
Genetic Testing in Patients with Hypertrophic Cardiomyopathy



Abstract
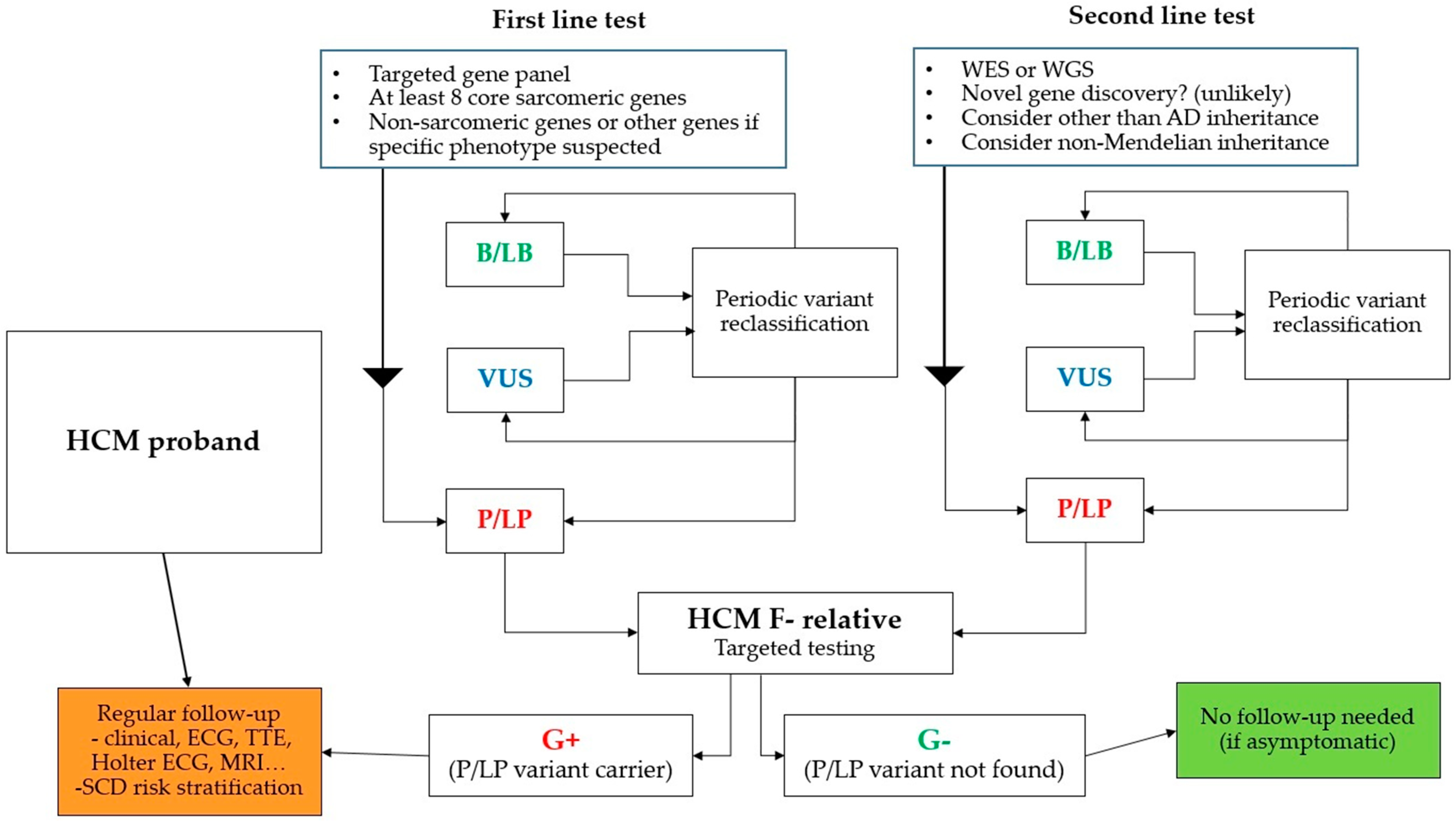
:1. Introduction
2. History of Finding the Cause of HCM
3. Identification of a Causative Mutation
- -
- -
- -
- In silico classification using software (e.g., Polyphen2, Sorting Intolerant From Tolerant) predicting the possible impact of the mutation on the structure and function of the final protein
- -
- Mutations in the so-called evolutionarily highly conserved functional domains of the target protein
- -
- Segregation analyses of genotype with phenotype in affected families (strong evidence)
- -
- Functional studies on animal models or in vitro (expensive, complex)
4. Genetic Screening
5. Diagnostic Yield of Molecular Genetic Testing
6. Genotype and Phenotype Correlation
7. Future
8. Take-Home Message
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Elliott, P.M.; Anastasakis, A.; Borger, M.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef] [Green Version]
- Veselka, J.; Anavekar, N.S.; Charron, P. Hypertrophic obstructive cardiomyopathy. Lancet 2016, 389, 1253–1267. [Google Scholar] [CrossRef]
- Richard, P.; Charron, P.; Carrier, L.; Ledeuil, C.; Cheav, T.; Pichereau, C.; Benaiche, A.; Isnard, R.; Dubourg, O.; Burban, M.; et al. Hypertrophic Cardiomyopathy. Circulation 2003, 107, 2227–2232. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Doerer, J.J.; Haas, T.S.; Tierney, D.; Mueller, F.O. Sudden Deaths in Young Competitive Athletes. Circulation 2009, 119, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Semsarian, C.; Ingles, J.; Maron, M.S.; Maron, B.J. New Perspectives on the Prevalence of Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2015, 65, 1249–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackerman, M.; Atkins, D.L.; Triedman, J.K. Sudden Cardiac Death in the Young. Circulation 2016, 133, 1006–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teare, D. Asymmetrical Hypertrophy of the Heart in Young Adults. Heart 1958, 20, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisterfer-Lowrance, A.A.; Kass, S.; Tanigawa, G.; Vosberg, H.-P.; McKenna, W.; Seidman, C.E.; Seidman, J. A molecular basis for familial hypertrophic cardiomyopathy: A β cardiac myosin heavy chain gene missense mutation. Cell 1990, 62, 999–1006. [Google Scholar] [CrossRef]
- Jarcho, J.A.; McKenna, W.J.; Pare, J.P.; Solomon, S.D.; Holcombe, R.F.; Dickie, S.; Levi, T.; Donis-Keller, H.; Seidman, J.; Seidman, C.E. Mapping a Gene for Familial Hypertrophic Cardiomyopathy to Chromosome 14q1. N. Engl. J. Med. 1989, 321, 1372–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, C.Y.; Charron, P.; Richard, P.; Girolami, F.; Van Spaendonck-Zwarts, K.Y.; Pinto, Y. Genetic advances in sarcomeric cardiomyopathies: State of the art. Cardiovasc. Res. 2015, 105, 397–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingles, J.; Goldstein, J.; Thaxton, C.; Caleshu, C.; Corty, E.W.; Crowley, S.B.; Dougherty, K.; Harrison, S.M.; McGlaughon, J.; Milko, L.V.; et al. Evaluating the Clinical Validity of Hypertrophic Cardiomyopathy Genes. Circ. Genom. Precis. Med. 2019, 12, e002460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, L.; Syrris, P.; Guttmann, O.P.; O’Mahony, C.; Tang, H.C.; Dalageorgou, C.; Jenkins, S.; Hubank, M.; Monserrat, L.; McKenna, W.J.; et al. Novel genotype–phenotype associations demonstrated by high-throughput sequencing in patients with hypertrophic cardiomyopathy. Heart 2014, 101, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Buchan, R.; Wilk, A.; John, S.; Felkin, L.E.; Thomson, K.; Chiaw, T.H.; Loong, C.C.W.; Pua, C.J.; Raphael, C.; et al. Defining the genetic architecture of hypertrophic cardiomyopathy: Re-evaluating the role of non-sarcomeric genes. Eur. Heart J. 2017, 38, 3461–3468. [Google Scholar] [CrossRef] [PubMed]
