The Resistance of Narrow-Leafed Lupin to Diaporthe toxica Is Based on the Rapid Activation of Defense Response Genes

 , , , , ,
, , , , ,  and
and
Abstract
:1. Introduction
2. Results
2.1. Isolates of Diaporthe toxica Represented a Diverse Gene Pool That Is Related to the Host Plant
2.2. PhtjR and Phr1 Alleles Conferred a High Level of Resistance to Diaporthe toxica
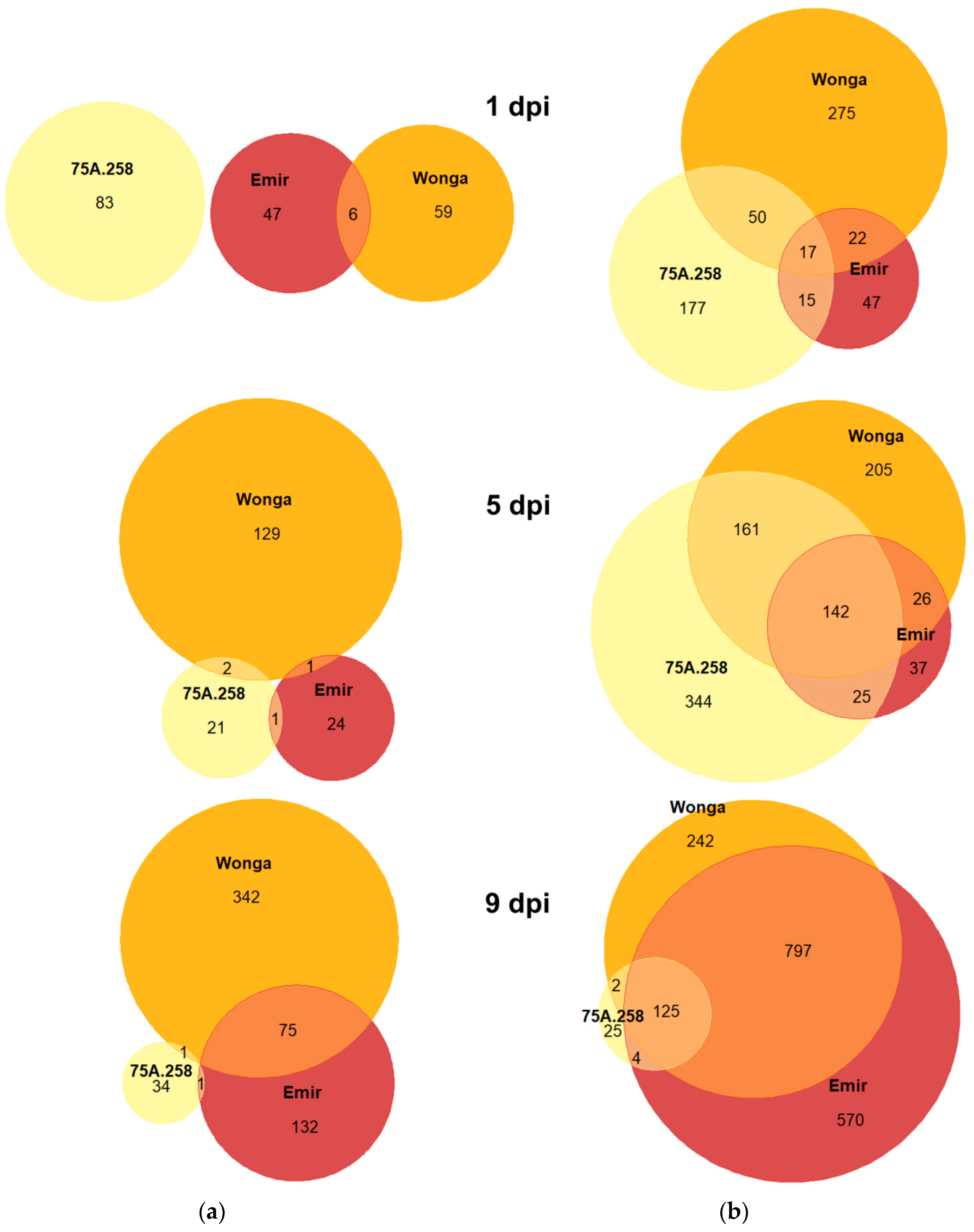
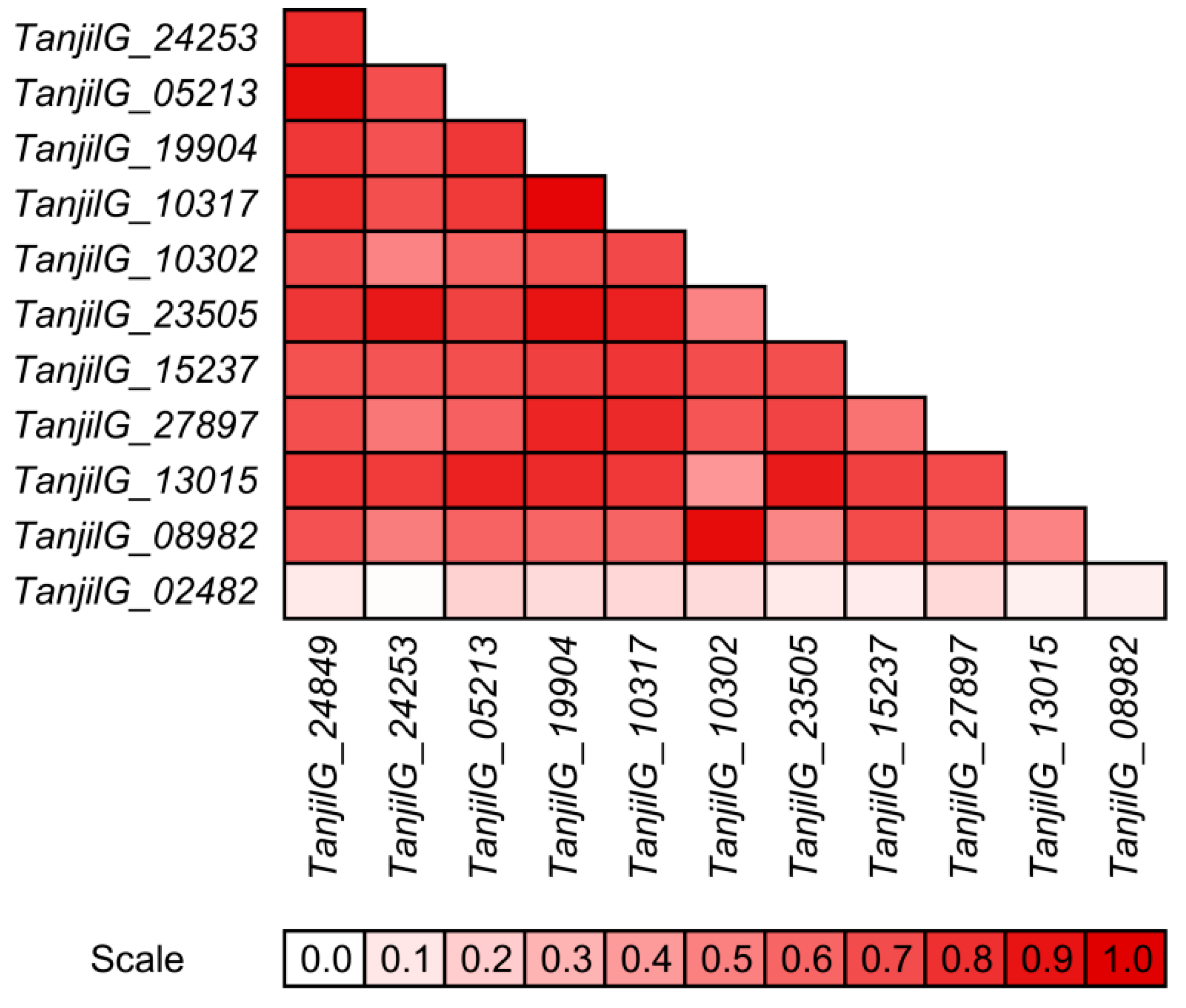
2.3. Resistance to D. toxica Was Associated with Rapid Transcriptome Reprogramming
2.4. Early Reaction of Resistant Lines Was Based on the Activation of the Defense Response and Oxylipin Biosynthesis Genes
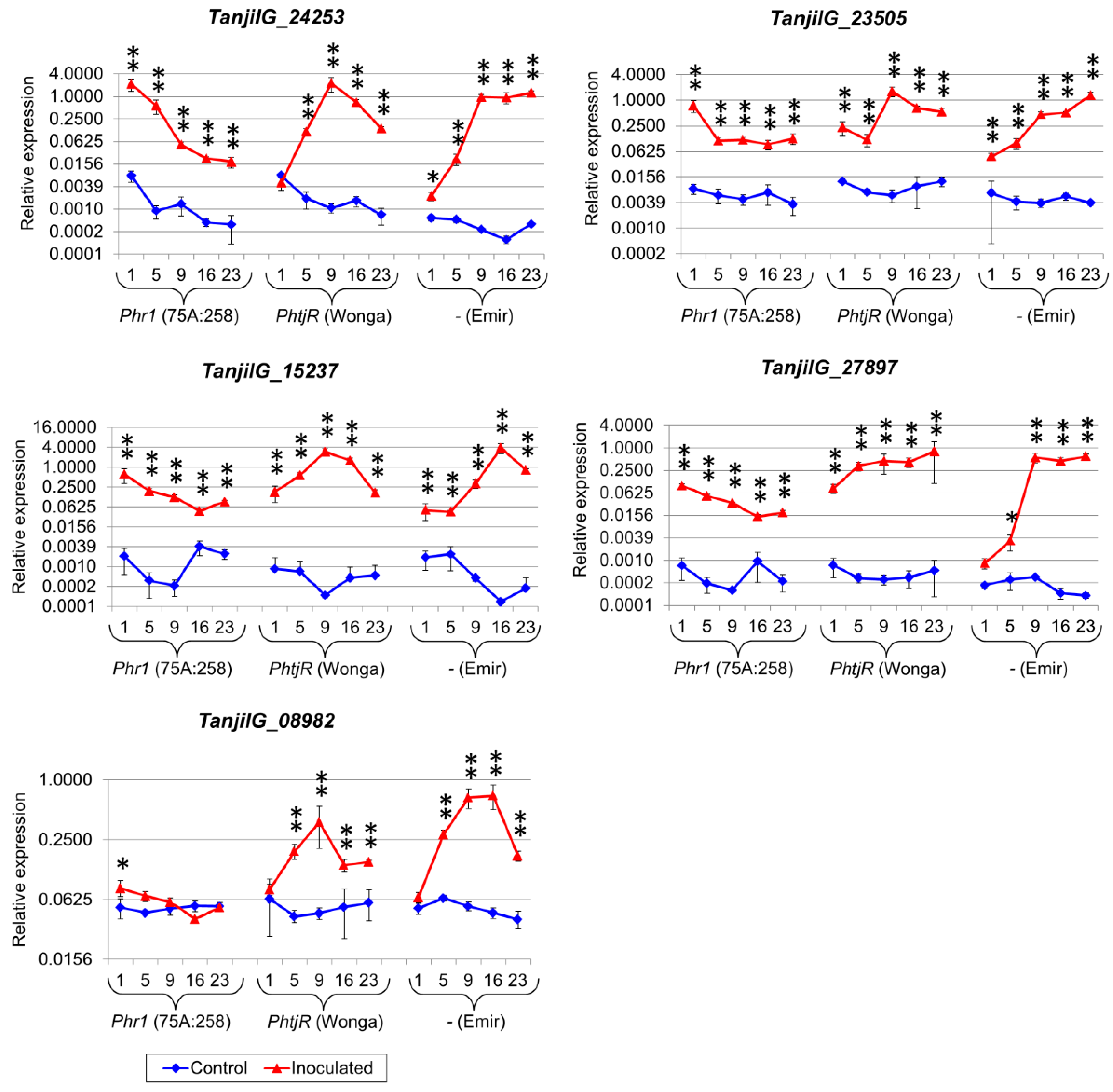
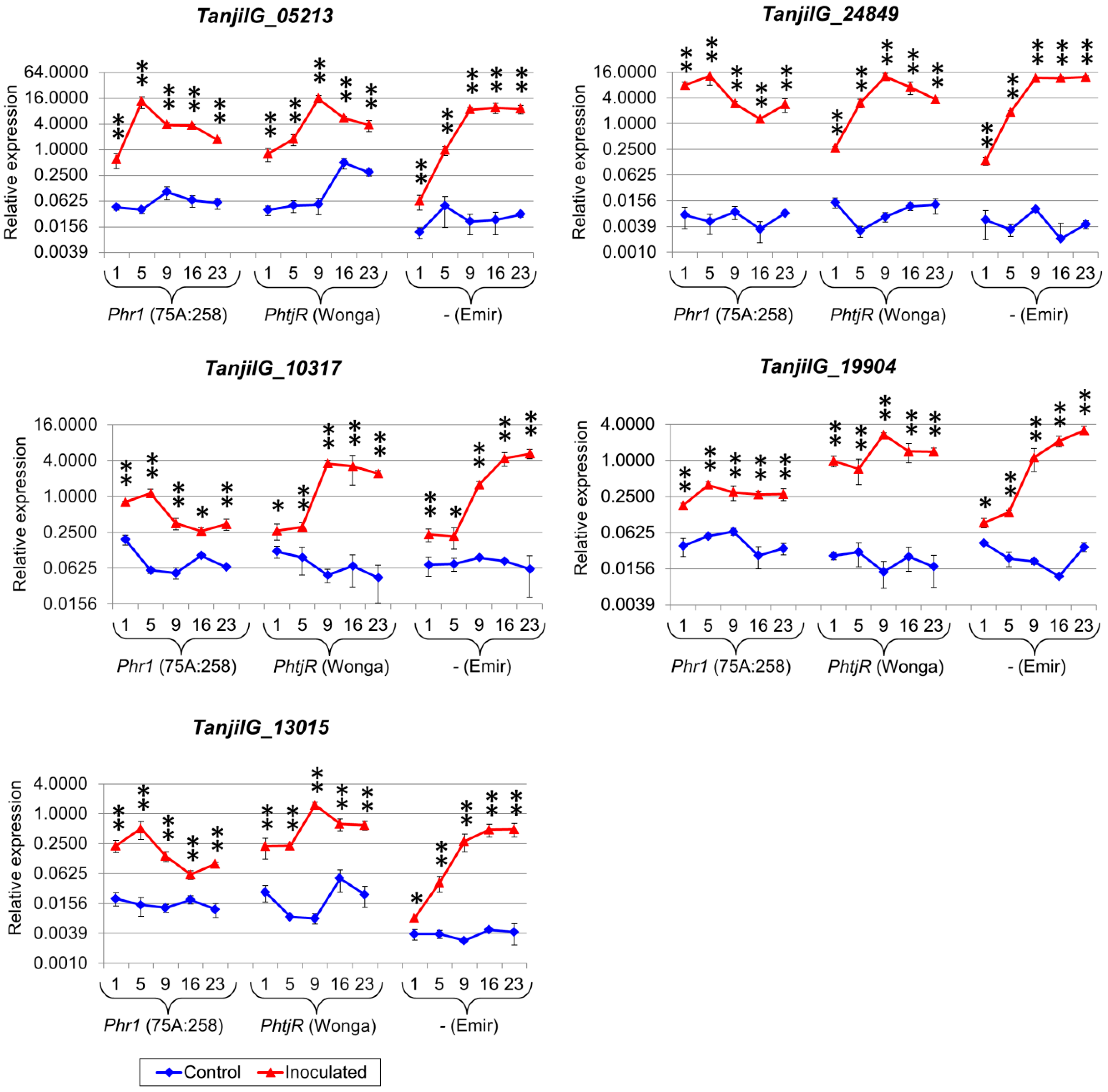
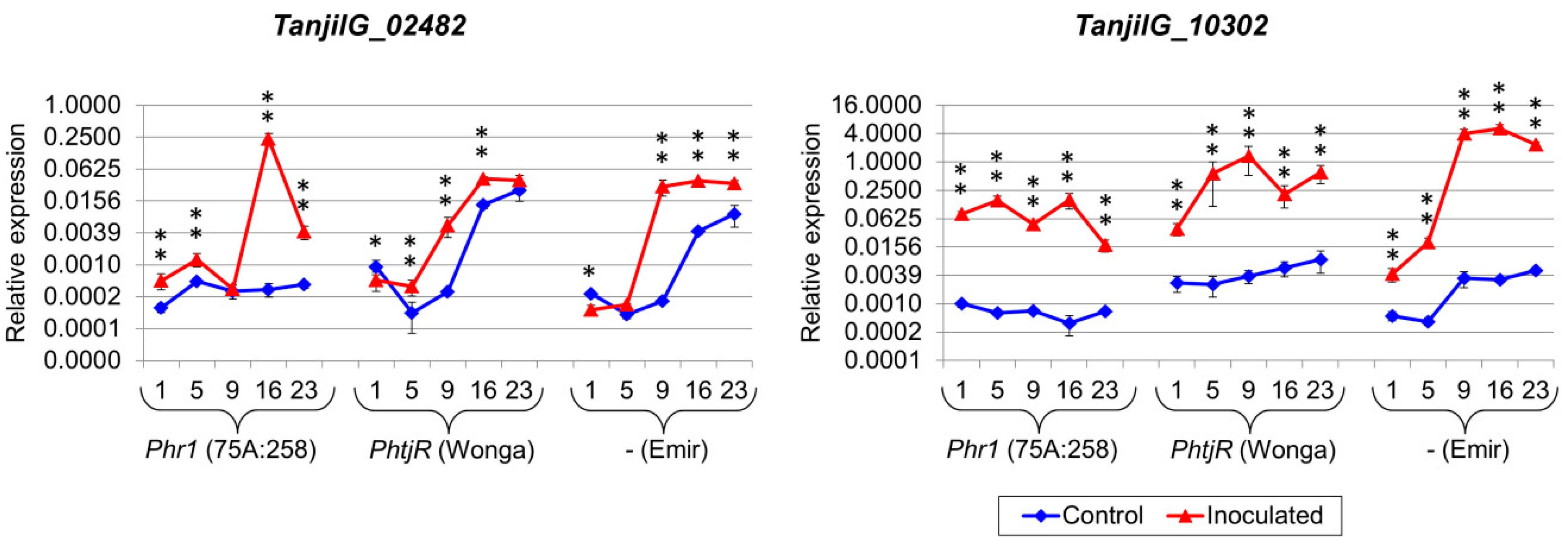
2.5. Genes from Defense Response Pathways Experienced up to a Thousandfold Upregulation in Inoculated Plants
3. Discussion
3.1. Genotype Profiling of Diaporthe toxica Isolates
3.2. Mechanisms Involved in Lupinus angustifolius’ Defense Response to Diaporthe toxica
3.2.1. Peroxidases and Reaction Oxygen Species
3.2.2. Glutathione S-Transferase-Like Genes
3.2.3. WRKY Transcription Factors
3.2.4. Isoflavonoid Biosynthesis Pathway
3.2.5. Lipoxygenase Pathway
3.2.6. Xyloglucan Endotransglucosylases/Hydrolases
3.2.7. Systemic Acquired Resistance Signaling
3.2.8. Pathogenesis-Related Protein Class PR10
3.2.9. Concluding Remarks
4. Materials and Methods
4.1. DNA Polymorphisms of D. toxica Isolates
4.2. Plant Material
4.3. Genotyping for the Phr1 and PhtjR Alleles
4.4. D. toxica Experiment in Controlled Conditions
4.5. RNA Isolation
4.6. RNA Sequencing and Data Analysis
4.7. Selection of Genes for Quantitative Expression Profiling
4.8. Quantitative Gene Expression Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| cv. | cultivar |
| DIR1 | defective in induced resistance 1 |
| dpi | day post inoculation |
| GST | glutathione S-transferase |
| ITS | internal transcribed spacer |
| Phr1 | allele conferring D. toxica resistance in the 75A:258 L. angustifolius line |
| Phr2 | allele conferring D. toxica resistance in the L. angustifolius cultivar Merrit |
| PhtjR | allele conferring D. toxica resistance in the L. angustifolius cultivar Wonga |
| RAPD | random amplification of polymorphic DNA |
| SAM22 | starvation-associated message 22 |
| SAR | systemic acquired resistance |
| WGCNA | weighted gene co-expression network analysis |
| XTH | xyloglucan endotransglucosylase/hydrolase |
References
- Lewis, G.; Schrirer, B.; Mackinder, B.; Lock, M. Legumes of the World; Royal Botanic Gardens: Kew, UK, 2005; p. 592. [Google Scholar]
- Cannon, S.B.; McKain, M.R.; Harkess, A.; Nelson, M.N.; Dash, S.; Deyholos, M.K.; Peng, Y.; Joyce, B.; Stewart, C.N.; Rolf, M.; et al. Multiple polyploidy events in the early radiation of nodulating and nonnodulating legumes. Mol. Biol. Evol. 2015, 32, 193–210. [Google Scholar] [CrossRef]
- Drummond, C.S.; Eastwood, R.J.; Miotto, S.T.S.; Hughes, C.E. Multiple continental radiations and correlates of diversification in Lupinus (Leguminosae): Testing for key innovation with incomplete taxon sampling. Syst. Biol. 2012, 61, 443–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, J.; Turner, G.L.; O’Connor, G.E.; Bergersen, F.J. Nitrogen fixation and accretion of soil nitrogen by field-grown lupins (Lupinus angustifolius). Field Crops Res. 1987, 16, 309–322. [Google Scholar] [CrossRef]
- Lambers, H.; Clements, J.C.; Nelson, M.N. How a phosphorus-acquisition strategy based on carboxylate exudation powers the success and agronomic potential of lupines (Lupinus, Fabaceae). Am. J. Bot. 2013, 100, 263–288. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.M.; Ganopoulos, I.; Madesis, P.; Mavromatis, A.; Mylona, P.; Nianiou-Obeidat, I.; Parissi, Z.; Polidoros, A.; Tani, E.; Vlachostergios, D. The use of lupin as a source of protein in animal feeding: Genomic tools and breeding approaches. Int. J. Mol. Sci. 2019, 20, 851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foyer, C.H.; Lam, H.M.; Nguyen, H.T.; Siddique, K.H.; Varshney, R.K.; Colmer, T.D.; Cowling, W.A.; Bramley, H.; Mori, T.A.; Hodgson, J.M.; et al. Neglecting legumes has compromised human health and sustainable food production. Nat. Plants 2016, 2, 16112. [Google Scholar] [CrossRef]
- Kouris-Blazos, A.; Belski, R. Health benefits of legumes and pulses with a focus on Australian sweet lupins. Asia Pac. J. Clin. Nutr. 2016, 25, 1–17. [Google Scholar]
- Lucas, M.M.; Stoddard, F.L.; Annicchiarico, P.; Frías, J.; Martínez-Villaluenga, C.; Sussmann, D.; Duranti, M.; Seger, A.; Zander, P.M.; Pueyo, J.J. The future of lupin as a protein crop in Europe. Front. Plant Sci. 2015, 6, 705. [Google Scholar] [CrossRef]
- Mattila, P.; Mäkinen, S.; Eurola, M.; Jalava, T.; Pihlava, J.M.; Hellström, J.; Pihlanto, A. Nutritional value of commercial protein-rich plant products. Plant Foods Hum. Nutr. 2018, 73, 108–115. [Google Scholar] [CrossRef] [Green Version]
- Halmemies-Beauchet-Filleau, A.; Rinne, M.; Lamminen, M.; Mapato, C.; Ampapon, T.; Wanapat, M.; Vanhatalo, A. Review: Alternative and novel feeds for ruminants: Nutritive value, product quality and environmental aspects. Animal 2018, 12, s295–s309. [Google Scholar] [CrossRef] [Green Version]
- Petterson, D.S. The use of lupins in feeding systems—Review. Asian-Australas. J. Anim. Sci. 2000, 13, 861–882. [Google Scholar] [CrossRef]
- Gardiner, M.R.; Parr, W.H. Pathogenesis of acute lupinosis of sheep. J. Comp. Pathol. 1967, 77, 51–62. [Google Scholar] [CrossRef]
- Gardiner, M.R. Lupinosis. Adv. Vet. Sci. 1967, 11, 85–138. [Google Scholar] [PubMed]
- Gardiner, M.R. The pathology of lupinosis of sheep. Gross- and histo-pathology. Pathol. Vet. 1965, 2, 417–445. [Google Scholar]
- Plumlee, K.H. Chapter 23—Mycotoxins. In Clinical Veterinary Toxicology; Plumlee, K.H., Ed.; Mosby: Saint Louis, MO, USA, 2004; pp. 231–281. [Google Scholar]
