
Impact of Posttranslational Modification in Pathogenesis of Rheumatoid Arthritis: Focusing on Citrullination, Carbamylation, and Acetylation


Abstract
:1. Introduction
2. Pathogenesis
3. Citrullination in RA
3.1. PAD Family
3.2. ACPAs, a Diagnostic and Prognostic Tool in RA
3.3. Citrullination and Cell Death Mechanism
3.4. Microorganisms Inducing Citrullination
4. Carbamylation in RA
4.1. Anti-Carbamylated Protein Antibodies, a Novel Hallmark for RA
4.2. Similarity between Anti-CarP Antibodies and ACPAs in RA
5. Citrullination and Carbamylation in Other Disease
6. Acetylation, Another Autoantibody-Inducing PTM Process
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAPAs | Anti-acetylated protein antibodies |
| Ab | Antibody |
| ACPAs | Anti-citrullinated protein/peptide antibodies |
| ACR | American College of Rheumatology |
| Ab | Antibody |
| Ag | Antigen |
| AMPAs | Anti-modified protein antibodies |
| Anti-CarP | Anti-carbamylated protein |
| Anti-MCV | Anti-mutated citrullinated vimentin antibodies |
| APC | Antigen presenting cell |
| Apo E | Apolipoprotein E |
| AutoAb | Autoantibody |
| AutoAg | Autoantigen |
| BIP | Immunoglobulin binding protein |
| BMI | Body mass index |
| CD | Cluster of differentiation |
| CD4+ T cell | Cluster of differentiation 4 positive T cell |
| CD40L | Cluster of differentiation 4 ligand |
| CEP-1 | Citrullinated enolase peptide-1 |
| c-LDL | Carbamylated LDL |
| CNS | Central nervous system |
| COPD | Chronic obstructive pulmonary disease |
| CRP | C-reactive protein |
| CTLA-4 | Cytotoxic T lymphocyte antigen 4 |
| DKK-1 | Dickkopf-1 |
| DMARDs | Disease-modifying antirheumatic drugs |
| EBV | Epstein–Barr virus |
| ECM | Extracellular matrix |
| EGF | Epidermal growth factor |
| EPO | Erythropoietin |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| ESR | Erythrocyte sedimentation rate |
| ESRD | End stage renal disease |
| EULAR | European League Against Rheumatism |
| Fc | Constant fragment |
| FLNA | Filamin A |
| FLS | Fibroblast-like synoviocyte |
| FOXP3 | Forkhead box P3 |
| FUSE-BP | Far upstream element-binding proteins |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| GNS | N-acetylglucosamine-6-sulfatase |
| GWAS | Genome-wide association studies |
| HIP-1 | Huntingtin-interacting protein-1 |
| HLA | Human leukocyte antigen |
| IBD | Inflammatory bowel disease |
| IC | Immune complex |
| IFN-γ | Interferon-γ |
| Ig | Immunoglobulin |
| IKK | Inhibitor of NF-κB kinase |
| IL | Interleukin |
| IP | Interphalangeal joint |
| IRAK1 | Interleukin-1 receptor-associated kinase 1 |
| ITP | Immune thrombocytopenia |
| IV | Intravenous |
| JAK | Janus kinase |
| JNK | c-Jun N-terminal kinase |
| LDL | Low-density lipoprotein |
| LTH | Leukotoxic hypercitrullination |
| LtxA | Leukotoxin A |
| MAC | Membrane attack complex |
| MAPKs | Mitogen-activated protein kinases |
| MCP | Metacarpophalangeal joint |
| MHC | Major histocompatibility complex |
| MMP | Matrix metalloproteinase |
| MNDA | Myeloid nuclear differentiation antigen |
| MPO | Myeloperoxidase |
| MS | Multiple sclerosis |
| MTP | Metatarsophalangeal joint |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NET | Neutrophil extracellular trap |
| NF | Nuclear factor |
| NTZ | Nitazoxanide |
| OA | Osteoarthritis |
| OB | Osteoblast |
| OC | Osteoclast |
| PAD | Peptidylarginine deiminase |
| pI | Iso-electric point |
| PIP | Proximal interphalangeal joint |
| PM | Particulate matter |
| PNS | Peripheral nervous system |
| PO | per os (orally) |
| PPAD | PAD of Porphyromonas gingivalis |
| PTM | Posttranslational modification |
| PTPN22 | Protein tyrosine phosphatase N22 |
| PVL | Panton–Valentine leukocidin |
| RA | Rheumatoid arthritis |
| RA-SF | Synovial fluid of rheumatoid arthritis patients |
| RF | Rheumatoid factor |
| SC | Subcutaneous |
| SE | Shared epitope |
| SF | Synovial fluid |
| SLE | Systemic lupus erythematosus |
| SLO | Streptolysin O |
| SNP | Single-nucleotide polymorphism |
| STAT | Signal transducers and activators of transcription |
| STEMI | ST-segment elevation myocardial infarction |
| Syk | Spleen tyrosine kinase |
| TCR | T cell receptor |
| Teff cell | Effector T cells |
| TGF-β | Transforming growth factor-β |
| TH1 cell | Helper T1 cell |
| TH17 cell | Helper T17 cell |
| TLR | Toll-like receptor |
| TNF | Tumor necrosis factor |
| TRAF1-C5 | TNF receptor associated factor 1-C5 |
| TReg cell | Regulatory T cell |
| Tyk | Tyrosine kinase |
| ULN | Upper limit of normal |
| FDA | US Food and Drug Administration |
References
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testa, D.; Calvacchi, S.; Petrelli, F.; Giannini, D.; Bilia, S.; Alunno, A.; Puxeddu, I. One year in review 2021: Pathogenesis of rheumatoid arthritis. Clin. Exp. Rheumatol. 2021, 39, 445–452. [Google Scholar] [PubMed]
- Shah, A.; Clair, E.W. Rheumatoid Arthritis. In Harrison’s Principles of Internal Medicine, 20e; Jameson, J.L., Fauci, A.S., Kasper, D.L., Hauser, S.L., Longo, D.L., Loscalzo, J., Eds.; McGraw-Hill Education: New York, NY, USA, 2018. [Google Scholar]
- England, B.R.; Hershberger, D. Management issues in rheumatoid arthritis-associated interstitial lung disease. Curr. Opin. Rheumatol. 2020, 32, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Conforti, A.; Di Cola, I.; Pavlych, V.; Ruscitti, P.; Berardicurti, O.; Ursini, F.; Giacomelli, R.; Cipriani, P. Beyond the joints, the extra-articular manifestations in rheumatoid arthritis. Autoimmun. Rev. 2020, 20, 102735. [Google Scholar] [CrossRef] [PubMed]
- Grassi, W.; De Angelis, R.; Lamanna, G.; Cervini, C. The clinical features of rheumatoid arthritis. Eur. J. Radiol. 1998, 27, S18–S24. [Google Scholar] [CrossRef]
- Lin, Y.-J.; Anzaghe, M.; Schülke, S. Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Arthritis. Cells 2020, 9, 880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imagama, T.; Tokushige, A.; Seki, K.; Taguchi, T. Weight Bearing Joints Destruction In Rheumatoid Arthritis. Curr. Rheumatol. Rev. 2017, 13, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Nell, V.P.K.; Machold, K.; Eberl, G.; Stamm, T.A.; Uffmann, M.; Smolen, J.S. Benefit of very early referral and very early therapy with disease-modifying anti-rheumatic drugs in patients with early rheumatoid arthritis. Rheumatology 2004, 43, 906–914. [Google Scholar] [CrossRef] [Green Version]
- Kay, J.; Upchurch, K.S. ACR/EULAR 2010 rheumatoid arthritis classification criteria. Rheumatology 2012, 51, vi5–vi9. [Google Scholar] [CrossRef] [Green Version]
- Alex, A.M.; Kunkel, G.; Sayles, H.; Arcos, J.D.F.; Mikuls, T.R.; Kerr, G.S. Exposure to ambient air pollution and autoantibody status in rheumatoid arthritis. Clin. Rheumatol. 2019, 39, 761–768. [Google Scholar] [CrossRef]
- Nemtsova, M.V.; Zaletaev, D.; Bure, I.V.; Mikhaylenko, D.; Kuznetsova, E.B.; Alekseeva, E.A.; Beloukhova, M.; Deviatkin, A.A.; Lukashev, A.N.; Zamyatnin, A. Epigenetic Changes in the Pathogenesis of Rheumatoid Arthritis. Front. Genet. 2019, 10, 570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, T.; Aune, D.; Heath, A.K. Adiposity and the risk of rheumatoid arthritis: A systematic review and meta-analysis of cohort studies. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.L.; Wolfe, F.; Huizinga, T.W. Rheumatoid arthritis. Lancet 2010, 376, 1094–1108. [Google Scholar] [CrossRef]
- Alamanos, Y.; Voulgari, P.V.; Drosos, A.A. Incidence and Prevalence of Rheumatoid Arthritis, Based on the 1987 American College of Rheumatology Criteria: A Systematic Review. Semin. Arthritis Rheum. 2006, 36, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, J.D.; Spruill, T.M.; Shan, Y.; Reed, G.; Kremer, J.M.; Potter, J.; Yazici, Y.; Ogedegbe, G.; Harrold, L.R. Racial and Ethnic Disparities in Disease Activity in Patients with Rheumatoid Arthritis. Am. J. Med. 2013, 126, 1089–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobon, G.; Youinou, P.; Saraux, A. The environment, geo-epidemiology, and autoimmune disease: Rheumatoid arthritis. J. Autoimmun. 2010, 35, 10–14. [Google Scholar] [CrossRef]
- Hughes, L.B.; Beasley, T.M.; Patel, H.; Tiwari, H.K.; Morgan, S.L.; Baggott, J.E.; Saag, K.G.; McNicholl, J.; Moreland, L.W.; Alarcon, G.S.; et al. Racial or ethnic differences in allele frequencies of single-nucleotide polymorphisms in the methylenetetrahydrofolate reductase gene and their influence on response to methotrexate in rheumatoid arthritis. Ann. Rheum. Dis. 2006, 65, 1213–1218. [Google Scholar] [CrossRef] [Green Version]
- del Rincón, I.; Battafarano, D.F.; Arroyo, R.A.; Murphy, F.T.; Fischbach, M.; Escalante, A. Ethnic variation in the clinical manifestations of rheumatoid arthritis: Role of HLA–DRB1 alleles. Arthritis Rheum. 2003, 49, 200–208. [Google Scholar] [CrossRef]
- Viatte, S.; Plant, D.; Raychaudhuri, S. Genetics and epigenetics of rheumatoid arthritis. Nat. Rev. Rheumatol. 2013, 9, 141–153. [Google Scholar] [CrossRef]
- Diaz-Gallo, L.-M.; Ramsköld, D.; Shchetynsky, K.; Folkersen, L.; Chemin, K.; Brynedal, B.; Uebe, S.; Okada, Y.; Alfredsson, L.; Klareskog, L.; et al. Systematic approach demonstrates enrichment of multiple interactions between non-HLA risk variants and HLA-DRB1 risk alleles in rheumatoid arthritis. Ann. Rheum. Dis. 2018, 77, 1454–1462. [Google Scholar] [CrossRef] [Green Version]
- Raychaudhuri, S.; Sandor, C.; Stahl, E.A.; Freudenberg, J.; Lee, H.-S.; Jia, X.; Alfredsson, L.; Padyukov, L.; Klareskog, L.; Worthington, J.; et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet. 2012, 44, 291–296. [Google Scholar] [CrossRef]
- Zhao, M.; Mauer, L.; Sayles, H.; Cannon, G.W.; Reimold, A.; Kerr, G.S.; Baker, J.F.; Thiele, G.M.; England, B.R.; Mikuls, T.R. HLA-DRB1 Haplotypes, Shared Epitope, and Disease Outcomes in US Veterans with Rheumatoid Arthritis. J. Rheumatol. 2019, 46, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Dedmon, L.E. The genetics of rheumatoid arthritis. Rheumatology 2020, 59, 2661–2670. [Google Scholar] [CrossRef] [PubMed]
- Hiwa, R.; Ikari, K.; Ohmura, K.; Nakabo, S.; Matsuo, K.; Saji, H.; Yurugi, K.; Miura, Y.; Maekawa, T.; Taniguchi, A.; et al. HLA-DRB1 Analysis Identified a Genetically Unique Subset within Rheumatoid Arthritis and Distinct Genetic Background of Rheumatoid Factor Levels from Anticyclic Citrullinated Peptide Antibodies. J. Rheumatol. 2018, 45, 470–480. [Google Scholar] [CrossRef] [PubMed]
- O’Dell, J.R.; Nepom, B.S.; Haire, C.; Gersuk, V.H.; Gaur, L.; Moore, G.F.; Drymalski, W.; Palmer, W.; Eckhoff, P.J.; Klassen, L.W.; et al. HLA-DRB1 typing in rheumatoid arthritis: Predicting response to specific treatments. Ann. Rheum. Dis. 1998, 57, 209–213. [Google Scholar] [CrossRef] [Green Version]
