
Complex Positive Effects of SGLT-2 Inhibitor Empagliflozin in the Liver, Kidney and Adipose Tissue of Hereditary Hypertriglyceridemic Rats: Possible Contribution of Attenuation of Cell Senescence and Oxidative Stress

 , , , ,
, , , ,
Abstract
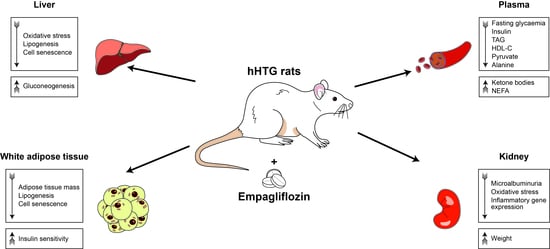
:
1. Introduction
2. Results
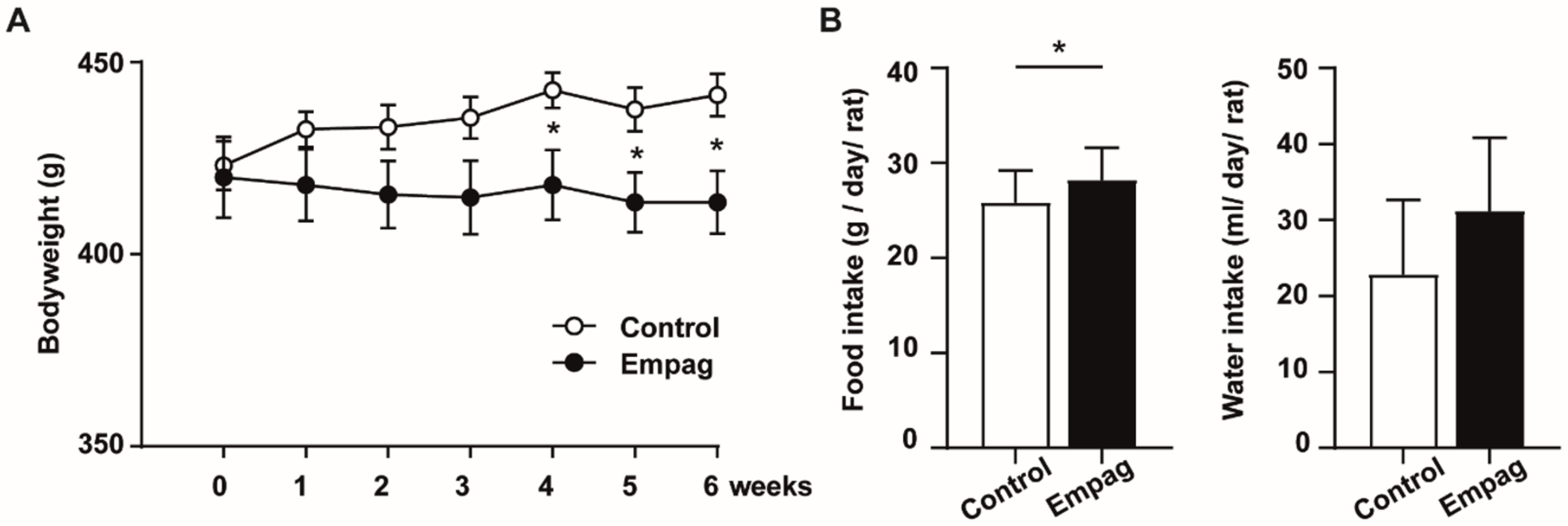
2.1. Empagliflozin Had Beneficial Effect on Body Weight, Glycaemia and Lipidaemia
2.2. Empagliflozin Reduced Oxidative Stress in the Liver and Modified Genes Involved in Lipid Metabolism Pathway
2.3. Empagliflozin Modulated Metabolomic Profiles in Plasma and Liver
2.4. Empagliflozin Improved White Adipose Tissue Insulin Sensitivity
2.5. Empagliflozin Reduced Oxidative Stress and Inflammation in Kidneys
2.6. Empagliflozin Reduced Palmitic Acid Oxidation in Myocardium but Had No Effect on Skeletal Muscle Glucose Metabolism
3. Discussion
4. Materials and Methods
4.1. Animals and Diets
4.2. Body Weight, Food and Water Consumption
4.3. Urinary Glucose and Microalbuminuria
4.4. Biochemical Analyses
4.5. Glucose Utilization in White Adipose Tissue, Glucose Oxidation and Glycogen Synthesis in Diaphragm
4.6. Palmitic Acid Oxidation in Isolated Myocardium
4.7. Lipolysis in Isolated Epididymal Adipose Tissue
4.8. Tissue Triglyceride Measurements
4.9. Anthrone Method (Polysacharide Quantification): Liver Glycogen
4.10. Oxidative Stress Parameters
4.11. Isolation and Determination of mRNA Level: TLDA (TaqMan Low Density Array)
4.12. NMR-Based Metabolomics in Plasma and Liver Samples
4.13. Cell Culture Analyses
4.14. Detection of Senescence-Associated β-Galactosidase Activity
4.15. Isolation and Quantification of Gene Expression in Cell Lines
4.16. Oil Red O Staining
4.17. Cell Viability
4.18. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Chobot, A.; Górowska-Kowolik, K.; Sokołowska, M.; Jarosz-Chobot, P. Obesity and diabetes-Not only a simple link between two epidemics. Diabetes Metab. Res. Rev. 2018, 34, e3042. [Google Scholar] [CrossRef] [Green Version]
- Shakeri, H.; Lemmens, K.; Gevaert, A.B.; De Meyer, G.; Segers, V.F.M. Cellular senescence links aging and diabetes in cardiovascular disease. Am. J. Physiol. Circ. Physiol. 2018, 315, H448–H462. [Google Scholar] [CrossRef]
- Hubackova, S.; Davidova, E.; Rohlenova, K.; Stursa, J.; Werner, L.; Andera, L.; Dong, L.; Terp, M.; Hodny, Z.; Ditzel, H.J.; et al. Selective elimination of senescent cells by mitochondrial targeting is regulated by ANT2. Cell Death Differ. 2018, 26, 276–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 485. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Tripolt, N.J.; Kolesnik, E.; Pferschy, P.N.; Verheyen, N.; Ablasser, K.; Sailer, S.; Alber, H.; Berger, R.; Kaulfersch, C.; Leitner, K.; et al. Impact of EMpagliflozin on cardiac function and biomarkers of heart failure in patients with acute MYocardial infarction—The EMMY trial. Am. Heart J. 2019, 221, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Baldi, S.; Frascerra, S.; Astiarraga, B.; Heise, T.; Bizzotto, R.; Mari, A.; Pieber, T.R.; Muscelli, E. Shift to Fatty Substrate Utilization in Response to Sodium–Glucose Cotransporter 2 Inhibition in Subjects Without Diabetes and Patients with Type 2 Diabetes. Diabetes 2016, 65, 1190–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, R.; Tanaka, Y.; Koiwai, K.; Ishida, K.; Salsali, A.; Kaspers, S.; Kohler, S.; Lund, S.S. Effect of Empagliflozin on Free Fatty Acids and Ketone Bodies in Japanese Patients with Type 2 Diabetes Mellitus: A Randomized Controlled Trial. Adv. Ther. 2019, 36, 2769–2782. [Google Scholar] [CrossRef] [PubMed]
