
Genetically Encoded Biosensors to Monitor Intracellular Reactive Oxygen and Nitrogen Species and Glutathione Redox Potential in Skeletal Muscle Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
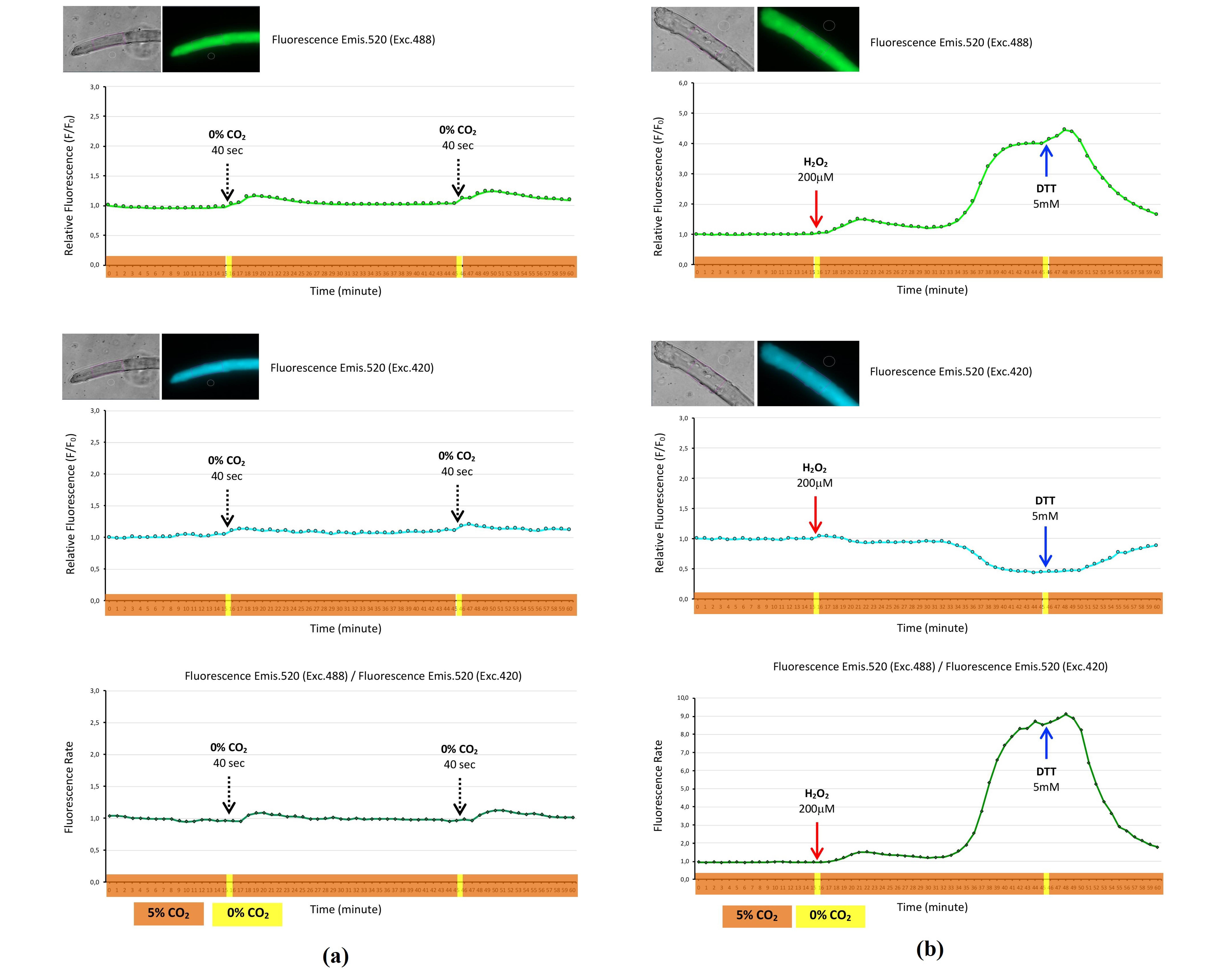
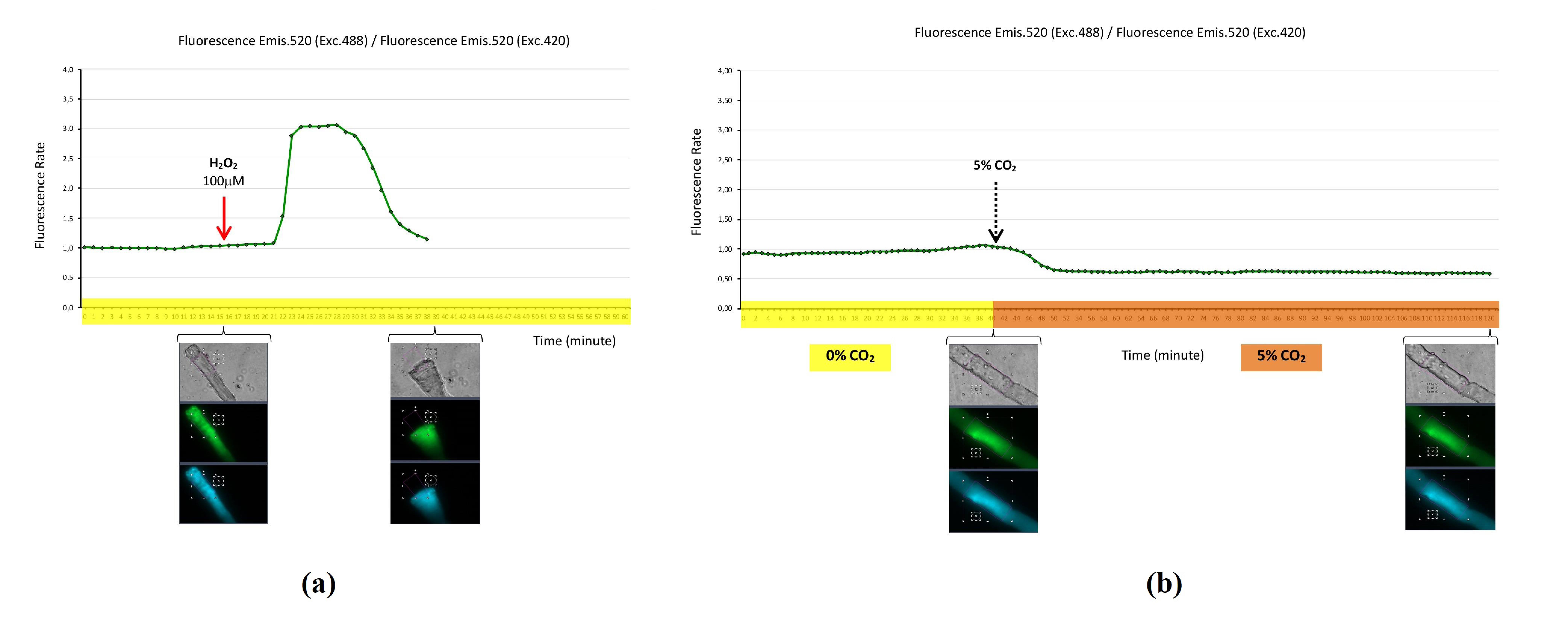
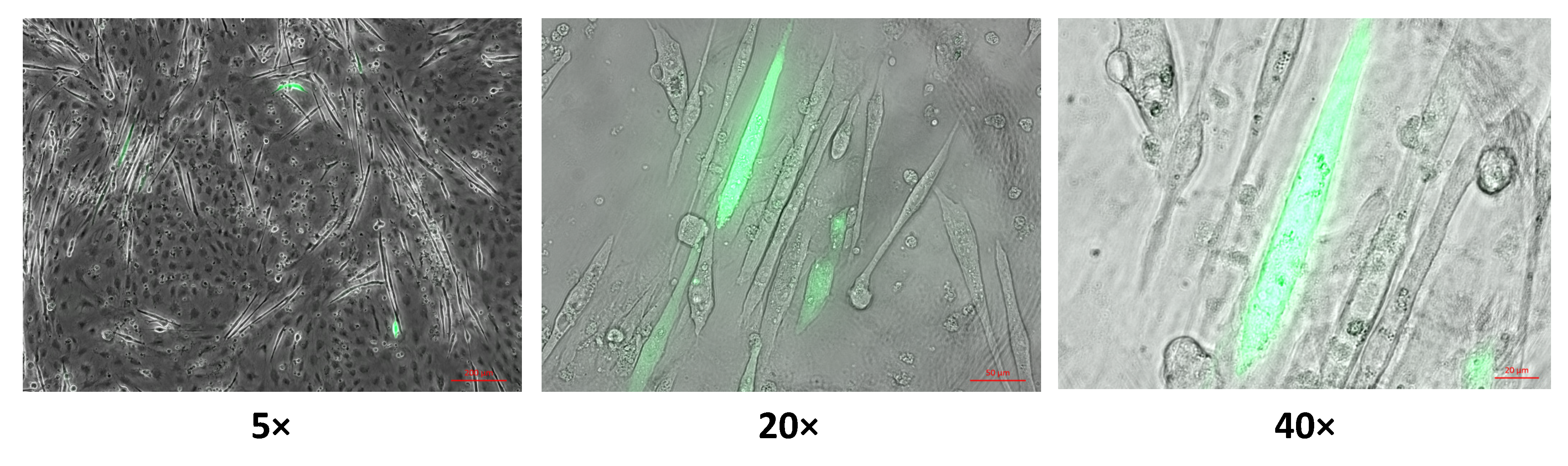
2.1. Hydrogen Peroxide Biosensor HyPer3 for the Detection of Cytosolic Hydrogen Peroxide in Skeletal Muscle Fibers
2.2. Mitochondrial Hydrogen Peroxide Biosensor HyPer-Mito for the Detection of Mitochondrial Hydrogen Peroxide in Skeletal Muscle Fibers
2.3. Nuclear Hydrogen Peroxide Biosensor HyPer-nuc for the Detection of Nuclear Hydrogen Peroxide in Skeletal Muscle Fibers
2.4. Hydrogen Peroxide Biosensor roGFP2-Orp1 for the Detection of Cytosolic H2O2 in Skeletal Muscle Fibers
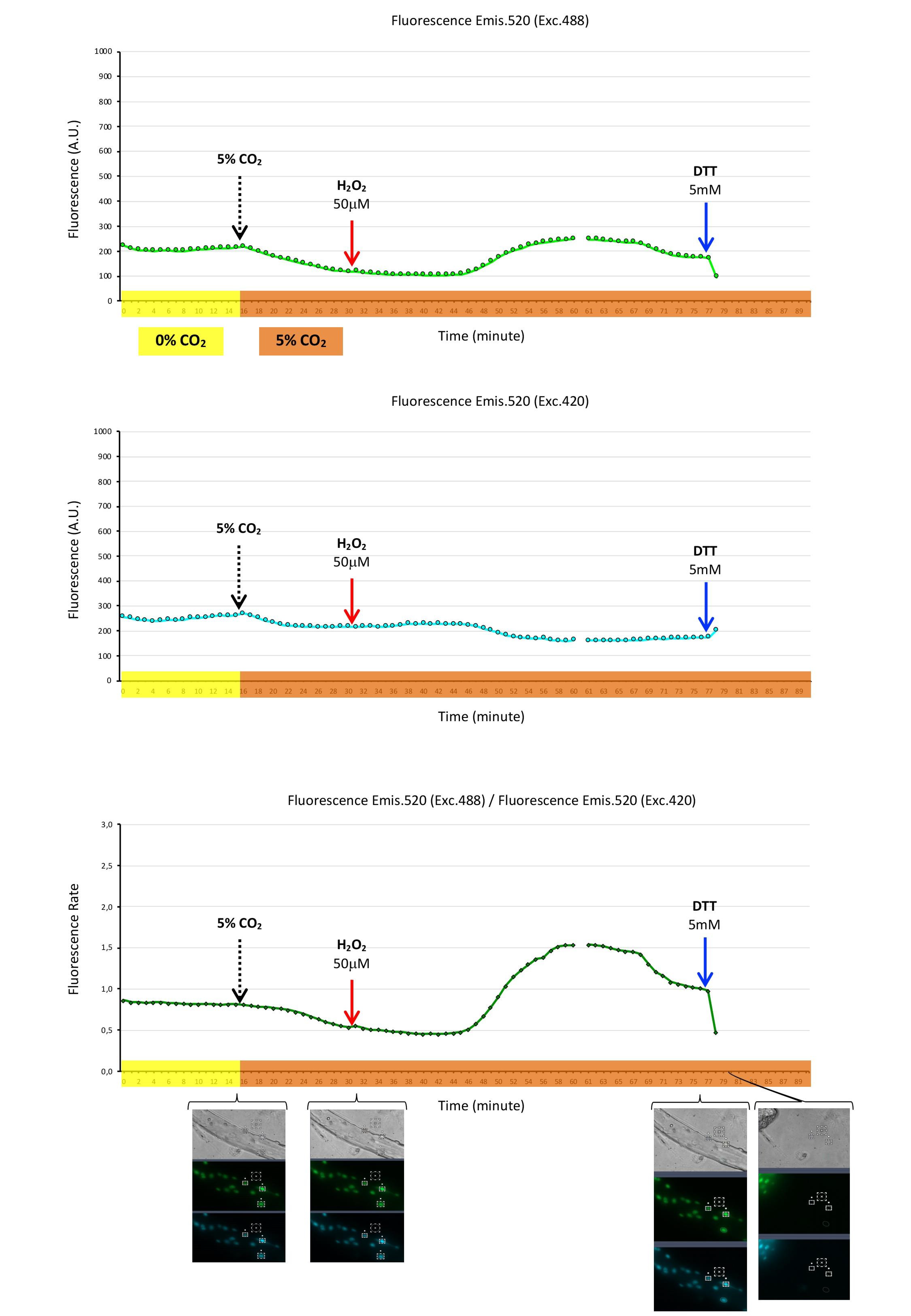
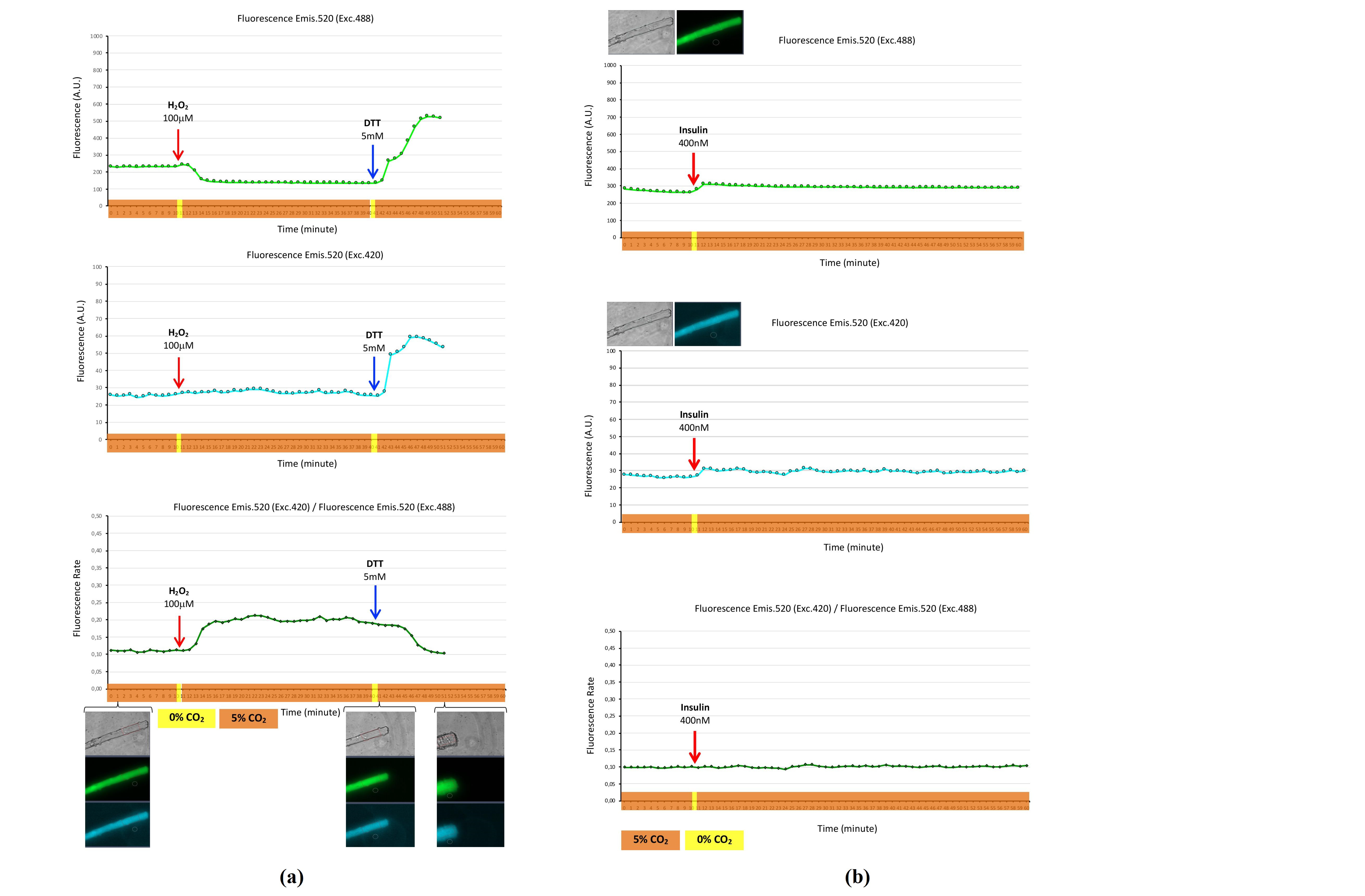
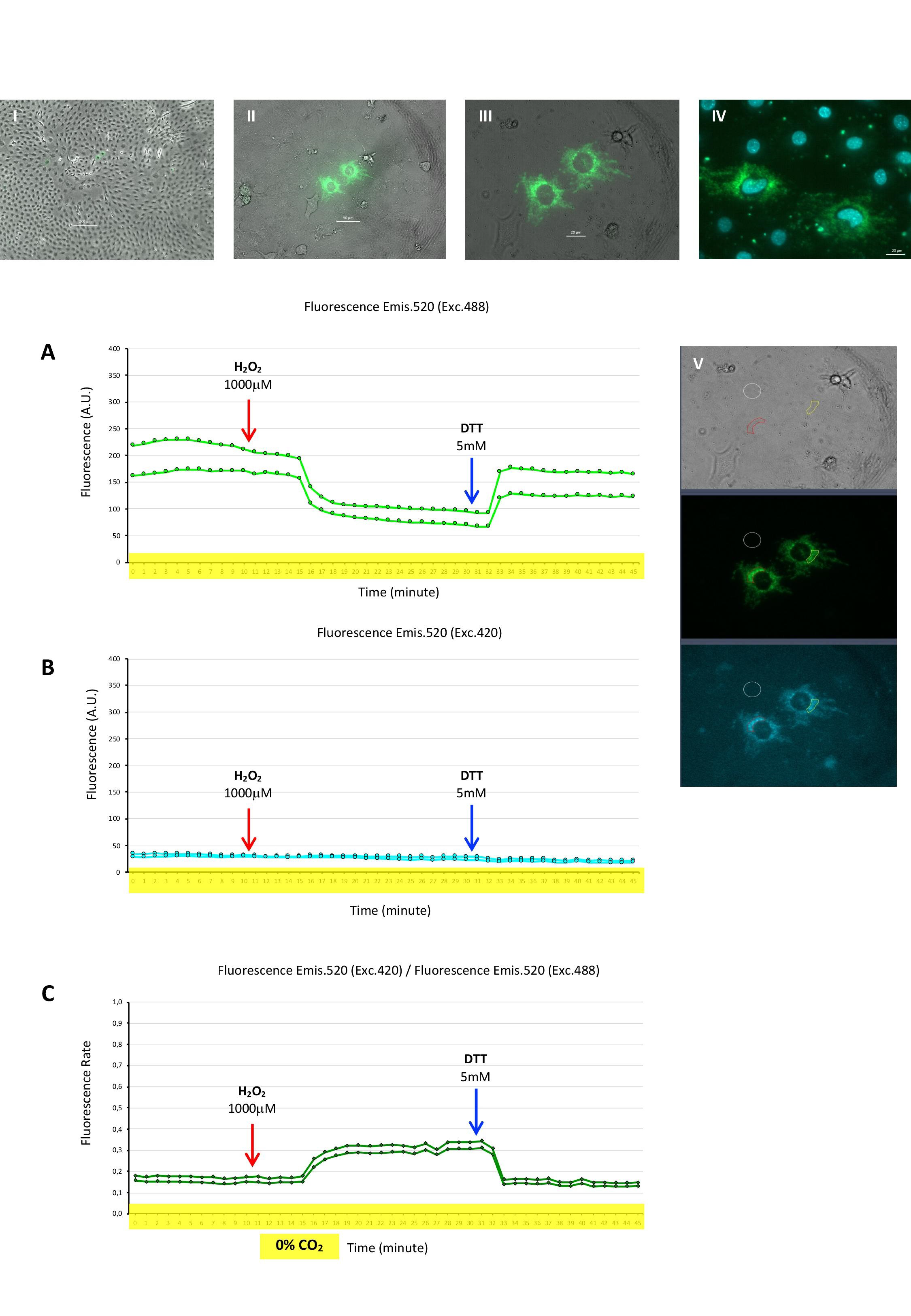
2.5. Mito-Grx1-roGFP2 Biosensor to Register Mitochondrial Glutathione Redox Potential (GSH/GSSG) in Myoblasts
2.6. Cyto-Grx1-roGFP2 Biosensor to Register Cytosolic Glutathione Redox Potential (GSH/GSSG) in Myotubes
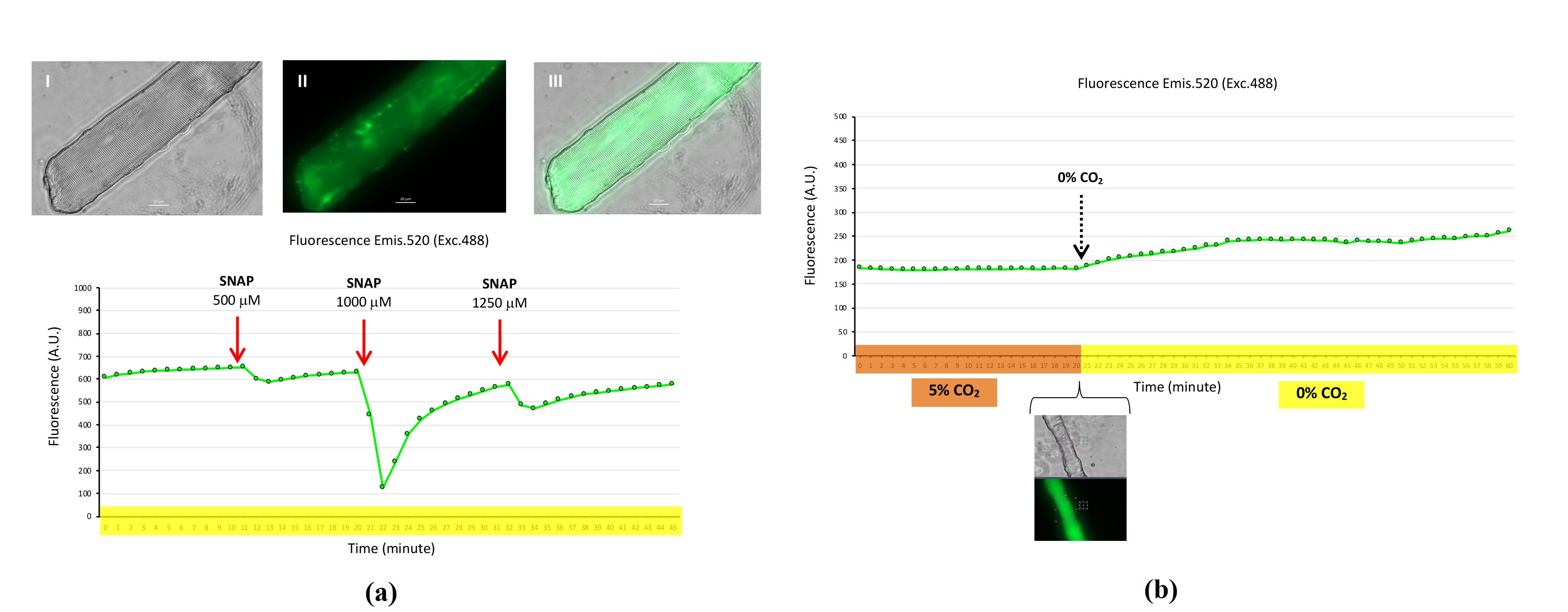
2.7. G-geNOp Biosensor to Detect Cytosolic Nitric Oxide in Isolated Skeletal Muscle Fibers
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Skeletal Muscle Cell Culture
4.3. Animals
4.4. Genetically Encoded Biosensors
4.5. Transfection of Biosensors into Flexor Digitorum Brevis (FDB) Mouse Muscle
4.6. Skeletal Muscle Fibers Isolation
4.7. Fluorescence Microscopy and Quantitative Image Analysis
5. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flück, M.; Hoppeler, H. Molecular basis of skeletal muscle plasticity-from gene to form and function. In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Berlin/Heidelberg, Germany, 2003; Volume 146, pp. 159–216. [Google Scholar] [CrossRef]
- Le Moal, E.; Pialoux, V.; Juban, G.; Groussard, C.; Zouhal, H.; Chazaud, B.; Mounier, R. Redox Control of Skeletal Muscle Regeneration. Antioxid. Redox Signal. 2017, 27, 276–310. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Jackson, M.J. Exercise-Induced Oxidative Stress: Cellular Mechanisms and Impact on Muscle Force Production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef] [PubMed]
- Palomero, J.; Jackson, M. Redox regulation in skeletal muscle during contractile activity and aging 1. J. Anim. Sci. 2010, 88, 1307–1313. [Google Scholar] [CrossRef]
- Szabó, C.; Ischiropoulos, H.; Radi, R. Peroxynitrite: Biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov. 2007, 6, 662–680. [Google Scholar] [CrossRef]
- Sakellariou, G.K.; Vasilaki, A.; Palomero, J.; Kayani, A.; Zibrik, L.; McArdle, A.; Jackson, M. Studies of Mitochondrial and Nonmitochondrial Sources Implicate Nicotinamide Adenine Dinucleotide Phosphate Oxidase(s) in the Increased Skeletal Muscle Superoxide Generation That Occurs During Contractile Activity. Antioxid. Redox Signal. 2013, 18, 603–621. [Google Scholar] [CrossRef]
- Powers, S.K.; Nelson, W.B.; Hudson, M. Exercise-induced oxidative stress in humans: Cause and consequences. Free Radic. Biol. Med. 2011, 51, 942–950. [Google Scholar] [CrossRef]
