
LEF1 Enhances the Progression of Colonic Adenocarcinoma via Remodeling the Cell Motility Associated Structures

 ,
,  and
and
Abstract
:1. Introduction
2. Results
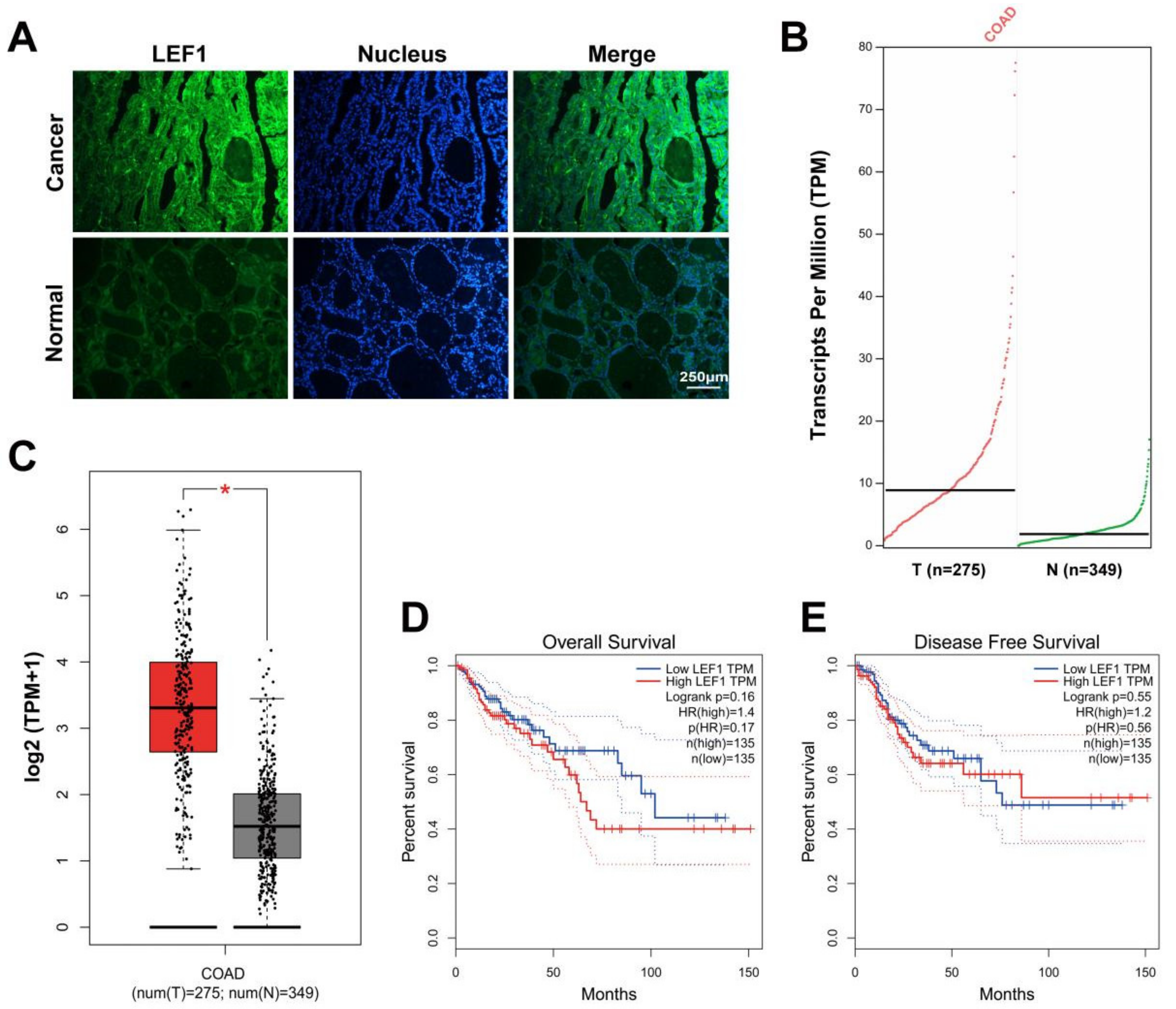
2.1. LEF1 Was Excessively Expressed in Colonic Adenocarcinoma Tissues and Enhanced Colonic Adenocarcinoma Progression
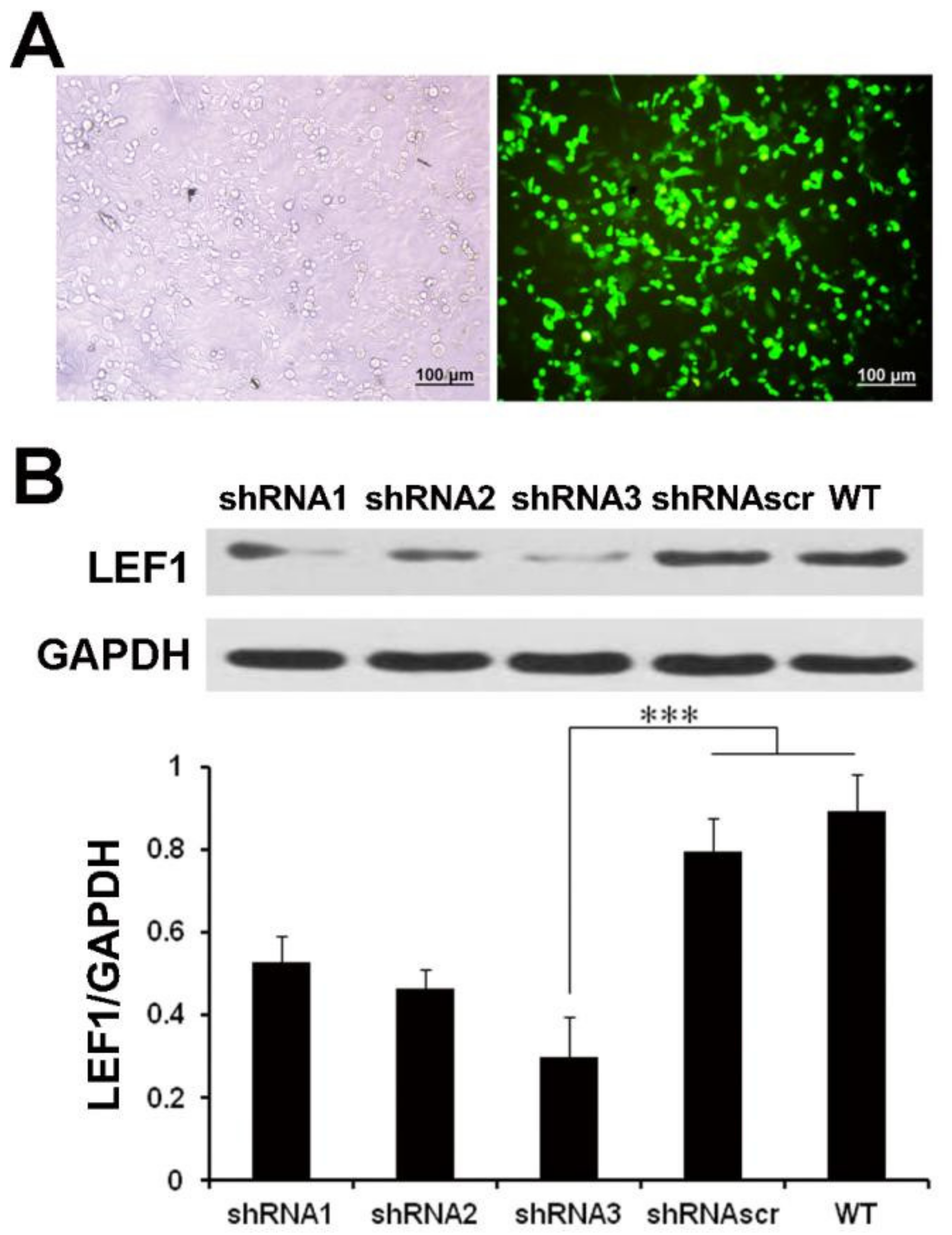
2.2. LEF1 Expression Was Efficiently Inhibited by Synthesized shRNA in Caco2 Cells
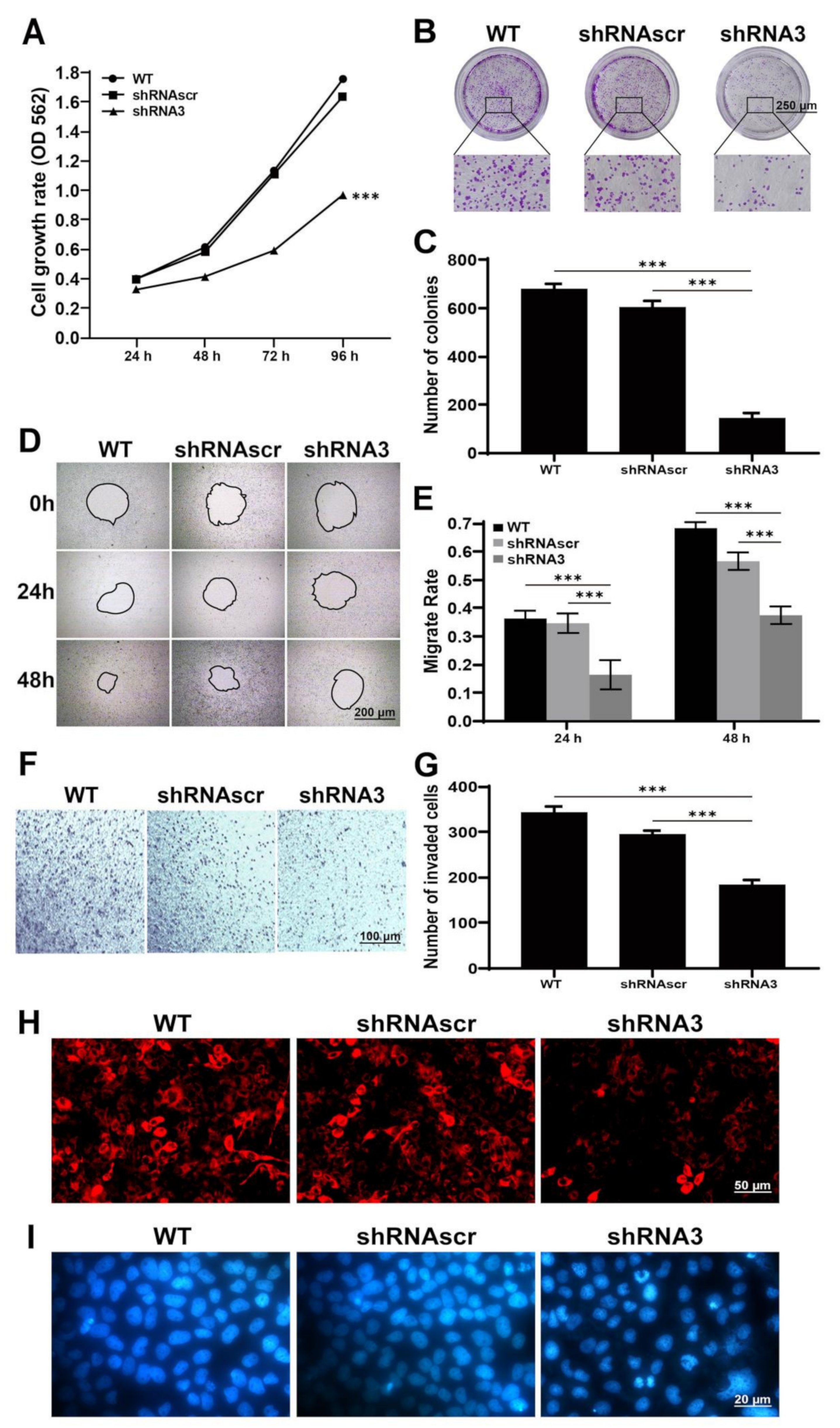
2.3. Downregulation of LEF1 Inhibited Cell Proliferation and Motility and Induced Apoptosis
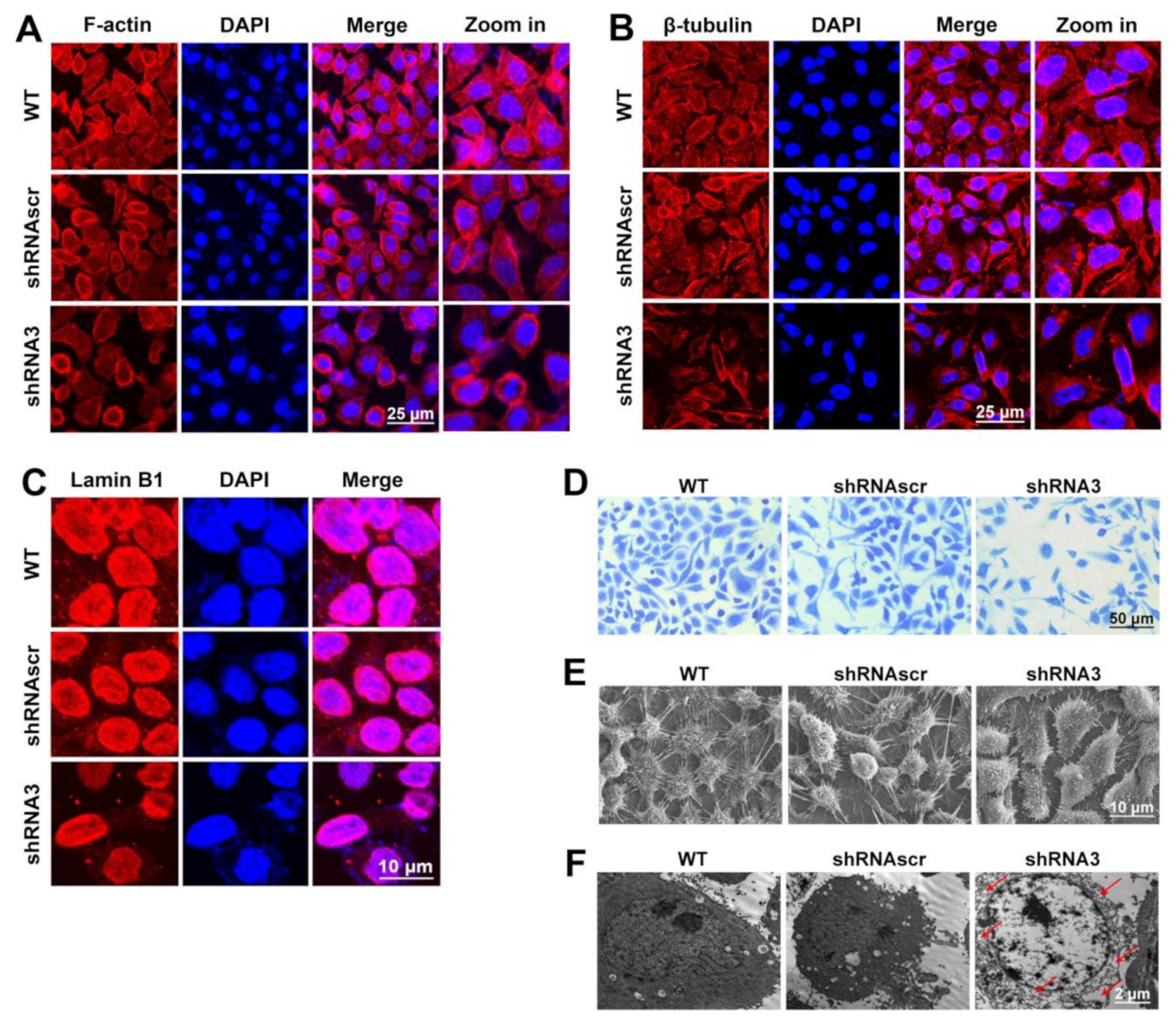
2.4. LEF1 Inhibition Remodeled Motility-Associated Microstructures
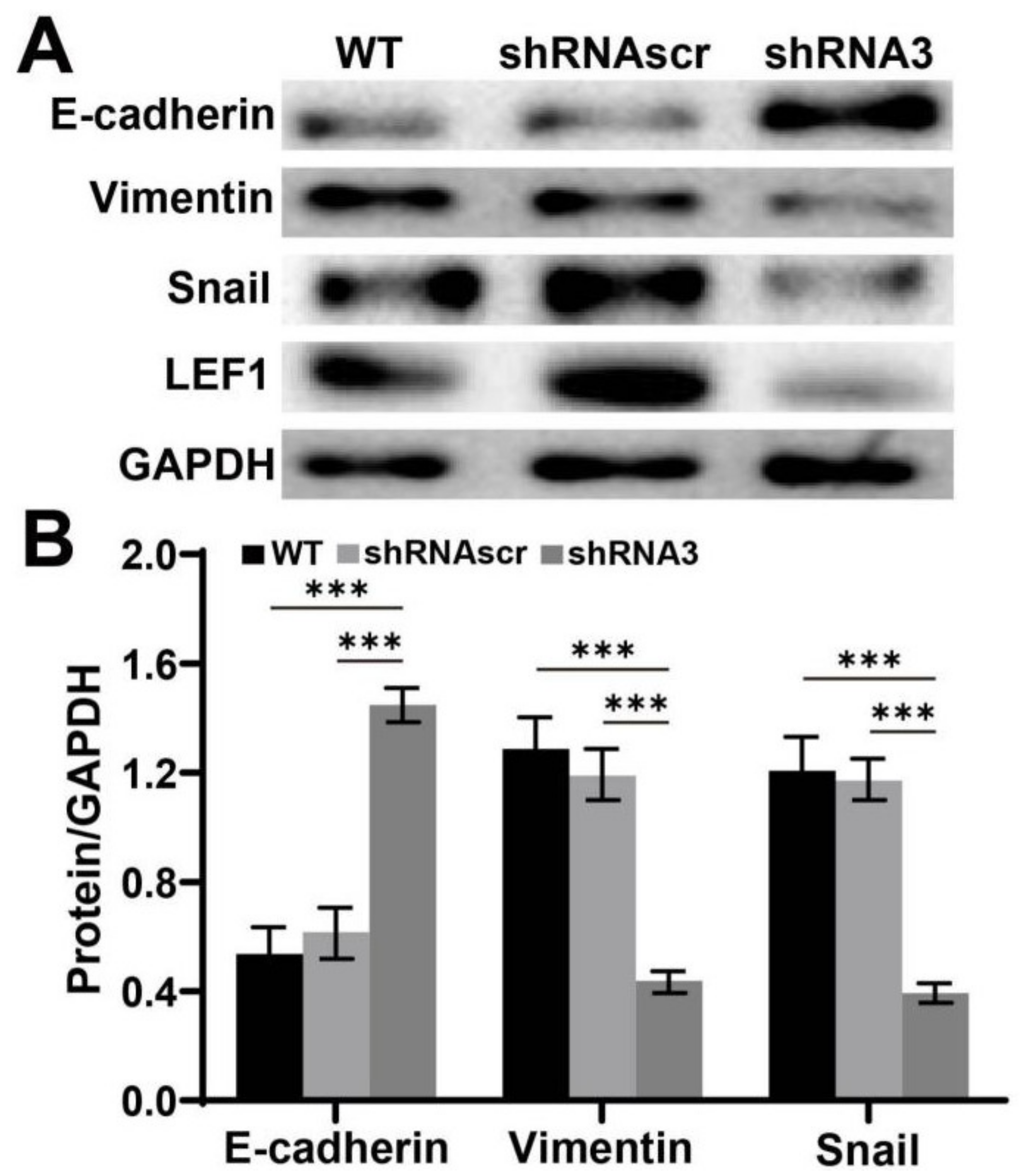
2.5. Downregulation of LEF1 Suppressed the Expression of EMT Relevant Genes in Caco2 Cells
3. Discussion
4. Materials and Methods
4.1. Colonic Adenocarcinoma Tissues
4.2. Immunofluorescence (IF)
4.3. GEPIA Data Collection
4.4. shRNA Plasmid Construction
4.5. Cell Culture and Transfection
4.6. Western Blot
4.7. MTT Assay
4.8. Colony Formation Assay
4.9. Wound Healing Assay
4.10. Transwell Assay
4.11. Apoptotic Morphology
4.12. Staining of the Microstructure-Associated Proteins
4.13. Coomassie Brilliant Blue Staining
4.14. Observation of the Microstructural Changes