- Mudaliar, S.; Alloju, S.; Henry, R.R. Can a Shift in Fuel Energetics Explain the Beneficial Cardiorenal Outcomes in the EMPA-REG OUTCOME Study? A Unifying Hypothesis. Diabetes Care 2016, 39, 1115–1122. [Google Scholar] [CrossRef] [Green Version]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV Protection in the EMPA-REG OUTCOME Trial: A “Thrifty Substrate” Hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.H.; Oh, T.J.; Lee, G.; Maeng, H.J.; Lee, D.H.; Kim, K.M.; Choi, S.H.; Jang, H.C.; Lee, H.S.; Park, K.S.; et al. The beneficial effects of empagliflozin, an sglt2 inhibitor, on atherosclerosis in apoe (−/−) mice fed a western diet. Diabetologia 2017, 60, 364–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoare, M.; Das, T.; Alexander, G. Ageing, telomeres, senescence, and liver injury. J. Hepatol. 2010, 53, 950–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Li, Q.; Huang, W.; Han, Y.; Tan, H.; An, M.; Xiang, Q.; Zhou, R.; Yang, L.; Cheng, Y. Dapagliflozin alleviates hepatic steatosis by restoring autophagy via the ampk-mtor pathway. Front. Pharmacol. 2021, 12, 589273. [Google Scholar] [CrossRef]
- Liao, X.; Wang, X.; Li, H.; Li, L.; Zhang, G.; Yang, M.; Yuan, L.; Liu, H.; Yang, G.; Gao, L. Sodium-Glucose Cotransporter 2 (SGLT2) Inhibitor Increases Circulating Zinc-A2-Glycoprotein Levels in Patients with Type 2 Diabetes. Sci. Rep. 2016, 6, 32887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, D.; Liao, L.; Wang, H.; Zhang, W.; Wang, T.; Xu, Z. Canagliflozin ameliorates obesity by improving mitochondrial function and fatty acid oxidation via pparalpha in vivo and in vitro. Life Sci. 2020, 247, 117414. [Google Scholar] [CrossRef] [PubMed]
- Skop, V.; Cahova, M.; Dankova, H.; Papackova, Z.; Palenickova, E.; Svoboda, P.; Zídková, J.; Kazdová, L. Autophagy inhibition in early but not in later stages prevents 3T3-L1 differentiation: Effect on mitochondrial remodeling. Differentiation 2014, 87, 220–229. [Google Scholar] [CrossRef]
- Gallo, L.A.; Wright, E.M.; Vallon, V. Probing SGLT2 as a therapeutic target for diabetes: Basic physiology and consequences. Diabetes Vasc. Dis. Res. 2015, 12, 78–89. [Google Scholar] [CrossRef] [Green Version]
- Pillon, N.J.; Loos, R.J.; Marshall, S.M.; Zierath, J.R. Metabolic consequences of obesity and type 2 diabetes: Balancing genes and environment for personalized care. Cell 2021, 184, 1530–1544. [Google Scholar] [CrossRef]
- Kiran, S.; Kumar, V.; Kumar, S.; Price, R.; Singh, U. Adipocyte, Immune Cells, and miRNA Crosstalk: A Novel Regulator of Metabolic Dysfunction and Obesity. Cells 2021, 10, 1004. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.W.; Yiu, W.H. Innate immunity in diabetic kidney disease. Nat. Rev. Nephrol. 2020, 16, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Verma, S. Mechanisms of cardiovascular benefits of sodium glucose co-transporter 2 (sglt2) inhibitors: A state-of-the-art review. JACC Basic Transl. Sci. 2020, 5, 632–644. [Google Scholar] [CrossRef]