- Abbasifard, M.; Imani, D.; Bagheri-Hosseinabadi, Z. PTPN22 gene polymorphism and susceptibility to rheumatoid arthritis (RA): Updated systematic review and meta-analysis. J. Gene Med. 2020, 22, e3204. [Google Scholar] [CrossRef]
- Rizvi, S.T.F.; Arif, A.; Azhar, A. TNF gene promoter region polymorphisms and association with young-onset rheumatoid arthritis. Pak. J. Pharm. Sci. 2019, 32, 2295–2297. [Google Scholar]
- Potter, C.; Eyre, S.; Cope, A.; Worthington, J.; Barton, A. Investigation of association between the TRAF family genes and RA susceptibility. Ann. Rheum. Dis. 2007, 66, 1322–1326. [Google Scholar] [CrossRef]
- Vossenaar, E.R.; Zendman, A.J.; van Venrooij, W.J.; Pruijn, G.J. PAD, a growing family of citrullinating enzymes: Genes, features and involvement in disease. BioEssays 2003, 25, 1106–1118. [Google Scholar] [CrossRef]
- Feitsma, A.L.; Toes, R.E.; Begovich, A.B.; Chokkalingam, A.P.; De Vries, R.R.P.; Huizinga, T.W.J.; van der Helm-van Mil, A.H. Risk of progression from undifferentiated arthritis to rheumatoid arthritis: The effect of the PTPN22 1858T-allele in anti-citrullinated peptide antibody positive patients. Rheumatology 2007, 46, 1092–1095. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.H.; Cho, C.L.; Tsai, W.C.; Ou, T.T.; Wu, C.C.; Yen, J.H.; Liu, H.W. Inhibitors of kB-like gene polymorphisms in rheumatoid arthritis. Immunol. Lett. 2006, 105, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Croia, C.; Bursi, R.; Sutera, D.; Petrelli, F.; Alunno, A.; Puxeddu, I. One year in review 2019: Pathogenesis of rheumatoid arthritis. Clin. Exp. Rheumatol. 2019, 37, 347–357. [Google Scholar]
- Giannini, D.; Antonucci, M.; Petrelli, F.; Bilia, S.; Alunno, A.; Puxeddu, I. One year in review 2020: Pathogenesis of rheumatoid arthritis. Clin. Exp. Rheumatol. 2020, 38, 387–397. [Google Scholar]
- Kwon, Y.-C.; Lim, J.; Bang, S.-Y.; Ha, E.; Hwang, M.Y.; Yoon, K.; Choe, J.-Y.; Yoo, D.-H.; Lee, S.-S.; Lee, J.; et al. Genome-wide association study in a Korean population identifies six novel susceptibility loci for rheumatoid arthritis. Ann. Rheum. Dis. 2020, 79, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Leng, R.-X.; Di, D.-S.; Ni, J.; Wu, X.-X.; Zhang, L.-L.; Wang, X.-F.; Liu, R.-S.; Huang, Q.; Fan, Y.-G.; Pan, H.-F.; et al. Identification of new susceptibility loci associated with rheumatoid arthritis. Ann. Rheum. Dis. 2020, 79, 1565–1571. [Google Scholar] [CrossRef] [PubMed]
- Alsalahy, M.M.; Nasser, H.S.; Hashem, M.M.; Elsayed, S.M. Effect of tobacco smoking on tissue protein citrullination and disease progression in patients with rheumatoid arthritis. Saudi Pharm. J. 2010, 18, 75–80. [Google Scholar] [CrossRef] [Green Version]
- Klareskog, L.; Stolt, P.; Lundberg, K.; Källberg, H.; Bengtsson, C.; Grunewald, J.; Rönnelid, J.; Harris, H.E.; Ulfgren, A.-K.; Rantapää-Dahlqvist, S.; et al. A new model for an etiology of rheumatoid arthritis: Smoking may trigger HLA–DR (shared epitope)–restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 2005, 54, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Hedström, A.K.; Rönnelid, J.; Klareskog, L.; Alfredsson, L. Complex Relationships of Smoking, HLA-DRB1 Genes, and Serologic Profiles in Patients With Early Rheumatoid Arthritis: Update From a Swedish Population-Based Case-Control Study. Arthritis Rheumatol. 2019, 71, 1504–1511. [Google Scholar] [CrossRef] [Green Version]
- Balandraud, N.; Roudier, J. Epstein-Barr virus and rheumatoid arthritis. Jt. Bone Spine 2018, 85, 165–170. [Google Scholar] [CrossRef]
- Silman, A.J.; Pearson, J.E. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res. 2002, 4 (Suppl. 3), S265–S272. [Google Scholar] [CrossRef] [Green Version]
- Newkirk, M.M.; Goldbach-Mansky, R.; Senior, B.W.; Klippel, J.; Schumacher, H.R.; El-Gabalawy, H.S. Elevated levels of IgM and IgA antibodies to Proteus mirabilis and IgM antibodies to Escherichia coli are associated with early rheumatoid factor (RF)-positive rheumatoid arthritis. Rheumatology 2005, 44, 1433–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Smit, M.J.; Brouwer, E.; Vissink, A.; van Winkelhoff, A.J. Rheumatoid arthritis and periodontitis; a possible link via citrullination. Anaerobe 2011, 17, 196–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maresz, K.J.; Hellvard, A.; Sroka, A.; Adamowicz, K.; Bielecka, E.; Koziel, J.; Gawron, K.; Mizgalska, D.; Marcinska, K.A.; Benedyk, M.; et al. Porphyromonas gingivalis Facilitates the Development and Progression of Destructive Arthritis through Its Unique Bacterial Peptidylarginine Deiminase (PAD). PLOS Pathog. 2013, 9, e1003627. [Google Scholar] [CrossRef] [PubMed]
- Pianta, A.; Arvikar, S.L.; Strle, K.; Drouin, E.E.; Wang, Q.; Costello, C.E.; Steere, A.C. Two rheumatoid arthritis–specific autoantigens correlate microbial immunity with autoimmune responses in joints. J. Clin. Investig. 2017, 127, 2946–2956. [Google Scholar] [CrossRef]
- Valesini, G.; Gerardi, M.C.; Iannuccelli, C.; Pacucci, V.A.; Pendolino, M.; Shoenfeld, Y. Citrullination and autoimmunity. Autoimmun. Rev. 2015, 14, 490–497. [Google Scholar] [CrossRef]
- Shoda, H. Citrullination and rheumatoid arthritis. Nihon Rinsho. Jpn. J. Clin. Med. 2016, 74, 902–906. [Google Scholar]
- Andrade, F.; Darrah, E.; Gucek, M.; Cole, R.N.; Rosen, A.; Zhu, X. Autocitrullination of human peptidyl arginine deiminase type 4 regulates protein citrullination during cell activation. Arthritis Rheum. 2010, 62, 1630–1640. [Google Scholar] [CrossRef] [Green Version]
- Kurowska, W.; Kuca-Warnawin, E.H.; Radzikowska, A.; Maśliński, W. The role of anti-citrullinated protein antibodies (ACPA) in the pathogenesis of rheumatoid arthritis. Cent. Eur. J. Immunol. 2017, 42, 390–398. [Google Scholar] [CrossRef]
- Cascone, P.; Vetrano, S.; Nicolai, G.; Fabiani, F. Temporomandibular joint biomechanical restrictions: The fluid and synovial membrane. J. Craniofac. Surg. 1999, 10, 301–307. [Google Scholar] [CrossRef]
- Barland, P.; Novikoff, A.B.; Hamerman, D. Electron microscopy of the human synovial membrane. J. Cell Biol. 1962, 14, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Tang, Y.; Song, B.; Yu, M.; Li, Q.; Zhang, C.; Hou, J.; Yang, R. Nomenclature clarification: Synovial fibroblasts and synovial mesenchymal stem cells. Stem Cell Res. Ther. 2019, 10, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falconer, J.; Murphy, A.N.; Young, S.P.; Clark, A.R.; Tiziani, S.; Guma, M.; Buckley, C.D. Review: Synovial Cell Metabolism and Chronic Inflammation in Rheumatoid Arthritis. Arthritis Rheumatol. 2018, 70, 984–999. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, M.F.; Garcia-Carbonell, R.; Whisenant, K.D.; Guma, M. Fibroblast-like synoviocyte metabolism in the pathogenesis of rheumatoid arthritis. Arthritis Res. 2017, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhu, Y.; Gao, Y.; Qi, D.; Zhao, L.; Zhao, L.; Liu, C.; Tao, T.; Zhou, C.; Sun, X.; et al. NR1D1 modulates synovial inflammation and bone destruction in rheumatoid arthritis. Cell Death Dis. 2020, 11, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, A.K.; Li, Y.; Dang, Q.; Yang, F. Monocytes in rheumatoid arthritis: Circulating precursors of macrophages and osteoclasts and, their heterogeneity and plasticity role in RA pathogenesis. Int. Immunopharmacol. 2018, 65, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-J.; Yoo, S.-A.; Kim, W.-U. Role of Endoplasmic Reticulum Stress in Rheumatoid Arthritis Pathogenesis. J. Korean Med Sci. 2014, 29, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Firestein, G.S.; Echeverri, F.; Yeo, M.; Zvaifler, N.J.; Green, D. Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc. Natl. Acad. Sci. USA 1997, 94, 10895–10900. [Google Scholar] [CrossRef] [Green Version]
- Kullmann, F.; Judex, M.; Neudecker, I.; Lechner, S.; Jüsten, H.P.; Green, D.; Wessinghage, D.; Firestein, G.S.; Gay, S.; Schölmerich, J.; et al. Analysis of the p53 tumor suppressor gene in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 1999, 42, 1594–1600. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Boyle, D.L.; Green, D.R.; Keystone, E.C.; Connor, A.; Zollman, S.; Firestein, G.S. p53 tumor suppressor gene mutations in fibroblast-like synoviocytes from erosion synovium and non-erosion synovium in rheumatoid arthritis. Arthritis Res. Ther. 2005, 7, R12–R18. [Google Scholar] [CrossRef] [Green Version]
- Baier, A.; Meineckel, I.; Gay, S.; Pap, T. Apoptosis in rheumatoid arthritis. Curr. Opin. Rheumatol. 2003, 15, 274–279. [Google Scholar] [CrossRef]
- Korb, A.; Pavenstädt, H.; Pap, T. Cell death in rheumatoid arthritis. Apoptosis 2009, 14, 447–454. [Google Scholar] [CrossRef]
- Vorobjeva, N.V.; Chernyak, B.V. NETosis: Molecular Mechanisms, Role in Physiology and Pathology. Biochemistry 2020, 85, 1178–1190. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2011, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Cahilog, Z.; Zhao, H.; Wu, L.; Alam, A.; Eguchi, S.; Weng, H.; Ma, D. The Role of Neutrophil NETosis in Organ Injury: Novel Inflammatory Cell Death Mechanisms. Inflammation 2020, 43, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Konig, M.F.; Andrade, F. A Critical Reappraisal of Neutrophil Extracellular Traps and NETosis Mimics Based on Differential Requirements for Protein Citrullination. Front. Immunol. 2016, 7, 461. [Google Scholar] [CrossRef] [Green Version]
- Darrah, E.; Andrade, F. Rheumatoid arthritis and citrullination. Curr. Opin. Rheumatol. 2018, 30, 72–78. [Google Scholar] [CrossRef]
- Roche, P.A.; Furuta, K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat. Rev. Immunol. 2015, 15, 203–216. [Google Scholar] [CrossRef]
- Yang, W.; Chen, X.; Hu, H. CD4+ T-Cell Differentiation In Vitro. Methods Mol. Biol. 2020, 2111, 91–99. [Google Scholar] [CrossRef]
- Ville, S.; Poirier, N.; Blancho, G.; Vanhove, B. Co-Stimulatory Blockade of the CD28/CD80-86/CTLA-4 Balance in Transplantation: Impact on Memory T Cells? Front. Immunol. 2015, 6, 411. [Google Scholar] [CrossRef] [Green Version]
- Korhonen, R.; Moilanen, E. Abatacept, a novel CD80/86-CD28 T cell co-stimulation modulator, in the treatment of rheumatoid arthritis. Basic Clin. Pharmacol. Toxicol. 2009, 104, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Kamali, A.N.; Noorbakhsh, S.M.; Hamedifar, H.; Jadidi-Niaragh, F.; Yazdani, R.; Bautista, J.M.; Azizi, G. A role for Th1-like Th17 cells in the pathogenesis of inflammatory and autoimmune disorders. Mol. Immunol. 2018, 105, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Samuels, J.; Ng, Y.-S.; Coupillaud, C.; Paget, D.; Meffre, E. Human B Cell Tolerance and Its Failure in Rheumatoid Arthritis. Ann. N. Y. Acad. Sci. 2005, 1062, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Calero, I.; Nieto, J.A.; Sanz, I. B Cell Therapies for Rheumatoid Arthritis: Beyond B cell Depletion. Rheum. Dis. Clin. N. Am. 2010, 36, 325–343. [Google Scholar] [CrossRef] [PubMed]
