
Adenovirus Vectors Expressing Eight Multiplex Guide RNAs of CRISPR/Cas9 Efficiently Disrupted Diverse Hepatitis B Virus Gene Derived from Heterogeneous Patient


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
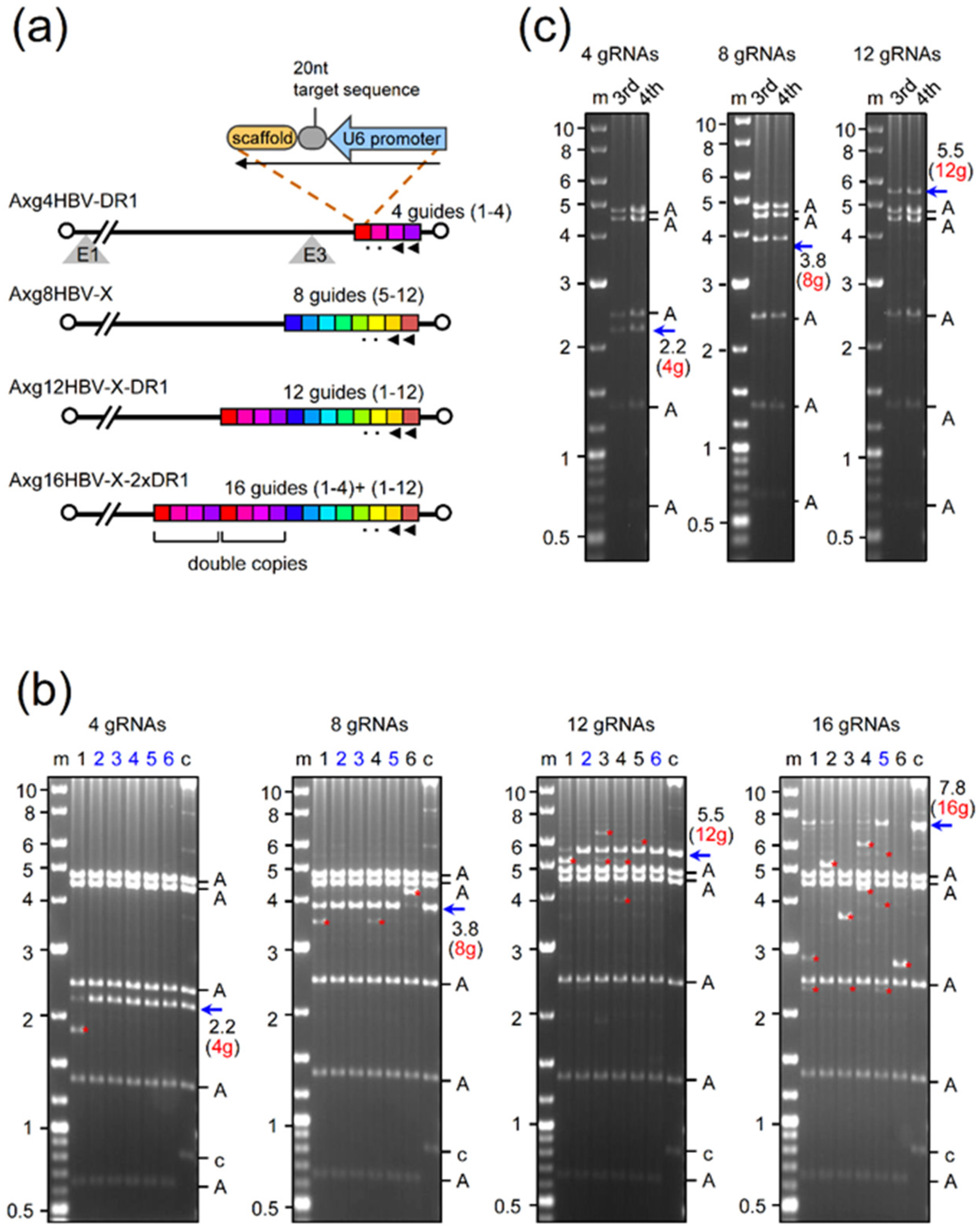
2.1. Production of AdVs Expressing Multiplex gRNAs
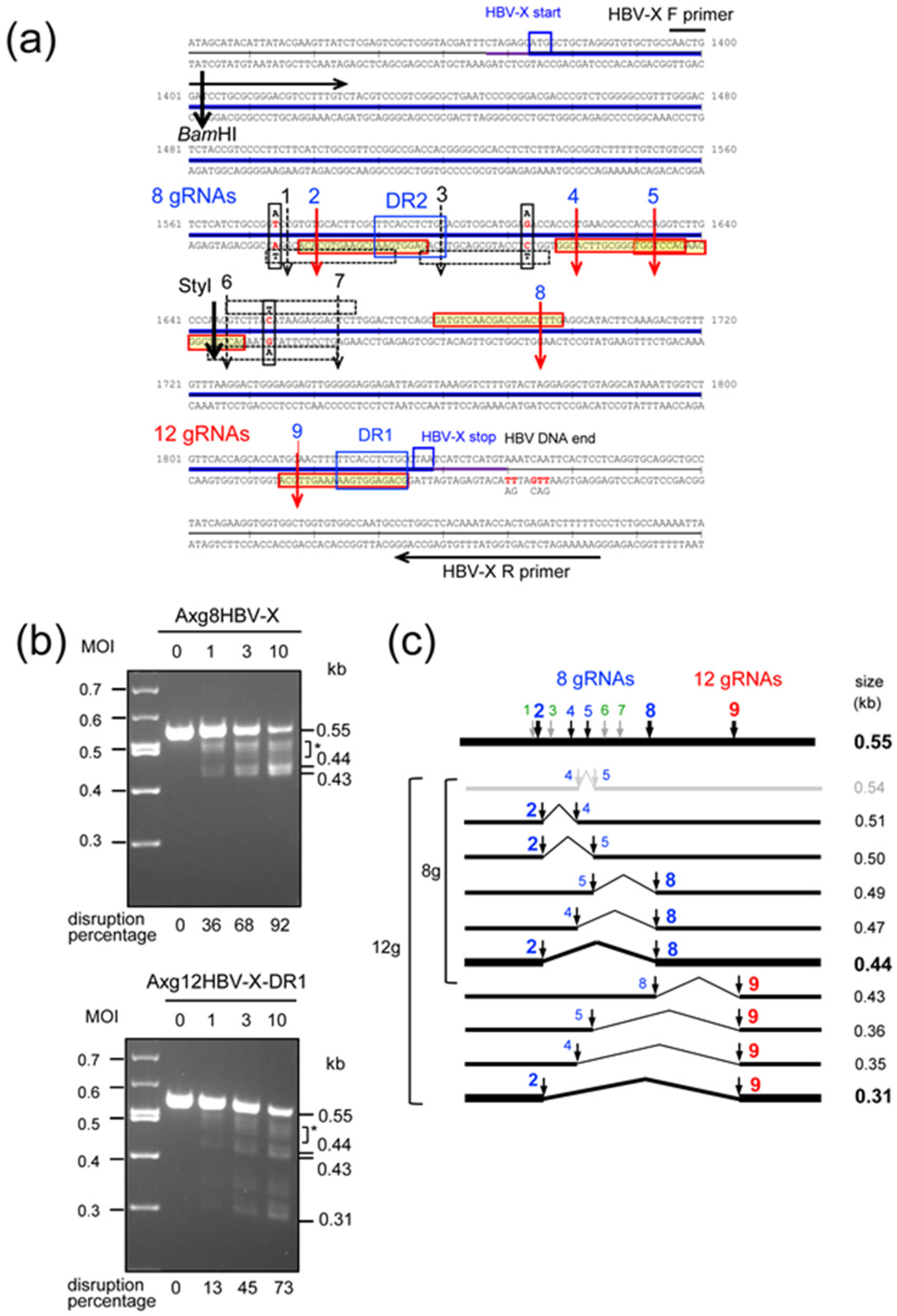
2.2. Disruption of HBV-X Gene Derived from a Heterogeneous Patient Using AdVs Expressing Eight and Twelve gRNAs
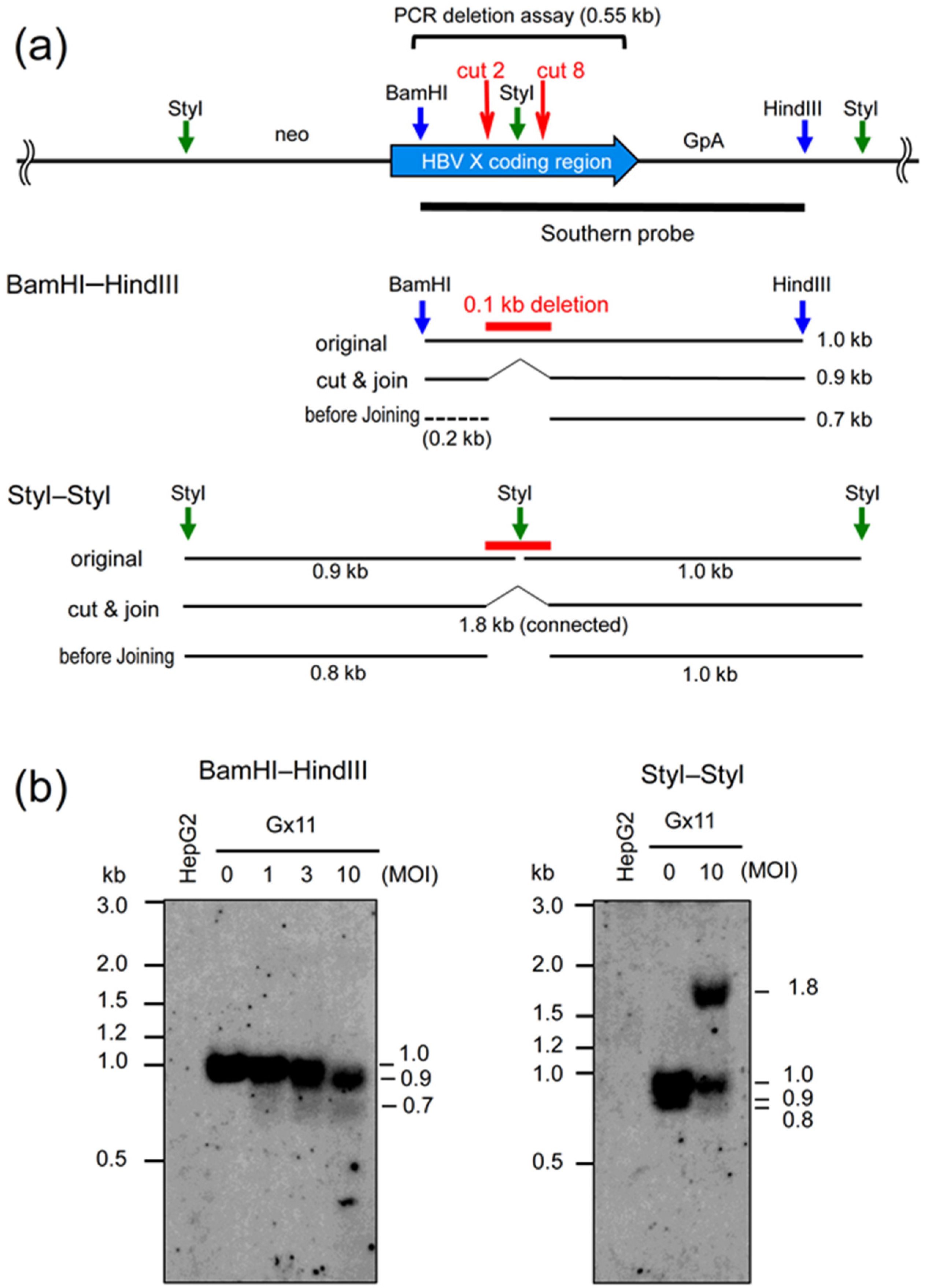
2.3. Southern Analyses Using the AdV Expressing Eight gRNAs Targeting HBV X Gene
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Construction of Cosmids Containing AdV Genome Bearing Multiplex gRNA Units
4.3. Production of AdVs
4.4. Conventional PCR
4.5. T7 Endonuclease Assay
4.6. Cellular DNA Isolation
4.7. Southern Blot Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273.e1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, K.B.; Kim, H.J. Management of antiviral drug resistance in chronic hepatitis B. World J. Gastroenterol. 2014, 20, 11641–11649. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T. Clinical features of hepatitis B virus-related hepatocellular carcinoma. World J. Gastroenterol. 2010, 16, 2463–2467. [Google Scholar] [CrossRef]
- Robinson, W.S. Molecular events in the pathogenesis of hepadnavirus-associated hepatocellular carcinoma. Annu. Rev. Med. 1994, 45, 297–323. [Google Scholar] [CrossRef] [PubMed]
- Buendia, M.A. Hepatitis B viruses and hepatocellular carcinoma. Adv. Cancer Res. 1992, 59, 167–226. [Google Scholar] [CrossRef]
- Kawai-Kitahata, F.; Asahina, Y.; Tanaka, S.; Kakinuma, S.; Murakawa, M.; Nitta, S.; Watanabe, T.; Otani, S.; Taniguchi, M.; Goto, F.; et al. Comprehensive analyses of mutations and hepatitis B virus integration in hepatocellular carcinoma with clinicopathological features. J. Gastroenterol. 2016, 51, 473–486. [Google Scholar] [CrossRef]
- Hai, H.; Tamori, A.; Kawada, N. Role of hepatitis B virus DNA integration in human hepatocarcinogenesis. World J. Gastroenterol. 2014, 20, 6236–6243. [Google Scholar] [CrossRef]
