
Molecular Modeling of Signal Peptide Recognition by Eukaryotic Sec Complexes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
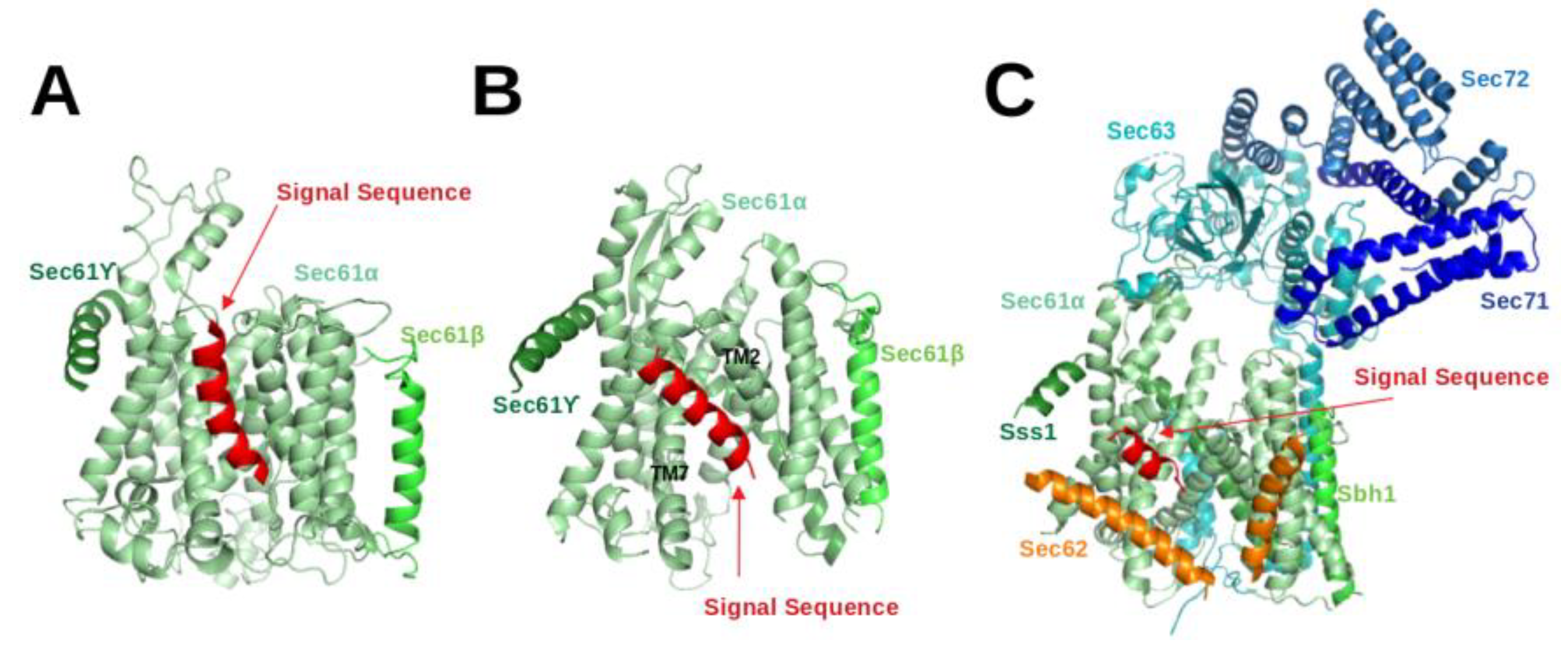
:1. Introduction
2. Properties of Signal Peptides
3. Structural Studies of Eukaryotic Sec61:SP Complexes
4. Conformational Dynamics of the Sec61 Translocon and Bound SPs in Molecular Dynamics Simulations
5. Molecular Docking Helps in Understanding the Interaction between Signal Peptide and Sec61 Translocon
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Park, E.; Rapoport, T.A. Mechanisms of Sec61/SecY-mediated protein translocation across membranes. Annu. Rev. Biophys. 2012, 41, 21–40. [Google Scholar] [CrossRef]
- Voorhees, R.M.; Hegde, R.S. Toward a structural understanding of co-translational protein translocation. Curr. Opin. Cell Biol. 2016, 41, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Ng, D.; Brown, J.D.; Walter, P. Signal sequences specify the targeting route to the endoplasmic reticulum membrane. J. Cell Biol. 1996, 134, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Voorhees, R.M.; Hegde, R.S. Structure of the Sec61 channel opened by a signal sequence. Science 2016, 351, 88–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, T.H.; Steinchen, W.; Beatrix, B.; Berninghausen, O.; Becker, T.; Bange, G.; Cheng, J.; Beckmann, R. Architecture of the active post-translational Sec translocon. Embo. J. 2021, 40, e105643. [Google Scholar] [CrossRef]
- Lang, S.; Pfeffer, S.; Lee, P.H.; Cavalié, A.; Helms, V.; Förster, F.; Zimmermann, R. An update on Sec61 channel functions, mechanisms, and related diseases. Front. Physiol. 2017, 8, 887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linxweiler, M.; Schick, B.; Zimmermann, R. Let’s talk about Secs: Sec61, Sec62 and Sec63 in signal transduction, oncology and personalized medicine. Signal Transduct. Target. Ther. 2017, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sicking, M.; Lang, S.; Bochen, F.; Roos, A.; Drenth, J.P.; Zakaria, M.; Zimmermann, R.; Linxweiler, M. Complexity and Specificity of Sec61-Channelopathies: Human Diseases Affecting Gating of the Sec61 Complex. Cells 2021, 10, 1036. [Google Scholar] [CrossRef]
- Hiss, J.A.; Schneider, G. Architecture, function and prediction of long signal peptides. Briefings Bioinform. 2009, 10, 569–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, S.; Dudek, J.; Schaffer, M.; Ng, B.G.; Albert, S.; Plitzko, J.M.; Baumeister, W.; Zimmermann, R.; Freeze, H.H.; Engel, B.D.; et al. Dissecting the molecular organization of the translocon-associated protein complex. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef]
- Wu, X.; Cabanos, C.; Rapoport, T.A. Structure of the post-translational protein translocation machinery of the ER membrane. Nature 2019, 566, 136–139. [Google Scholar] [CrossRef]
- Itskanov, S.; Park, E. Structure of the posttranslational Sec protein-translocation channel complex from yeast. Science 2019, 363, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Itskanov, S.; Kuo, K.M.; Gumbart, J.C.; Park, E. Stepwise gating of the Sec61 protein-conducting channel by Sec63 and Sec62. Nat. Struct. Mol. Biol. 2021, 28, 162–172. [Google Scholar] [CrossRef]