- Weyand, C.M.; Goronzy, J.J. The immunology of rheumatoid arthritis. Nat. Immunol. 2020, 22, 10–18. [Google Scholar] [CrossRef]
- Bartok, B.; Firestein, G.S. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol. Rev. 2009, 233, 233–255. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Kwon, J.-E.; Lee, S.-Y.; Lee, E.-J.; Kim, D.S.; Moon, S.-J.; Lee, J.; Kwok, S.-K.; Park, S.-H.; Cho, M.-L. IL-17-mediated mitochondrial dysfunction impairs apoptosis in rheumatoid arthritis synovial fibroblasts through activation of autophagy. Cell Death Dis. 2017, 8, e2565. [Google Scholar] [CrossRef] [PubMed]
- Lubberts, E. The IL-23–IL-17 axis in inflammatory arthritis. Nat. Rev. Rheumatol. 2015, 11, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Pfeifle, R.; Rothe, T.; Ipseiz, N.; Scherer, H.U.; Culemann, S.; Harre, U.; Ackermann, J.A.; Seefried, M.; Kleyer, A.; Uderhardt, S.; et al. Regulation of autoantibody activity by the IL-23-T(H)17 axis determines the onset of autoimmune disease. Nat. Immunol. 2017, 18, 104–113. [Google Scholar] [CrossRef]
- Bazzazi, H.; Aghaei, M.; Memarian, A.; Asgarian-Omran, H.; Behnampour, N.; Yazdani, Y. Th1-Th17 Ratio as a New Insight in Rheumatoid Arthritis Disease. Iran. J. Allergy Asthma Immunol. 2018, 17, 68–77. [Google Scholar]
- Malemud, C.J. The role of the JAK/STAT signal pathway in rheumatoid arthritis. Ther. Adv. Musculoskelet. Dis. 2018, 10, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Massalska, M.; Maslinski, W.; Ciechomska, M. Small Molecule Inhibitors in the Treatment of Rheumatoid Arthritis and Beyond: Latest Updates and Potential Strategy for Fighting COVID-19. Cells 2020, 9, 1876. [Google Scholar] [CrossRef] [PubMed]
- Fraenkel, L.; Bathon, J.M.; England, B.R.; St. Clair, E.W.; Arayssi, T.; Carandang, K.; Deane, K.D.; Genovese, M.; Huston, K.K.; Kerr, G.; et al. 2021 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Rheumatol. 2021, 73, 1108–1123. [Google Scholar] [CrossRef] [PubMed]
- Kunwar, S.; Devkota, A.R.; Ghimire, D.K.C. Fostamatinib, an oral spleen tyrosine kinase inhibitor, in the treatment of rheumatoid arthritis: A meta-analysis of randomized controlled trials. Rheumatol. Int. 2016, 36, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Jiang, X.; Qin, D.; Wang, L.; Yang, J.; Wu, A.; Huang, F.; Ye, Y.; Wu, J. Efficacy and Safety of Multiple Dosages of Fostamatinib in Adult Patients With Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Front. Pharmacol. 2019, 10, 897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Millson, D.; Iwata, S.; Nakayamada, S. Safety and efficacy of fostamatinib in rheumatoid arthritis patients with an inadequate response to methotrexate in phase II OSKIRA-ASIA-1 and OSKIRA-ASIA-1X study. Rheumatology 2020, 60, 2884–2895. [Google Scholar] [CrossRef] [PubMed]
- Malemud, C.J. Defective T-Cell Apoptosis and T-Regulatory Cell Dysfunction in Rheumatoid Arthritis. Cells 2018, 7, 223. [Google Scholar] [CrossRef] [Green Version]
- Flores-Borja, F.; Jury, E.C.; Mauri, C.; Ehrenstein, M.R. Defects in CTLA-4 are associated with abnormal regulatory T cell function in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2008, 105, 19396–19401. [Google Scholar] [CrossRef] [Green Version]
- Radner, H.; Aletaha, D. Anti-TNF in rheumatoid arthritis: An overview. Wien. Med. Wochenschr. 2015, 165, 3–9. [Google Scholar] [CrossRef]
- Dai, Y.; Ding, J.; Yin, W.; He, Y.; Yu, F.; Ye, C.; Hu, S.; Yu, Y. Increased Autophagy Enhances the Resistance to Tumor Necrosis Factor-Alpha Treatment in Rheumatoid Arthritis Human Fibroblast-Like Synovial Cell. BioMed Res. Int. 2018, 2018, 4941027. [Google Scholar] [CrossRef]
- Noack, M.; Miossec, P. Selected cytokine pathways in rheumatoid arthritis. Semin. Immunopathol. 2017, 39, 365–383. [Google Scholar] [CrossRef] [PubMed]
- Kaneshiro, K.; Sakai, Y.; Suzuki, K.; Uchida, K.; Tateishi, K.; Terashima, Y.; Kawasaki, Y.; Shibanuma, N.; Yoshida, K.; Hashiramoto, A. Interleukin-6 and tumour necrosis factor-α cooperatively promote cell cycle regulators and proliferate rheumatoid arthritis fibroblast-like synovial cells. Scand. J. Rheumatol. 2019, 48, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Cici, D.; Corrado, A.; Rotondo, C.; Cantatore, F.P. Wnt Signaling and Biological Therapy in Rheumatoid Arthritis and Spondyloarthritis. Int. J. Mol. Sci. 2019, 20, 5552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marahleh, A.; Kitaura, H.; Ohori, F.; Kishikawa, A.; Ogawa, S.; Shen, W.-R.; Qi, J.; Noguchi, T.; Nara, Y.; Mizoguchi, I. TNF-α Directly Enhances Osteocyte RANKL Expression and Promotes Osteoclast Formation. Front. Immunol. 2019, 10, 2925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitaura, H.; Marahleh, A.; Ohori, F.; Noguchi, T.; Shen, W.-R.; Qi, J.; Nara, Y.; Pramusita, A.; Kinjo, R.; Mizoguchi, I. Osteocyte-Related Cytokines Regulate Osteoclast Formation and Bone Resorption. Int. J. Mol. Sci. 2020, 21, 5169. [Google Scholar] [CrossRef]
- Wang, T.; He, C. TNF-α and IL-6: The Link between Immune and Bone System. Curr. Drug Targets 2020, 21, 213–227. [Google Scholar] [CrossRef]
- Ai, R.; Laragione, T.; Hammaker, D.; Boyle, D.L.; Wildberg, A.; Maeshima, K.; Palescandolo, E.; Krishna, V.; Pocalyko, D.; Whitaker, J.W.; et al. Comprehensive epigenetic landscape of rheumatoid arthritis fibroblast-like synoviocytes. Nat. Commun. 2018, 9, 1921. [Google Scholar] [CrossRef]
- Laragione, T.; Brenner, M.; Lahiri, A.; Gao, E.; Harris, C.; Gulko, P.S. Huntingtin-interacting protein 1 (HIP1) regulates arthritis severity and synovial fibroblast invasiveness by altering PDGFR and Rac1 signalling. Ann. Rheum. Dis. 2018, 77, 1627–1635. [Google Scholar] [CrossRef]
- Di, D.; Zhang, L.; Wu, X.; Leng, R. Long-term exposure to outdoor air pollution and the risk of development of rheumatoid arthritis: A systematic review and meta-analysis. Semin. Arthritis Rheum. 2019, 50, 266–275. [Google Scholar] [CrossRef]
- Chu, X.-J.; Cao, N.-W.; Zhou, H.-Y.; Meng, X.; Guo, B.; Zhang, H.-Y.; Li, B.-Z. The oral and gut microbiome in rheumatoid arthritis patients: A systematic review. Rheumatology 2020, 60, 1054–1066. [Google Scholar] [CrossRef]
- Ferro, M.; Charneca, S.; Dourado, E.; Guerreiro, C.S.; Fonseca, J.E. Probiotic Supplementation for Rheumatoid Arthritis: A Promising Adjuvant Therapy in the Gut Microbiome Era. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef]
- Kishikawa, T.; Maeda, Y.; Nii, T.; Motooka, D.; Matsumoto, Y.; Matsushita, M.; Matsuoka, H.; Yoshimura, M.; Kawada, S.; Teshigawara, S.; et al. Metagenome-wide association study of gut microbiome revealed novel aetiology of rheumatoid arthritis in the Japanese population. Ann. Rheum. Dis. 2019, 79, 103–111. [Google Scholar] [CrossRef]
- Yao, Y.; Cai, X.; Fei, W.; Ren, F.; Wang, F.; Luan, X.; Chen, F.; Zheng, C. Regulating Gut Microbiome: Therapeutic Strategy for Rheumatoid Arthritis During Pregnancy and Lactation. Front. Pharmacol. 2020, 11, 594042. [Google Scholar] [CrossRef]
- György, B.; Tóth, E.; Tarcsa, E.; Falus, A.; Buzás, E.I. Citrullination: A posttranslational modification in health and disease. Int. J. Biochem. Cell Biol. 2006, 38, 1662–1677. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, M.; Takahara, H.; Nishi, Y.; Sugawara, K.; Sato, C. Ca2+-dependent deimination-induced disassembly of intermediate filaments involves specific modification of the amino-terminal head domain. J. Biol. Chem. 1989, 264, 18119–18127. [Google Scholar] [CrossRef]
- Orgován, G.; Noszál, B. The complete microspeciation of arginine and citrulline. J. Pharm. Biomed. Anal. 2011, 54, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Marín, E.; Paz-Miguel, J.E.; López-Mato, P.; Alvarez-Domínguez, C.; Leyva-Cobián, F. Oxidation of defined antigens allows protein unfolding and increases both proteolytic processing and exposes peptide epitopes which are recognized by specific T cells. Immunology 1998, 95, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Proost, P.; Loos, T.; Mortier, A.; Schutyser, E.; Gouwy, M.; Noppen, S.; Dillen, C.; Ronsse, I.; Conings, R.; Struyf, S.; et al. Citrullination of CXCL8 by peptidylarginine deiminase alters receptor usage, prevents proteolysis, and dampens tissue inflammation. J. Exp. Med. 2008, 205, 2085–2097. [Google Scholar] [CrossRef] [Green Version]
- Tarcsa, E.; Marekov, L.N.; Mei, G.; Melino, G.; Lee, S.C.; Steinert, P.M. Protein unfolding by peptidylarginine deiminase. Substrate specificity and structural relationships of the natural substrates trichohyalin and filaggrin. J. Biol. Chem. 1996, 271, 30709–30716. [Google Scholar] [CrossRef] [Green Version]
- Romero, V.; Fert-Bober, J.; Nigrovic, P.A.; Darrah, E.; Haque, U.J.; Lee, D.M.; Van Eyk, J.; Rosen, A.; Andrade, F. Immune-Mediated Pore-Forming Pathways Induce Cellular Hypercitrullination and Generate Citrullinated Autoantigens in Rheumatoid Arthritis. Sci. Transl. Med. 2013, 5, 209ra150. [Google Scholar] [CrossRef] [Green Version]
- Ting, Y.T.; Petersen, J.; Ramarathinam, S.H.; Scally, S.W.; Loh, K.L.; Thomas, R.; Suri, A.; Baker, D.G.; Purcell, A.W.; Reid, H.H.; et al. The interplay between citrullination and HLA-DRB1 polymorphism in shaping peptide binding hierarchies in rheumatoid arthritis. J. Biol. Chem. 2018, 293, 3236–3251. [Google Scholar] [CrossRef] [Green Version]
- Bicker, K.L.; Thompson, P.R. The protein arginine deiminases: Structure, function, inhibition, and disease. Biopolymers 2012, 99, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Ottaviano, F.G.; Handy, D.E.; Loscalzo, J. Redox Regulation in the Extracellular Environment. Circ. J. 2008, 72, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Cordova, K.N.; Willis, V.C.; Haskins, K.; Holers, V.M. A Citrullinated Fibrinogen-Specific T Cell Line Enhances Autoimmune Arthritis in a Mouse Model of Rheumatoid Arthritis. J. Immunol. 2013, 190, 1457–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brentville, V.A.; Vankemmelbeke, M.; Metheringham, R.L.; Durrant, L.G. Post-translational modifications such as citrullination are excellent targets for cancer therapy. Semin. Immunol. 2020, 47, 101393. [Google Scholar] [CrossRef] [PubMed]