- Gomez-Cabrera, M.-C.; Borrás, C.; Pallardó, F.V.; Sastre, J.; Ji, L.L.; Viña, J. Decreasing xanthine oxidase-mediated oxidative stress prevents useful cellular adaptations to exercise in rats. J. Physiol. 2005, 567, 113–120. [Google Scholar] [CrossRef]
- Jackson, M.J. Control of Reactive Oxygen Species Production in Contracting Skeletal Muscle. Antioxid. Redox Signal. 2011, 15, 2477–2486. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Valle, N.R.-D.; Huang, P. Redox Regulation of Cell Survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Pouvreau, S. Genetically encoded reactive oxygen species (ROS) and redox indicators. Biotechnol. J. 2014, 9, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, E.; Gottschalk, B.; Charoensin, S.; Blass, S.; Bischof, H.; Rost, R.; Madreiter-Sokolowski, C.T.; Pelzmann, B.; Bernhart, E.; Sattler, W.; et al. Development of novel FP-based probes for live-cell imaging of nitric oxide dynamics. Nat. Commun. 2016, 7, 10623. [Google Scholar] [CrossRef] [PubMed]
- Belousov, V.V.; Fradkov, A.F.; Lukyanov, K.; Staroverov, D.; Shakhbazov, K.S.; Terskikh, A.V.; Lukyanov, S. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat. Methods 2006, 3, 281–286. [Google Scholar] [CrossRef]
- Bilan, D.; Belousov, V.V. In Vivo Imaging of Hydrogen Peroxide with HyPer Probes. Antioxid. Redox Signal. 2018, 29, 569–584. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, A.; Campos, C.; Díaz-Vegas, A.; Galgani, J.E.; Juretic, N.; Osorio-Fuentealba, C.; Bucarey, J.L.; Tapia, G.; Valenzuela, R.; Contreras-Ferrat, A.; et al. Insulin-Dependent H2O2 Production Is Higher in Muscle Fibers of Mice Fed with a High-Fat Diet. Int. J. Mol. Sci. 2013, 14, 15740–15754. [Google Scholar] [CrossRef]