- Vrána, A.; Kazdová, L. The hereditary hypertriglyceridemic nonobese rat: An experimental model of human hypertriglyceridemia. Transplant. Proc. 1990, 22, 2579. [Google Scholar]
- Zicha, J.; Pechanova, O.; Cacanyiova, S.; Cebova, M.; Kristek, F.; Torok, J.; Simko, F.; Dobesova, Z.; Kunes, J. Hereditary hypertriglyceridemic rat: A suitable model of cardiovascular disease and metabolic syndrome? Physiol. Res. 2006, 55, S49–S63. [Google Scholar]
- Klimes, I.; Vrána, A.; Kunes, J.; Seböková, E.; Dobesová, Z.; Stolba, P.; Zicha, J. Hereditary hypertriglyceridemic rat: A new animal model of metabolic alterations in hypertension. Blood Press. 1995, 4, 137–142. [Google Scholar] [CrossRef]
- Yokono, M.; Takasu, T.; Hayashizaki, Y.; Mitsuoka, K.; Kihara, R.; Muramatsu, Y.; Miyoshi, S.; Tahara, A.; Kurosaki, E.; Li, Q.; et al. SGLT2 selective inhibitor ipragliflozin reduces body fat mass by increasing fatty acid oxidation in high-fat diet-induced obese rats. Eur. J. Pharmacol. 2014, 727, 66–74. [Google Scholar] [CrossRef]
- Devenny, J.J.; Godonis, H.E.; Harvey, S.J.; Rooney, S.; Cullen, M.J.; Pelleymounter, M.A. Weight Loss Induced by Chronic Dapagliflozin Treatment Is Attenuated by Compensatory Hyperphagia in Diet-Induced Obese (DIO) Rats. Obesity 2012, 20, 1645–1652. [Google Scholar] [CrossRef] [Green Version]
- Obata, A.; Kubota, N.; Kubota, T.; Iwamoto, M.; Sato, H.; Sakurai, Y.; Takamoto, I.; Katsuyama, H.; Suzuki, Y.; Fukazawa, M.; et al. Tofogliflozin Improves Insulin Resistance in Skeletal Muscle and Accelerates Lipolysis in Adipose Tissue in Male Mice. Endocrinology 2015, 157, 1029–1042. [Google Scholar] [CrossRef]
- Suzuki, M.; Takeda, M.; Kito, A.; Fukazawa, M.; Yata, T.; Yamamoto, M.; Nagata, T.; Fukuzawa, T.; Yamane, M.; Honda, K.; et al. Tofogliflozin, a sodium/glucose cotransporter 2 inhibitor, attenuates body weight gain and fat accumulation in diabetic and obese animal models. Nutr. Diabetes 2014, 4, e125. [Google Scholar] [CrossRef] [PubMed]
- Mosley, J.F., 2nd; Smith, L.; Everton, E.; Fellner, C. Sodium-glucose linked transporter 2 (sglt2) inhibitors in the management of type-2 diabetes: A drug class overview. Pharm. Ther. 2015, 40, 451–462. [Google Scholar]
- Ndefo, U.A.; Anidiobi, N.O.; Basheer, E.; Eaton, A.T. Empagliflozin (Jardiance): A Novel SGLT2 Inhibitor for the Treatment of Type-2 Diabetes. Pharm. Ther. 2015, 40, 364–368. [Google Scholar]
- Lin, Y.; Berg, A.H.; Iyengar, P.; Lam, T.K.; Giacca, A.; Combs, T.P.; Rajala, M.W.; Du, X.; Rollman, B.; Li, W.; et al. The hyperglycemia-induced inflammatory response in adipocytes: The role of reactive oxygen species. J. Biol. Chem. 2005, 280, 4617–4626. [Google Scholar] [CrossRef] [Green Version]
- Daniele, G.; Xiong, J.; Solis-Herrera, C.; Merovci, A.; Eldor, R.; Tripathy, D.; DeFronzo, R.A.; Norton, L.; Abdul-Ghani, M. Dapagliflozin Enhances Fat Oxidation and Ketone Production in Patients with Type 2 Diabetes. Diabetes Care 2016, 39, 2036–2041. [Google Scholar] [CrossRef] [Green Version]