- Schorr, S.; Nguyen, D.; Haßdenteufel, S.; Nagaraj, N.; Cavalié, A.; Greiner, M.; Weissgerber, P.; Loi, M.; Paton, A.W.; Paton, J.C.; et al. Identification of signal peptide features for substrate specificity in human Sec62/Sec63-dependent ER protein import. FEBS J. 2020, 287, 4612–4640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhadra, P.; Yadhanapudi, L.; Römisch, K.; Helms, V. How does Sec63 affect the conformation of Sec61 in yeast? PLoS Comput. Biol. 2021, 17, e1008855. [Google Scholar] [CrossRef] [PubMed]
- Clérico, E.M.; Maki, J.L.; Gierasch, L.M. Use of synthetic signal sequences to explore the protein export machinery. Pept. Sci. 2008, 90, 307–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hainzl, T.; Sauer-Eriksson, A.E. Signal-sequence induced conformational changes in the signal recognition particle. Nat. Commun. 2015, 6, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, G.S.; Choi, B.S.; Kim, H. Structures of wild-type and mutant signal sequences of Escherichia coli ribose binding protein. Biophys. J. 1994, 66, 1604–1611. [Google Scholar] [CrossRef] [Green Version]
- Chi, S.W.; Yi, G.S.; Suh, J.Y.; Choi, B.S.; Kim, H. Structures of revertant signal sequences of Escherichia coli ribose binding protein. Biophys. J. 1995, 69, 2703–2709. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.; Stutz, R.; Schorr, S.; Lang, S.; Pfeffer, S.; Freeze, H.H.; Förster, F.; Helms, V.; Dudek, J.; Zimmermann, R. Proteomics reveals signal peptide features determining the client specificity in human TRAP-dependent ER protein import. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Lassmann, T.; Sonnhammer, E.L. Kalign–an accurate and fast multiple sequence alignment algorithm. BMC Bioinform. 2005, 6, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Rheede, T.; Smolenaars, M.M.; Madsen, O.; de Jong, W.W. Molecular evolution of the mammalian prion protein. Mol. Biol. Evol. 2003, 20, 111–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Stothard, P. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 2000, 28, 1102–1104. [Google Scholar] [CrossRef] [Green Version]
- Käll, L.; Krogh, A.; Sonnhammer, E.L. Advantages of combined transmembrane topology and signal peptide prediction—the Phobius web server. Nucleic Acids Res. 2007, 35, W429–W432. [Google Scholar] [CrossRef] [Green Version]
- Ménétret, J.F.; Hegde, R.S.; Aguiar, M.; Gygi, S.P.; Park, E.; Rapoport, T.A.; Akey, C.W. Single copies of Sec61 and TRAP associate with a nontranslating mammalian ribosome. Structure 2008, 16, 1126–1137. [Google Scholar] [CrossRef] [Green Version]
- Becker, T.; Bhushan, S.; Jarasch, A.; Armache, J.P.; Funes, S.; Jossinet, F.; Gumbart, J.; Mielke, T.; Berninghausen, O.; Schulten, K.; et al. Structure of monomeric yeast and mammalian Sec61 complexes interacting with the translating ribosome. Science 2009, 326, 1369–1373. [Google Scholar] [CrossRef] [Green Version]
- Gogala, M.; Becker, T.; Beatrix, B.; Armache, J.P.; Barrio-Garcia, C.; Berninghausen, O.; Beckmann, R. Structures of the Sec61 complex engaged in nascent peptide translocation or membrane insertion. Nature 2014, 506, 107–110. [Google Scholar] [CrossRef]
- Voorhees, R.M.; Fernández, I.S.; Scheres, S.H.; Hegde, R.S. Structure of the mammalian ribosome-Sec61 complex to 3.4 Å resolution. Cell 2014, 157, 1632–1643. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, S.; Burbaum, L.; Unverdorben, P.; Pech, M.; Chen, Y.; Zimmermann, R.; Beckmann, R.; Förster, F. Structure of the native Sec61 protein-conducting channel. Nat. Commun. 2015, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Gérard, S.F.; Hall, B.S.; Zaki, A.M.; Corfield, K.A.; Mayerhofer, P.U.; Costa, C.; Whelligan, D.K.; Biggin, P.C.; Simmonds, R.E.; Higgins, M.K. Structure of the inhibited state of the Sec translocon. Mol. Cell 2020, 79, 406–415. [Google Scholar] [CrossRef]