- Thomson, K.L.; NIHR BioResource—Rare Diseases Consortium; Ormondroyd, E.; Harper, A.R.; Dent, T.; McGuire, K.; Baksi, J.; Blair, E.; Brennan, P.; Buchan, R.; et al. Analysis of 51 proposed hypertrophic cardiomyopathy genes from genome sequencing data in sarcomere negative cases has negligible diagnostic yield. Genet. Med. 2018, 21, 1576–1584. [Google Scholar] [CrossRef] [Green Version]
- Mazzarotto, F.; Olivotto, I.; Boschi, B.; Girolami, F.; Poggesi, C.; Barton, P.; Walsh, R. Contemporary Insights Into the Genetics of Hypertrophic Cardiomyopathy: Toward a New Era in Clinical Testing? J. Am. Heart Assoc. 2020, 9, e015473. [Google Scholar] [CrossRef] [PubMed]
- Lopes, L.R.; Garcia-Hernández, S.; Lorenzini, M.; Futema, M.; Chumakova, O.; Zateyshchikov, D.; Isidoro-Garcia, M.; Villacorta, E.; Escobar-Lopez, L.; Garcia-Pavia, P.; et al. Alpha-protein kinase 3 (ALPK3) truncating variants are a cause of autosomal dominant hypertrophic cardiomyopathy. Eur. Heart J. 2021, 42, 3063–3073. [Google Scholar] [CrossRef] [PubMed]
- Harper, A.R.; HCMR Investigators; Goel, A.; Grace, C.; Thomson, K.L.; Petersen, S.E.; Xu, X.; Waring, A.; Ormondroyd, E.; Kramer, C.M.; et al. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat. Genet. 2021, 53, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, J.P.; Sabater-Molina, M.; García-Pinilla, J.M.; Mogensen, J.; Restrepo-Córdoba, A.; Palomino-Doza, J.; Villacorta, E.; Martinez-Moreno, M.; Ramos-Maqueda, J.; Zorio, E.; et al. Formin Homology 2 Domain Containing 3 (FHOD3) Is a Genetic Basis for Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2018, 72, 2457–2467. [Google Scholar] [CrossRef]
- Walsh, R.; Offerhaus, J.A.; Tadros, R.; Bezzina, C.R. Minor hypertrophic cardiomyopathy genes, major insights into the genetics of cardiomyopathies. Nat. Rev. Cardiol. 2021. [Google Scholar] [CrossRef]
- Tadros, R.; Francis, C.; Xu, X.; Vermeer, A.M.C.; Harper, A.R.; Huurman, R.; Bisabu, K.K.; Walsh, R.; Hoorntje, E.T.; Rijdt, W.P.T.; et al. Shared genetic pathways contribute to risk of hypertrophic and dilated cardiomyopathies with opposite directions of effect. Nat. Genet. 2021, 53, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Maron, M.S.; Semsarian, C. Genetics of Hypertrophic Cardiomyopathy After 20 Years. J. Am. Coll. Cardiol. 2012, 60, 705–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfares, A.A.; Kelly, M.A.; McDermott, G.; Funke, B.H.; Lebo, M.S.; Baxter, S.B.; Shen, J.; McLaughlin, H.M.; Clark, E.H.; Babb, L.J.; et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: Expanded panels offer limited additional sensitivity. Genet. Med. 2015, 17, 880–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingles, J.; Burns, C.; Barratt, A.; Semsarian, C. Application of Genetic Testing in Hypertrophic Cardiomyopathy for Preclinical Disease Detection. Circ. Cardiovasc. Genet. 2015, 8, 852–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabater-Molina, M.; Pérez-Sánchez, I.; Del Rincón, J.H.; Gimeno, J. Genetics of hypertrophic cardiomyopathy: A review of current state. Clin. Genet. 2017, 93, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man. Available online: www.omim.org (accessed on 28 August 2021).