- Hattori, S. Anti-inflammatory effects of empagliflozin in patients with type 2 diabetes and insulin resistance. Diabetol. Metab. Syndr. 2018, 10, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiner, Z. Hypertriglyceridaemia and risk of coronary artery disease. Nat. Rev. Cardiol. 2017, 14, 401–411. [Google Scholar] [CrossRef]
- Bence, K.K.; Birnbaum, M.J. Metabolic drivers of non-alcoholic fatty liver disease. Mol. Metab. 2020, 50, 101143. [Google Scholar] [CrossRef] [PubMed]
- Su, R.C.; Lad, A.; Breidenbach, J.D.; Blomquist, T.M.; Gunning, W.T.; Dube, P.; Kleinhenz, A.L.; Malhotra, D.; Haller, S.T.; Kennedy, D.J. Hyperglycemia induces key genetic and phenotypic changes in human liver epithelial HepG2 cells which parallel the Leprdb/J mouse model of non-alcoholic fatty liver disease (NAFLD). PLoS ONE 2019, 14, e0225604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asmat, U.; Abad, K.; Ismail, K. Diabetes mellitus and oxidative stress—A concise review. Saudi Pharm. J. 2015, 24, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef] [PubMed]
- Kohjima, M.; Enjoji, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2007, 20, 351–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jojima, T.; Wakamatsu, S.; Kase, M.; Iijima, T.; Maejima, Y.; Shimomura, K.; Kogai, T.; Tomaru, T.; Usui, I.; Aso, Y. The SGLT2 Inhibitor Canagliflozin Prevents Carcinogenesis in a Mouse Model of Diabetes and Non-Alcoholic Steatohepatitis-Related Hepatocarcinogenesis: Association with SGLT2 Expression in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 5237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaji, K.; Nishimura, N.; Seki, K.; Sato, S.; Saikawa, S.; Nakanishi, K.; Furukawa, M.; Kawaratani, H.; Kitade, M.; Moriya, K.; et al. Sodium glucose cotransporter 2 inhibitor canagliflozin attenuates liver cancer cell growth and angiogenic activity by inhibiting glucose uptake. Int. J. Cancer 2017, 142, 1712–1722. [Google Scholar] [CrossRef] [Green Version]
- Obara, K.; Shirakami, Y.; Maruta, A.; Ideta, T.; Miyazaki, T.; Kochi, T.; Sakai, H.; Tanaka, T.; Seishima, M.; Shimizu, M. Preventive effects of the sodium glucose cotransporter 2 inhibitor tofogliflozin on diethylnitrosamine-induced liver tumorigenesis in obese and diabetic mice. Oncotarget 2017, 8, 58353–58363. [Google Scholar] [CrossRef]
- Wang, L.; Liu, M.; Yin, F.; Wang, Y.; Li, X.; Wu, Y.; Ye, C.; Liu, J. Trilobatin, a Novel SGLT1/2 Inhibitor, Selectively Induces the Proliferation of Human Hepatoblastoma Cells. Molecules 2019, 24, 3390. [Google Scholar] [CrossRef] [Green Version]
- Locatelli, F.; Pozzoni, P.; Del Vecchio, L. Renal Manifestations in the Metabolic Syndrome: Table 1. J. Am. Soc. Nephrol. 2006, 17, S81–S85. [Google Scholar] [CrossRef] [Green Version]
- Leoncini, G.; Ratto, E.; Viazzi, F.; Vaccaro, V.; Parodi, D.; Parodi, A.; Falqui, V.; Tomolillo, C.; Deferrari, G.; Pontremoli, R. Metabolic syndrome is associated with early signs of organ damage in nondiabetic, hypertensive patients. J. Intern. Med. 2005, 257, 454–460. [Google Scholar] [CrossRef]
- Forbes, J.; Coughlan, M.; Cooper, M.E. Oxidative Stress as a Major Culprit in Kidney Disease in Diabetes. Diabetes 2008, 57, 1446–1454. [Google Scholar] [CrossRef] [Green Version]
- Duran-Salgado, M.B.; Rubio-Guerra, A.F. Diabetic nephropathy and inflammation. World J. Diabetes 2014, 5, 393–398. [Google Scholar] [CrossRef]