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, B.K.; Ward, E.; Kohler, B.A.; Eheman, C.; Zauber, A.G.; Anderson, R.N.; Jemal, A.; Schymura, M.J.; Lansdorp-Vogelaar, I.; Seeff, L.C.; et al. Annual report to the nation on the status of cancer, 1975–2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer 2010, 116, 544–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, R.J.; Zhou, D.; Zhou, L.; Liu, Y. Wnt/β-catenin signaling and kidney fibrosis. Kidney Int. Suppl. 2014, 4, 84–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrens, J.; von Kries, J.P.; Kühl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Petropoulos, K.; Arseni, N.; Schessl, C.; Stadler, C.R.; Rawat, V.P.; Deshpande, A.J.; Heilmeier, B.; Hiddemann, W.; Quintanilla-Martinez, L.; Bohlander, S.K.; et al. A novel role for Lef-1, a central transcription mediator of Wnt signaling, in leukemogenesis. J. Exp. Med. 2008, 205, 515–522. [Google Scholar] [CrossRef] [Green Version]
- Xing, S.; Li, F.; Zeng, Z.; Zhao, Y.; Yu, S.; Shan, Q.; Li, Y.; Phillips, F.C.; Maina, P.K.; Qi, H.H.; et al. Tcf1 and Lef1 transcription factors establish CD8(+) T cell identity through intrinsic HDAC activity. Nat. Immunol. 2016, 17, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Tang, T.; Pang, J.; Xu, J.; Yang, X.; Wang, L.; Huang, Y.; Huang, Z.; Liu, G.; Tong, D.; et al. LSD1 Promotes Bladder Cancer Progression by Upregulating LEF1 and Enhancing EMT. Front. Oncol. 2020, 10, 1234. [Google Scholar] [CrossRef]