- Soler Rodriguez, F.; Miguez Santiyan, M.P.; Pedrera Zamorano, J.D.; Roncero Cordero, V. An outbreak of lupinosis in sheep. Vet. Hum. Toxicol. 1991, 33, 492–494. [Google Scholar]
- Culvenor, C.C.; Beck, A.B.; Clarke, M.; Cockrum, P.A.; Edgar, J.A.; Frahn, J.L.; Jago, M.V.; Lanigan, G.W.; Payne, A.L.; Peterson, J.E.; et al. Isolation of toxic metabolites of Phomopsis leptostromiformis responsible for lupinosis. Aust. J. Biol. Sci. 1977, 30, 269–277. [Google Scholar] [CrossRef] [Green Version]
- Van Warmelo, K.T.; Marasas, W.F. Phomopsis leptostromiformis: The causal fungus of lupinosis, a mycotoxicosis, in sheep. Mycologia 1972, 64, 316–324. [Google Scholar] [CrossRef]
- Williamson, P.M.; Highet, A.S.; Gams, W.; Sivasithamparam, K.; Cowling, W.A. Diaporthe toxica sp. nov. the cause of lupinosis in sheep. Mycol. Res. 1994, 98, 1364–1368. [Google Scholar] [CrossRef]
- Williamson, P.; Sivasithamparam, K. Factors influencing the establishment of latent infection of narrow-leafed lupins by Diaporthe toxica. Aust. J. Agric. Res. 1994, 45, 1387–1394. [Google Scholar] [CrossRef]
- Williamson, P.M.; Sivasithamparam, K.; Cowling, W.A. Formation of subcuticular coralloid hyphae by Phomopsis leptostromiformis upon latent infection of narrow-leafed lupins. Plant Dis. 1991, 75, 1023–1026. [Google Scholar] [CrossRef]
- Cowling, W.A.; Hamblin, J.; Wood, P.M.; Gladstones, J.S. Resistance to Phomopsis stem blight in Lupinus angustifolius L. Crop Sci. 1987, 27, 648–652. [Google Scholar] [CrossRef]
- Stefanova, K.T.; Buirchell, B. Multiplicative mixed models for genetic gain assessment in lupin breeding. Crop Sci. 2010, 50, 880–891. [Google Scholar] [CrossRef]
- Yang, H.; Jian, J.; Li, X.; Renshaw, D.; Clements, J.; Sweetingham, M.W.; Tan, C.; Li, C. Application of whole genome re-sequencing data in the development of diagnostic DNA markers tightly linked to a disease-resistance locus for marker-assisted selection in lupin (Lupinus angustifolius). BMC Genom. 2015, 16, 660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Shankar, M.; Buirchell, J.; Sweetingham, W.; Caminero, C.; Smith, C. Development of molecular markers using MFLP linked to a gene conferring resistance to Diaporthe toxica in narrow-leafed lupin (Lupinus angustifolius L.). Theor. Appl. Genet. 2002, 105, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Tao, Y.; Zheng, Z.; Shao, D.; Li, Z.; Sweetingham, M.W.; Buirchell, B.J.; Li, C. Rapid development of molecular markers by next-generation sequencing linked to a gene conferring phomopsis stem blight disease resistance for marker-assisted selection in lupin (Lupinus angustifolius L.) breeding. Theor. Appl. Genet. 2013, 126, 511–522. [Google Scholar] [CrossRef]
- Shankar, M.; Cowling, W.A.; Sweetingham, M.W. The expression of resistance to latent stem infection by Diaporthe toxica in narrow-leafed lupins. Phytopathology 1996, 86, 692–697. [Google Scholar] [CrossRef]
- Shankar, M.; Sweetingham, M.W.; Cowling, W.A. Identification of alleles at two loci controlling resistance to Phomopsis stem blight in narrow-leafed lupin (Lupinus angustifolius L.). Euphytica 2002, 125, 35–44. [Google Scholar] [CrossRef]
- Książkiewicz, M.; Wójcik, K.; Irzykowski, W.; Bielski, W.; Rychel, S.; Kaczmarek, J.; Plewiński, P.; Rudy, E.; Jędryczka, M. Validation of Diaporthe toxica resistance markers in European Lupinus angustifolius germplasm and identification of novel resistance donors for marker-assisted selection. J. Appl. Genet. 2020, 61, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Tao, Y.; Zheng, Z.; Zhang, Q.; Zhou, G.; Sweetingham, M.W.; Howieson, J.G.; Li, C. Draft genome sequence, and a sequence-defined genetic linkage map of the legume crop species Lupinus angustifolius L. PLoS ONE 2013, 8, e64799. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M. Zur Entwiklungsgeschichte des Cryptosporium leptostromiforme. J. Kuhn. Bot. Zent. 1893, 54, 289. [Google Scholar]
- Lind, J.V.A. Danish Fungi as Represented in the Herbarium of E. Rostrup; Gyldendalske Boghandel—Nordisk Forlag: Copenhagen, Denmark, 1913; p. 680. [Google Scholar]
- Marcinkowska, J. Reappearance of Phomopsis leptostromiformis on yellow lupine in Poland. Phytopathol. Pol. 2007, 45, 67–69. [Google Scholar]
- Lewartowska, E.; Jędryczka, M.; Frencel, I.; Pieczyrak, J. Seed-borne fungi of Lupinus angustifolius L. cultivars. Phytopathol. Pol. 1994, 7, 123–130. [Google Scholar]
- NOAA National Centers for Environmental information, Climate at a Glance: Global Time Series. Available online: https://www.ncdc.noaa.gov/cag/ (accessed on 28 April 2020).
- Hulke, B.S.; Markell, S.G.; Kane, N.C.; Mathew, F.M. Phomopsis stem canker of sunflower in North America: Correlation with climate and solutions through breeding and management. OCL 2019, 26, 13. [Google Scholar] [CrossRef]
- Hane, J.K.; Ming, Y.; Kamphuis, L.G.; Nelson, M.N.; Garg, G.; Atkins, C.A.; Bayer, P.E.; Bravo, A.; Bringans, S.; Cannon, S.; et al. A comprehensive draft genome sequence for lupin (Lupinus angustifolius), an emerging health food: Insights into plant-microbe interactions and legume evolution. Plant Biotechnol. J. 2017, 15, 318–330. [Google Scholar] [CrossRef]
- Zhou, G.; Jian, J.; Wang, P.; Li, C.; Tao, Y.; Li, X.; Renshaw, D.; Clements, J.; Sweetingham, M.; Yang, H. Construction of an ultra-high density consensus genetic map, and enhancement of the physical map from genome sequencing in Lupinus angustifolius. Theor. Appl. Genet. 2018, 131, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowling, W.A.; Gladstones, J.S. Lupin Breeding in Australia. In Linking Research and Marketing Opportunities for Pulses in the 21st Century: Proceedings of the Third International Food Legumes Research Conference, Adelaide, Australia, 22–26 September 1997; Knight, R., Ed.; Springer: Dordrecht, The Netherlands, 2000; pp. 541–547. [Google Scholar]