- James, E.A.; Rieck, M.; Pieper, J.; Gebe, J.A.; Yue, B.B.; Tatum, M.; Peda, M.; Sandin, C.; Klareskog, L.; Malmström, V.; et al. Citrulline-specific Th1 cells are increased in rheumatoid arthritis and their frequency is influenced by disease duration and therapy. Arthritis Rheumatol. 2014, 66, 1712–1722. [Google Scholar] [CrossRef] [PubMed]
- Cianciotti, B.C.; Ruggiero, E.; Campochiaro, C.; Oliveira, G.; Magnani, Z.I.; Baldini, M.; Doglio, M.; Tassara, M.; Manfredi, A.A.; Baldissera, E.; et al. CD4+ Memory Stem T Cells Recognizing Citrullinated Epitopes Are Expanded in Patients With Rheumatoid Arthritis and Sensitive to Tumor Necrosis Factor Blockade. Arthritis Rheumatol. 2019, 72, 565–575. [Google Scholar] [CrossRef]
- Lundberg, K.; Kinloch, A.; Fisher, B.A.; Wegner, N.; Wait, R.; Charles, P.; Mikuls, T.R.; Venables, P.J. Antibodies to citrullinated α-enolase peptide 1 are specific for rheumatoid arthritis and cross-react with bacterial enolase. Arthritis Rheum. 2008, 58, 3009–3019. [Google Scholar] [CrossRef]
- Makrygiannakis, D.; Revu, S.; Engström, M.; Klint, E.A.; Nicholas, A.P.; Pruijn, G.J.; Catrina, A.I. Local administration of glucocorticoids decreases synovial citrullination in rheumatoid arthritis. Arthritis Res. Ther. 2012, 14, R20. [Google Scholar] [CrossRef] [Green Version]
- Fisher, B.A.; Venables, P.J. Inhibiting citrullination in rheumatoid arthritis: Taking fuel from the fire. Arthritis Res. Ther. 2012, 14, 108. [Google Scholar] [CrossRef] [Green Version]
- Milara, J.; Peiró, T.; Serrano, A.; Cortijo, J. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax 2013, 68, 410–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerardi, M.C.; De Luca, N.; Alessandri, C.; Iannuccelli, C.; Valesini, G.; Di Franco, M. Frequency of Antibodies to Mutated Citrullinated Vimentin in Chronic Obstructive Pulmonary Disease: Comment on the Article by Demoruelle et al. Arthritis Rheum. 2013, 65, 1672–1673. [Google Scholar] [CrossRef]
- Makrygiannakis, D.; Hermansson, M.; Ulfgren, A.-K.; Nicholas, A.P.; Zendman, A.J.W.; Eklund, A.; Grunewald, J.; Skold, C.M.; Klareskog, L.; Catrina, A.I. Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in BAL cells. Ann. Rheum. Dis. 2008, 67, 1488–1492. [Google Scholar] [CrossRef] [PubMed]
- Damgaard, D.; Nielsen, M.F.B.; Gaunsbaek, M.Q.; Palarasah, Y.; Svane-Knudsen, V.; Nielsen, C.H. Smoking is associated with increased levels of extracellular peptidylarginine deiminase 2 (PAD2) in the lungs. Clin. Exp. Rheumatol. 2015, 33, 405–408. [Google Scholar] [PubMed]
- Odoardi, F.; Sie, C.; Streyl, K.; Ulaganathan, V.K.; Schläger, C.; Lodygin, D.; Heckelsmiller, K.; Nietfeld, W.; Ellwart, J.; Klinkert, W.E.F.; et al. T cells become licensed in the lung to enter the central nervous system. Nature 2012, 488, 675–679. [Google Scholar] [CrossRef]
- McGraw, W.T.; Potempa, J.; Farley, D.; Travis, J. Purification, Characterization, and Sequence Analysis of a Potential Virulence Factor from Porphyromonas gingivalis, Peptidylarginine Deiminase. Infect. Immun. 1999, 67, 3248–3256. [Google Scholar] [CrossRef] [Green Version]
- Quirke, A.M.; Lugli, E.B.; Wegner, N.; Hamilton, B.C.; Charles, P.; Chowdhury, M.; Ytterberg, A.J.; Zubarev, R.A.; Potempa, J.; Culshaw, S.; et al. Heightened immune response to autocitrullinated Porphyromonas gingivalis peptidylarginine deiminase: A potential mechanism for breaching immunologic tolerance in rheumatoid arthritis. Ann. Rheum. Dis. 2014, 73, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Abdullah, S.-N.; Farmer, E.-A.; Spargo, L.; Logan, R.; Gully, N. Porphyromonas gingivalis peptidylarginine deiminase substrate specificity. Anaerobe 2013, 23, 102–108. [Google Scholar] [CrossRef]
- Olsen, I.; Singhrao, S.K.; Potempa, J. Citrullination as a plausible link to periodontitis, rheumatoid arthritis, atherosclerosis and Alzheimer’s disease. J. Oral Microbiol. 2018, 10, 1487742. [Google Scholar] [CrossRef] [Green Version]
- Ishida-Yamamoto, A.; Senshu, T.; Eady, R.A.; Takahashi, H.; Shimizu, H.; Akiyama, M.; Iizuka, H. Sequential Reorganization of Cornified Cell Keratin Filaments Involving Filaggrin-Mediated Compaction and Keratin 1 Deimination. J. Investig. Dermatol. 2002, 118, 282–287. [Google Scholar] [CrossRef] [Green Version]
- Qin, H.; Liu, X.; Li, F.; Miao, L.; Li, T.; Xu, B.; An, X.; Muth, A.; Thompson, P.R.; Coonrod, S.A.; et al. PAD1 promotes epithelial-mesenchymal transition and metastasis in triple-negative breast cancer cells by regulating MEK1-ERK1/2-MMP2 signaling. Cancer Lett. 2017, 409, 30–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, R.A. RNA polymerase II. Annu. Rev. Biochem. 1991, 60, 689–715. [Google Scholar] [CrossRef]
- Qu, Y.; Olsen, J.R.; Yuan, X.; Cheng, P.F.; Levesque, M.P.; Brokstad, K.A.; Hoffman, P.S.; Oyan, A.M.; Zhang, W.; Kalland, K.-H.; et al. Small molecule promotes β-catenin citrullination and inhibits Wnt signaling in cancer. Nat. Chem. Biol. 2017, 14, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-Y.; Wang, D.; Wilhelm, M.; Zolg, D.P.; Schmidt, T.; Schnatbaum, K.; Reimer, U.; Pontén, F.; Uhlén, M.; Hahne, H.; et al. Mining the Human Tissue Proteome for Protein Citrullination. Mol. Cell. Proteom. 2018, 17, 1378–1391. [Google Scholar] [CrossRef] [Green Version]
- Damgaard, D.; Bawadekar, M.; Senolt, L.; Stensballe, A.; Shelef, M.A.; Nielsen, C.H. Relative efficiencies of peptidylarginine deiminase 2 and 4 in generating target sites for anti-citrullinated protein antibodies in fibrinogen, alpha-enolase and histone H3. PLoS ONE 2018, 13, e0203214. [Google Scholar] [CrossRef] [PubMed]
- Rogers, G.; Winter, B.; McLaughlan, C.; Powell, B.; Nesci, T. Peptidylarginine Deiminase of the Hair Follicle: Characterization, Localization, and Function in Keratinizing Tissues. J. Investig. Dermatol. 1997, 108, 700–707. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.; Fast, W. Citrullination of Inhibitor of Growth 4 (ING4) by Peptidylarginine Deminase 4 (PAD4) Disrupts the Interaction between ING4 and p53. J. Biol. Chem. 2011, 286, 17069–17078. [Google Scholar] [CrossRef] [Green Version]
- Stadler, S.C.; Vincent, C.T.; Fedorov, V.D.; Patsialou, A.; Cherrington, B.D.; Wakshlag, J.J.; Mohanan, S.; Zee, B.M.; Zhang, X.; Garcia, B.A.; et al. Dysregulation of PAD4-mediated citrullination of nuclear GSK3β activates TGF-β signaling and induces epithelial-to-mesenchymal transition in breast cancer cells. Proc. Natl. Acad. Sci. USA 2013, 110, 11851–11856. [Google Scholar] [CrossRef] [Green Version]
- Esposito, G.; Vitale, A.M.; Leijten, F.P.J.; Strik, A.M.; Koonen-Reemst, A.M.C.B.; Yurttas, P.; Robben, T.J.A.A.; Coonrod, S.; Gossen, J.A. Peptidylarginine deiminase (PAD) 6 is essential for oocyte cytoskeletal sheet formation and female fertility. Mol. Cell. Endocrinol. 2007, 273, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Wegner, N.; Wait, R.; Sroka, A.; Eick, S.; Nguyen, K.-A.; Lundberg, K.; Kinloch, A.; Culshaw, S.; Potempa, J.; Venables, P.J. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and α-enolase: Implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2662–2672. [Google Scholar] [CrossRef]
- Konig, M.F.; Paracha, A.S.; Moni, M.; Bingham, C.O., 3rd; Andrade, F. Defining the role of Porphyromonas gingivalis peptidylarginine deiminase (PPAD) in rheumatoid arthritis through the study of PPAD biology. Ann. Rheum. Dis. 2015, 74, 2054–2061. [Google Scholar] [CrossRef] [Green Version]
- Witalison, E.E.; Thompson, P.R.; Hofseth, L.J. Protein Arginine Deiminases and Associated Citrullination: Physiological Functions and Diseases Associated with Dysregulation. Curr. Drug Targets 2015, 16, 700–710. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Y. Peptidylarginine deiminases in citrullination, gene regulation, health and pathogenesis. Biochim. Biophys. Acta Bioenerg. 2013, 1829, 1126–1135. [Google Scholar] [CrossRef] [Green Version]
- Slack, J.L.; Causey, C.P.; Thompson, P.R. Protein arginine deiminase 4: A target for an epigenetic cancer therapy. Experientia 2010, 68, 709–720. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wichapong, K.; Lamers, S.; Reutelingsperger, C.P.M.; Nicolaes, G.A.F. Autocitrullination of PAD4 does not alter its enzymatic activity: In vitro and in silico studies. Int. J. Biochem. Cell Biol. 2021, 134, 105938. [Google Scholar] [CrossRef]
- Laugisch, O.; Wong, A.; Sroka, A.; Kantyka, T.; Koziel, J.; Neuhaus, K.; Sculean, A.; Venables, P.J.; Potempa, J.; Möller, B.; et al. Citrullination in the periodontium—a possible link between periodontitis and rheumatoid arthritis. Clin. Oral Investig. 2015, 20, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Arévalo-Caro, C.; Romero-Sánchez, C.; Garavito-Rodríguez, E. Relation between anti-Porphyromonas gingivalis antibody titers and HLA-DRB1 neutral alleles in individuals with rheumatoid arthritis. Acta Odontol. Scand. 2021, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Vossenaar, E.R.; Radstake, T.R.D.; van der Heijden, A.; van Mansum, M.A.M.; Dieteren, C.; De Rooij, D.-J.; Barrera, P.; Zendman, A.J.W.; van Venrooij, W.J. Expression and activity of citrullinating peptidylarginine deiminase enzymes in monocytes and macrophages. Ann. Rheum. Dis. 2004, 63, 373–381. [Google Scholar] [CrossRef] [Green Version]
- Vossenaar, E.R.; Nijenhuis, S.; Helsen, M.M.A.; Van Der Heijden, A.; Senshu, T.; van den Berg, W.B.; Van Venrooij, W.J.; Joosten, L.A.B. Citrullination of synovial proteins in murine models of rheumatoid arthritis. Arthritis Rheum. 2003, 48, 2489–2500. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhao, Y.; He, J.; Jia, R.; Li, Z. Prevalence and significance of anti-peptidylarginine deiminase 4 antibodies in rheumatoid arthritis. J. Rheumatol. 2008, 35, 969–974. [Google Scholar] [PubMed]
- Trouw, L.A.; Huizinga, T.W.J.; Toes, R.E. Autoimmunity in rheumatoid arthritis: Different antigens—common principles. Ann. Rheum. Dis. 2012, 72, ii132–ii136. [Google Scholar] [CrossRef]
- Ge, C.; Xu, B.; Liang, B.; Lönnblom, E.; Lundström, S.L.; Zubarev, R.A.; Ayoglu, B.; Nilsson, P.; Skogh, T.; Kastbom, A.; et al. Structural Basis of Cross-Reactivity of Anti-Citrullinated Protein Antibodies. Arthritis Rheumatol. 2019, 71, 210–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Yu, Y.; Yue, Y.; Liao, H.; Xie, W.; Thai, J.; Mikuls, T.R.; Thiele, G.M.; Duryee, M.J.; Sayles, H.; et al. Autoantibodies From Single Circulating Plasmablasts React With Citrullinated Antigens and Porphyromonas gingivalis in Rheumatoid Arthritis. Arthritis Rheumatol. 2015, 68, 614–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, R.; Liao, K.; Nair, R.; Ringold, S.; Costenbander, K.H. Anti-citrullinated peptide antibody assays and their role in the diagnosis of rheumatoid arthritis. Arthritis Rheum. 2009, 61, 1472–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatfield, S.M.; Wicks, I.P.; Sturgess, A.D.; Roberts, L.J. Anti-citrullinated peptide antibody: Death of the rheumatoid factor? Med. J. Aust. 2009, 190, 693–695. [Google Scholar] [CrossRef] [PubMed]