- Xing, S.; Gai, K.; Li, X.; Shao, P.; Zeng, Z.; Zhao, X.; Zhao, X.; Chen, X.; Paradee, W.J.; Meyerholz, D.K.; et al. Tcf1 and Lef1 are required for the immunosuppressive function of regulatory T cells. J. Exp. Med. 2019, 216, 847–866. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhu, J.; Shi, B.; Wang, X.; Lu, Q.; Li, C.; Chen, H. The transcription factor LEF1 promotes tumorigenicity and activates the TGF-β signaling pathway in esophageal squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 304. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Son, Y.; Kang, C.J. A strong promoter activity of pre-B cell stage-specific Crlz1 gene is caused by one distal LEF-1 and multiple proximal Ets sites. Mol. Cell. 2011, 32, 67–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeppner, L.H.; Secreto, F.J.; Razidlo, D.F.; Whitney, T.J.; Westendorf, J.J. Lef1DeltaN binds beta-catenin and increases osteoblast activity and trabecular bone mass. J. Biol. Chem. 2011, 286, 10950–10959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago, L.; Daniels, G.; Wang, D.; Deng, F.M.; Lee, P. Wnt signaling pathway protein LEF1 in cancer, as a biomarker for prognosis and a target for treatment. Am. J. Cancer. Res. 2017, 7, 1389–1406. [Google Scholar]