- Akchurin, O.M.; Kaskel, F. Update on Inflammation in Chronic Kidney Disease. Blood Purif. 2015, 39, 84–92. [Google Scholar] [CrossRef]
- Wanner, C. Empa-reg outcome: The nephrologist’s point of view. Am. J. Cardiol. 2017, 120, S59–S67. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.M.; Tang, P.M.; Li, J.; Lan, H.Y. Tgf-beta/smad signaling in renal fibrosis. Front. Physiol. 2015, 6, 82. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.; Nick, H.S. Renal response to tissue injury: Lessons from heme oxygenase-1 GeneAblation and expression. J. Am. Soc. Nephrol. 2000, 11, 965–973. [Google Scholar] [CrossRef]
- Lever, J.; Boddu, R.; George, J.F.; Agarwal, A. Heme Oxygenase-1 in Kidney Health and Disease. Antioxid. Redox Signal. 2016, 25, 165–183. [Google Scholar] [CrossRef] [Green Version]
- Devarajan, P. Neutrophil gelatinase-associated lipocalin (NGAL): A new marker of kidney disease. Scand. J. Clin. Lab. Investig. 2008, 68, 89–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallon, V.; Rose, M.; Gerasimova, M.; Satriano, J.; Platt, K.A.; Koepsell, H.; Cunard, R.; Sharma, K.; Thomson, S.C.; Rieg, T. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am. J. Physiol. Physiol. 2013, 304, F156–F167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Nagata, N.; Nagashimada, M.; Zhuge, F.; Ni, Y.; Chen, G.; Mayoux, E.; Kaneko, S.; Ota, T. SGLT2 Inhibition by Empagliflozin Promotes Fat Utilization and Browning and Attenuates Inflammation and Insulin Resistance by Polarizing M2 Macrophages in Diet-induced Obese Mice. EBioMedicine 2017, 20, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Pandey, G.; Shankar, K.; Makhija, E.; Gaikwad, A.; Ecelbarger, C.; Mandhani, A.; Srivastava, A.; Tiwari, S. Reduced Insulin Receptor Expression Enhances Proximal Tubule Gluconeogenesis. J. Cell. Biochem. 2016, 118, 276–285. [Google Scholar] [CrossRef]
- Wilding, J.P. The role of the kidneys in glucose homeostasis in type 2 diabetes: Clinical implications and therapeutic significance through sodium glucose co-transporter 2 inhibitors. Metabolism 2014, 63, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Rieg, T.; Masuda, T.; Gerasimova, M.; Mayoux, E.; Platt, K.; Powell, D.R.; Thomson, S.C.; Koepsell, H.; Vallon, V. Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am. J. Physiol. Physiol. 2014, 306, F188–F193. [Google Scholar] [CrossRef] [PubMed]
- Merovci, A.; Solis-Herrera, C.; Daniele, G.; Eldor, R.; Fiorentino, T.V.; Tripathy, D.; Xiong, J.; Perez, Z.; Norton, L.; Abdul-Ghani, M.A.; et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J. Clin. Investig. 2014, 124, 509–514. [Google Scholar] [CrossRef]
- Neschen, S.; Scheerer, M.; Seelig, A.; Huypens, P.; Schultheiss, J.; Wu, M.; Wurst, W.; Rathkolb, B.; Suhre, K.; Wolf, E.; et al. Metformin Supports the Antidiabetic Effect of a Sodium Glucose Cotransporter 2 Inhibitor by Suppressing Endogenous Glucose Production in Diabetic Mice. Diabetes 2014, 64, 284–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besseiche, A.; Riveline, J.; Gautier, J.-F.; Breant, B.; Blondeau, B. Metabolic roles of PGC-1α and its implications for type 2 diabetes. Diabetes Metab. 2015, 41, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Welsh, P.; Rankin, N.; Li, Q.; Mark, P.; Würtz, P.; Ala-Korpela, M.; Marre, M.; Poulter, N.; Hamet, P.; Chalmers, J.; et al. Circulating amino acids and the risk of macrovascular, microvascular and mortality outcomes in individuals with type 2 diabetes: Results from the ADVANCE trial. Diabetologia 2018, 61, 1581–1591. [Google Scholar] [CrossRef] [Green Version]