- Rönnelid, J.; Hansson, M.; Mathsson-Alm, L.; Cornillet, M.; Reed, E.; Jakobsson, P.-J.; Alfredsson, L.; Holmdahl, R.; Skriner, K.; Serre, G.; et al. Anticitrullinated protein/peptide antibody multiplexing defines an extended group of ACPA-positive rheumatoid arthritis patients with distinct genetic and environmental determinants. Ann. Rheum. Dis. 2017, 77, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Sugiyama, D.; Kogata, Y.; Tsuji, G.; Nakazawa, T.; Kawano, S.; Saigo, K.; Morinobu, A.; Koshiba, M.; Kuntz, K.M.; et al. Meta-analysis: Diagnostic Accuracy of Anti–Cyclic Citrullinated Peptide Antibody and Rheumatoid Factor for Rheumatoid Arthritis. Ann. Intern. Med. 2007, 146, 797–808. [Google Scholar] [CrossRef]
- Szekanecz, Z.; Szabo, Z.; Zeher, M.; Soós, L.; Dankó, K.; Horváth, I.; Lakos, G. Superior performance of the CCP3.1 test compared to CCP2 and MCV in the rheumatoid factor-negative RA population. Immunol. Res. 2013, 56, 439–443. [Google Scholar] [CrossRef]
- van Delft, M.A.M.; Huizinga, T.W.J. An overview of autoantibodies in rheumatoid arthritis. J. Autoimmun. 2020, 110, 102392. [Google Scholar] [CrossRef]
- Kurowska, W.; Slowinska, I.; Krogulec, Z.; Syrowka, P.; Maslinski, W. Antibodies to Citrullinated Proteins (ACPA) Associate with Markers of Osteoclast Activation and Bone Destruction in the Bone Marrow of Patients with Rheumatoid Arthritis. J. Clin. Med. 2021, 10, 1778. [Google Scholar] [CrossRef]
- Van Der Woude, D.; Syversen, S.W.; Van der Voort, E.I.; Verpoort, K.N.; Goll, G.L.; Van Der Linden, M.P.M.; van der Helm-van Mil, A.H.; Van Der Heijde, D.M.; Huizinga, T.W.J.; Kvien, T.K.; et al. The ACPA isotype profile reflects long-term radiographic progression in rheumatoid arthritis. Ann. Rheum. Dis. 2010, 69, 1110–1116. [Google Scholar] [CrossRef] [Green Version]
- Koga, T.; Okada, A.; Fukuda, T.; Hidaka, T.; Ishii, T.; Ueki, Y.; Kodera, T.; Nakashima, M.; Takahashi, Y.; Honda, S.; et al. Anti-citrullinated peptide antibodies are the strongest predictor of clinically relevant radiographic progression in rheumatoid arthritis patients achieving remission or low disease activity: A post hoc analysis of a nationwide cohort in Japan. PLoS ONE 2017, 12, e0175281. [Google Scholar] [CrossRef]
- Arlestig, L.; Mullazehi, M.; Kokkonen, H.; Rocklöv, J.; Rönnelid, J.; Dahlqvist, S.R. Antibodies against cyclic citrullinated peptides of IgG, IgA and IgM isotype and rheumatoid factor of IgM and IgA isotype are increased in unaffected members of multicase rheumatoid arthritis families from northern Sweden. Ann. Rheum. Dis. 2011, 71, 825–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohmi, Y.; Ise, W.; Harazono, A.; Takakura, D.; Fukuyama, H.; Baba, Y.; Narazaki, M.; Shoda, H.; Takahashi, N.; Ohkawa, Y.; et al. Sialylation converts arthritogenic IgG into inhibitors of collagen-induced arthritis. Nat. Commun. 2016, 7, 11205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherer, H.U.; van der Woude, D.; Ioan-Facsinay, A.; El Bannoudi, H.; Trouw, L.A.; Wang, J.; Häupl, T.; Burmester, G.-R.; Deelder, A.M.; Huizinga, T.W.J.; et al. Glycan profiling of anti-citrullinated protein antibodies isolated from human serum and synovial fluid. Arthritis Rheum. 2010, 62, 1620–1629. [Google Scholar] [CrossRef]
- Rombouts, Y.; Ewing, E.; Van De Stadt, L.A.; Selman, M.H.J.; Trouw, L.A.; Deelder, A.M.; Huizinga, T.W.J.; Wuhrer, M.; Van Schaardenburg, D.; Toes, R.; et al. Anti-citrullinated protein antibodies acquire a pro-inflammatory Fc glycosylation phenotype prior to the onset of rheumatoid arthritis. Ann. Rheum. Dis. 2013, 74, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Lundström, S.L.; Fernandes-Cerqueira, C.; Ytterberg, A.J.; Ossipova, E.; Hensvold, A.H.; Jakobsson, P.J.; Malmström, V.; Catrina, A.I.; Klareskog, L.; Lundberg, K.; et al. IgG antibodies to cyclic citrullinated peptides exhibit profiles specific in terms of IgG subclasses, Fc-glycans and a fab-Peptide sequence. PLoS ONE 2014, 9, e113924. [Google Scholar]
- Hafkenscheid, L.; Bondt, A.; Scherer, H.U.; Huizinga, T.W.J.; Wuhrer, M.; Toes, R.; Rombouts, Y. Structural Analysis of Variable Domain Glycosylation of Anti-Citrullinated Protein Antibodies in Rheumatoid Arthritis Reveals the Presence of Highly Sialylated Glycans. Mol. Cell. Proteom. 2017, 16, 278–287. [Google Scholar] [CrossRef] [Green Version]
- Shoda, H.; Fujio, K.; Shibuya, M.; Okamura, T.; Sumitomo, S.; Okamoto, A.; Sawada, T.; Yamamoto, K. Detection of autoantibodies to citrullinated BiP in rheumatoid arthritis patients and pro-inflammatory role of citrullinated BiP in collagen-induced arthritis. Arthritis Res. Ther. 2011, 13, R191. [Google Scholar] [CrossRef] [Green Version]
- Anquetil, F.; Clavel, C.; Offer, G.; Serre, G.; Sebbag, M. IgM and IgA Rheumatoid Factors Purified from Rheumatoid Arthritis Sera Boost the Fc Receptor– and Complement-Dependent Effector Functions of the Disease-Specific Anti–Citrullinated Protein Autoantibodies. J. Immunol. 2015, 194, 3664–3674. [Google Scholar] [CrossRef] [Green Version]
- Kempers, A.C.; Nejadnik, M.R.; Rombouts, Y.; Ioan-Facsinay, A.; Van Oosterhout, M.; Jiskoot, W.; Huizinga, T.W.J.; Toes, R.E.M.; Scherer, H.U. Fc gamma receptor binding profile of anti-citrullinated protein antibodies in immune complexes suggests a role for FcγRI in the pathogenesis of synovial inflammation. Clin. Exp. Rheumatol. 2018, 36, 284–293. [Google Scholar]
- Sokolove, J.; Zhao, X.; Chandra, P.E.; Robinson, W.H. Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcγ receptor. Arthritis Rheum. 2010, 63, 53–62. [Google Scholar] [CrossRef]
- Kempers, A.C.; Hafkenscheid, L.; Dorjée, A.L.; Moutousidou, E.; van de Bovenkamp, F.S.; Rispens, T.; Trouw, L.A.; van Oosterhout, M.; Huizinga, T.W.; Toes, R.; et al. The extensive glycosylation of the ACPA variable domain observed for ACPA-IgG is absent from ACPA-IgM. Ann. Rheum. Dis. 2018, 77, 1087–1088. [Google Scholar] [CrossRef]
- Rombouts, Y.; Willemze, A.; van Beers, J.J.; Shi, J.; Kerkman, P.F.; van Toorn, L.; Janssen, G.M.; Zaldumbide, A.; Hoeben, R.C.; Pruijn, G.J.; et al. Extensive glycosylation of ACPA-IgG variable domains modulates binding to citrullinated antigens in rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 578–585. [Google Scholar] [CrossRef]
- Hafkenscheid, L.; De Moel, E.; Smolik, I.; Tanner, S.; Meng, X.; Jansen, B.C.; Bondt, A.; Wuhrer, M.; Huizinga, T.W.J.; Toes, R.E.M.; et al. N-Linked Glycans in the Variable Domain of IgG Anti–Citrullinated Protein Antibodies Predict the Development of Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 1626–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Too, C.L.; Murad, S.; Hansson, M.; Alm, L.M.; Dhaliwal, J.S.; Holmdahl, R.; Jakobsson, P.-J.; Alfredsson, L.; Klareskog, L.; Rönnelid, J.; et al. Differences in the Spectrum of Anti–Citrullinated Protein Antibody Fine Specificities Between Malaysian and Swedish Patients With Rheumatoid Arthritis: Implications for Disease Pathogenesis. Arthritis Rheumatol. 2016, 69, 58–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Beers, J.J.; Willemze, A.; Jansen, J.J.; Engbers, G.H.; Salden, M.; Raats, J.; Drijfhout, J.W.; van der Helm-van Mil, A.H.; Toes, R.E.; Pruijn, G.J. ACPA fine-specificity profiles in early rheumatoid arthritis patients do not correlate with clinical features at baseline or with disease progression. Arthritis Res. Ther. 2013, 15, R140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masson-Bessiere, C.; Sebbag, M.; Girbal-Neuhauser, E.; Nogueira, L.; Vincent, C.; Senshu, T.; Serre, G. The major synovial targets of the rheumatoid arthritis-specific antifilaggrin autoantibodies are deiminated forms of the alpha- and beta-chains of fibrin. J. Immunol. 2001, 166, 4177–4184. [Google Scholar] [CrossRef] [Green Version]
- Ioan-Facsinay, A.; Willemze, A.; Robinson, D.B.; Peschken, C.A.; Markland, J.; van der Woude, D.; Elias, B.; Ménard, H.A.; Newkirk, M.; Fritzler, M.J.; et al. Marked differences in fine specificity and isotype usage of the anti-citrullinated protein antibody in health and disease. Arthritis Rheum. 2008, 58, 3000–3008. [Google Scholar] [CrossRef]
- Snir, O.; Widhe, M.; Von Spee, C.; Lindberg, J.; Padyukov, L.; Lundberg, K.; Engström, A.; Venables, P.J.; Lundeberg, J.; Holmdahl, R.; et al. Multiple antibody reactivities to citrullinated antigens in sera from patients with rheumatoid arthritis: Association with HLA-DRB1 alleles. Ann. Rheum. Dis. 2008, 68, 736–743. [Google Scholar] [CrossRef]
- Vossenaar, E.R.; Després, N.; Lapointe, E.; Van Der Heijden, A.; Lora, M.; Senshu, T.; Van Venrooij, W.J.; Ménard, H.A. Rheumatoid arthritis specific anti-Sa antibodies target citrullinated vimentin. Arthritis Res. 2004, 6, R142–R150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.; Subramanian, V.; et al. NETs Are a Source of Citrullinated Autoantigens and Stimulate Inflammatory Responses in Rheumatoid Arthritis. Sci. Transl. Med. 2013, 5, 178ra40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roland, P.N.; Mignot, S.G.; Bruns, A.; Hurtado, M.; Palazzo, E.; Hayem, G.; Dieudé, P.; Meyer, O.; Martin, S.C. Antibodies to mutated citrullinated vimentin for diagnosing rheumatoid arthritis in anti-CCP-negative patients and for monitoring infliximab therapy. Arthritis Res. Ther. 2008, 10, R142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinloch, A.; Tatzer, V.; Wait, R.; Peston, D.; Lundberg, K.; Donatien, P.; Moyes, D.; Taylor, P.C.; Venables, P.J. Identification of citrullinated α-enolase as a candidate autoantigen in rheumatoid arthritis. Arthritis Res. 2005, 7, R1421–R1429. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Kang, M.J.; Choi, J.Y.; Park, J.S.; Park, J.K.; Lee, E.Y.; Lee, E.B.; Pap, T.; Yi, E.C.; Song, Y.W. Apolipoprotein B binds to enolase-1 and aggravates inflammation in rheumatoid arthritis. Ann. Rheum. Dis. 2018, 77, 1480–1489. [Google Scholar] [CrossRef]
- Sofat, N.; Wait, R.; Robertson, S.D.; Baines, D.L.; Baker, E.H. Interaction between extracellular matrix molecules and microbial pathogens: Evidence for the missing link in autoimmunity with rheumatoid arthritis as a disease model. Front. Microbiol. 2015, 5, 783. [Google Scholar] [CrossRef]