- Han, W.; He, L.; Cao, B.; Zhao, X.; Zhang, K.; Li, Y.; Beck, P.; Zhou, Z.; Tian, Y.; Cheng, S.; et al. Differential expression of LEF1/TCFs family members in colonic carcinogenesis. Mol. Carcinog. 2017, 56, 2372–2381. [Google Scholar] [CrossRef]
- Yokoyama, N.N.; Pate, K.T.; Sprowl, S.; Waterman, M.L. A role for YY1 in repression of dominant negative LEF-1 expression in colon cancer. Nucleic Acids Res. 2010, 38, 6375–6388. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.H.; Lafita-Navarro, M.C.; Zacharias, L.; Borenstein-Auerbach, N.; Kim, M.; Barnes, S.; Kim, J.; Shay, J.; DeBerardinis, R.J.; Conacci-Sorrell, M. Induction of LEF1 by MYC activates the WNT pathway and maintains cell proliferation. Cell Commun. Signal. 2019, 17, 129. [Google Scholar] [CrossRef] [Green Version]
- Fife, C.M.; McCarroll, J.A.; Kavallaris, M. Movers and shakers: Cell cytoskeleton in cancer metastasis. Br. J. Pharmacol. 2014, 171, 5507–5523. [Google Scholar] [CrossRef] [Green Version]
- Sanz-Moreno, V.; Marshall, C.J. The plasticity of cytoskeletal dynamics underlying neoplastic cell migration. Curr. Opin. Cell Biol. 2010, 22, 690–696. [Google Scholar] [CrossRef]
- Rottner, K.; Stradal, T.E. Actin dynamics and turnover in cell motility. Curr. Opin. Cell Biol. 2011, 23, 569–578. [Google Scholar] [CrossRef]