- Guerrieri, F.; Belloni, L.; Pediconi, N.; Levrero, M. Molecular mechanisms of HBV-associated hepatocarcinogenesis. Semin. Liver Dis. 2013, 33, 147–156. [Google Scholar] [CrossRef]
- Kim, C.M.; Koike, K.; Saito, I.; Miyamura, T.; Jay, G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 1991, 351, 317–320. [Google Scholar] [CrossRef]
- Kew, M.C. Hepatitis B virus x protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2011, 26 (Suppl. 1), 144–152. [Google Scholar] [CrossRef]
- Ahodantin, J.; Lekbaby, B.; Bou Nader, M.; Soussan, P.; Kremsdorf, D. Hepatitis B virus X protein enhances the development of liver fibrosis and the expression of genes associated with epithelial-mesenchymal transitions and tumor progenitor cells. Carcinogenesis 2020, 41, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Stone, D.; Niyonzima, N.; Jerome, K.R. Genome editing and the next generation of antiviral therapy. Hum. Genet. 2016, 135, 1071–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, N.D.; Aubert, M.; Dang, C.H.; Stone, D.; Jerome, K.R. DNA cleavage enzymes for treatment of persistent viral infections: Recent advances and the pathway forward. Virology 2014, 454–455, 353–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revill, P.A.; Tu, T.; Netter, H.J.; Yuen, L.K.W.; Locarnini, S.A.; Littlejohn, M. The evolution and clinical impact of hepatitis B virus genome diversity. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 618–634. [Google Scholar] [CrossRef] [PubMed]
- Glebe, D.; Goldmann, N.; Lauber, C.; Seitz, S. HBV evolution and genetic variability: Impact on prevention, treatment and development of antivirals. Antivir. Res. 2021, 186, 104973. [Google Scholar] [CrossRef] [PubMed]
- Rajoriya, N.; Combet, C.; Zoulim, F.; Janssen, H.L.A. How viral genetic variants and genotypes influence disease and treatment outcome of chronic hepatitis B. Time for an individualised approach? J. Hepatol. 2017, 67, 1281–1297. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.A.S.; Chang, H.; Shim, H.; Heo, J.; Cho, M.; Moon, B.; Moon, Y.; Paik, Y.; Lee, K. 563 hepatitis b virus quasispecies in the polymerase gene in treatment-naive chronic hepatitis b patients. J. Hepatol. 2008, 48, S211. [Google Scholar] [CrossRef]