- Cannon, K.S.; Or, E.; Clemons Jr, W.M.; Shibata, Y.; Rapoport, T.A. Disulfide bridge formation between SecY and a translocating polypeptide localizes the translocation pore to the center of SecY. J. Cell Biol. 2005, 169, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Mothes, W.; Prehn, S.; Rapoport, T.A. Systematic probing of the environment of a translocating secretory protein during translocation through the ER membrane. EMBO J. 1994, 13, 3973–3982. [Google Scholar] [CrossRef]
- Plath, K.; Mothes, W.; Wilkinson, B.M.; Stirling, C.J.; Rapoport, T.A. Signal sequence recognition in posttranslational protein transport across the yeast ER membrane. Cell 1998, 94, 795–807. [Google Scholar] [CrossRef] [Green Version]
- Plath, K.; Wilkinson, B.M.; Stirling, C.J.; Rapoport, T.A. Interactions between Sec complex and prepro-α-factor during posttranslational protein transport into the endoplasmic reticulum. Mol. Biol. Cell 2004, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gumbart, J.; Schulten, K. Structural determinants of lateral gate opening in the protein translocon. Biochemistry 2007, 46, 11147–11157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gumbart, J.; Schulten, K. The roles of pore ring and plug in the SecY protein-conducting channel. J. Gen. Physiol. 2008, 132, 709–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gumbart, J.; Trabuco, L.G.; Schreiner, E.; Villa, E.; Schulten, K. Regulation of the protein-conducting channel by a bound ribosome. Structure 2009, 17, 1453–1464. [Google Scholar] [CrossRef] [Green Version]
- Mori, T.; Ishitani, R.; Tsukazaki, T.; Nureki, O.; Sugita, Y. Molecular mechanisms underlying the early stage of protein translocation through the Sec translocon. Biochemistry 2010, 49, 945–950. [Google Scholar] [CrossRef]
- Sun, S.; Wang, S.; Tong, Z.; Yao, X.; Gao, J. A molecular dynamics study on the resilience of Sec61 channel from open to closed state. RSC Adv. 2019, 9, 14876–14883. [Google Scholar] [CrossRef] [Green Version]
- Demirci, E.; Junne, T.; Baday, S.; Bernèche, S.; Spiess, M. Functional asymmetry within the Sec61p translocon. Proc. Natl. Acad. Sci. USA 2013, 110, 18856–18861. [Google Scholar] [CrossRef] [Green Version]
- Rychkova, A.; Warshel, A. Exploring the nature of the translocon-assisted protein insertion. Proc. Natl. Acad. Sci. USA 2013, 110, 495–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niesen, M.J.; Wang, C.Y.; Van Lehn, R.C.; Miller III, T.F. Structurally detailed coarse-grained model for Sec-facilitated co-translational protein translocation and membrane integration. PLoS Comput. Biol. 2017, 13, e1005427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niesen, M.J.; Zimmer, M.H.; Miller III, T.F. Dynamics of co-translational membrane protein integration and translocation via the Sec translocon. J. Am. Chem. Soc. 2020, 142, 5449–5460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Miller, T.F. Hydrophobically stabilized open state for the lateral gate of the Sec translocon. Proc. Natl. Acad. Sci. USA 2010, 107, 5399–5404. [Google Scholar] [CrossRef] [Green Version]