- Hall, A. The cytoskeleton and cancer. Cancer Metastasis Rev. 2009, 28, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Schenk, M.; Aykut, B.; Teske, C.; Giese, N.A.; Weitz, J.; Welsch, T. Salinomycin inhibits growth of pancreatic cancer and cancer cell migration by disruption of actin stress fiber integrity. Cancer Lett. 2015, 358, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Okada, N.; Sato, M. Spatiotemporal regulation of nuclear transportmachinery and microtuble organization. Cells 2015, 4, 406–426. [Google Scholar] [CrossRef] [Green Version]
- Hutchison, C.J. B-type lamins in health and disease. Semin. Cell Dev. Biol. 2014, 29, 158–163. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Mi, Y.; Ma, Y.; Jin, W. LEF1 regulates glioblastoma cell proliferation, migration, invasion, and cancer stem-like cell self-renewal. Tumour Biol. 2014, 35, 11505–11511. [Google Scholar] [CrossRef]
- Yuan, M.; Zhang, X.; Zhang, J.; Wang, K.; Zhang, Y.; Shang, W.; Zhang, Y.; Cui, J.; Shi, X.; Na, H.; et al. DC-SIGN-LEF1/TCF1-miR-185 feedback loop promotes colorectal cancer invasion and metastasis. Cell Death Differ. 2020, 27, 379–395. [Google Scholar] [CrossRef]
- Kim, G.H.; Fang, X.Q.; Lim, W.J.; Park, J.; Kang, T.B.; Kim, J.H.; Lim, J.H. Cinobufagin Suppresses Melanoma Cell Growth by Inhibiting LEF1. Int. J. Mol. Sci. 2020, 21, 6706. [Google Scholar] [CrossRef]
