basis Dis. 2014, 1842, 1295–1301. [Google Scholar] [CrossRef] [Green Version]
- Killackey, S.A.; Philpott, D.J.; Girardin, S. Mitophagy pathways in health and disease. J. Cell. Biol. 2020, 219, e202004029. [Google Scholar] [CrossRef]
- Yamada, T.; Dawson, T.M.; Yanagawa, T.; Iijima, M.; Sesaki, H. SQSTM1/p62 promotes mitochondrial ubiquitination independently of PINK1 and PRKN/parkin in mitophagy. Autophagy 2019, 15, 2012–2018. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.E.; Iyer, S.; Thangavel, R.; Kempuraj, D.; Selvakumar, G.P.; Raikwar, S.P.; Zaheer, S.; Zaheer, A. Co-Localization of Glia Maturation Factor with NLRP3 Inflammasome and Autophagosome Markers in Human Alzheimer’s Disease Brain. J. Alzheimers Dis. 2017, 60, 1143–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Lee, D.; Song, J.C.; Cho, S.-J.; Yun, S.-M.; Koh, Y.H.; Song, J.; Johnson, G.V.W.; Jo, C. NDP52 associates with phosphorylated tau in brains of an Alzheimer disease mouse model. Biochem. Biophys. Res. Commun. 2014, 454, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Aman, Y.; Adriaanse, B.A.; Cader, M.Z.; Plun-Favreau, H.; Xiao, J.; Fang, E.F. Culprit or Bystander: Defective Mitophagy in Alzheimer’s Disease. Front. Cell Dev. Biol. 2020, 7, 391. [Google Scholar] [CrossRef] [PubMed]
- Weil, R.; Laplantine, E.; Curic, S.; Génin, P. Role of Optineurin in the Mitochondrial Dysfunction: Potential Implications in Neurodegenerative Diseases and Cancer. Front. Immunol. 2018, 9, 1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Toth, R.P.; Atkin, J.D. Dysfunction of Optineurin in Amyotrophic Lateral Sclerosis and Glaucoma. Front. Immunol. 2018, 9, 1017. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Re, D.; Nagata, T.; Chalazonitis, A.; Jessell, T.M.; Wichterle, H.; Przedborski, S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 2007, 10, 615–622. [Google Scholar] [CrossRef] [Green Version]
- Madill, M.; McDonagh, K.; Ma, J.; Vajda, A.; McLoughlin, P.; O’Brien, T.; Hardiman, O.; Shen, S. Amyotrophic lateral sclerosis patient iPSC-derived astrocytes impair autophagy via non-cell autonomous mechanisms. Mol. Brain 2017, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Ugarteburu, O.; Sánchez-Vilés, M.; Ramos, J.; Barcos-Rodríguez, T.; Garrabou, G.; García-Villoria, J.; Ribes, A.; Tort, F. Physiopathological Bases of the Disease Caused by HACE1 Mutations: Alterations in Autophagy, Mitophagy and Oxidative Stress Response. J. Clin. Med. 2020, 9, 913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrnhoefer, D.E.; Southwell, A.L.; Sivasubramanian, M.; Qiu, X.; Villanueva, E.B.; Xie, Y.; Waltl, S.; Anderson, L.; Fazeli, A.; Casal, L.; et al. HACE1 is essential for astrocyte mitochondrial function and influences Huntington disease phenotypes In Vivo. Hum. Mol. Genet. 2018, 27, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Bruno, C.; Sieverding, K.; Freischmidt, A.; Satoh, T.; Walther, P.; Mayer, B.; Ludolph, A.C.; Akira, S.; Yilmazer-Hanke, D.; Danzer, K.M.; et al. Haploinsufficiency of TANK-binding kinase 1 prepones age-associated neuroinflammatory changes without causing motor neuron degeneration in aged mice. Brain Commun. 2020, 2, fcaa133. [Google Scholar] [CrossRef]
- Singh, A.; Azad, M.; Shymko, M.D.; Henson, E.S.; Katyal, S.; Eisenstat, D.D.; Gibson, S.B. The BH3 only Bcl-2 family member BNIP3 regulates cellular proliferation. PLoS ONE 2018, 13, e0204792. [Google Scholar] [CrossRef]
- Van Humbeeck, C.; Cornelissen, T.; Hofkens, H.; Mandemakers, W.; Gevaert, K.; De Strooper, B.; Vandenberghe, W. Parkin Interacts with Ambra1 to Induce Mitophagy. J. Neurosci. 2011, 31, 10249–10261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 Complexes Mediate mTOR Signaling to the Autophagy Machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latacz, A.; Russell, J.A.; Ocłoń, E.; Zubel-Łojek, J.; Pierzchała-Koziec, K. mTOR Pathway—Novel Modulator of Astrocyte Activity. Folia Biol. 2015, 63, 95–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.; Patric, I.R.P.; Patil, V.; Shwetha, S.D.; Hegde, A.S.; Chandramouli, B.A.; Arivazhagan, A.; Santosh, V.; Somasundaram, K. Methylation Silencing of ULK2, an Autophagy Gene, Is Essential for Astrocyte Transformation and Tumor Growth. J. Biol. Chem. 2014, 289, 22306–22318. [Google Scholar] [CrossRef] [Green Version]
- Farina, V.; Lepore, G.; Biagi, F.; Carcupino, M.; Zedda, M. Autophagic processes increase during senescence in cultured sheep neurons and astrocytes. Eur. J. Histochem. 2018, 62, 2891. [Google Scholar] [CrossRef] [Green Version]