- Gomes, R.R.; Glienke, C.; Videira, S.I.R.; Lombard, L.; Groenewald, J.Z.; Crous, P.W. Diaporthe: A genus of endophytic, saprobic and plant pathogenic fungi. Persoonia 2013, 31, 1–41. [Google Scholar] [CrossRef] [Green Version]
- Abramczyk, B.; Król, E. Use of RAPD-PCR and its markers for identification of Diaporthe/Phomopsis from fruit trees in south-eastern Poland. Acta Sci. Pol. Hortorum Cultus 2016, 15, 15. [Google Scholar]
- Michalecka, M.; Bryk, H.; Seliga, P. Identification and characterization of Diaporthe vaccinii Shear causing upright dieback and viscid rot of cranberry in Poland. Eur. J. Plant Pathol. 2017, 148, 595–605. [Google Scholar] [CrossRef] [Green Version]
- Pioli, R.N.; Morandi, E.N.; Martinez, M.C.; Lucca, F.; Tozzini, A.; Bisaro, V.; Hopp, H.E. Morphologic, molecular, and pathogenic characterization of Diaporthe phaseolorum variability in the core soybean-producing area of Argentina. Phytopathology 2003, 93, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.W.; Hartman, G.L.; Riccioni, L.; Chen, W.D.; Ma, R.Z.; Pedersen, W.L. Using PCR to distinguish Diaporthe phaseolorum and Phomopsis longicolla from other soybean fungal pathogens and to detect them in soybean tissues. Plant Dis. 1997, 81, 1143–1149. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, T.T.; de Souza Leite, T.; de Queiroz, C.B.; de Araujo, E.F.; Pereira, O.L.; de Queiroz, M.V. High genetic variability in endophytic fungi from the genus Diaporthe isolated from common bean (Phaseolus vulgaris L.) in Brazil. J. Appl. Microbiol. 2016, 120, 388–401. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.W.; Riccioni, L.; Pedersen, W.L.; Kollipara, K.P.; Hartman, G.L. Molecular identification and phylogenetic grouping of Diaporthe phaseolorum and Phomopsis longicolla isolates from soybean. Phytopathology 1998, 88, 1306–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Rensburg, J.C.J.; Lamprecht, S.C.; Groenewald, J.Z.; Castlebury, L.A.; Crous, P.W. Characterisation of Phomopsis spp. associated with die-back of rooibos (Aspalathus linearis) in South Africa. Stud. Mycol. 2006, 55, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Niekerk, J.M.; Groenewald, J.Z.; Farr, D.F.; Fourie, P.H.; Halleer, F.; Crous, P.W. Reassessment of Phomopsis species on grapevines. Australas. Plant Path. 2005, 34, 27–39. [Google Scholar] [CrossRef]
- Książkiewicz, M.; Yang, H. Molecular marker resources supporting the Australian lupin breeding program. In The Lupin Genome; Singh, K.B., Kamphuis, L.G., Nelson, M.N., Eds.; Springer International Publishing: Cham, Germany, 2020; pp. 73–86. [Google Scholar]
- Abdelrahman, M.; Suzumura, N.; Mitoma, M.; Matsuo, S.; Ikeuchi, T.; Mori, M.; Murakami, K.; Ozaki, Y.; Matsumoto, M.; Uragami, A.; et al. Comparative de novo transcriptome profiles in Asparagus officinalis and A. kiusianus during the early stage of Phomopsis asparagi infection. Sci. Rep. 2017, 7, 2608. [Google Scholar] [CrossRef] [Green Version]
- Almagro, L.; Gómez Ros, L.V.; Belchi-Navarro, S.; Bru, R.; Ros Barceló, A.; Pedreño, M.A. Class III peroxidases in plant defence reactions. J. Exp. Bot. 2008, 60, 377–390. [Google Scholar] [CrossRef] [Green Version]
- Vance, C.P.; Kirk, T.K.; Sherwood, R.T. Lignification as a mechanism of disease resistance. Annu. Rev. Phytopathol. 1980, 18, 259–288. [Google Scholar] [CrossRef]
- Nicholson, R.L.; Hammerschmidt, R. Phenolic compounds and their role in disease resistance. Annu. Rev. Phytopathol. 1992, 30, 369–389. [Google Scholar] [CrossRef]
- Bhuiyan, N.H.; Selvaraj, G.; Wei, Y.; King, J. Role of lignification in plant defense. Plant Signal. Behav. 2009, 4, 158–159. [Google Scholar] [CrossRef] [Green Version]
- Barba, P.; Lillis, J.; Luce, R.S.; Travadon, R.; Osier, M.; Baumgartner, K.; Wilcox, W.F.; Reisch, B.I.; Cadle-Davidson, L. Two dominant loci determine resistance to Phomopsis cane lesions in F1 families of hybrid grapevines. Theor. Appl. Genet. 2018, 131, 1173–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Østergaard, L.; Teilum, K.; Mirza, O.; Mattsson, O.; Petersen, M.; Welinder, K.G.; Mundy, J.; Gajhede, M.; Henriksen, A. Arabidopsis ATP A2 peroxidase. Expression and high-resolution structure of a plant peroxidase with implications for lignification. Plant Mol. Biol. 2000, 44, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Bolwell, G.P.; Bindschedler, L.V.; Blee, K.A.; Butt, V.S.; Davies, D.R.; Gardner, S.L.; Gerrish, C.; Minibayeva, F. The apoplastic oxidative burst in response to biotic stress in plants: A three-component system. J. Exp. Bot. 2002, 53, 1367–1376. [Google Scholar] [PubMed]
- Torres, M.A.; Jones, J.D.G.; Dangl, J.L. Reactive oxygen species signaling in response to pathogens. Plant Physiol. 2006, 141, 373–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wally, O.; Punja, Z.K. Enhanced disease resistance in transgenic carrot (Daucus carota L.) plants over-expressing a rice cationic peroxidase. Planta 2010, 232, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Coego, A.; Ramirez, V.; Gil, M.J.; Flors, V.; Mauch-Mani, B.; Vera, P. An Arabidopsis homeodomain transcription factor, OVEREXPRESSOR OF CATIONIC PEROXIDASE 3, mediates resistance to infection by necrotrophic pathogens. Plant Cell 2005, 17, 2123–2137. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, A.; Okumura, T.; Kaminaka, H.; Sumi, K.; Tanaka, K. Structure and differential response to abscisic acid of two promoters for the cytosolic copper/zinc-superoxide dismutase genes, SodCc1 and SodCc2, in rice protoplasts. FEBS Lett. 1995, 358, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Nowogórska, A.; Patykowski, J. Selected reactive oxygen species and antioxidant enzymes in common bean after Pseudomonas syringae pv. phaseolicola and Botrytis cinerea infection. Acta Physiol. Plant. 2014, 37, 1725. [Google Scholar]