- Blazquez, R.; Rietkötter, E.; Wenske, B.; Wlochowitz, D.; Sparrer, D.; Vollmer, E.; Müller, G.; Seegerer, J.; Sun, X.; Dettmer, K.; et al. LEF1 supports metastatic brain colonization by regulating glutathione metabolism and increasing ROS resistance in breast cancer. Int. J. Cancer 2020, 146, 3170–3183. [Google Scholar] [CrossRef] [Green Version]
- Radspieler, M.M.; Schindeldecker, M.; Stenzel, P.; Försch, S.; Tagscherer, K.E.; Herpel, E.; Hohenfellner, M.; Hatiboglu, G.; Roth, W.; Macher-Goeppinger, S. Lamin-B1 is a senescence-associated biomarker in clear-cell renal cell carcinoma. Oncol. Lett. 2019, 18, 2654–2660. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal 2014, 7, re8. [Google Scholar] [CrossRef] [Green Version]
- Yook, J.I.; Li, X.Y.; Ota, I.; Fearon, E.R.; Weiss, S.J. Wnt-dependent regulation of the E-cadherin repressor snail. J. Biol. Chem. 2005, 280, 11740–11748. [Google Scholar] [CrossRef] [Green Version]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef]
- Ye, Y.C.; Zhao, J.L.; Lu, Y.T.; Gao, C.C.; Yang, Y.; Liang, S.Q.; Lu, Y.Y.; Wang, L.; Yue, S.Q.; Dou, K.F.; et al. NOTCH Signaling via WNT Regulates the Proliferation of Alternative, CCR2-Independent Tumor-Associated Macrophages in Hepatocellular Carcinoma. Cancer Res. 2019, 79, 4160–4172. [Google Scholar] [CrossRef]