- Holeček, M. Branched-chain amino acids in health and disease: Metabolism, alterations in blood plasma, and as supplements. Nutr. Metab. 2018, 15, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, M.-S. The Emerging Role of Branched-Chain Amino Acids in Insulin Resistance and Metabolism. Nutrients 2016, 8, 405. [Google Scholar] [CrossRef] [Green Version]
- Holeček, M. The BCAA–BCKA cycle: Its relation to alanine and glutamine synthesis and protein balance. Nutrition 2001, 17, 70. [Google Scholar] [CrossRef]
- Stumvoll, M.; Perriello, G.; Meyer, C.; Gerich, J. Role of glutamine in human carbohydrate metabolism in kidney and other tissues. Kidney Int. 1999, 55, 778–792. [Google Scholar] [CrossRef] [Green Version]
- Felig, P. The glucose-alanine cycle. Metabolism 1973, 22, 179–207. [Google Scholar] [CrossRef]
- Oshima, H.; Miki, T.; Kuno, A.; Mizuno, M.; Sato, T.; Tanno, M.; Yano, T.; Nakata, K.; Kimura, Y.; Abe, K.; et al. Empagliflozin, an SGLT2 Inhibitor, Reduced the Mortality Rate after Acute Myocardial Infarction with Modification of Cardiac Metabolomes and Antioxidants in Diabetic Rats. J. Pharmacol. Exp. Ther. 2018, 368, 524–534. [Google Scholar] [CrossRef] [Green Version]
- Steven, S.; Oelze, M.; Hanf, A.; Kröller-Schön, S.; Kashani, F.; Roohani, S.; Welschof, P.; Kopp, M.; Gödtel-Armbrust, U.; Xia, N.; et al. The SGLT2 inhibitor empagliflozin improves the primary diabetic complications in ZDF rats. Redox Biol. 2017, 13, 370–385. [Google Scholar] [CrossRef] [PubMed]
- Michel, M.C.; Mayoux, E.; Vallon, V. A comprehensive review of the pharmacodynamics of the SGLT2 inhibitor empagliflozin in animals and humans. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2015, 388, 801–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vickers, S.P.; Cheetham, S.; Headland, K.; Dickinson, K.; Grempler, R.; Mayoux, E.; Mark, M.; Klein, T. Combination of the sodium-glucose cotransporter-2 inhibitor empagliflozin with orlistat or sibutramine further improves the body-weight reduction and glucose homeostasis of obese rats fed a cafeteria diet. Diabetes Metab. Syndr. Obes. Targets Ther. 2014, 7, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Contois, J.H.; Hartigan, C.; Rao, L.V.; Snyder, L.M.; Thompson, M.J. Analytical validation of an HPLC assay for urinary albumin. Clin. Chim. Acta 2006, 367, 150–155. [Google Scholar] [CrossRef]
- Trnovská, J.; Šilhavý, J.; Kuda, O.; Landa, V.; Zídek, V.; Mlejnek, P.; Šimáková, M.; Strnad, H.; Skop, V.; Oliyarnyk, O.; et al. Salsalate ameliorates metabolic disturbances by reducing inflammation in spontaneously hypertensive rats expressing human C-reactive protein and by activating brown adipose tissue in nontransgenic controls. PLoS ONE 2017, 12, e0179063. [Google Scholar] [CrossRef]
- Vrána, A.; Kazdová, L. Increased adipose tissue lipolysis in a hypertriglyceridemic rat line. Ann. N. Y. Acad. Sci. 1997, 827, 510–513. [Google Scholar] [CrossRef]