- Kim, D.Y.; Chang, H.Y.; Lim, S.M.; Kim, S.U.; Park, J.Y.; Kim, J.K.; Lee, K.S.; Han, K.H.; Chon, C.Y.; Ahn, S.H. Quasispecies and pre-existing drug-resistant mutations of hepatitis B virus in patients with chronic hepatitis B. Gut Liver 2013, 7, 329–334. [Google Scholar] [CrossRef] [Green Version]
- Lebbink, R.J.; de Jong, D.C.; Wolters, F.; Kruse, E.M.; van Ham, P.M.; Wiertz, E.J.; Nijhuis, M. A combinational CRISPR/Cas9 gene-editing approach can halt HIV replication and prevent viral escape. Sci. Rep. 2017, 7, 41968. [Google Scholar] [CrossRef]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. A Combinatorial CRISPR-Cas9 Attack on HIV-1 DNA Extinguishes All Infectious Provirus in Infected T Cell Cultures. Cell Rep. 2016, 17, 2819–2826. [Google Scholar] [CrossRef] [Green Version]
- De Silva Feelixge, H.S.; Stone, D.; Roychoudhury, P.; Aubert, M.; Jerome, K.R. CRISPR/Cas9 and Genome Editing for Viral Disease-Is Resistance Futile? ACS Infect. Dis. 2018, 4, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Bishop, E.S.; Zhang, R.; Yu, X.; Farina, E.M.; Yan, S.; Zhao, C.; Zheng, Z.; Shu, Y.; Wu, X.; et al. Adenovirus-Mediated Gene Delivery: Potential Applications for Gene and Cell-Based Therapies in the New Era of Personalized Medicine. Genes Dis. 2017, 4, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Holkers, M.; Maggio, I.; Henriques, S.F.; Janssen, J.M.; Cathomen, T.; Goncalves, M.A. Adenoviral vector DNA for accurate genome editing with engineered nucleases. Nat. Methods 2014, 11, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Nakai, M.; Komiya, K.; Murata, M.; Kimura, T.; Kanaoka, M.; Kanegae, Y.; Saito, I. Expression of pIX gene induced by transgene promoter: Possible cause of host immune response in first-generation adenoviral vectors. Hum. Gene Ther. 2007, 18, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Chiyo, T.; Sekiguchi, S.; Hayashi, M.; Tobita, Y.; Kanegae, Y.; Saito, I.; Kohara, M. Conditional gene expression in hepatitis C virus transgenic mice without induction of severe liver injury using a non-inflammatory Cre-expressing adenovirus. Virus Res. 2011, 160, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, C.T.; Deshpande, P.S.; Dever, D.P.; Camarena, J.; Lemgart, V.T.; Cromer, M.K.; Vakulskas, C.A.; Collingwood, M.A.; Zhang, L.; Bode, N.M.; et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019, 25, 249–254. [Google Scholar] [CrossRef]