- Van Beers, J.J.; Willemze, A.; Stammen-Vogelzangs, J.; Drijfhout, J.W.; Toes, R.E.; Pruijn, G.J.M. Anti-citrullinated fibronectin antibodies in rheumatoid arthritis are associated with human leukocyte antigen-DRB1 shared epitope alleles. Arthritis Res. Ther. 2012, 14, R35. [Google Scholar] [CrossRef] [Green Version]
- Kuusela, P.; Ruoslahti, E.; Engvall, E.; Vaheri, A. Immunological interspecies cross-reactions of fibroblast surface antigen (fibronectin). Immunochemistry 1976, 13, 639–642. [Google Scholar] [CrossRef]
- Johansson, L.; Pratesi, F.; Brink, M.; Arlestig, L.; D’Amato, C.; Bartaloni, D.; Migliorini, P.; Rantapa-Dahlqvist, S. Antibodies directed against endogenous and exogenous citrullinated antigens pre-date the onset of rheumatoid arthritis. Arthritis Res. 2016, 18, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Ezzati, P.; Smolik, I.; Bernstein, C.N.; Hitchon, C.A.; El-Gabalawy, H.S. Characterization of Autoantigens Targeted by Anti-Citrullinated Protein Antibodies In Vivo: Prominent Role for Epitopes Derived from Histone 4 Proteins. PLoS ONE 2016, 11, e0165501. [Google Scholar] [CrossRef]
- Panayi, G.S.; Corrigall, V.M. Immunoglobulin heavy-chain-binding protein (BiP): A stress protein that has the potential to be a novel therapy for rheumatoid arthritis. Biochem. Soc. Trans. 2014, 42, 1752–1755. [Google Scholar] [CrossRef] [PubMed]
- Schwenzer, A.; Jiang, X.; Mikuls, T.R.; Payne, J.; Sayles, H.R.; Quirke, A.-M.; Kessler, B.; Fischer, R.; Venables, P.J.; Lundberg, K.; et al. Identification of an immunodominant peptide from citrullinated tenascin-C as a major target for autoantibodies in rheumatoid arthritis. Ann. Rheum. Dis. 2015, 75, 1876–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutolo, M.; Soldano, S.; Paolino, S. Potential roles for tenascin in (very) early diagnosis and treatment of rheumatoid arthritis. Ann. Rheum. Dis. 2019, 79, e42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Union, A.; Meheus, L.; Humbel, R.L.; Conrad, K.; Steiner, G.; Moereels, H.; Pottel, H.; Serre, G.; De Keyser, F. Identification of citrullinated rheumatoid arthritis-specific epitopes in natural filaggrin relevant for antifilaggrin autoantibody detection by line immunoassay. Arthritis Rheum. 2002, 46, 1185–1195. [Google Scholar] [CrossRef] [PubMed]
- Schellekens, G.A.; De Jong, B.A.; van den Hoogen, F.H.; Van De Putte, L.B.; Van Venrooij, W.J. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J. Clin. Investig. 1998, 101, 273–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Beers, J.J.B.C.; Schwarte, C.M.; Stammen-Vogelzangs, J.; Oosterink, E.; Božič, B.; Pruijn, G.J.M. The rheumatoid arthritis synovial fluid citrullinome reveals novel citrullinated epitopes in apolipoprotein E, myeloid nuclear differentiation antigen, and β-actin. Arthritis Rheum. 2012, 65, 69–80. [Google Scholar] [CrossRef]
- Vogt, L.M.; Kwasniewicz, E.; Talens, S.; Scavenius, C.; Bielecka, E.; Ekdahl, K.N.; Enghild, J.J.; Mörgelin, M.; Saxne, T.; Potempa, J.; et al. Apolipoprotein E Triggers Complement Activation in Joint Synovial Fluid of Rheumatoid Arthritis Patients by Binding C1q. J. Immunol. 2020, 204, 2779–2790. [Google Scholar] [CrossRef]
- De Bont, C.M.; Eerden, N.; Boelens, W.C.; Pruijn, G.J.M.; Bont, C.M. Neutrophil proteases degrade autoepitopes of NET-associated proteins. Clin. Exp. Immunol. 2019, 199, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Goëb, V.; Thomas-L’Otellier, M.; Daveau, R.; Charlionet, R.; Fardellone, P.; Le Loët, X.; Tron, F.; Gilbert, D.; Vittecoq, O. Candidate autoantigens identified by mass spectrometry in early rheumatoid arthritis are chaperones and citrullinated glycolytic enzymes. Arthritis Res. Ther. 2009, 11, R38. [Google Scholar] [CrossRef] [Green Version]
- Konig, M.F.; Giles, J.T.; Nigrovic, P.A.; Andrade, F. Antibodies to native and citrullinated RA33 (hnRNP A2/B1) challenge citrullination as the inciting principle underlying loss of tolerance in rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 2022–2028. [Google Scholar] [CrossRef]
- Willemze, A.; Shi, J.; Mulder, M.; Stoeken-Rijsbergen, G.; Drijfhout, J.W.; Huizinga, T.W.J.; Trouw, L.A.; Toes, R.E. The concentration of anticitrullinated protein antibodies in serum and synovial fluid in relation to total immunoglobulin concentrations. Ann. Rheum. Dis. 2013, 72, 1059–1063. [Google Scholar] [CrossRef]
- Lu, M.C.; Lai, N.S.; Yin, W.Y.; Yu, H.C.; Huang, H.B.; Tung, C.H.; Huang, K.Y.; Yu, C.L. Anti-citrullinated protein antibodies activated ERK1/2 and JNK mitogen-activated protein kinases via binding to surface-expressed citrullinated GRP78 on mononuclear cells. J. Clin. Immunol. 2013, 33, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Chirivi, R.G.S.; van Rosmalen, J.W.G.; van der Linden, M.; Euler, M.; Schmets, G.; Bogatkevich, G.; Kambas, K.; Hahn, J.; Braster, Q.; Soehnlein, O.; et al. Therapeutic ACPA inhibits NET formation: A potential therapy for neutrophil-mediated inflammatory diseases. Cell. Mol. Immunol. 2020, 18, 1528–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, P.; Kim, Y.; Jung, H.; Rim, Y.A.; Sohn, D.H.; Robinson, W.H.; Moon, S.-J.; Ju, J.H. Pathogenic Role of Circulating Citrullinated Antigens and Anti-Cyclic Monoclonal Citrullinated Peptide Antibodies in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 2389. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Alghamdi, M.; Alasmari, D.; Assiri, A.; Mattar, E.; AlJaddawi, A.A.; Alattas, S.G.; Redwan, E.M. An Overview of the Intrinsic Role of Citrullination in Autoimmune Disorders. J. Immunol. Res. 2019, 2019, 1–39. [Google Scholar] [CrossRef] [Green Version]
- Blachère, N.E.; Parveen, S.; Fak, J.; Frank, M.O.; Orange, D.E. Inflammatory but not apoptotic death of granulocytes citrullinates fibrinogen. Arthritis Res. Ther. 2015, 17, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Spengler, J.; Lugonja, B.A.; Ytterberg, A.J.; Zubarev, R.; Creese, A.J.; Pearson, M.J.; Grant, M.M.; Milward, M.; Lundberg, K.; Buckley, C.D.; et al. Release of Active Peptidyl Arginine Deiminases by Neutrophils Can Explain Production of Extracellular Citrullinated Autoantigens in Rheumatoid Arthritis Synovial Fluid. Arthritis Rheumatol. 2015, 67, 3135–3145. [Google Scholar] [CrossRef] [Green Version]
- Pratesi, F.; Dioni, I.; Tommasi, C.; Alcaro, M.C.; Paolini, I.; Barbetti, F.; Boscaro, F.; Panza, F.; Puxeddu, I.; Rovero, P.; et al. Antibodies from patients with rheumatoid arthritis target citrullinated histone 4 contained in neutrophils extracellular traps. Ann. Rheum. Dis. 2013, 73, 1414–1422. [Google Scholar] [CrossRef]
- Tegla, C.A.; Cudrici, C.; Patel, S.; Trippe, R., 3rd; Rus, V.; Niculescu, F.; Rus, H. Membrane attack by complement: The assembly and biology of terminal complement complexes. Immunol. Res. 2011, 51, 45–60. [Google Scholar] [CrossRef] [Green Version]
- Podack, E.R.; Hengartner, H.; Lichtenheld, M.G. A central role of perforin in cytolysis? Annu. Rev. Immunol. 1991, 9, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Morgan, P.; Luzio, J.P.; Campbell, A.K. Intracellular Ca2+ and cell injury: A paradoxical role of Ca2+ in complement membrane attack. Cell Calcium 1986, 7, 399–411. [Google Scholar] [CrossRef]
- Douda, D.N.; Khan, M.A.; Grasemann, H.; Palaniyar, N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc. Natl. Acad. Sci. USA 2015, 112, 2817–2822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, H.; Dragunow, M.; Hampton, M.B.; Kettle, T.; Winterbourn, C.C. Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J. Leukoc. Biol. 2012, 92, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Konig, M.F.; Abusleme, L.; Reinholdt, J.; Palmer, R.J.; Teles, R.P.; Sampson, K.; Rosen, A.; Nigrovic, P.A.; Sokolove, J.; Giles, J.T.; et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci. Transl. Med. 2016, 8, 369ra176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagasawa, T.; Kato, S.; Furuichi, Y. Evaluation of the Virulence of Aggregatibacter actinomycetemcomitans Through the Analysis of Leukotoxin. Methods Mol. Biol. 2020, 2210, 185–193. [Google Scholar] [CrossRef]
- Vega, B.A.; Schober, L.T.; Kim, T.; Belinka, B.A.; Kachlany, S.C. Aggregatibacter actinomycetemcomitans Leukotoxin (LtxA) Requires Death Receptor Fas, in Addition to LFA-1, To Trigger Cell Death in T Lymphocytes. Infect. Immun. 2019, 87, e00309-19. [Google Scholar] [CrossRef] [Green Version]
- Brusca, S.B.; Abramson, S.B.; Scher, J.U. Microbiome and mucosal inflammation as extra-articular triggers for rheumatoid arthritis and autoimmunity. Curr. Opin. Rheumatol. 2014, 26, 101–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dal Peraro, M.; van der Goot, F.G. Pore-forming toxins: Ancient, but never really out of fashion. Nat. Rev. Microbiol. 2016, 14, 77–92. [Google Scholar] [CrossRef]
- Los, F.C.O.; Randis, T.M.; Aroian, R.V.; Ratner, A. Role of Pore-Forming Toxins in Bacterial Infectious Diseases. Microbiol. Mol. Biol. Rev. 2013, 77, 173–207. [Google Scholar] [CrossRef] [Green Version]
- Grace, L.E.; Bukhari, M.; Lauder, R.M.; Bishop, L.A.; Taylor, A.M. AB0097 The Presence of Staphylococcal Toxins in The Urine of Patients with Rheumatoid Arthritis. Ann. Rheum. Dis. 2016, 75, 930. [Google Scholar] [CrossRef]
- Pavone, B.; Sirolli, V.; Giardinelli, A.; Bucci, S.; Forlì, F.; Di Cesare, M.; Sacchetta, P.; Di Pietro, N.; Pandolfi, A.; Urbani, A.; et al. Plasma protein carbonylation in chronic uremia. J. Nephrol. 2011, 24, 453–464. [Google Scholar] [CrossRef]
- Kalim, S.; Karumanchi, S.A.; Thadhani, R.I.; Berg, A.H. Protein Carbamylation in Kidney Disease: Pathogenesis and Clinical Implications. Am. J. Kidney Dis. 2014, 64, 793–803. [Google Scholar] [CrossRef] [Green Version]
- Long, J.; Parada, X.V.; Kalim, S. Protein Carbamylation in Chronic Kidney Disease and Dialysis. Adv. Clin. Chem. 2018, 87, 37–67. [Google Scholar] [CrossRef]
- Jaisson, S.; Pietrement, C.; Gillery, P. Carbamylation-Derived Products: Bioactive Compounds and Potential Biomarkers in Chronic Renal Failure and Atherosclerosis. Clin. Chem. 2011, 57, 1499–1505. [Google Scholar] [CrossRef] [Green Version]
- Verbrugge, F.H.; Tang, W.H.W.; Hazen, S.L. Protein carbamylation and cardiovascular disease. Kidney Int. 2015, 88, 474–478. [Google Scholar] [CrossRef] [Green Version]
- Flückiger, R.; Harmon, W.; Meier, W.; Loo, S.; Gabbay, K.H. Hemoglobin Carbamylation in Uremia. N. Engl. J. Med. 1981, 304, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C.L. Role of cyanate in the induction of vascular dysfunction during uremia: More than protein carbamylation? Kidney Int. 2014, 86, 875–877. [Google Scholar] [CrossRef] [Green Version]