- Gumbart, J.; Schulten, K. Molecular dynamics studies of the archaeal translocon. Biophys. J. 2006, 90, 2356–2367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Miller III, T.F. Long-timescale dynamics and regulation of Sec-facilitated protein translocation. Cell Rep. 2012, 2, 927–937. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Miller III, T.F. Direct simulation of early-stage Sec-facilitated protein translocation. J. Am. Chem. Soc. 2012, 134, 13700–13707. [Google Scholar] [CrossRef] [Green Version]
- Van den Berg, B.; Clemons, W.M.; Collinson, I.; Modis, Y.; Hartmann, E.; Harrison, S.C.; Rapoport, T.A. X-ray structure of a protein-conducting channel. Nature 2004, 427, 36–44. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Tian, P.; Andricioaei, I. Size, motion, and function of the SecY translocon revealed by molecular dynamics simulations with virtual probes. Biophys. J. 2006, 90, 2718–2730. [Google Scholar] [CrossRef] [Green Version]
- Gumbart, J.C.; Teo, I.; Roux, B.; Schulten, K. Reconciling the roles of kinetic and thermodynamic factors in membrane–protein insertion. J. Am. Chem. Soc. 2013, 135, 2291–2297. [Google Scholar] [CrossRef] [PubMed]
- Gumbart, J.C.; Chipot, C. Decrypting protein insertion through the translocon with free-energy calculations. Biochim. Biophys. Acta-(Bba)-Biomembr. 2016, 1858, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Capponi, S.; Heyden, M.; Bondar, A.N.; Tobias, D.J.; White, S.H. Anomalous behavior of water inside the SecY translocon. Proc. Natl. Acad. Sci. USA 2015, 112, 9016–9021. [Google Scholar] [CrossRef] [Green Version]
- Salmaso, V.; Moro, S. Bridging molecular docking to molecular dynamics in exploring ligand-protein recognition process: An overview. Front. Pharmacol. 2018, 9, 923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Sanner, M.F. AutoDock CrankPep: Combining folding and docking to predict protein–peptide complexes. Bioinformatics 2019, 35, 5121–5127. [Google Scholar] [CrossRef] [PubMed]
- Reithinger, J.H.; Yim, C.; Kim, S.; Lee, H.; Kim, H. Structural and functional profiling of the lateral gate of the Sec61 translocon. J. Biol. Chem. 2014, 289, 15845–15855. [Google Scholar] [CrossRef] [Green Version]
- Santos, L.H.; Ferreira, R.S.; Caffarena, E.R. Integrating molecular docking and molecular dynamics simulations. In Docking Screens for Drug Discovery; Springer: Berlin/Heidelberg, Germany, 2019; pp. 13–34. [Google Scholar]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolym. Orig. Res. Biomol. 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Elia, F.; Yadhanapudi, L.; Tretter, T.; Römisch, K. The N-terminus of Sec61p plays key roles in ER protein import and ERAD. PLoS ONE 2019, 14, e0215950. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhadra, P.; Helms, V. Molecular Modeling of Signal Peptide Recognition by Eukaryotic Sec Complexes. Int. J. Mol. Sci. 2021, 22, 10705. https://doi.org/10.3390/ijms221910705
Bhadra P, Helms V. Molecular Modeling of Signal Peptide Recognition by Eukaryotic Sec Complexes. International Journal of Molecular Sciences. 2021; 22(19):10705. https://doi.org/10.3390/ijms221910705
Chicago/Turabian StyleBhadra, Pratiti, and Volkhard Helms. 2021. "Molecular Modeling of Signal Peptide Recognition by Eukaryotic Sec Complexes" International Journal of Molecular Sciences 22, no. 19: 10705. https://doi.org/10.3390/ijms221910705
APA StyleBhadra, P., & Helms, V. (2021). Molecular Modeling of Signal Peptide Recognition by Eukaryotic Sec Complexes. International Journal of Molecular Sciences, 22(19), 10705. https://doi.org/10.3390/ijms221910705