- McNally, E.; Dellefave, L. Sarcomere Mutations in Cardiogenesis and Ventricular Noncompaction. Trends Cardiovasc. Med. 2009, 19, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, J.; Kubo, T.; Duque, M.; Uribe, W.; Shaw, A.; Murphy, R.; Gimeno, J.R.; Elliott, P.; McKenna, W.J. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J. Clin. Investig. 2003, 111, 209–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimiotti, D.; Budde, H.; Hassoun, R.; Jaquet, K. Genetic Restrictive Cardiomyopathy: Causes and Consequences—An Integrative Approach. Int. J. Mol. Sci. 2021, 22, 558. [Google Scholar] [CrossRef] [PubMed]
- Bortot, B.; Athanasakis, E.; Brun, F.; Rizzotti, D.; Mestroni, L.; Sinagra, G.; Severini, G.M. High-throughput Genotyping Robot-assisted Method for Mutation Detection in Patients With Hypertrophic Cardiomyopathy. Diagn. Mol. Pathol. 2011, 20, 175–179. [Google Scholar] [CrossRef]
- Fokstuen, S.; Munoz, A.; Melacini, P.; Iliceto, S.; Perrot, A.; Ozcelik, C.; Jeanrenaud, X.; Rieubland, C.; Farr, M.; Faber, L.; et al. Rapid detection of genetic variants in hypertrophic cardiomyopathy by custom DNA resequencing array in clinical practice. J. Med. Genet. 2011, 48, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Meder, B.; Haas, J.; Keller, A.; Heid, C.; Just, S.; Borries, A.; Boisguerin, V.; Scharfenberger-Schmeer, M.; Stähler, P.; Beier, M.; et al. Targeted Next-Generation Sequencing for the Molecular Genetic Diagnostics of Cardiomyopathies. Circ. Cardiovasc. Genet. 2011, 4, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Charron, P.; Villard, E.; Sébillon, P.; Laforêt, P.; Maisonobe, T.; Duboscq-Bidot, L.; Romero, N.; Drouin-Garraud, V.; Frébourg, T.; Richard, P.; et al. Danon’s disease as a cause of hypertrophic cardiomyopathy: A systematic survey. Heart 2004, 90, 842–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, H.S.; Bishop, D.F.; Astrin, K.H.; Kornreich, R.; Eng, C.M.; Sakuraba, H.; Desnick, R.J. Fabry disease: Six gene rearrangements and an exonic point mutation in the alpha-galactosidase gene. J. Clin. Investig. 1989, 83, 1390–1399. [Google Scholar] [CrossRef] [PubMed]
- Martiniuk, F.; Mehler, M.; Bodkin, M.; Tzall, S.; Hirschhorn, K.; Zhong, N.; Hirschhorn, R. Identification of a Missense Mutation in an Adult-Onset Patient with Glycogenosis Type II Expressing Only One Allele. DNA Cell Biol. 1991, 10, 681–687. [Google Scholar] [CrossRef]
- Martiniuk, F.; Mehler, M.; Pellicer, A.; Tzall, S.; La Badie, G.; Hobart, C.; Ellenbogen, A.; Hirschhorn, R. Isolation of a cDNA for human acid alpha-glucosidase and detection of genetic heterogeneity for mRNA in three alpha-glucosidase-deficient patients. Proc. Natl. Acad. Sci. USA 1986, 83, 9641–9644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Ploeg, A.T.; Hoefsloot, L.H.; Hoogeveen-Westerveld, M.; Petersen, E.M.; Reuser, A.J. Glycogenosis type II: Protein and DNA analysis in five South African families from various ethnic origins. Am. J. Hum. Gen. 1989, 44, 787–793. [Google Scholar]
- Genomes Project. Available online: http://www.internationalgenome.org/ (accessed on 28 August 2021).