- Stark, G.R.; Stein, W.H.; Moore, S. Reactions of the Cyanate Present in Aqueous Urea with Amino Acids and Proteins. J. Biol. Chem. 1960, 235, 3177–3181. [Google Scholar] [CrossRef]
- Wang, Z.; Nicholls, S.; Rodriguez, E.R.; Kummu, O.; Hörkkö, S.; Barnard, J.W.; Reynolds, W.F.; Topol, E.; A DiDonato, J.; Hazen, S.L. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat. Med. 2007, 13, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Ospelt, C.; Bang, H.; Feist, E.; Camici, G.; Keller, S.; Detert, J.; Krämer, A.; Gay, S.; Ghannam, K.; Burmester, G.R. Carbamylation of vimentin is inducible by smoking and represents an independent autoantigen in rheumatoid arthritis. Ann. Rheum. Dis. 2017, 76, 1176–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manganelli, V.; Recalchi, S.; Capozzi, A.; Riitano, G.; Mattei, V.; Longo, A.; Di Franco, M.; Alessandri, C.; Bombardieri, M.; Valesini, G.; et al. Autophagy induces protein carbamylation in fibroblast-like synoviocytes from patients with rheumatoid arthritis. Rheumatology 2018, 57, 2032–2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apostolov, E.O.; Shah, S.V.; Ok, E.; Basnakian, A.G. Quantification of Carbamylated LDL in Human Sera by a New Sandwich ELISA. Clin. Chem. 2005, 51, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Stim, J.; Shaykh, M.; Anwar, F.; Ansari, A.; Arruda, J.A.; Dunea, G. Factors determining hemoglobin carbamylation in renal failure. Kidney Int. 1995, 48, 1605–1610. [Google Scholar] [CrossRef] [Green Version]
- Davenport, A.; Jones, S.R.; Goel, S.; Astley, J.P.; Hartog, M. Differentiation of acute from chronic renal impairment by detection of carbamylated haemoglobin. Lancet 1993, 341, 1614–1617. [Google Scholar] [CrossRef]
- Wynckel, A.; Randoux, C.; Millart, H.; Desroches, C.; Gillery, P.; Canivet, E.; Chanard, J. Kinetics of carbamylated haemoglobin in acute renal failure. Nephrol. Dial. Transplant. 2000, 15, 1183–1188. [Google Scholar] [CrossRef]
- Ok, E.; Basnakian, A.G.; Apostolov, E.O.; Barri, Y.M.; Shah, S.V. Carbamylated low-density lipoprotein induces death of endothelial cells: A link to atherosclerosis in patients with kidney disease. Kidney Int. 2005, 68, 173–178. [Google Scholar] [CrossRef] [Green Version]
- Bright, R.; Proudman, S.M.; Rosenstein, E.D.; Bartold, P.M. Is there a link between carbamylation and citrullination in periodontal disease and rheumatoid arthritis? Med. Hypotheses 2015, 84, 570–576. [Google Scholar] [CrossRef]
- Reed, E.; Jiang, X.; Kharlamova, N.; Ytterberg, A.J.; Catrina, A.I.; Israelsson, L.; Mathsson-Alm, L.; Hansson, M.; Alfredsson, L.; Rönnelid, J.; et al. Antibodies to carbamylated α-enolase epitopes in rheumatoid arthritis also bind citrullinated epitopes and are largely indistinct from anti-citrullinated protein antibodies. Arthritis Res. 2016, 18, 1–9. [Google Scholar] [CrossRef]
- Shi, J.; Van Steenbergen, H.W.; Van Nies, J.A.B.; Levarht, E.W.N.; Huizinga, T.W.J.; van der Helm-van Mil, A.H.; Toes, R.E.M.; Trouw, L.A. The specificity of anti-carbamylated protein antibodies for rheumatoid arthritis in a setting of early arthritis. Arthritis Res. Ther. 2015, 17, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Verpoort, K.N.; van Gaalen, F.A.; van der Helm-van Mil, A.H.; Schreuder, G.M.T.; Breedveld, F.C.; Huizinga, T.W.J.; de Vries, R.R.P.; Toes, R.E.M. Association of HLA-DR3 with anti-cyclic citrullinated peptide antibody-negative rheumatoid arthritis. Arthritis Rheum. 2005, 52, 3058–3062. [Google Scholar] [CrossRef] [PubMed]
- Irigoyen, P.; Lee, A.T.; Wener, M.H.; Li, W.; Kern, M.; Batliwalla, F.; Lum, R.F.; Massarotti, E.; Weisman, M.; Bombardier, C.; et al. Regulation of anti-cyclic citrullinated peptide antibodies in rheumatoid arthritis: Contrasting effects of HLA-DR3 and the shared epitope alleles. Arthritis Rheum. 2005, 52, 3813–3818. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Trouw, L.A.; van Wesemael, T.J.; Shi, J.; Bengtsson, C.; Källberg, H.; Malmström, V.; Israelsson, L.; Hreggvidsdottir, H.; Verduijn, W.; et al. Anti-CarP antibodies in two large cohorts of patients with rheumatoid arthritis and their relationship to genetic risk factors, cigarette smoking and other autoantibodies. Ann. Rheum. Dis. 2014, 73, 1761–1768. [Google Scholar] [CrossRef] [PubMed]
- Regueiro, C.; Rodriguez-Rodriguez, L.; Triguero-Martinez, A.; Nuño, L.; Castaño-Nuñez, A.L.; Villalva, A.; Perez-Pampin, E.; Lopez-Golan, Y.; Abasolo, L.; Ortiz, A.M.; et al. Specific Association of HLA—DRB 1*03 With Anti–Carbamylated Protein Antibodies in Patients With Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Kummu, O.; Turunen, S.P.; Wang, C.; Lehtimäki, J.; Veneskoski, M.; Kastarinen, H.; Koivula, M.K.; Risteli, J.; Kesäniemi, Y.A.; Hörkkö, S. Carbamyl adducts on low-density lipoprotein induce IgG response in LDLR-/- mice and bind plasma autoantibodies in humans under enhanced carbamylation. Antioxid. Redox. Signal. 2013, 19, 1047–1062. [Google Scholar] [CrossRef] [PubMed]
- van Delft, M.A.M.; Verheul, M.K.; Burgers, L.E.; Derksen, V.F.A.M.; van der Helm-van Mil, A.H.; van der Woude, D.; Huizinga, T.W.J.; Toes, R.E.M.; Trouw, L.A. The isotype and IgG subclass distribution of anti-carbamylated protein antibodies in rheumatoid arthritis patients. Arthritis Res. 2017, 19, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verheul, M.K.; Van Erp, S.J.H.; Van Der Woude, D.; Levarht, E.W.N.; Mallat, M.J.K.; Verspaget, H.W.; Stolk, J.; Toes, R.E.; van der Meulen-de, A.E.; Hiemstra, P.; et al. Anti-carbamylated protein antibodies: A specific hallmark for rheumatoid arthritis. Comparison to conditions known for enhanced carbamylation; renal failure, smoking and chronic inflammation. Ann. Rheum. Dis. 2016, 75, 1575–1576. [Google Scholar] [CrossRef]
- Shi, J.; Knevel, R.; Suwannalai, P.; van der Linden, M.P.; Janssen, G.M.C.; van Veelen, P.; Levarht, N.E.W.; van der Helm-van Mil, A.H.; Cerami, A.; Huizinga, T.W.J.; et al. Autoantibodies recognizing carbamylated proteins are present in sera of patients with rheumatoid arthritis and predict joint damage. Proc. Natl. Acad. Sci. USA 2011, 108, 17372–17377. [Google Scholar] [CrossRef] [Green Version]
- Ajeganova, S.; van Steenbergen, H.W.; Verheul, M.K.; Forslind, K.; Hafström, I.; Toes, R.E.; Huizinga, T.W.; Svensson, B.; Trouw, L.A.; van der Helm-van Mil, A.H. The association between anti-carbamylated protein (anti-CarP) antibodies and radiographic progression in early rheumatoid arthritis: A study exploring replication and the added value to ACPA and rheumatoid factor. Ann. Rheum. Dis. 2017, 76, 112–118. [Google Scholar] [CrossRef]
- Truchetet, M.E.; Dublanc, S.; Barnetche, T.; Vittecoq, O.; Mariette, X.; Richez, C.; Blanco, P.; Mahler, M.; Contin-Bordes, C.; Schaeverbeke, T. Association of the Presence of Anti-Carbamylated Protein Antibodies in Early Arthritis With a Poorer Clinical and Radiologic Outcome: Data From the French ESPOIR Cohort. Arthritis Rheumatol. 2017, 69, 2292–2302. [Google Scholar] [CrossRef] [Green Version]
- Verheul, M.K.; Böhringer, S.; van Delft, M.A.M.; Jones, J.D.; Rigby, W.F.C.; Gan, R.W.; Holers, V.M.; Edison, J.D.; Deane, K.D.; Janssen, K.M.J.; et al. Triple Positivity for Anti-Citrullinated Protein Autoantibodies, Rheumatoid Factor, and Anti-Carbamylated Protein Antibodies Conferring High Specificity for Rheumatoid Arthritis: Implications for Very Early Identification of At-Risk Individuals. Arthritis Rheumatol. 2018, 70, 1721–1731. [Google Scholar] [CrossRef] [Green Version]
- Verheul, M.; Böhringer, S.; van Delft, M.; Jones, J.; Rigby, W.; Gan, R.; Holers, V.; Edison, J.; Deane, K.; Janssen, K.; et al. The combination of three autoantibodies, ACPA, RF and anti-CarP antibodies is highly specific for rheumatoid arthritis: Implications for very early identification of individuals at risk to develop rheumatoid arthritis. Arthritis Rheumatol. 2018, 70, 1721–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, R.W.; Trouw, L.A.; Shi, J.; Toes, R.E.; Huizinga, T.W.; Demoruelle, M.K.; Kolfenbach, J.R.; Zerbe, G.O.; Deane, K.D.; Edison, J.D.; et al. Anti-carbamylated Protein Antibodies Are Present Prior to Rheumatoid Arthritis and Are Associated with Its Future Diagnosis. J. Rheumatol. 2015, 42, 572–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruijn, G.J.M. Citrullination and Carbamylation in the Pathophysiology of Rheumatoid Arthritis. Front. Immunol. 2015, 6, 192. [Google Scholar] [CrossRef] [Green Version]
- Regueiro, C.; Rodriguez-Rodriguez, L.; Lopez-Mejias, R.; Nuno, L.; Triguero-Martinez, A.; Perez-Pampin, E.; Corrales, A.; Villalba, A.; Lopez-Golan, Y.; Abasolo, L.; et al. A predominant involvement of the triple seropositive patients and others with rheumatoid factor in the association of smoking with rheumatoid arthritis. Sci. Rep. 2020, 10, 3355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brink, M.; Hansson, M.; Mathsson-Alm, L.; Wijayatunga, P.; Verheul, M.K.; Trouw, L.A.; Holmdahl, R.; Rönnelid, J.; Klareskog, L.; Rantapää-Dahlqvist, S. Rheumatoid factor isotypes in relation to antibodies against citrullinated peptides and carbamylated proteins before the onset of rheumatoid arthritis. Arthritis Res. 2016, 18, 1–11. [Google Scholar] [CrossRef] [Green Version]
- van Wesemael, T.; Ajeganova, S.; Humphreys, J.; Terao, C.; Muhammad, A.; Symmons, D.P.M.; MacGregor, A.J.; Hafström, I.; Trouw, L.A.; Mil, A.H.M.V.D.H.-V.; et al. Smoking is associated with the concurrent presence of multiple autoantibodies in rheumatoid arthritis rather than with anti-citrullinated protein antibodies per se: A multicenter cohort study. Arthritis Res. 2016, 18, 285. [Google Scholar] [CrossRef] [Green Version]
- Brink, M.; Verheul, M.K.; Rönnelid, J.; Berglin, E.; Holmdahl, R.; Toes, R.E.; Klareskog, L.; Trouw, L.A.; Rantapää-Dahlqvist, S. Anti-carbamylated protein antibodies in the pre-symptomatic phase of rheumatoid arthritis, their relationship with multiple anti-citrulline peptide antibodies and association with radiological damage. Arthritis Res. Ther. 2015, 17, 25–28. [Google Scholar] [CrossRef] [Green Version]
- Bos, W.H.; Wolbink, G.J.; Boers, M.; Tijhuis, G.J.; de Vries, N.; Van Der Horst-Bruinsma, I.E.; Tak, P.P.; Van De Stadt, R.J.; Van Der Laken, C.J.; Dijkmans, B.A.C.; et al. Arthritis development in patients with arthralgia is strongly associated with anti-citrullinated protein antibody status: A prospective cohort study. Ann. Rheum. Dis. 2009, 69, 490–494. [Google Scholar] [CrossRef]