- Díaz-Vegas, A.; Campos, C.A.; Contreras-Ferrat, A.; Casas, M.; Buvinic, S.; Jaimovich, E.; Espinosa, A. ROS Production via P2Y1-PKC-NOX2 Is Triggered by Extracellular ATP after Electrical Stimulation of Skeletal Muscle Cells. PLoS ONE 2015, 10, e0129882. [Google Scholar] [CrossRef]
- Pearson, T.; Kabayo, T.; Ng, R.; Chamberlain, J.; McArdle, A.; Jackson, M.J. Skeletal Muscle Contractions Induce Acute Changes in Cytosolic Superoxide, but Slower Responses in Mitochondrial Superoxide and Cellular Hydrogen Peroxide. PLoS ONE 2014, 9, e96378. [Google Scholar] [CrossRef]
- Fernández-Puente, E.; Sánchez-Martín, M.A.; De Andrés, J.; Rodríguez-Izquierdo, L.; Méndez, L.; Palomero, J. Expression and functional analysis of the hydrogen peroxide biosensors HyPer and HyPer2 in C2C12 myoblasts/myotubes and single skeletal muscle fibres. Sci. Rep. 2020, 10, 871. [Google Scholar] [CrossRef]
- Bilan, D.S.; Pase, L.; Joosen, L.; Gorokhovatsky, A.Y.; Ermakova, Y.G.; Gadella, T.W.J.; Grabher, C.; Schultz, C.; Lukyanov, S.; Belousov, V.V. HyPer-3: A Genetically Encoded H2O2 Probe with Improved Performance for Ratiometric and Fluorescence Lifetime Imaging. ACS Chem. Biol. 2013, 8, 535–542. [Google Scholar] [CrossRef]
- Quatresous, E.; Legrand, C.; Pouvreau, S. Mitochondria-targeted cpYFP: pH or superoxide sensor? J. Gen. Physiol. 2012, 140, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Malinouski, M.; Zhou, Y.; Belousov, V.V.; Hatfield, L.L.; Gladyshev, V.N. Hydrogen Peroxide Probes Directed to Different Cellular Compartments. PLoS ONE 2011, 6, e14564. [Google Scholar] [CrossRef] [PubMed]