- Exome Aggregation Consortium. Available online: http://exac.broadinstitute.org/ (accessed on 28 August 2021).
- Exome Sequencing Project. Available online: http://evs.gs.washington.edu/EVS/ (accessed on 28 August 2021).
- ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar (accessed on 28 August 2021).
- Human Gene Mutation Database. Available online: http://www.hgmd.cf.ac.uk/ac/index.php (accessed on 28 August 2021).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Lahrouchi, N.; Raju, H.; Lodder, E.M.; Papatheodorou, E.; Miles, C.; Ware, J.S.; Papadakis, M.; Tadros, R.; Cole, D.; Skinner, J.R.; et al. The yield of postmortem genetic testing in sudden death cases with structural findings at autopsy. Eur. J. Hum. Genet. 2019, 28, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Writing Committee Members; Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients with Hypertrophic Cardiomyopathy. Circulation 2020, 142. [Google Scholar] [CrossRef]
- Cardoso, B.; Gomes, I.; Loureiro, P.; Trigo, C.; Pinto, F.F. Diagnóstico clínico e genético de miocardiopatia hipertrófica familiar: Resultados em cardiologia pediátrica. Rev. Port. Cardiol. 2017, 36, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.K.; Havndrup, O.; Christiansen, M.; Andersen, P.S.; Diness, B.; Axelsson, A.; Skovby, F.; Køber, L.; Bundgaard, H. Penetrance of Hypertrophic Cardiomyopathy in Children and Adolescents. Circulation 2013, 127, 48–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.; Vatta, M.; Ware, S.M. Genetic Evaluation of Cardiomyopathy—A Heart Failure Society of America Practice Guideline. J. Card. Fail. 2018, 24, 281–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, B.; Semsarian, C.; Chan, K.H.; Sy, R.W. Sudden Cardiac Death and Ventricular Arrhythmias in Hypertrophic Cardiomyopathy. Heart Lung Circ. 2018, 28, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Europace 2015, 17, 1601–1687. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, I.; Girolami, F.; Ackerman, M.J.; Nistri, S.; Bos, J.M.; Zachara, E.; Ommen, S.R.; Theis, J.L.; Vaubel, R.A.; Re, F.; et al. Myofilament Protein Gene Mutation Screening and Outcome of Patients With Hypertrophic Cardiomyopathy. Mayo Clin. Proc. 2008, 83, 630–638. [Google Scholar] [CrossRef]
- Varnava, A.M.; Elliott, P.M.; Baboonian, C.; Davison, F.; Davies, M.J.; McKenna, W.J. Hypertrophic Cardiomyopathy. Circulation 2001, 104, 1380–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maron, B.J.; Yeates, L.; Semsarian, C. Clinical Challenges of Genotype Positive (+)–Phenotype Negative (−) Family Members in Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2011, 107, 604–608. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y. Genetics and Clinical Destiny: Improving Care in Hypertrophic Cardiomyopathy. Circulation 2010, 122, 2430–2440. [Google Scholar] [CrossRef] [PubMed]
- Maurizi, N.; Michels, M.; Rowin, E.J.; Semsarian, C.; Girolami, F.; Tomberli, B.; Cecchi, F.; Maron, M.S.; Olivotto, I.; Maron, B.J. Clinical Course and Significance of Hypertrophic Cardiomyopathy Without Left Ventricular Hypertrophy. Circulation 2019, 139, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Charron, P.; Carrier, L.; Dubourg, O.; Tesson, F.; Desnos, M.; Richard, P.; Bonne, G.; Guicheney, P.; Hainque, B.; Bouhour, J.B.; et al. Penetrance of familial hypertrophic cardiomyopathy. Genet. Couns. 1997, 8, 107–114. [Google Scholar]