- Gullner, G.; Komives, T.; Király, L.; Schröder, P. Glutathione S-Transferase Enzymes in Plant-Pathogen Interactions. Front. Plant Sci. 2018, 9, 1836. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.M.; Liu, H.Y.; Xu, H.M.; Li, M.; Kang, Z.S. Analysis of differential transcriptional profiling in wheat infected by Blumeria graminis f. sp. tritici using GeneChip. Mol. Biol. Rep. 2012, 39, 381–387. [Google Scholar] [CrossRef]
- Pei, D.; Ma, H.; Zhang, Y.; Ma, Y.; Wang, W.; Geng, H.; Wu, J.; Li, C. Virus-induced gene silencing of a putative glutathione S-transferase gene compromised Ol-1-mediated resistance against powdery mildew in tomato. Plant Mol. Biol. Rep. 2011, 29, 972–978. [Google Scholar] [CrossRef]
- Panthee, D.R.; Yuan, J.S.; Wright, D.L.; Marois, J.J.; Mailhot, D.; Stewart, C.N. Gene expression analysis in soybean in response to the causal agent of Asian soybean rust (Phakopsora pachyrhizi Sydow) in an early growth stage. Funct. Integr. Genom. 2007, 7, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Soria-Guerra, R.E.; Rosales-Mendoza, S.; Chang, S.; Haudenshield, J.S.; Padmanaban, A.; Rodriguez-Zas, S.; Hartman, G.L.; Ghabrial, S.A.; Korban, S.S. Transcriptome analysis of resistant and susceptible genotypes of Glycine tomentella during Phakopsora pachyrhizi infection reveals novel rust resistance genes. Theor. Appl. Genet. 2010, 120, 1315–1333. [Google Scholar] [CrossRef] [PubMed]
- Hahn, K.; Strittmatter, G. Pathogen-defence gene prp1–1 from potato encodes an auxin-responsive glutathione S-transferase. Eur. J. Biochem. 1994, 226, 619–626. [Google Scholar] [CrossRef]
- Gardiner, S.A.; Boddu, J.; Berthiller, F.; Hametner, C.; Stupar, R.M.; Adam, G.; Muehlbauer, G.J. Transcriptome analysis of the barley–deoxynivalenol interaction: Evidence for a role of glutathione in deoxynivalenol detoxification. Mol. Plant-Microbe Interact. 2010, 23, 962–976. [Google Scholar] [CrossRef] [Green Version]
- Dean, J.D.; Goodwin, P.H.; Hsiang, T. Induction of glutathione S-transferase genes of Nicotiana benthamiana following infection by Colletotrichum destructivum and C. orbiculare and involvement of one in resistance. J. Exp. Bot. 2005, 56, 1525–1533. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhu, F.; Liu, H.; Chu, A.; Lo, C. Isolation and expression analysis of defense-related genes in sorghum—Colletotrichum sublineolum interaction. Physiol. Mol. Plant Pathol. 2013, 84, 123–130. [Google Scholar] [CrossRef]
- Han, Q.; Chen, R.; Yang, Y.; Cui, X.; Ge, F.; Chen, C.; Liu, D. A glutathione S-transferase gene from Lilium regale Wilson confers transgenic tobacco resistance to Fusarium oxysporum. Sci. Hortic. 2016, 198, 370–378. [Google Scholar] [CrossRef]
- Piślewska-Bednarek, M.; Nakano, R.T.; Hiruma, K.; Pastorczyk, M.; Sanchez-Vallet, A.; Singkaravanit-Ogawa, S.; Ciesiołka, D.; Takano, Y.; Molina, A.; Schulze-Lefert, P.; et al. Glutathione transferase U13 functions in pathogen-triggered glucosinolate metabolism. Plant Physiol. 2018, 176, 538–551. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sun, S.; Ge, W.; Zhao, L.; Hou, B.; Wang, K.; Lyu, Z.; Chen, L.; Xu, S.; Guo, J.; et al. Horizontal gene transfer of Fhb7 from fungus underlies Fusarium head blight resistance in wheat. Science 2020, 368, 6493. [Google Scholar] [CrossRef]
- Mulema, J.M.K.; Okori, P.; Denby, K.J. Proteomic analysis of the Arabidopsis thaliana-Botrytis cinerea interaction using two-dimensional liquid chromatography. Afr. J. Biotechnol. 2011, 10, 17551–17563. [Google Scholar]
- Soon Young, A.; Seon Ae, K.; Hae Keun, Y. Glutathione S-Transferase genes differently expressed by pathogen-infection in Vitis flexuosa. Plant Breed. Biotechnol. 2016, 4, 61–70. [Google Scholar]
- Agudelo-Romero, P.; Erban, A.; Rego, C.; Carbonell-Bejerano, P.; Nascimento, T.; Sousa, L.; Martínez-Zapater, J.M.; Kopka, J.; Fortes, A.M. Transcriptome and metabolome reprogramming in Vitis vinifera cv. Trincadeira berries upon infection with Botrytis cinerea. J. Exp. Bot. 2015, 66, 1769–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenk, P.M.; Kazan, K.; Wilson, I.; Anderson, J.P.; Richmond, T.; Somerville, S.C.; Manners, J.M. Coordinated plant defense responses in Arabidopsis revealed by microarray analysis. Proc. Natl. Acad. Sci. USA 2000, 97, 11655–11660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, A.K.; Carp, M.J.; Zuchman, R.; Ziv, T.; Horwitz, B.A.; Gepstein, S. Proteomics of the response of Arabidopsis thaliana to infection with Alternaria brassicicola. J. Proteom. 2010, 73, 709–720. [Google Scholar] [CrossRef] [PubMed]
- De Vos, M.; Van Oosten, V.R.; Van Poecke, R.M.; Van Pelt, J.A.; Pozo, M.J.; Mueller, M.J.; Buchala, A.J.; Métraux, J.P.; Van Loon, L.C.; Dicke, M.; et al. Signal signature and transcriptome changes of Arabidopsis during pathogen and insect attack. Mol. Plant-Microbe Interact. 2005, 18, 923–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Buchwaldt, L.; Rimmer, S.R.; Sharpe, A.; McGregor, L.; Bekkaoui, D.; Hegedus, D. Patterns of differential gene expression in Brassica napus cultivars infected with Sclerotinia sclerotiorum. Mol. Plant Pathol. 2009, 10, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, J.; An, L.; Doerge, R.W.; Chen, Z.J.; Grau, C.R.; Meng, J.; Osborn, T.C. Analysis of gene expression profiles in response to Sclerotinia sclerotiorum in Brassica napus. Planta 2007, 227, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Garg, H.; Li, H.; Sivasithamparam, K.; Barbetti, M.J. Differentially expressed proteins and associated histological and disease progression changes in cotyledon tissue of a resistant and susceptible genotype of Brassica napus infected with Sclerotinia sclerotiorum. PLoS ONE 2013, 8, e65205. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Jian, H.; Lu, K.; Filardo, F.; Yin, N.; Liu, L.; Qu, C.; Li, W.; Du, H.; Li, J. Genome-wide association analysis and differential expression analysis of resistance to Sclerotinia stem rot in Brassica napus. Plant Biotechnol. J. 2016, 14, 1368–1380. [Google Scholar] [CrossRef] [Green Version]