- Malinska, H.; Hüttl, M.; Oliyarnyk, O.; Markova, I.; Poruba, M.; Racova, Z.; Kazdova, L.; Vecera, R. Beneficial effects of troxerutin on metabolic disorders in non-obese model of metabolic syndrome. PLoS ONE 2019, 14, e0220377. [Google Scholar] [CrossRef] [Green Version]
- Trachta, P.; Drápalová, J.; Kaválková, P.; Toušková, V.; Cinkajzlová, A.; Lacinová, Z.; Matoulek, M.; Zelinka, T.; Widimský, J.; Mráz, M.; et al. Three months of regular aerobic exercise in patients with obesity improve systemic subclinical inflammation without major influence on blood pressure and endocrine production of subcutaneous fat. Physiol. Res. 2014, 63, S299–S308. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S. Using MetaboAnalyst 3.0 for Comprehensive Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2016, 55, 14.10.1–14.10.91. [Google Scholar] [CrossRef] [PubMed]
- Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H. Probabilistic Quotient Normalization as Robust Method to Account for Dilution of Complex Biological Mixtures. Application in 1H NMR Metabonomics. Anal. Chem. 2006, 78, 4281–4290. [Google Scholar] [CrossRef]
- Svoboda, P.; Křížová, E.; Čeňková, K.; Vápenková, K.; Zídková, J.; Zídek, V.; Skop, V. Visfatin Is Actively Secreted In Vitro From U-937 Macrophages, but Only Passively Released From 3T3-L1 Adipocytes and HepG2 Hepatocytes. Physiol. Res. 2017, 66, 709–714. [Google Scholar] [CrossRef]
- Svoboda, P.; Krizova, E.; Sestakova, S.; Vapenkova, K.; Knejzlik, Z.; Rimpelova, S.; Rayova, D.; Volfova, N.; Krizova, I.; Rumlova, M.; et al. Nuclear transport of nicotinamide phosphoribosyltransferase is cell cycle-dependent in mammalian cells, and its inhibition slows cell growth. J. Biol. Chem. 2019, 294, 8676–8689. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Kraus, N.A.; Ehebauer, F.; Zapp, B.; Rudolphi, B.; Kraus, B.J.; Kraus, D. Quantitative assessment of adipocyte differentiation in cell culture. Adipocyte 2016, 5, 351–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Allison, P.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2017, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Units | Control | Empagliflozin |
|---|---|---|---|
| Urinary glucose | mmol/L | 0.9 ± 0.2 | 93.7 ± 12.2 *** |
| Glucose fasted | mmol/L | 5.314 ± 0.347 | 4.407 ± 0.131 * |
| Glucose postprandial | mmol/L | 8.296 ± 0.347 | 7.969 ± 0.248 |
| Insulin | nmol/L | 0.221 ± 0.024 | 0.142 ± 0.024 * |
| TAG | mmol/L | 4.750 ± 0.294 | 3.12 ± 0.226 *** |
| CHOL | mmol/L | 1.567 ± 0.014 | 1.536 ± 0.032 |
| HDL-C | mmol/L | 0.760 ± 0.021 | 0.913 ± 0.037 ** |
| NEFA | mmol/L | 0.426 ± 0.027 | 0.666 ± 0.083 ** |
| Glycerol | mmol/L | 0.192 ± 0.013 | 0.203 ± 0.013 |
| Leptin | pg/mL | 4425 ± 220 | 3508 ± 245 * |
| MCP-1 | pg/mL | 149.62 ± 67.25 | 123.1 ± 15.5 |
| Parameter | Units | Control | Empagliflozin |
|---|---|---|---|
| SOD | U/mg | 0.139 ± 0.008 | 0.126 ± 0.006 |
| GSH-Px | µM NADPH/min/mg | 322 ± 21 | 380 ± 17 * |
| GR | nM NADPH/min/mg | 87 ± 4 | 101 ± 3 ** |
| CAT | µM H2O2/min/mg | 1556 ± 194 | 2074 ± 110 * |
| CD | nM/mg | 36.9 ± 1.6 | 41.1 ± 3.3 |
| TBARS | nM/mg | 1.91 ± 0.12 | 1.50 ± 0.11 * |
| Metabolite | NMR Signal Used for Quantitation [ppm] | (Empag-Control)/ Control [%] | p-Value |