- Bos, J.M.; Will, M.L.; Gersh, B.J.; Kruisselbrink, T.M.; Ommen, S.R.; Ackerman, M.J. Characterization of a Phenotype-Based Genetic Test Prediction Score for Unrelated Patients With Hypertrophic Cardiomyopathy. Mayo Clin. Proc. 2014, 89, 727–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaventura, J.; Norambuena, P.; Tomašov, P.; Jindrová, D.; Šedivá, H.; Jr, M.M.; Veselka, J.; Macek, M. The utility of the Mayo Score for predicting the yield of genetic testing in patients with hypertrophic cardiomyopathy. Arch. Med Sci. 2019, 15, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.S.; Havndrup, O.; Hougs, L.; Sørensen, K.M.; Jensen, M.K.; Larsen, L.A.; Hedley, P.; Thomsen, A.; Moolman-Smook, J.; Christiansen, M.; et al. Diagnostic yield, interpretation, and clinical utility of mutation screening of sarcomere encoding genes in Danish hypertrophic cardiomyopathy patients and relatives. Hum. Mutat. 2008, 30, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Van Driest, S.L.; Ommen, S.R.; Tajik, A.J.; Gersh, B.J.; Ackerman, M.J. Yield of Genetic Testing in Hypertrophic Cardiomyopathy. Mayo Clin. Proc. 2005, 80, 739–744. [Google Scholar] [CrossRef]
- Murphy, S.L.; Anderson, J.; Kapplinger, J.D.; Kruisselbrink, T.M.; Gersh, B.J.; Ommen, S.R.; Ackerman, M.J.; Bos, J.M. Evaluation of the Mayo Clinic Phenotype-Based Genotype Predictor Score in Patients with Clinically Diagnosed Hypertrophic Cardiomyopathy. J. Cardiovasc. Transl. Res. 2016, 9, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, S.P.; Kounas, S.; Syrris, P.; Christiansen, M.; Frank-Hansen, R.; Andersen, P.S.; Elliott, P.M.; McKenna, W.J. Cardiac Myosin Binding Protein-C Mutations in Families with Hypertrophic Cardiomyopathy. Circ. Cardiovasc. Genet. 2012, 5, 156–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidman, C.E.; Seidman, J. Identifying Sarcomere Gene Mutations in Hypertrophic Cardiomyopathy. Circ. Res. 2011, 108, 743–750. [Google Scholar] [CrossRef] [Green Version]
- Van Velzen, H.G.; Vriesendorp, P.A.; Oldenburg, R.A.; Van Slegtenhorst, M.A.; Van Der Velden, J.; Schinkel, A.F.; Michels, M. Value of Genetic Testing for the Prediction of Long-Term Outcome in Patients with Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2016, 118, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Mathew, J.; Zahavich, L.; Lafreniere-Roula, M.; Wilson, J.; George, K.; Benson, L.; Bowdin, S.; Mital, S. Utility of genetics for risk stratification in pediatric hypertrophic cardiomyopathy. Clin. Genet. 2017, 93, 310–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, L.R.; Brito, D.; Belo, A.; Cardim, N. Genetic characterization and genotype-phenotype associations in a large cohort of patients with hypertrophic cardiomyopathy—An ancillary study of the Portuguese registry of hypertrophic cardiomyopathy. Int. J. Cardiol. 2018, 278, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Day, S.M.; Ashley, E.A.; Michels, M.; Pereira, A.C.; Jacoby, D.; Cirino, A.L.; Fox, J.C.; Lakdawala, N.K.; Ware, J.; et al. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy. Circulation 2018, 138, 1387–1398. [Google Scholar] [CrossRef]
- Bonaventura, J.; Veselka, J. Genetic testing in patients with hypertrophic cardiomyopathy. Vnitrni Lek. 2019, 65, 652–658. [Google Scholar] [CrossRef]
- Ingles, J.; Doolan, A.; Chiu, C.L.; Seidman, J.; Seidman, C.; Semsarian, C. Compound and double mutations in patients with hypertrophic cardiomyopathy: Implications for genetic testing and counselling. J. Med. Genet. 2005, 42, e59. [Google Scholar] [CrossRef] [Green Version]