- Stephen, T.-L.; Gupta-Agarwal, S.; Kittler, J.T. Mitochondrial dynamics in astrocytes. Biochem. Soc. Trans. 2014, 42, 1302–1310. [Google Scholar] [CrossRef] [Green Version]
- Eshraghi, M.; Adlimoghaddam, A.; Mahmoodzadeh, A.; Sharifzad, F.; Yasavoli-Sharahi, H.; Lorzadeh, S.; Albensi, B.C.; Ghavami, S. Alzheimer’s Disease Pathogenesis: Role of Autophagy and Mitophagy Focusing in Microglia. Int. J. Mol. Sci. 2021, 22, 3330. [Google Scholar] [CrossRef]
- Chakravorty, A.; Jetto, C.T.; Manjithaya, R. Dysfunctional Mitochondria and Mitophagy as Drivers of Alzheimer’s Disease Pathogenesis. Front. Aging Neurosci. 2019, 11, 311. [Google Scholar] [CrossRef]
- Lautrup, S.; Lou, G.; Aman, Y.; Nilsen, H.; Tao, J.; Fang, E.F. Microglial mitophagy mitigates neuroinflammation in Alzheimer’s disease. Neurochem. Int. 2019, 129, 104469. [Google Scholar] [CrossRef]
- Yazdankhah, M.; Ghosh, S.; Shang, P.; Stepicheva, N.; Hose, S.; Liu, H.; Chamling, X.; Tian, S.; Sullivan, M.L.G.; Calderon, M.J.; et al. BNIP3L-mediated mitophagy is required for mitochondrial remodeling during thedifferentiation of optic nerve oligodendrocytes. Autophagy 2021, 1–20. [Google Scholar] [CrossRef]
- Lahiri, V.; Klionsky, D.J. PHB2/prohibitin 2: An inner membrane mitophagy receptor. Cell Res. 2017, 27, 311–312. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.-G.; Keilhoff, G.; Dobrowolny, H.; Steiner, J. Enhanced mitochondrial autophagy (mitophagy) in oligodendrocytes might play a role in white matter pathology in schizophrenia. Med. Hypotheses 2020, 134, 109443. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.H.; Vázquez de la Torre, A.; Hoshikawa, T.; Briston, T. Targeting mitophagy in Parkinson’s disease. J. Biol. Chem. 2021, 296, 100209. [Google Scholar] [CrossRef]
- Jayatunga, D.P.W.; Hone, E.; Bharadwaj, P.; Garg, M.; Verdile, G.; Guillemin, G.J.; Martins, R.N. Targeting Mitophagy in Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 78, 1273–1297. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Protein Abbreviation | Full Name | AD | PD | HT | ALS | Aging | Citation | |
|---|---|---|---|---|---|---|---|---|
| Non-receptor- mediated mitophagy | PINK1 | Phosphatase and tensin homolog (PTEN)-induced kinase | + | + | + | [12,13,23,24,25,38,68,85] | ||
| USP30 | Ubiquitin carboxyl-terminal hydrolase 30 | + | [5,41,42,84] | |||||
| ULK1 | Unc-51-like autophagy activating kinase 1 | + | + | [48,51,52,93,102,104] | ||||
| TBK1 | TANK-binding kinase 1 | + | + | [5,42,48,92] | ||||
| Drp1 | Dynamin-related protein 1 (Drp1) | + | + | [40] | ||||
| p62/SQSTM1 | Sequestosome 1 | + | + | + | [14,73,87,88,96,99] | |||
| NDP52 | Nuclear dot protein 52 kDa | + | [90,92] | |||||
| Optn | Optineurin | + | + | [48,94] | ||||
| Receptor-mediated mitophagy | FUNDC1 | FUN14 domain-containing protein 1 (phosphorylation) | + | [48,54,55,56] | ||||
| AMBRA1 | Autophagy and Beclin 1 regulator 1 | + | + | [43,101] | ||||
| BNIP3 | BCL2-interacting protein 3 (phosphorylation) | + | [96] | |||||
| Autophagosome-forming proteins | GABARP | γ-aminobutyric-acid type A receptor-associated proteins | + | [12,14,86] | ||||
| LC3-II | Microtubule-associated protein 1A/1B-light chain | + | + | + | [14,87,88,89,102,105] | |||
| BECN1 | Beclin-1 | + | + | [44] | ||||
| ATG5/ATG7 | Autophagy-related genes, associated with LC3 modification | + | [68,72,102,104] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sukhorukov, V.; Voronkov, D.; Baranich, T.; Mudzhiri, N.; Magnaeva, A.; Illarioshkin, S. Impaired Mitophagy in Neurons and Glial Cells during Aging and Age-Related Disorders. Int. J. Mol. Sci. 2021, 22, 10251. https://doi.org/10.3390/ijms221910251
Sukhorukov V, Voronkov D, Baranich T, Mudzhiri N, Magnaeva A, Illarioshkin S. Impaired Mitophagy in Neurons and Glial Cells during Aging and Age-Related Disorders. International Journal of Molecular Sciences. 2021; 22(19):10251. https://doi.org/10.3390/ijms221910251
Chicago/Turabian StyleSukhorukov, Vladimir, Dmitry Voronkov, Tatiana Baranich, Natalia Mudzhiri, Alina Magnaeva, and Sergey Illarioshkin. 2021. "Impaired Mitophagy in Neurons and Glial Cells during Aging and Age-Related Disorders" International Journal of Molecular Sciences 22, no. 19: 10251. https://doi.org/10.3390/ijms221910251
APA StyleSukhorukov, V., Voronkov, D., Baranich, T., Mudzhiri, N., Magnaeva, A., & Illarioshkin, S. (2021). Impaired Mitophagy in Neurons and Glial Cells during Aging and Age-Related Disorders. International Journal of Molecular Sciences, 22(19), 10251. https://doi.org/10.3390/ijms221910251