|---|---|---|---|
| Acetone | 2.24 (s) | 138.5 | 0.002 |
| β-Hydroxybutyrate | 1.20 (d) | 66.7 | 0.043 |
| Leucine | 0.97 (m) | 12.6 | 0.028 |
| Pyruvate | 2.38 (s) | −33.3 | 0.007 |
| Alanine | 1.49 (d) | −14.2 | 0.003 |
| Tyrosine | 6.91 (m) | −20.0 | 0.034 |
| Threonine | 4.25 (dd) | −11.5 | 0.008 |
| Cytidine | 6.07 (d) | −9.6 | 0.021 |
| Tryptophan | 7.29 (m) | −8.1 | 0.058 |
| Metabolite | NMR Signal Used for Quantitation [ppm] | (Empag-Control)/ Control [%] | p-Value |
|---|---|---|---|
| Glutamine | 2.45 (m) | 23.6 | 0.014 |
| Leucine | 1.71 (m) | 12.3 | 0.032 |
| Valine | 0.99 (d) | 10.6 | 0.028 |
| Uracil | 7.54 (d) | 11.5 | 0.017 |
| Glutathione (reduced) | 4.57 (dd) | −43.1 | 0.020 |
| Glycogen | 5.41 (m) | −22.0 | 0.001 |
| Xanthosine | 5.86 (d) | −18.9 | 0.027 |
| Parameter | Units | Control | Empagliflozin |
|---|---|---|---|
| SOD | U/mg | 0.067 ± 0.01 | 0.088 ± 0.01 * |
| GSH-Px | µM NADPH/min/mg | 128 ± 12 | 186 ± 15 ** |
| GR | nM NADPH/min/mg | 54.4 ± 3.2 | 57.3 ± 3.7 |
| CAT | µM H2O2/min/mg | 17.7 ± 1.42 | 20.1 ± 1.09 |
| CD | nM/mg | 24.3 ± 1.34 | 22.3 ± 1.93 |
| TBARS | nM/mg | 0.681 ± 0.02 | 0.560 ± 0.03 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trnovska, J.; Svoboda, P.; Pelantova, H.; Kuzma, M.; Kratochvilova, H.; Kasperova, B.J.; Dvorakova, I.; Rosolova, K.; Malinska, H.; Huttl, M.; et al. Complex Positive Effects of SGLT-2 Inhibitor Empagliflozin in the Liver, Kidney and Adipose Tissue of Hereditary Hypertriglyceridemic Rats: Possible Contribution of Attenuation of Cell Senescence and Oxidative Stress. Int. J. Mol. Sci. 2021, 22, 10606. https://doi.org/10.3390/ijms221910606
Trnovska J, Svoboda P, Pelantova H, Kuzma M, Kratochvilova H, Kasperova BJ, Dvorakova I, Rosolova K, Malinska H, Huttl M, et al. Complex Positive Effects of SGLT-2 Inhibitor Empagliflozin in the Liver, Kidney and Adipose Tissue of Hereditary Hypertriglyceridemic Rats: Possible Contribution of Attenuation of Cell Senescence and Oxidative Stress. International Journal of Molecular Sciences. 2021; 22(19):10606. https://doi.org/10.3390/ijms221910606
Chicago/Turabian StyleTrnovska, Jaroslava, Petr Svoboda, Helena Pelantova, Marek Kuzma, Helena Kratochvilova, Barbora Judita Kasperova, Iveta Dvorakova, Katerina Rosolova, Hana Malinska, Martina Huttl, and et al. 2021. "Complex Positive Effects of SGLT-2 Inhibitor Empagliflozin in the Liver, Kidney and Adipose Tissue of Hereditary Hypertriglyceridemic Rats: Possible Contribution of Attenuation of Cell Senescence and Oxidative Stress" International Journal of Molecular Sciences 22, no. 19: 10606. https://doi.org/10.3390/ijms221910606
APA StyleTrnovska, J., Svoboda, P., Pelantova, H., Kuzma, M., Kratochvilova, H., Kasperova, B. J., Dvorakova, I., Rosolova, K., Malinska, H., Huttl, M., Markova, I., Oliyarnyk, O., Melcova, M., Skop, V., Mraz, M., Stemberkova-Hubackova, S., & Haluzik, M. (2021). Complex Positive Effects of SGLT-2 Inhibitor Empagliflozin in the Liver, Kidney and Adipose Tissue of Hereditary Hypertriglyceridemic Rats: Possible Contribution of Attenuation of Cell Senescence and Oxidative Stress. International Journal of Molecular Sciences, 22(19), 10606. https://doi.org/10.3390/ijms221910606