- Blankenburg, R.; Hackert, K.; Wurster, S.; Deenen, R.; Seidman, J.; Seidman, C.E.; Lohse, M.J.; Schmitt, J.P. β-Myosin Heavy Chain Variant Val606Met Causes Very Mild Hypertrophic Cardiomyopathy in Mice, but Exacerbates HCM Phenotypes in Mice Carrying Other HCM Mutations. Circ. Res. 2014, 115, 227–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorn, G.W.; McNally, E.M. Two Strikes and You’re Out. Circ. Res. 2014, 115, 208–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiffin, N.; Minikel, E.V.; Walsh, R.; O’Donnell-Luria, A.; Karczewski, K.; Ing, A.Y.; Barton, P.; Funke, B.; A Cook, S.; MacArthur, D.; et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet. Med. 2017, 19, 1151–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiffin, N.; Walsh, R.; Govind, R.; Edwards, M.; Ahmad, M.; Zhang, X.; Tayal, U.; Buchan, R.; Midwinter, W.; E Wilk, A.; et al. CardioClassifier: Disease- and gene-specific computational decision support for clinical genome interpretation. Genet. Med. 2018, 20, 1246–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baulina, N.M.; Kiselev, I.S.; Chumakova, O.S.; Favorova, O.O. Hypertrophic Cardiomyopathy as an Oligogenic Disease: Transcriptomic Arguments. Mol. Biol. 2020, 54, 840–850. [Google Scholar] [CrossRef]
- Aurigemma, G.P.; de Simone, G.; Fitzgibbons, T. Cardiac Remodeling in Obesity. Circ. Cardiovasc. Imaging 2013, 6, 142–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, J.; Lindgren, M.; Schaufelberger, M.; Adiels, M.; Björck, L.; Lundberg, C.E.; Sattar, N.; Rosengren, A.; Aberg, M. Body Mass Index in Young Women and Risk of Cardiomyopathy. Circulation 2020, 141, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, C.; Maurizi, N.; Day, S.M.; Ashley, E.A.; Michels, M.; Colan, S.D.; Jacoby, D.; Marchionni, N.; Vincent-Tompkins, J.; Ho, C.Y.; et al. Association of Obesity With Adverse Long-term Outcomes in Hypertrophic Cardiomyopathy. JAMA Cardiol. 2020, 5, 65–68. [Google Scholar] [CrossRef]
- Nollet, E.E.; Westenbrink, B.D.; de Boer, R.A.; Kuster, D.W.D.; van der Velden, J. Unraveling the Genotype-Phenotype Relationship in Hypertrophic Cardiomyopathy: Obesity-Related Cardiac Defects as a Major Disease Modifier. J. Am. Heart Assoc. 2020, 9, e018641. [Google Scholar] [CrossRef] [PubMed]
- Tini, G.; Autore, C.; Musumeci, B. The Many Faces of Arterial Hypertension in Hypertrophic Cardiomyopathy and Its Phenocopies: Bystander, Consequence, Modifier. High Blood Press. Cardiovasc. Prev. 2021, 28, 327–329. [Google Scholar] [CrossRef] [PubMed]
- Siontis, K.C.; Ommen, S.R.; Geske, J.B. Sex, Survival, and Cardiomyopathy: Differences Between Men and Women With Hypertrophic Cardiomyopathy. J. Am. Heart Assoc. 2019, 8, e014448. [Google Scholar] [CrossRef] [PubMed]
- Michels, M.; Soliman, O.I.; Phefferkorn, J.; Hoedemaekers, Y.M.; Kofflard, M.J.; Dooijes, D.; Majoor-Krakauer, D.; Cate, F.J.T. Disease penetrance and risk stratification for sudden cardiac death in asymptomatic hypertrophic cardiomyopathy mutation carriers. Eur. Heart J. 2009, 30, 2593–2598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzini, M.; Norrish, G.; Field, E.; Ochoa, J.P.; Cicerchia, M.; Akhtar, M.M.; Syrris, P.; Lopes, L.R.; Kaski, J.P.; Elliott, P.M. Penetrance of Hypertrophic Cardiomyopathy in Sarcomere Protein Mutation Carriers. J. Am. Coll. Cardiol. 2020, 76, 550–559. [Google Scholar] [CrossRef]
- Geske, J.B.; Ong, K.C.; Siontis, K.C.; Hebl, V.B.; Ackerman, M.J.; O Hodge, D.; Miller, V.M.; A Nishimura, R.; Oh, J.K.; Schaff, H.; et al. Women with hypertrophic cardiomyopathy have worse survival. Eur. Heart J. 2017, 38, 3434–3440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veselka, J.; Faber, L.; Liebregts, M.; Cooper, R.; Kashtanov, M.; Hansen, P.R.; Bonaventura, J.; Polakova, E.; Hansvenclova, E.; Bundgaard, H.; et al. Sex-Related Differences in Outcomes of Alcohol Septal Ablation for Hypertrophic Obstructive Cardiomyopathy. JACC Cardiovasc. Interv. 2021, 14, 1390–1392. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, C.; Olivotto, I. The Importance of Sex Differences in Patients With Hypertrophic Cardiomyopathy—Tailoring Management and Future Perspectives. Am. J. Med. Sci. 2020, 360, 433–434. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, H.-W.; Wang, C.-F.; Meng, Q.-K.; Cui, C.-S.; Zhang, X.-J.; Zhu, Y.; Fan, C.-Y.; Luo, D.-F.; Chen, B.-J.; et al. Gender Disparities in Clinical Outcome After Alcohol Septal Ablation for Hypertrophic Obstructive Cardiomyopathy in the Chinese Han Population: A Cohort Study. Heart Lung Circ. 2020, 29, 1856–1864. [Google Scholar] [CrossRef] [PubMed]