- Rusk, N. Human immunity to Cas9. Nat. Methods 2019, 16, 286. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.L.; Amini, L.; Wendering, D.J.; Burkhardt, L.M.; Akyuz, L.; Reinke, P.; Volk, H.D.; Schmueck-Henneresse, M. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med. 2019, 25, 242–248. [Google Scholar] [CrossRef]
- Nakanishi, T.; Maekawa, A.; Suzuki, M.; Tabata, H.; Sato, K.; Mori, M.; Saito, I. Construction of adenovirus vectors simultaneously expressing four multiplex, double-nicking guide RNAs of CRISPR/Cas9 and in vivo genome editing. Sci. Rep. 2021, 11, 3961. [Google Scholar] [CrossRef]
- Shintani, Y.; Yotsuyanagi, H.; Moriya, K.; Fujie, H.; Tsutsumi, T.; Kanegae, Y.; Kimura, S.; Saito, I.; Koike, K. Induction of apoptosis after switch-on of the hepatitis B virus X gene mediated by the Cre/loxP recombination system. J. Gen. Virol. 1999, 80 Pt. 12, 3257–3265. [Google Scholar] [CrossRef] [Green Version]
- Kabadi, A.M.; Ousterout, D.G.; Hilton, I.B.; Gersbach, C.A. Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res. 2014, 42, e147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, C.; Zhang, T.; Qu, X.; Zhang, Y.; Putatunda, R.; Xiao, X.; Li, F.; Xiao, W.; Zhao, H.; Dai, S.; et al. In Vivo Excision of HIV-1 Provirus by saCas9 and Multiplex Single-Guide RNAs in Animal Models. Mol. Ther. 2017, 25, 1168–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakanishi, T.; Maekawa, A.; Tabata, H.; Yoshioka, T.; Pei, Z.; Sato, K.; Mori, M.; Kato, M.; Saito, I. Highly multiplex guide RNA expression units of CRISPR/Cas9 were completely stable using cosmid amplification in a novel polygonal structure. J. Gene Med. 2019, 21, e3115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, I.; Stark, G.R. Charomids: cosmid vectors for efficient cloning and mapping of large or small restriction fragments. Proc. Natl. Acad. Sci. USA 1986, 83, 8664–8668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, H.; Terashima, M.; Koshikawa, M.; Kanegae, Y.; Saito, I. Possible mechanism of adenovirus generation from a cloned viral genome tagged with nucleotides at its ends. Microbiol. Immunol. 