- Plecitá-Hlavatá, L.; Engstová, H.; Holendová, B.; Tauber, J.; Špaček, T.; Petrásková, L.; Křen, V.; Špačková, J.; Gotvaldová, K.; Ježek, J.; et al. Mitochondrial Superoxide Production Decreases on Glucose-Stimulated Insulin Secretion in Pancreatic β Cells Due to Decreasing Mitochondrial Matrix NADH/NAD + Ratio. Antioxid. Redox Signal. 2020, 33, 789–815. [Google Scholar] [CrossRef]
- Matsushima, S.; Kuroda, J.; Ago, T.; Zhai, P.; Park, J.Y.; Xie, L.-H.; Tian, B.; Sadoshima, J. Increased Oxidative Stress in the Nucleus Caused by Nox4 Mediates Oxidation of HDAC4 and Cardiac Hypertrophy. Circ. Res. 2013, 112, 651–663. [Google Scholar] [CrossRef]
- Gutscher, M.; Sobotta, M.C.; Wabnitz, G.H.; Ballikaya, S.; Meyer, A.J.; Samstag, Y.; Dick, T.P. Proximity-based Protein Thiol Oxidation by H2O2-scavenging Peroxidases. J. Biol. Chem. 2009, 284, 31532–31540. [Google Scholar] [CrossRef]
- Morgan, B.; Sobotta, M.C.; Dick, T.P. Measuring EGSH and H2O2 with roGFP2-based redox probes. Free Radic. Biol. Med. 2011, 51, 1943–1951. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Schneider, J.F.; Degrossoli, A.; Lupilova, N.; Dick, T.P.; Leichert, L. Systematic in vitro assessment of responses of roGFP2-based probes to physiologically relevant oxidant species. Free Radic. Biol. Med. 2017, 106, 329–338. [Google Scholar] [CrossRef]
- Deglasse, J.-P.; Roma, L.P.; Pastor-Flores, D.; Gilon, P.; Dick, T.P.; Jonas, J.-C. Glucose Acutely Reduces Cytosolic and Mitochondrial H2O2 in Rat Pancreatic Beta Cells. Antioxid. Redox Signal. 2019, 30, 297–313. [Google Scholar] [CrossRef] [PubMed]