- Phukan, U.J.; Jeena, G.S.; Shukla, R.K. WRKY transcription factors: Molecular regulation and stress responses in plants. Front. Plant Sci. 2016, 7, 760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Li, C.; Wang, H.; Guo, Z. WRKY transcription factors: Evolution, binding, and action. Phytopathol. Res. 2019, 1, 13. [Google Scholar] [CrossRef]
- Eulgem, T.; Somssich, I.E. Networks of WRKY transcription factors in defense signaling. Curr. Opin. Plant Biol. 2007, 10, 366–371. [Google Scholar] [CrossRef] [Green Version]
- Pandey, S.P.; Somssich, I.E. The role of WRKY transcription factors in plant immunity. Plant Physiol. 2009, 150, 1648–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, R.A.; Harrison, M.J.; Paiva, N.L. The isoflavonoid phytoalexin pathway: From enzymes to genes to transcription factors. Physiol. Plant. 1995, 93, 385–392. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.A.; Banks, S.W. Biosynthesis, elicitation and biological activity of isoflavonoid phytoalexins. Phytochemistry 1986, 25, 979–995. [Google Scholar] [CrossRef]
- Schröder, G.; Zähringer, U.; Heller, W.; Ebel, J.; Grisebach, H. Biosynthesis of antifungal isoflavonoids in Lupinus albus. Enzymatic prenylation of genistein and 2′-hydroxygenistein. Arch. Biochem. Biophys. 1979, 194, 635–636. [Google Scholar] [CrossRef]
- Muth, D.; Kachlicki, P.; Krajewski, P.; Przystalski, M.; Stobiecki, M. Differential metabolic response of narrow leafed lupine (Lupinus angustifolius) leaves to infection with Colletotrichum lupini. Metabolomics 2009, 5, 354–362. [Google Scholar] [CrossRef]
- Jung, W.; Yu, O.; Lau, S.M.; O’Keefe, D.P.; Odell, J.; Fader, G.; McGonigle, B. Identification and expression of isoflavone synthase, the key enzyme for biosynthesis of isoflavones in legumes. Nat. Biotechnol. 2000, 18, 208–212. [Google Scholar] [CrossRef]
- Narożna, D.; Książkiewicz, M.; Przysiecka, Ł.; Króliczak, J.; Wolko, B.; Naganowska, B.; Mądrzak, C.J. Legume isoflavone synthase genes have evolved by whole-genome and local duplications yielding transcriptionally active paralogs. Plant Sci. 2017, 264, 149–167. [Google Scholar] [CrossRef]
- Subramanian, S.; Graham, M.Y.; Yu, O.; Graham, T.L. RNA interference of soybean isoflavone synthase genes leads to silencing in tissues distal to the transformation site and to enhanced susceptibility to Phytophthora sojae. Plant Physiol. 2005, 137, 1345–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, S.; Tiemann, K.; Wittkampf, U.; Bless, W.; Hinderer, W.; Barz, W. Elicitor-induced metabolic changes in cell cultures of chickpea (Cicer arietinum L.) cultivars resistant and susceptible to Ascochyta rabiei: I. Investigations of enzyme activities involved in isoflavone and pterocarpan phytoalexin biosynthesis. Planta 1990, 182, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Gunia, W.; Hinderer, W.; Wittkampf, U.; Barz, W. Elicitor induction of cytochrome P-450 monooxygenases in cell suspension cultures of chickpea (Cicer arietinum L.) and their involvement in pterocarpan phytoalexin biosynthesis. Z. Nat. C 1991, 46, 58–66. [Google Scholar] [CrossRef]
- Edwards, R.; Dixon, R.A. Isoflavone O-methyltransferase activities in elicitor-treated cell suspension cultures of Medicago sativa. Phytochemistry 1991, 30, 2597–2606. [Google Scholar] [CrossRef]
- He, X.Z.; Dixon, R.A. Genetic manipulation of isoflavone 7-O-methyltransferase enhances biosynthesis of 4′-O-methylated isoflavonoid phytoalexins and disease resistance in alfalfa. Plant Cell 2000, 12, 1689–1702. [Google Scholar]
- Wasternack, C. Jasmonates: An update on biosynthesis, signal transduction and action in plant stress response, growth and development. Ann. Bot. 2007, 100, 681–697. [Google Scholar] [CrossRef] [Green Version]
- Vicente, J.; Cascón, T.; Vicedo, B.; García-Agustín, P.; Hamberg, M.; Castresana, C. Role of 9-lipoxygenase and α-dioxygenase oxylipin pathways as modulators of local and systemic defense. Mol. Plant 2012, 5, 914–928. [Google Scholar] [CrossRef] [Green Version]
- Blée, E. Impact of phyto-oxylipins in plant defense. Trends Plant Sci. 2002, 7, 315–322. [Google Scholar] [CrossRef]
- Genva, M.; Obounou Akong, F.; Andersson, M.X.; Deleu, M.; Lins, L.; Fauconnier, M.L. New insights into the biosynthesis of esterified oxylipins and their involvement in plant defense and developmental mechanisms. Phytochem. Rev. 2019, 18, 343–358. [Google Scholar] [CrossRef] [Green Version]
- Mena, E.; Stewart, S.; Montesano, M.; Ponce de León, I. Soybean stem canker caused by Diaporthe caulivora; pathogen diversity, colonization process, and plant defense activation. Front. Plant Sci. 2020, 10, 1733. [Google Scholar] [CrossRef]
- López, M.A.; Vicente, J.; Kulasekaran, S.; Vellosillo, T.; Martínez, M.; Irigoyen, M.L.; Cascón, T.; Bannenberg, G.; Hamberg, M.; Castresana, C. Antagonistic role of 9-lipoxygenase-derived oxylipins and ethylene in the control of oxidative stress, lipid peroxidation and plant defence. Plant J. 2011, 67, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Vellosillo, T.; Martínez, M.; López, M.A.; Vicente, J.; Cascón, T.; Dolan, L.; Hamberg, M.; Castresana, C. Oxylipins produced by the 9-lipoxygenase pathway in Arabidopsis regulate lateral root development and defense responses through a specific signaling cascade. Plant Cell 2007, 19, 831–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marmey, P.; Jalloul, A.; Alhamdia, M.; Assigbetse, K.; Cacas, J.L.; Voloudakis, A.E.; Champion, A.; Clerivet, A.; Montillet, J.L.; Nicole, M. The 9-lipoxygenase GhLOX1 gene is associated with the hypersensitive reaction of cotton Gossypium hirsutum to Xanthomonas campestris pv malvacearum. Plant Physiol. Biochem. 2007, 45, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.S.; Hwang, B.K. The pepper 9-lipoxygenase gene CaLOX1 functions in defense and cell death responses to microbial pathogens. Plant Physiol. 2010, 152, 948–967. [Google Scholar] [CrossRef] [Green Version]