- Meghji, Z.; Nguyen, A.; Fatima, B.; Geske, J.B.; Nishimura, R.A.; Ommen, S.R.; Lahr, B.D.; Dearani, J.A.; Schaff, H.V. Survival Differences in Women and Men After Septal Myectomy for Obstructive Hypertrophic Cardiomyopathy. JAMA Cardiol. 2019, 4, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Rigopoulos, A.G.; Ali, M.; Abate, E.; Torky, A.-R.; Matiakis, M.; Mammadov, M.; Melnyk, H.; Vogt, A.; De Vecchis, R.; Bigalke, B.; et al. Advances in the diagnosis and treatment of transthyretin amyloidosis with cardiac involvement. Heart Fail. Rev. 2019, 24, 521–533. [Google Scholar] [CrossRef]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Chien, C.-S.; Chiang, C.-E.; Chen, C.-H.; Cheng, H.-M. From Genetic Mutations to Molecular Basis of Heart Failure Treatment: An Overview of the Mechanism and Implication of the Novel Modulators for Cardiac Myosin. Int. J. Mol. Sci. 2021, 22, 6617. [Google Scholar] [CrossRef] [PubMed]
- Prondzynski, M.; Mearini, G.; Carrier, L. Gene therapy strategies in the treatment of hypertrophic cardiomyopathy. Pflügers Arch. Eur. J. Physiol. 2018, 471, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, C.; De Gregorio, M.G.; Zampieri, M.; Fedele, E.; Tomberli, A.; Chiriatti, C.; Marchi, A.; Olivotto, I. Targeted Medical Therapies for Hypertrophic Cardiomyopathy. Curr. Cardiol. Rep. 2020, 22, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sewanan, L.R.; Jacoby, D.L. Novel Myosin-Based Therapies in Hypertrophic Cardiomyopathy. Curr. Treat. Options Cardiovasc. Med. 2021, 23, 1–12. [Google Scholar] [CrossRef]
- Jiang, J.; Wakimoto, H.; Seidman, J.G.; Seidman, C.E. Allele-Specific Silencing of MutantMyh6Transcripts in Mice Suppresses Hypertrophic Cardiomyopathy. Science 2013, 342, 111–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannon, L.; Yu, Z.-Y.; Marciniec, T.; Waardenberg, A.J.; Iismaa, S.E.; Nikolova-Krstevski, V.; Neist, E.; Ohanian, M.; Qiu, M.R.; Rainer, S.; et al. Irreversible Triggers for Hypertrophic Cardiomyopathy Are Established in the Early Postnatal Period. J. Am. Coll. Cardiol. 2015, 65, 560–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, P.; Lander, E.S.; Zhang, F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Marti-Gutierrez, N.; Park, S.-W.; Wu, J.; Lee, Y.; Suzuki, K.; Koski, A.; Jianhui, G.; Hayama, T.; Ahmed, R.; et al. Correction of a pathogenic gene mutation in human embryos. Nat. Cell Biol. 2017, 548, 413–419. [Google Scholar] [CrossRef]
- Maron, B.J. Clinical Course and Management of Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2018, 379, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.; Arscott, P.; Concannon, M.; Saberi, S.; Day, S.M.; Yashar, B.M.; Helms, A. Genetic testing impacts the utility of prospective familial screening in hypertrophic cardiomyopathy through identification of a nonfamilial subgroup. Genet. Med. 2017, 20, 69–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingles, J.; Burns, C.; Bagnall, R.D.; Lam, L.; Yeates, L.; Sarina, T.; Puranik, R.; Briffa, T.; Atherton, J.J.; Driscoll, T.; et al. Nonfamilial Hypertrophic Cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaventura, J.; Norambuena, P.; Votýpka, P.; Hnátová, H.; Adlová, R.; Macek, M.; Veselka, J.; Jr, M.M. Patients with hypertrophic obstructive cardiomyopathy after alcohol septal ablation have favorable long-term outcome irrespective of their genetic background. Cardiovasc. Diagn. Ther. 2020, 10, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Gruner, C.; Ivanov, J.; Care, M.; Williams, L.; Moravsky, G.; Yang, H.; Laczay, B.; Siminovitch, K.; Woo, A.; Rakowski, H. Toronto Hypertrophic Cardiomyopathy Genotype Score for Prediction of a Positive Genotype in Hypertrophic Cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.W.; Fifer, M.A.; Hasegawa, K.; Maurer, M.S.; Reilly, M.P.; Shimada, Y.J. Prediction of Genotype Positivity in Patients with Hypertrophic Cardiomyopathy Using Machine Learning. Circ. Genom. Precis. Med. 2021, 14. [Google Scholar] [CrossRef] [PubMed]