- Pouvreau, S. Beyond the Cuvette: Redox Indicators in Biological Experiments. Antioxid. Redox Signal. 2016, 25, 517–519. [Google Scholar] [CrossRef]
- Lukyanov, K.; Belousov, V.V. Genetically encoded fluorescent redox sensors. Biochim. Biophys. Acta (BBA) Gen. Subj. 2014, 1840, 745–756. [Google Scholar] [CrossRef]
- Sies, H. Glutathione and its role in cellular functions. Free Radic. Biol. Med. 1999, 27, 916–921. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive oxygen species in living systems: Source, biochemistry, and role in human disease. Am. J. Med. 1991, 91, S14–S22. [Google Scholar] [CrossRef]
- Palomero, J.; Vasilaki, A.; Pye, D.; McArdle, A.; Jackson, M.J. Aging increases the oxidation of dichlorohydrofluorescein in single isolated skeletal muscle fibers at rest, but not during contractions. Am. J. Physiol. Integr. Comp. Physiol. 2013, 305, R351–R358. [Google Scholar] [CrossRef]
- Gutscher, M.; Pauleau, A.-L.; Marty, L.; Brach, T.; Wabnitz, G.H.; Samstag, Y.; Meyer, A.J.; Dick, T.P. Real-time imaging of the intracellular glutathione redox potential. Nat. Methods 2008, 5, 553–559. [Google Scholar] [CrossRef]
- Nanadikar, M.S.; Leon, A.M.V.; Borowik, S.; Hillemann, A.; Zieseniss, A.; Belousov, V.V.; Bogeski, I.; Rehling, P.; Dudek, J.; Katschinski, D.M. O2 affects mitochondrial functionality ex vivo. Redox Biol. 2019, 22, 101152. [Google Scholar] [CrossRef] [PubMed]
- Trautsch, I.; Heta, E.; Soong, P.L.; Levent, E.; Nikolaev, V.O.; Bogeski, I.; Katschinski, D.M.; Mayr, M.; Zimmermann, W.-H. Optogenetic Monitoring of the Glutathione Redox State in Engineered Human Myocardium. Front. Physiol. 2019, 10, 272. [Google Scholar] [CrossRef] [PubMed]