- Christensen, S.A.; Huffaker, A.; Kaplan, F.; Sims, J.; Ziemann, S.; Doehlemann, G.; Ji, L.; Schmitz, R.J.; Kolomiets, M.V.; Alborn, H.T.; et al. Maize death acids, 9-lipoxygenase–derived cyclopente(a)nones, display activity as cytotoxic phytoalexins and transcriptional mediators. Proc. Natl. Acad. Sci. USA 2015, 112, 11407–11412. [Google Scholar] [CrossRef] [Green Version]
- Constantino, N.; Mastouri, F.; Damarwinasis, R.; Borrego, E.; Moran-Diez, M.; Kenerley, C.; Gao, X.; Kolomiets, M. Root-expressed maize lipoxygenase 3 negatively regulates induced systemic resistance to Colletotrichum graminicola in shoots. Front. Plant Sci. 2013, 4, 510. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.D.; Borrego, E.J.; Kenerley, C.M.; Kolomiets, M.V. Oxylipins other than jasmonic acid are xylem-resident signals regulating systemic resistance induced by Trichoderma virens in maize. Plant Cell 2020, 32, 166–185. [Google Scholar] [CrossRef]
- Carella, P. Xylem-mobile oxylipins are critical regulators of induced systemic resistance in maize. Plant Cell 2020, 32, 13–14. [Google Scholar] [CrossRef] [Green Version]
- McCann, M.C.; Wells, B.; Roberts, K. Complexity in the spatial localization and length distribution of plant cell-wall matrix polysaccharides. J. Microsc. 1992, 166, 123–136. [Google Scholar] [CrossRef]
- Hayashi, T. Xyloglucans in the primary cell wall. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1989, 40, 139–168. [Google Scholar] [CrossRef]
- Rose, J.K.; Braam, J.; Fry, S.C.; Nishitani, K. The XTH family of enzymes involved in xyloglucan endotransglucosylation and endohydrolysis: Current perspectives and a new unifying nomenclature. Plant Cell Physiol. 2002, 43, 1421–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, M.J.; Eklöf, J.M.; Michel, G.; Kallas, A.M.; Teeri, T.T.; Czjzek, M.; Brumer, H. Structural evidence for the evolution of xyloglucanase activity from xyloglucan endo-transglycosylases: Biological implications for cell wall metabolism. Plant Cell 2007, 19, 1947–1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houston, K.; Tucker, M.R.; Chowdhury, J.; Shirley, N.; Little, A. The plant cell wall: A complex and dynamic structure as revealed by the responses of genes under stress conditions. Front. Plant Sci. 2016, 7, 984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novaković, L.; Guo, T.; Bacic, A.; Sampathkumar, A.; Johnson, K.L. Hitting the wall-sensing and signaling pathways involved in plant cell wall remodeling in response to abiotic stress. Plants 2018, 7, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasidharan, R.; Voesenek, L.A.C.J.; Pierik, R. Cell wall modifying proteins mediate plant acclimatization to biotic and abiotic stresses. Crit. Rev. Plant Sci. 2011, 30, 548–562. [Google Scholar] [CrossRef]
- Miedes, E.; Lorences, E.P. The implication of xyloglucan endotransglucosylase/hydrolase (XTHs) in tomato fruit infection by Penicillium expansum Link. A. J. Agric. Food Chem. 2007, 55, 9021–9026. [Google Scholar] [CrossRef]
- Scotti, R.; D’Agostino, N.; Zaccardelli, M. Gene expression profiling of tomato roots interacting with Pseudomonas fluorescens unravels the molecular reprogramming that occurs during the early phases of colonization. Symbiosis 2019, 78, 177–192. [Google Scholar] [CrossRef]
- Maldonado-Mendoza, I.E.; Dewbre, G.R.; Blaylock, L.; Harrison, M.J. Expression of a xyloglucan endotransglucosylase/hydrolase gene, Mt-XTH1, from Medicago truncatula is induced systemically in mycorrhizal roots. Gene 2005, 345, 191–197. [Google Scholar] [CrossRef]
- Panthapulakkal Narayanan, S.; Liao, P.; Taylor, P.W.J.; Lo, C.; Chye, M.L. Overexpression of a monocot acyl-CoA-binding protein confers broad-spectrum pathogen protection in a dicot. Proteomics 2019, 19, 1800368. [Google Scholar] [CrossRef]
- Sharmin, S.; Azam, M.S.; Islam, M.S.; Sajib, A.A.; Mahmood, N.; Hasan, A.M.; Ahmed, R.; Sultana, K.; Khan, H. Xyloglucan endotransglycosylase/hydrolase genes from a susceptible and resistant jute species show opposite expression pattern following Macrophomina phaseolina infection. Commun. Integr. Biol. 2012, 5, 598–606. [Google Scholar] [CrossRef]
- Bedre, R.; Irigoyen, S.; Schaker, P.D.C.; Monteiro-Vitorello, C.B.; Da Silva, J.A.; Mandadi, K.K. Genome-wide alternative splicing landscapes modulated by biotrophic sugarcane smut pathogen. Sci. Rep. 2019, 9, 8876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Divol, F.; Vilaine, F.; Thibivilliers, S.; Kusiak, C.; Sauge, M.H.; Dinant, S. Involvement of the xyloglucan endotransglycosylase/hydrolases encoded by celery XTH1 and Arabidopsis XTH33 in the phloem response to aphids. Plant Cell Environ. 2007, 30, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Divol, F.; Vilaine, F.; Thibivilliers, S.; Amselem, J.; Palauqui, J.C.; Kusiak, C.; Dinant, S. Systemic response to aphid infestation by Myzus persicae in the phloem of Apium graveolens. Plant Mol. Biol. 2005, 57, 517–540. [Google Scholar] [CrossRef]
- Lascombe, M.B.; Bakan, B.; Buhot, N.; Marion, D.; Blein, J.P.; Larue, V.; Lamb, C.; Prangé, T. The structure of “defective in induced resistance” protein of Arabidopsis thaliana, DIR1, reveals a new type of lipid transfer protein. Protein Sci. 2008, 17, 1522–1530. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, A.M.; Doerner, P.; Dixon, R.A.; Lamb, C.J.; Cameron, R.K. A putative lipid transfer protein involved in systemic resistance signalling in Arabidopsis. Nature 2002, 419, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Carella, P.; Kempthorne, C.J.; Wilson, D.C.; Isaacs, M.; Cameron, R.K. Exploring the role of DIR1, DIR1-like and other lipid transfer proteins during systemic immunity in Arabidopsis. Physiol. Mol. Plant Pathol. 2017, 97, 49–57. [Google Scholar] [CrossRef]