2006, 50, 643–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grable, M.; Hearing, P. cis and trans requirements for the selective packaging of adenovirus type 5 DNA. J. Virol. 1992, 66, 723–731. [Google Scholar] [CrossRef] [Green Version]
- Bett, A.J.; Haddara, W.; Prevec, L.; Graham, F.L. An efficient and flexible system for construction of adenovirus vectors with insertions or deletions in early regions 1 and 3. Proc. Natl. Acad. Sci. USA 1994, 91, 8802–8806. [Google Scholar] [CrossRef] [Green Version]
- Mizuguchi, H.; Kay, M.A. A simple method for constructing E1- and E1/E4-deleted recombinant adenoviral vectors. Hum. Gene Ther. 1999, 10, 2013–2017. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Wang, J.; Shen, B.; Chen, L.; Su, Y.; Yang, J.; Zhang, W.; Tian, X.; Huang, X. Dual sgRNAs facilitate CRISPR/Cas9-mediated mouse genome targeting. FEBS J. 2014, 281, 1717–1725. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.Q.; Wyvekens, N.; Khayter, C.; Foden, J.A.; Thapar, V.; Reyon, D.; Goodwin, M.J.; Aryee, M.J.; Joung, J.K. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat. Biotechnol. 2014, 32, 569–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalappa, R.; Suresh, B.; Ramakrishna, S.; Kim, H.H. Paired D10A Cas9 nickases are sometimes more efficient than individual nucleases for gene disruption. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurihara, T.; Fukuhara, T.; Ono, C.; Yamamoto, S.; Uemura, K.; Okamoto, T.; Sugiyama, M.; Motooka, D.; Nakamura, S.; Ikawa, M.; et al. Suppression of HBV replication by the expression of nickase- and nuclease dead-Cas9. Sci. Rep. 2017, 7, 6122. [Google Scholar] [CrossRef] [PubMed]
- Haeussler, M.; Concordet, J.P. Genome Editing with CRISPR-Cas9: Can It Get Any Better? J. Genet. Genomics 2016, 43, 239–250. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. CRISPR-Cas based antiviral strategies against HIV-1. Virus Res. 2018, 244, 321–332. [Google Scholar] [CrossRef]