- Swain, L.; Kesemeyer, A.; Meyer-Roxlau, S.; Vettel, C.; Zieseniss, A.; Güntsch, A.; Jatho, A.; Becker, A.; Nanadikar, M.S.; Morgan, B.; et al. Redox Imaging Using Cardiac Myocyte-Specific Transgenic Biosensor Mice. Circ. Res. 2016, 119, 1004–1016. [Google Scholar] [CrossRef] [PubMed]
- Hearon, C.M., Jr.; Dinenno, F.A. Regulation of skeletal muscle blood flow during exercise in ageing humans. J. Physiol. 2015, 8, 2261–2273. [Google Scholar] [CrossRef]
- Kobayashi, J.; Uchida, H.; Kofuji, A.; Ito, J.; Shimizu, M.; Kim, H.; Sekiguchi, Y.; Kushibe, S. Molecular regulation of skeletal muscle mass and the contribution of nitric oxide: A review. FASEB BioAdv. 2019, 1, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Pye, D.; Palomero, J.; Kabayo, T.; Jackson, M.J. Real-time measurement of nitric oxide in single mature mouse skeletal muscle fibres during contractions. J. Physiol. 2007, 581, 309–318. [Google Scholar] [CrossRef]
- Palomero, J.; Pye, D.; Kabayo, T.; Jackson, M.J. Effect of passive stretch on intracellular nitric oxide and superoxide activities in single skeletal muscle fibres: Influence of ageing. Free Radic. Res. 2011, 46, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Bryan, N.S.; Grisham, M.B. Methods to detect nitric oxide and its metabolites in biological samples. Free Radic. Biol. Med. 2007, 43, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, E.; Charoensin, S.; Bischof, H.; Ramadani-Muja, J.; Gottschalk, B.; Depaoli, M.R.; Waldeck-Weiermair, M.; Graier, W.; Malli, R. Genetic biosensors