- Lascombe, M.B.; Buhot, N.; Bakan, B.; Marion, D.; Blein, J.P.; Lamb, C.J.; Prangé, T. Crystallization of DIR1, a LTP2-like resistance signalling protein from Arabidopsis thaliana. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 702–704. [Google Scholar] [CrossRef]
- Carella, P.; Isaacs, M.; Cameron, R.K. Plasmodesmata-located protein overexpression negatively impacts the manifestation of systemic acquired resistance and the long-distance movement of Defective in Induced Resistance1 in Arabidopsis. Plant Biol. 2015, 17, 395–401. [Google Scholar] [CrossRef]
- Champigny, M.J.; Isaacs, M.; Carella, P.; Faubert, J.; Fobert, P.R.; Cameron, R.K. Long distance movement of DIR1 and investigation of the role of DIR1-like during systemic acquired resistance in Arabidopsis. Front. Plant Sci. 2013, 4, 230. [Google Scholar] [CrossRef] [Green Version]
- Cameron, R.K.; Carella, P.; Isaacs, M.; Champigny, M.; Merl-Pham, J.; Dey, S.; Vlot, A.C. Using DIR1 to investigate long-distance signal movement during systemic acquired resistance. Can. J. Plant Pathol. 2016, 38, 19–24. [Google Scholar] [CrossRef]
- Champigny, M.J.; Shearer, H.; Mohammad, A.; Haines, K.; Neumann, M.; Thilmony, R.; He, S.Y.; Fobert, P.; Dengler, N.; Cameron, R.K. Localization of DIR1 at the tissue, cellular and subcellular levels during systemic acquired resistance in Arabidopsis using DIR1:GUS and DIR1:EGFP reporters. BMC Plant Biol. 2011, 11, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanda, B.; Xia, Y.; Mandal, M.K.; Yu, K.; Sekine, K.T.; Gao, Q.M.; Selote, D.; Hu, Y.; Stromberg, A.; Navarre, D.; et al. Glycerol-3-phosphate is a critical mobile inducer of systemic immunity in plants. Nat. Genet. 2011, 43, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, R.; Venables, B.; Petros, R.A.; Nalam, V.; Li, M.; Wang, X.; Takemoto, L.J.; Shah, J. An abietane diterpenoid is a potent activator of systemic acquired resistance. Plant J. 2012, 71, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.W.; Tschaplinski, T.J.; Wang, L.; Glazebrook, J.; Greenberg, J.T. Priming in systemic plant immunity. Science 2009, 324, 89–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.P.; von Dahl, C.C.; Klessig, D.F. The extent to which methyl salicylate is required for signaling systemic acquired resistance is dependent on exposure to light after infection. Plant Physiol. 2011, 157, 2216–2226. [Google Scholar] [CrossRef] [Green Version]
- Isaacs, M.; Carella, P.; Faubert, J.; Champigny, M.J.; Rose, J.K.C.; Cameron, R.K. Orthology analysis and in vivo complementation studies to elucidate the role of DIR1 during systemic acquired resistance in Arabidopsis thaliana and Cucumis sativus. Front. Plant Sci. 2016, 7, 566. [Google Scholar] [CrossRef] [Green Version]
- Sikorski, M.M.; Szlagowska, A.E.; Legocki, A.B. Structure of Lupinus luteus genes Ypr10.1a and Ypr10.1b encoding two homologs of pathogenesis-related proteins of PR10 class (Accession Nos. AF002277 and AF002278) (PGR98-045). Plant Physiol. 1998, 116, 1192–1193. [Google Scholar]
- Biesiadka, J.; Sikorski, M.M.; Bujacz, G.; Jaskolski, M. Crystallization and preliminary X-ray structure determination of Lupinus luteus PR10 protein. Acta Crystallogr. Sect. D 1999, 55, 1925–1927. [Google Scholar] [CrossRef]
- Hashimoto, M.; Kisseleva, L.; Sawa, S.; Furukawa, T.; Komatsu, S.; Koshiba, T. A novel rice PR10 protein, RSOsPR10, specifically induced in roots by biotic and abiotic stresses, possibly via the jasmonic acid signaling pathway. Plant Cell Physiol. 2004, 45, 550–559. [Google Scholar] [CrossRef] [Green Version]
- Handschuh, L.; Femiak, I.; Kasperska, A.; Figlerowicz, M.; Sikorski, M.M. Structural and functional characteristics of two novel members of pathogensis-related multigene family of class 10 from yellow lupine+. Acta Biochim. Pol. 2007, 54, 783–796. [Google Scholar] [CrossRef] [Green Version]
- Crowell, D.N.; Amasino, R.M. Induction of specific mRNAs in cultured soybean cells during cytokinin or auxin starvation. Plant Physiol. 1991, 95, 711–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowell, D.N.; John, M.E.; Russell, D.; Amasino, R.M. Characterization of a stress-induced, developmentally regulated gene family from soybean. Plant Mol. Biol. 1992, 18, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Irzykowski, W.; Sun, J.; Li, Q.; Gao, T.; Hou, S.; Águedo, A.; Jędryczka, M. DNA polymorphism in Sclerotinia sclerotiorum isolates from oilseed rape in China. IOBC-WPRS Bull. 2004, 27, 67–76. [Google Scholar]
- Van de Peer, Y.; De Wachter, R. TREECON for Windows: A software package for the construction and drawing of evolutionary trees for the Microsoft Windows environment. Comput. Applic. Biosci. 1994, 10, 569–570. [Google Scholar] [CrossRef] [PubMed]
- Balesdent, M.H.; Jedryczka, M.; Jain, L.; Mendes-Pereira, E.; Bertrandy, J.; Rouxel, T. Conidia as a Substrate for Internal Transcribed Spacer-Based PCR Identification of Members of the Leptosphaeria maculans Species Complex. Phytopathology 1998, 88, 1210–1217. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The ClustalX windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 24, 4876–4882. [Google Scholar] [CrossRef] [Green Version]
- Cowling, W.A. Pedigrees and characteristics of narrow-leafed lupin cultivars released in Australia from 1967 to 1998. Bull. Agric. West. Aust. 1999, 4365, 4–11. [Google Scholar]
- Kurlovich, B.S. Lupins: Geography, Classification, Genetic Resources and Breeding; Intan: St. Petersburg, FL, USA, 2002. [Google Scholar]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grote, S. GOfuncR: Gene Ontology Enrichment Using FUNC. R Package Version 1.10.0. 2020. Available online: https://bioconductor.riken.jp/packages/3.12/bioc/html/GOfuncR.html (accessed on 5 January 2021).