- Li, C.; Lieber, A. Adenovirus vectors in hematopoietic stem cell genome editing. FEBS Lett. 2019, 593, 3623–3648. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Kondo, S.; Pei, Z.; Maekawa, A.; Saito, I.; Kanegae, Y. Preferable sites and orientations of transgene inserted in the adenovirus vector genome: The E3 site may be unfavorable for transgene position. Gene Ther. 2015, 22, 421–429. [Google Scholar] [CrossRef]
- Pei, Z.; Kondo, S.; Kanegae, Y.; Saito, I. Copy number of adenoviral vector genome transduced into target cells can be measured using quantitative PCR: application to vector titration. Biochem. Biophys. Res. Commun. 2012, 417, 945–950. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kato, Y.; Tabata, H.; Sato, K.; Nakamura, M.; Saito, I.; Nakanishi, T. Adenovirus Vectors Expressing Eight Multiplex Guide RNAs of CRISPR/Cas9 Efficiently Disrupted Diverse Hepatitis B Virus Gene Derived from Heterogeneous Patient. Int. J. Mol. Sci. 2021, 22, 10570. https://doi.org/10.3390/ijms221910570
Kato Y, Tabata H, Sato K, Nakamura M, Saito I, Nakanishi T. Adenovirus Vectors Expressing Eight Multiplex Guide RNAs of CRISPR/Cas9 Efficiently Disrupted Diverse Hepatitis B Virus Gene Derived from Heterogeneous Patient. International Journal of Molecular Sciences. 2021; 22(19):10570. https://doi.org/10.3390/ijms221910570
Chicago/Turabian StyleKato, Yuya, Hirotaka Tabata, Kumiko Sato, Mariko Nakamura, Izumu Saito, and Tomoko Nakanishi. 2021. "Adenovirus Vectors Expressing Eight Multiplex Guide RNAs of CRISPR/Cas9 Efficiently Disrupted Diverse Hepatitis B Virus Gene Derived from Heterogeneous Patient" International Journal of Molecular Sciences 22, no. 19: 10570. https://doi.org/10.3390/ijms221910570
APA StyleKato, Y., Tabata, H., Sato, K., Nakamura, M., Saito, I., & Nakanishi, T. (2021). Adenovirus Vectors Expressing Eight Multiplex Guide RNAs of CRISPR/Cas9 Efficiently Disrupted Diverse Hepatitis B Virus Gene Derived from Heterogeneous Patient. International Journal of Molecular Sciences, 22(19), 10570. https://doi.org/10.3390/ijms221910570