for imaging nitric oxide in single cells. Free Radic. Biol. Med. 2018, 128, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Palomero, J.; Pye, D.; Kabayo, T.; Spiller, D.; Jackson, M.J. In Situ Detection and Measurement of Intracellular Reactive Oxygen Species in Single Isolated Mature Skeletal Muscle Fibers by Real Time Fluorescence Microscopy. Antioxid. Redox Signal. 2008, 10, 1463–1474. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Puente, E.; Palomero, J. Genetically Encoded Biosensors to Monitor Intracellular Reactive Oxygen and Nitrogen Species and Glutathione Redox Potential in Skeletal Muscle Cells. Int. J. Mol. Sci. 2021, 22, 10876. https://doi.org/10.3390/ijms221910876
Fernández-Puente E, Palomero J. Genetically Encoded Biosensors to Monitor Intracellular Reactive Oxygen and Nitrogen Species and Glutathione Redox Potential in Skeletal Muscle Cells. International Journal of Molecular Sciences. 2021; 22(19):10876. https://doi.org/10.3390/ijms221910876
Chicago/Turabian StyleFernández-Puente, Escarlata, and Jesús Palomero. 2021. "Genetically Encoded Biosensors to Monitor Intracellular Reactive Oxygen and Nitrogen Species and Glutathione Redox Potential in Skeletal Muscle Cells" International Journal of Molecular Sciences 22, no. 19: 10876. https://doi.org/10.3390/ijms221910876
APA StyleFernández-Puente, E., & Palomero, J. (2021). Genetically Encoded Biosensors to Monitor Intracellular Reactive Oxygen and Nitrogen Species and Glutathione Redox Potential in Skeletal Muscle Cells. International Journal of Molecular Sciences, 22(19), 10876. https://doi.org/10.3390/ijms221910876