- Kaemmerer, E.; Jeon, M.K.; Berndt, A.; Liedtke, C.; Gassler, N. Targeting Wnt Signaling via Notch in Intestinal Carcinogenesis. Cancers 2019, 11, 555. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Khan, S.K.; Gvozdenovic-Jeremic, J.; Kim, Y.; Dahlman, J.; Kim, H.; Park, O.; Ishitani, T.; Jho, E.H.; Gao, B.; et al. Hippo signaling interactions with Wnt/β-catenin and Notch signaling repress liver tumorigenesis. J. Clin. Invest. 2017, 127, 137–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The Varied Roles of Notch in Cancer. Annu. Rev. Pathol. 2017, 12, 245–275. [Google Scholar] [CrossRef] [Green Version]
- Majumder, S.; Crabtree, J.S.; Golde, T.E.; Minter, L.M.; Osborne, B.A.; Miele, L. Targeting Notch in oncology: The path forward. Nat. Rev. Drug Discov. 2021, 20, 125–144. [Google Scholar] [CrossRef] [PubMed]
- Ruland, J. Colon Cancer: Epithelial Notch Signaling Recruits Neutrophils to Drive Metastasis. Cancer Cell 2019, 36, 213–214. [Google Scholar] [CrossRef] [PubMed]
- Spuul, P.; Daubon, T.; Pitter, B.; Alonso, F.; Fremaux, I.; Kramer, I.; Montanez, E.; Génot, E. VEGF-A/Notch-Induced Podosomes Proteolyse Basement Membrane Collagen-IV during Retinal Sprouting Angiogenesis. Cell Rep. 2016, 17, 484–500. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| shRNAs | Sequences (5′-3′) |
|---|---|
| shRNA1 | GCGATTTAGCTGACATCAA |
| shRNA2 | AGATGTCAACTCCAAACAA |
| shRNA3 | GTTGCTGAGTGTACTCTAA |