- van Delft, M.A.M.; Verheul, M.K.; E Burgers, L.; Rantapää-Dahlqvist, S.; van der Helm-van Mil, A.H.; Huizinga, T.W.J.; Toes, R.E.M.; A Trouw, L. The anti-carbamylated protein antibody response is of overall low avidity despite extensive isotype switching. Rheumatology 2018, 57, 1583–1591. [Google Scholar] [CrossRef] [Green Version]
- Khumaedi, A.I.; Purnamasari, D.; Wijaya, I.P.; Soeroso, Y. The relationship of diabetes, periodontitis and cardiovascular disease. Diabetes Metab. Syndr. 2019, 13, 1675–1678. [Google Scholar] [CrossRef] [PubMed]
- Sokolove, J.; Brennan, M.J.; Sharpe, O.; Lahey, L.J.; Kao, A.H.; Krishnan, E.; Edmundowicz, D.; Lepus, C.M.; Wasko, M.C.; Robinson, W.H. Brief Report: Citrullination Within the Atherosclerotic Plaque: A Potential Target for the Anti-Citrullinated Protein Antibody Response in Rheumatoid Arthritis. Arthritis Rheum. 2013, 65, 1719–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinelli, F.R.; Pecani, A.; Conti, F.; Mancini, R.; Alessandri, C.; Valesini, G. Post-translational modifications in rheumatoid arthritis and atherosclerosis: Focus on citrullination and carbamylation. J. Int. Med Res. 2016, 44, 81–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermans, M.P.J.; van der Velden, D.; Cabezas, J.M.M.; Putter, H.; Huizinga, T.W.J.; Kuiper, J.; Toes, R.E.M.; Schalij, M.J.; Jukema, J.W.; van der Woude, D. Long-term mortality in patients with ST-segment elevation myocardial infarction is associated with anti-citrullinated protein antibodies. Int. J. Cardiol. 2017, 240, 20–24. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.L.; Sodre, F.M.C.; Mamula, M.J.; Overbergh, L. Citrullination and PAD Enzyme Biology in Type 1 Diabetes—Regulators of Inflammation, Autoimmunity, and Pathology. Front. Immunol. 2021, 12, 678953. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Tan, D.; Piao, H. Myelin Basic Protein Citrullination in Multiple Sclerosis: A Potential Therapeutic Target for the Pathology. Neurochem. Res. 2016, 41, 1845–1856. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Thompson, P.R. Protein Arginine Deiminases (PADs): Biochemistry and Chemical Biology of Protein Citrullination. Accounts Chem. Res. 2019, 52, 818–832. [Google Scholar] [CrossRef]
- Caprariello, A.V.; Rogers, J.A.; Morgan, M.L.; Hoghooghi, V.; Plemel, J.; Koebel, A.; Tsutsui, S.; Dunn, J.F.; Kotra, L.P.; Ousman, S.S.; et al. Biochemically altered myelin triggers autoimmune demyelination. Proc. Natl. Acad. Sci. USA 2018, 115, 5528–5533. [Google Scholar] [CrossRef] [Green Version]
- Bisset, G.W.; Poisner, A.M.; Smyth, D.G. Carbamylation of Oxytocin and Arginine-Vasopressin. Nature 1963, 199, 69–70. [Google Scholar] [CrossRef]
- Oimomi, M.; Hatanaka, H.; Yoshimura, Y.; Yokono, K.; Baba, S.; Taketomi, Y. Carbamylation of Insulin and Its Biological Activity. Nephron 1987, 46, 63–66. [Google Scholar] [CrossRef]
- Kraus, L.M.; Traxinger, R.; Kraus, A.P.; Kraus, R.T.L.M. Uremia and insulin resistance: N-carbamoyl-asparagine decreases insulin-sensitive glucose uptake in rat adipocytes. Kidney Int. 2004, 65, 881–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.-D.; Mun, K.-C.; Chang, E.-J.; Park, S.-B.; Kim, H.-C. Inhibition of erythropoietin activity by cyanate. Scand. J. Urol. Nephrol. 2004, 38, 69–72. [Google Scholar] [CrossRef]
- Satake, R.; Kozutsumi, H.; Takeuchi, M.; Asano, K. Chemical modification of erythropoietin: An increase in in vitro activity by guanidination. Biochim. Biophys. Acta Protein Struct. Mol. Enzym. 1990, 1038, 125–129. [Google Scholar] [CrossRef]
- Hörkkö, S.; Huttunen, K.; Kervinen, K.; Kesäniemi, Y.A. Decreased clearance of uraemic and mildly carbamylated low-density lipoprotein. Eur. J. Clin. Investig. 1994, 24, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Weisgraber, K.H.; Innerarity, T.L.; Mahley, R.W. Role of lysine residues of plasma lipoproteins in high affinity binding to cell surface receptors on human fibroblasts. J. Biol. Chem. 1978, 253, 9053–9062. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Melnichenko, A.A.; Orekhov, A.N.; Bobryshev, Y.V. How do macrophages sense modified low-density lipoproteins? Int. J. Cardiol. 2017, 230, 232–240. [Google Scholar] [CrossRef]
- Asci, G.; Basci, A.; Shah, S.V.; Basnakian, A.; Töz, H.; Ozkahya, M.; Duman, S.; Ok, E. Carbamylated low-density lipoprotein induces proliferation and increases adhesion molecule expression of human coronary artery smooth muscle cells. Nephrology 2008, 13, 480–486. [Google Scholar] [CrossRef]
- Verdin, E.; Ott, M. 50 years of protein acetylation: From gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2014, 16, 258–264. [Google Scholar] [CrossRef]
- Narita, T.; Weinert, B.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2018, 20, 156–174. [Google Scholar] [CrossRef]
- Silva, R.D.; Martinho, R.G. Developmental roles of protein N-terminal acetylation. Proteomics 2015, 15, 2402–2409. [Google Scholar] [CrossRef] [PubMed]
- Arnesen, T.; Van Damme, P.; Polevoda, B.; Helsens, K.; Evjenth, R.; Colaert, N.; Varhaug, J.E.; Vandekerckhove, J.; Lillehaug, J.R.; Sherman, F.; et al. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8157–8162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, K.A.; Wigerblad, G.; Sahlström, P.; Garimella, M.G.; Chemin, K.; Steen, J.; Titcombe, P.J.; Marklein, B.; Zhou, D.; Stålesen, R.; et al. Differential ACPA Binding to Nuclear Antigens Reveals a PAD-Independent Pathway and a Distinct Subset of Acetylation Cross-Reactive Autoantibodies in Rheumatoid Arthritis. Front. Immunol. 2018, 9, 3033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kampstra, A.S.B.; Dekkers, J.S.; Volkov, M.; Dorjée, A.L.; Hafkenscheid, L.; Kempers, A.C.; Van Delft, M.; Kissel, T.; Reijm, S.; Janssen, G.M.C.; et al. Different classes of anti-modified protein antibodies are induced on exposure to antigens expressing only one type of modification. Ann. Rheum. Dis. 2019, 78, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Jing, J.; Li, W.; Ma, J.; Zhang, X.; Wang, Z.; Zhou, Z.; Dai, L.; Shao, L. Impaired Tip60-mediated Foxp3 acetylation attenuates regulatory T cell development in rheumatoid arthritis. J. Autoimmun. 2019, 100, 27–39. [Google Scholar] [CrossRef]



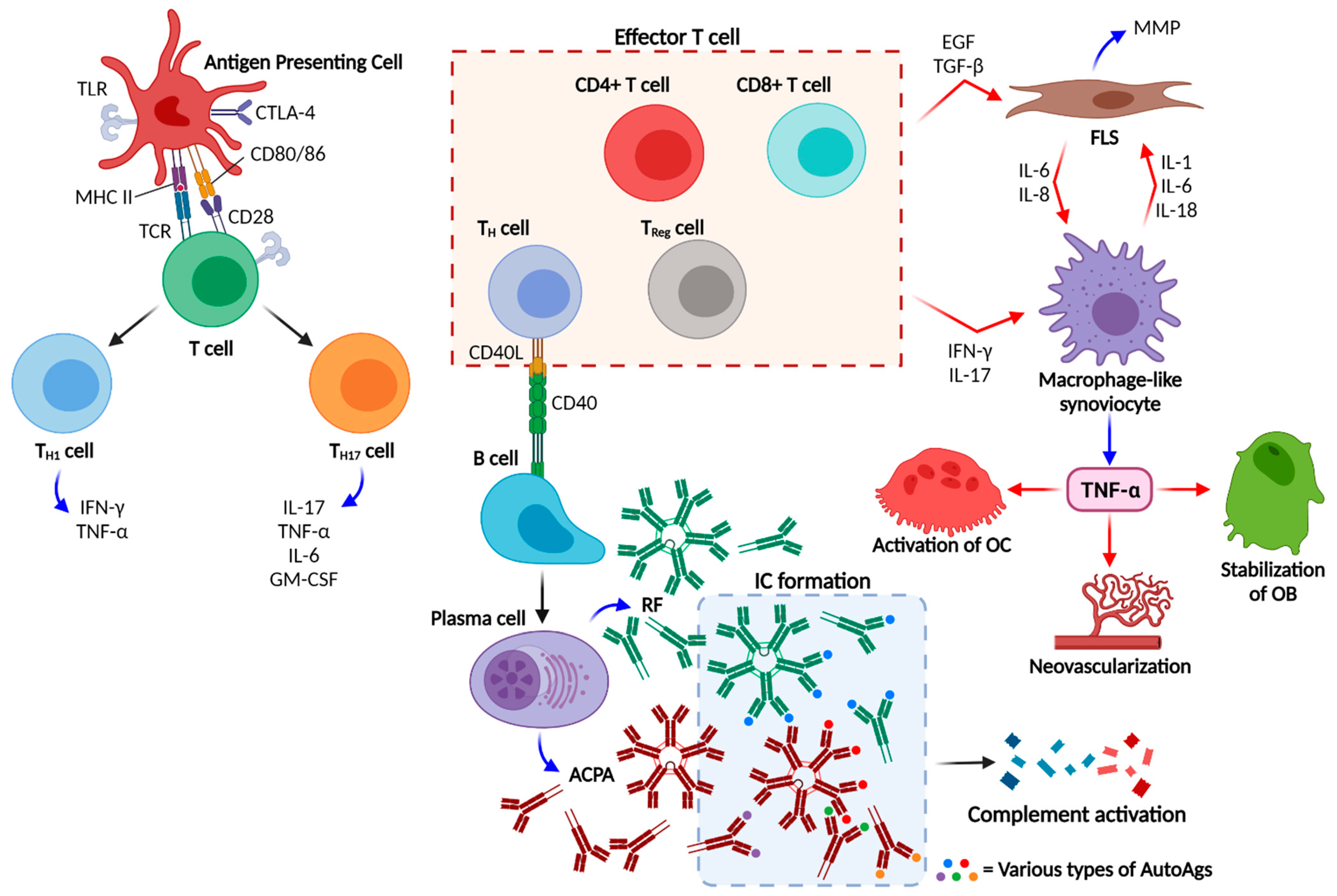{kind=link}
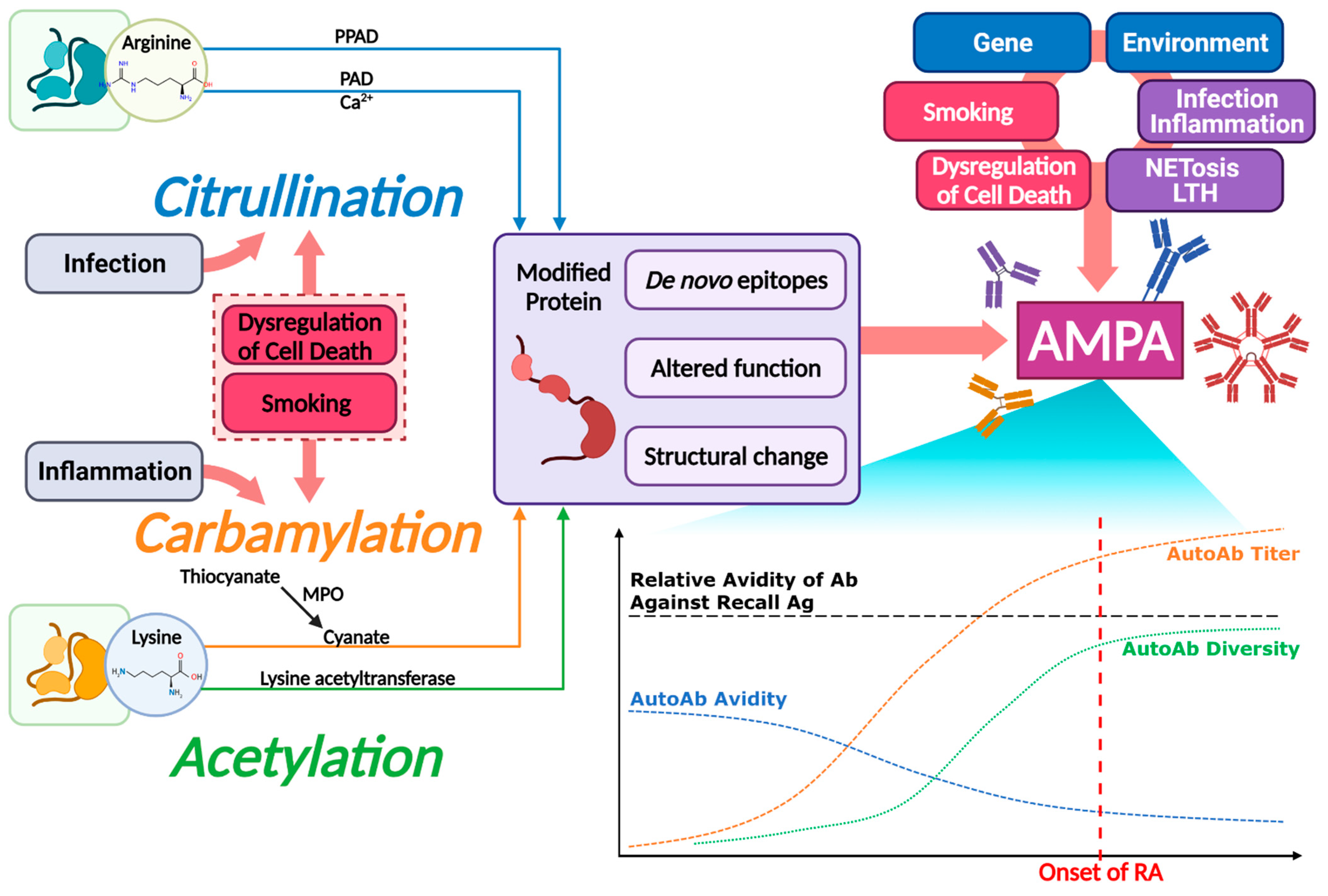{kind=link}
| Domain | Category | Score |
|---|---|---|
| Joint involvement | 1 large joint (Shoulder, elbow, hip, knee, ankle) | 0 |
| 2–10 large joints | 1 | |
| 1–3 small joints (MCP, PIP, thumb IP, MTP, wrists) | 2 | |
| 4–10 small joints | 3 | |
| More than 10 joints (Including at least 1 small joint) | 5 | |
| Serologic study | Negative RF and negative ACPAs (Under ULN) | 0 |
| Low-positive 1 RF or low-positive ACPAs | 2 | |
| High-positive 2 RF or high positive ACPAs | 3 | |
| Acute phase reactants | Normal CRP and normal ESR | 0 |
| Abnormal CRP or abnormal ESR | 1 | |
| Duration of symptoms | <6 weeks | 0 |
| ≥6 weeks | 1 |
| DMARDs | Mechanism | Route of Administration |
|---|---|---|
| Abatacept | Fusion protein consists of extracellular domain of CTLA-4 and Fc region of IgG1, binding to CD80/86 | IV, SC |
| Adalimumab | TNF-α inhibitor | SC |
| Infliximab | IV | |
| Certolizumab pegol | SC | |
| Golimumab | SC | |
| Etanercept | SC | |
| Rituximab | Monoclonal antibody against CD20 | IV |
| Tocilizumab | Humanized monoclonal antibody against IL-6 receptor | IV, SC |
| Sarilumab | SC | |
| Tofacitinib | JAK1/JAK3 inhibitor | PO |
| Baricitinib | JAK1/JAK2 inhibitor | PO |
| Upadacitinib | JAK1 inhibitor | PO |
| Filgotinib | PO | |
| Peficitinib | Pan-JAK(JAK1/JAK2/JAK3/Tyk2) inhibitor | PO |
| Type | Site of Expression | Substrate | Reference |
|---|---|---|---|
| PAD1 | Epidermis Uterus Hair follicle, sweat gland Stomach | Filaggrin Keratin-associated protein MEK1 | [131,132] |
| PAD2 | CNS, PNS Skeletal muscle Immune cells, spleen Skin, secretory gland Uterus, pancreas, kidney, inner ear | Myelin basic protein in CNS Histones Actin Vimentin RNAP2 β-catenin Enolase Fibrinogen | [133,134,135,136] |
| PAD3 | Hair follicle Keratinocyte Peripheral nerve | Vimentin Filaggrin Trichohyalin Apoptosis-inducing factors | [137] |
| PAD4 | Immune cells, spleen Secretory gland Brain Uterus Joints | Histones and nucleophosmin/B23 Type I collagen ING4 p300, p21, p53 Lamin C GSK3β PAD4 | [138,139] |
| PAD6 | Egg, ovary, testis, early embryo | Uncertain | [30,140] |
| PPAD | Enzyme of P. gingivalis | Fibrinogen α-enolase Collagen type II PPAD | [44,119,141,142] |
| Origin | Candidate Protein/Peptide | Site of Expression in Human | Reference |
|---|---|---|---|
| Endogenous | Fibrinogen | Inflamed joint | [179,180,181] |
| Vimentin | Inflamed joint | [180,182,183,184] | |
| α-enolase | Joint tissue, inflammatory cells ¶ | [180,181,185,186] | |
| Fibronectin | Plasma, synovium, SF | [187,188,189] | |
| Type II collagen | Joint tissue | [181] | |
| Histone § | Nucleus | [190,191] | |
| BIP § | ER | [170,192] | |
| Tenascin-C | ECM of joint | [193,194] | |
| Filaggrin | Epithelium | [195,196] | |
| Apo E | Plasma, CNS, RA-SF | [197,198] | |
| MNDA | Inflammatory cells ¶ | [197,199] | |
| β-actin | Inflammatory cells ¶ | [197,199] | |
| FUSE-BP § | Nucleus | [200] | |
| hnRNP § | Nucleus, RA-SF | [201] | |
| Exogenous | Viral citrullinated peptides from EBV antigen | — | [190] |
| α-enolase from P. gingivalis | [190] | ||
| GNS sequence homologue of surface proteins of the P. copri | [45] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, E.-J.; Ju, J.H. Impact of Posttranslational Modification in Pathogenesis of Rheumatoid Arthritis: Focusing on Citrullination, Carbamylation, and Acetylation. Int. J. Mol. Sci. 2021, 22, 10576. https://doi.org/10.3390/ijms221910576
Kwon E-J, Ju JH. Impact of Posttranslational Modification in Pathogenesis of Rheumatoid Arthritis: Focusing on Citrullination, Carbamylation, and Acetylation. International Journal of Molecular Sciences. 2021; 22(19):10576. https://doi.org/10.3390/ijms221910576
Chicago/Turabian StyleKwon, Eui-Jong, and Ji Hyeon Ju. 2021. "Impact of Posttranslational Modification in Pathogenesis of Rheumatoid Arthritis: Focusing on Citrullination, Carbamylation, and Acetylation" International Journal of Molecular Sciences 22, no. 19: 10576. https://doi.org/10.3390/ijms221910576
APA StyleKwon, E. -J., & Ju, J. H. (2021). Impact of Posttranslational Modification in Pathogenesis of Rheumatoid Arthritis: Focusing on Citrullination, Carbamylation, and Acetylation. International Journal of Molecular Sciences, 22(19), 10576. https://doi.org/10.3390/ijms221910576