- Aziz, A.; Musiol, S.K.; Moody, W.E.; Pickup, L.; Cooper, R.; Lip, G.Y.H. Clinical prediction of genotypes in hypertrophic cardiomyopathy: A systematic review. Eur. J. Clin. Investig. 2021, e13593. [Google Scholar] [CrossRef]
- Zhou, H.; Li, L.; Liu, Z.; Zhao, K.; Chen, X.; Lu, M.; Yin, G.; Song, L.; Zhao, S.; Zheng, H.; et al. Deep learning algorithm to improve hypertrophic cardiomyopathy mutation prediction using cardiac cine images. Eur. Radiol. 2020, 31, 3931–3940. [Google Scholar] [CrossRef]


{kind=link}
| Gene | Protein | Year of Discovery | Frequency (%) * | Inheritance | Most Common Pathogenic Variant |
|---|---|---|---|---|---|
| Thick filament | |||||
| MYH7 | Beta-myosin heavy chain | 1989 | 20–30 | AD | c.1988G>A |
| MYL2 | Regulatory myosin light chain | 1998 | 2–4 | AD | c.173G>A |
| MYL3 | Essential myosin light chain | 1996 | 1–2 | AD | c.281G>A |
| Thin filament | |||||
| TNNT2 | Cardiac troponin T | 1993 | 10 | AD | c.236T>A |
| TNNI3 | Cardiac troponin I | 1997 | 7 | AD | c.433C>T |
| TPM1 | Alpha tropomyosin | 1993 | <1 | AD | c.574G>A |
| ACTC1 | Alpha cardiac actin | 1999 | <1 | AD | c.301G>A |
| Intermediate filament | |||||
| MYBPC3 | Myosin-binding protein C | 1993 | 30–40 | AD | c.1504C>T |
| Gene | Protein | Phenotype | Prevalence * | Inheritance | Frequency (%) ** |
|---|---|---|---|---|---|
| PRKAG2 | Protein kinase, AMP-activated, gamma 2 subunit | Wolff–Parkinson–White syndrome | 1/4000 | AD | 0.2–1.0 |
| LAMP2 | Protein kinase, AMP-activated, gamma 2 subunit | Danon disease | 1/100,000 | X | 0.1–0.2 |
| GLA | Galactosidase, alpha | Fabry disease | 1/40,000 | X | 0.5–1.0 |
| FHL1 | Four and a half LIM domains 1 | Emery–Dreifuss myopathy | 1/100,000 | X | 0.1–0.5 |
| TTR | Transthyretin | Amyloidosis *** | 1/100,000 | AD | 0.8–5 |
| GAA | Glucosidase, alpha | Pompe disease | 1/40,000 | AR | 0.01–0.1 |
| PTPN11 | Protein tyrosine phosphatase, non-receptor type 11 | Noonan syndrome LEOPARD | 1/2000 | AD | 1–5 |
| FXN | Frataxin | Friedreich ataxia | 1/20,000 | AR | 0.05–0.2 |
| Clinical Variable | Points |
|---|---|
| Age < 45 years | 1 |
| Left ventricular wall thickness > 20 mm | 1 |
| Family history of HCM | 1 |
| Family history of sudden cardiac death | 1 |
| Reverse septal shape | 1 |
| Arterial hypertension | −1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonaventura, J.; Polakova, E.; Vejtasova, V.; Veselka, J. Genetic Testing in Patients with Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2021, 22, 10401. https://doi.org/10.3390/ijms221910401
Bonaventura J, Polakova E, Vejtasova V, Veselka J. Genetic Testing in Patients with Hypertrophic Cardiomyopathy. International Journal of Molecular Sciences. 2021; 22(19):10401. https://doi.org/10.3390/ijms221910401
Chicago/Turabian StyleBonaventura, Jiri, Eva Polakova, Veronika Vejtasova, and Josef Veselka. 2021. "Genetic Testing in Patients with Hypertrophic Cardiomyopathy" International Journal of Molecular Sciences 22, no. 19: 10401. https://doi.org/10.3390/ijms221910401
APA StyleBonaventura, J., Polakova, E., Vejtasova, V., & Veselka, J. (2021). Genetic Testing in Patients with Hypertrophic Cardiomyopathy. International Journal of Molecular Sciences, 22(19), 10401. https://doi.org/10.3390/ijms221910401