| shRNAscr | TTCTCCGAACGTGTCACGT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, L.; Zhang, C.; Li, X.; Jia, C.; Chen, L.; Yuan, Y.; Gao, Q.; Lu, Z.; Feng, Y.; Zhao, R.; et al. LEF1 Enhances the Progression of Colonic Adenocarcinoma via Remodeling the Cell Motility Associated Structures. Int. J. Mol. Sci. 2021, 22, 10870. https://doi.org/10.3390/ijms221910870
Xiao L, Zhang C, Li X, Jia C, Chen L, Yuan Y, Gao Q, Lu Z, Feng Y, Zhao R, et al. LEF1 Enhances the Progression of Colonic Adenocarcinoma via Remodeling the Cell Motility Associated Structures. International Journal of Molecular Sciences. 2021; 22(19):10870. https://doi.org/10.3390/ijms221910870
Chicago/Turabian StyleXiao, Li, Caixia Zhang, Xinyao Li, Chenshuang Jia, Lirong Chen, Yue Yuan, Qian Gao, Zheng Lu, Yang Feng, Ruixia Zhao, and et al. 2021. "LEF1 Enhances the Progression of Colonic Adenocarcinoma via Remodeling the Cell Motility Associated Structures" International Journal of Molecular Sciences 22, no. 19: 10870. https://doi.org/10.3390/ijms221910870
APA StyleXiao, L., Zhang, C., Li, X., Jia, C., Chen, L., Yuan, Y., Gao, Q., Lu, Z., Feng, Y., Zhao, R., Zhao, X., Cheng, S., Shu, Z., Xu, J., Duan, W., Nie, G., & Hou, Y. (2021). LEF1 Enhances the Progression of Colonic Adenocarcinoma via Remodeling the Cell Motility Associated Structures. International Journal of Molecular Sciences, 22(19), 10870. https://doi.org/10.3390/ijms221910870