- Langfelder, P.; Horvath, S. Fast R functions for robust correlations and hierarchical clustering. J. Stat. Softw. 2012, 46, i11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osuna-Cruz, C.M.; Paytuvi-Gallart, A.; Di Donato, A.; Sundesha, V.; Andolfo, G.; Aiese Cigliano, R.; Sanseverino, W.; Ercolano, M.R. PRGdb 3.0: A comprehensive platform for prediction and analysis of plant disease resistance genes. Nucleic Acids Res. 2018, 46, D1197–D1201. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.M.; Jost, R.; Erskine, W.; Nelson, M.N. Identifying stable reference genes for qRT-PCR normalisation in gene expression studies of narrow-leafed lupin (Lupinus angustifolius L.). PLoS ONE 2016, 11, e0148300. [Google Scholar] [CrossRef] [Green Version]
- Taylor, C.M.; Kamphuis, L.G.; Zhang, W.; Garg, G.; Berger, J.D.; Mousavi-Derazmahalleh, M.; Bayer, P.E.; Edwards, D.; Singh, K.B.; Cowling, W.A.; et al. INDEL variation in the regulatory region of the major flowering time gene LanFTc1 is associated with vernalization response and flowering time in narrow-leafed lupin (Lupinus angustifolius L.). Plant Cell Environ. 2019, 42, 174–187. [Google Scholar] [CrossRef] [Green Version]
- Nelson, M.N.; Książkiewicz, M.; Rychel, S.; Besharat, N.; Taylor, C.M.; Wyrwa, K.; Jost, R.; Erskine, W.; Cowling, W.A.; Berger, J.D.; et al. The loss of vernalization requirement in narrow-leafed lupin is associated with a deletion in the promoter and de-repressed expression of a Flowering Locus T (FT) homologue. New Phytol. 2017, 213, 220–232. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [Green Version]
- Svec, D.; Tichopad, A.; Novosadova, V.; Pfaffl, M.W.; Kubista, M. How good is a PCR efficiency estimate: Recommendations for precise and robust qPCR efficiency assessments. Biomol. Detect. Quantif. 2015, 3, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress update—From bulk to single-cell expression data. Nucleic Acids Res. 2018, 47, D711–D715. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Line | Ph25M1 Phr1 | Ph258M2 Phr1 | PhtjM7 PhtjR | InDel2 PhtjR | InDel10 PhtjR | Disease Index 2017 1 | Disease Index 2018 2 | Resistance Genes |
|---|---|---|---|---|---|---|---|---|---|
| 96191 | Wonga | S 3 | S | R | R | R | 1.8 ± 1.3 | 1.3 ± 0.4 | PhtjR |
| 96214 | Tanjil | S | S | R | R | R | 2.2 ± 1.4 | 2.2 ± 1.4 | PhtjR |
| 26979 | 75A:258 | R 4 | R | S | S | S | 1.7 ± 1.3 | 1.7 ± 1.3 | Phr1 |
| 96102 | Unicrop | S | S | S | S | S | 3.4 ± 1.7 | 3.4 ± 1.5 | - |
| 96121 | Emir | S | S | S | S | S | 5.2 ± 1.8 | 4.1 ± 1.1 | - |
| 96210 | Baron | S | S | S | S | S | 4.3 ± 1.3 | 5.2 ± 1.4 | - |
| Line | Response | 1 dpi 1 | 5 dpi | 9 dpi | 16 dpi | 23 dpi |
|---|---|---|---|---|---|---|
| 75A:258 (Phr1, phtjR) | Repression | 83 | 24 | 36 | 86 | 153 |
| Induction | 259 | 672 | 156 | 418 | 691 | |
| Wonga (phr1, PhtjR) | Repression | 65 | 132 | 418 | - 2 | - |
| Induction | 364 | 534 | 1166 | |||
| Emir (phr1, phtjR) | Repression | 53 | 26 | 208 | - | - |
| Induction | 101 | 230 | 1496 |
| Line. | 75A:258 | Wonga | Emir | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Days Post Inoculation | 1 1 | 5 | 9 | 1 | 5 | 9 | 1 | 5 | 9 |
| Defense response | 2.4 2 | >3 | >3 | 2.7 | >3 | >3 | >3 | >3 | |
| Response to stress | >3 | >3 | >3 | >3 | >3 | 1.8 | |||
| Oxidation-reduction process | >3 | >3 | 2.7 | >3 | >3 | ||||
| Antibiotic catabolic process | >3 | >3 | >3 | >3 | |||||
| Cofactor catabolic process | 2.7 | >3 | 3.0 | >3 | |||||
| Drug catabolic process | 2.7 | >3 | 3.0 | >3 | |||||
| Hydrogen peroxide catabolic process | >3 | >3 | >3 | >3 | |||||
| Hydrogen peroxide metabolic process | >3 | >3 | >3 | >3 | |||||
| Nucleic acid-templated transcription | 2.2 | 1.9 | 2.5 | 2.0 | |||||
| Reactive oxygen species metabolic process | >3 | >3 | >3 | >3 | |||||
| Regulation of biosynthetic process | 2.7 | 2.5 | >3 | >3 | |||||
| Regulation of cellular biosynthetic process | 2.7 | 2.5 | >3 | >3 | |||||
| Regulation of cellular macromolecule biosynthetic process | 3.0 | 3.0 | >3 | >3 | |||||
| Regulation of cellular metabolic process | 1.8 | 1.7 | 2.5 | 2.0 | |||||
| Regulation of gene expression | 2.2 | 2.5 | 2.5 | 2.1 | |||||
| Regulation of macromolecule biosynthetic process | 2.7 | 2.7 | >3 | >3 | |||||
| Regulation of macromolecule metabolic process | 1.4 | 1.7 | 1.9 | 1.5 | |||||
| Regulation of nitrogen compound metabolic process | 2.2 | 1.9 | 3.0 | 2.5 | |||||
| Regulation of nucleic acid-templated transcription | 3.0 | >3 | >3 | >3 | |||||
| Regulation of nucleobase-containing compound metabolic process | 2.7 | 2.7 | >3 | >3 | |||||
| Regulation of primary metabolic process | 2.0 | 1.8 | 3.0 | 2.0 | |||||
| Regulation of RNA biosynthetic process | 3.0 | >3 | >3 | >3 | |||||
| Regulation of RNA metabolic process | 2.7 | 3.0 | >3 | >3 | |||||
| Regulation of transcription, DNA-templated | 3.0 | >3 | >3 | >3 | |||||
| Response to oxidative stress | 1.7 | 3.0 | 3.0 | 1.7 | |||||
| RNA biosynthetic process | 2.2 | 1.9 | 2.5 | 1.9 | |||||
| Transcription, DNA-templated | 2.2 | 2.0 | 2.5 | 1.9 | |||||
| Antibiotic metabolic process | 2.7 | 1.8 | 2.0 | ||||||
| Oxylipin biosynthetic process | >3 | >3 | |||||||
| Oxylipin metabolic process | >3 | >3 | |||||||
| Regulation of metabolic process | 1.4 | 1.5 | |||||||
| Response to drug | 2.2 | 2.7 | |||||||
| Response to stimulus | 1.8 | 1.5 | |||||||
| Line | 75A:258 | Wonga | Emir | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Days Post Inoculation | 1 1 | 5 | 9 | 1 | 5 | 9 | 1 | 5 | 9 |
| Chitin binding | >3 2 | 2.7 | >3 | 1.5 | >3 | 1.4 | |||
| Heme binding | 2.3 | >3 | >3 | 2.1 | >3 | >3 | |||
| Oxidoreductase activity | >3 | >3 | >3 | 2.2 | >3 | >3 | |||
| Sequence-specific DNA binding | 1.4 | >3 | >3 | >3 | 2.5 | >3 | |||
| Tetrapyrrole binding | 2.2 | >3 | >3 | 1.6 | >3 | >3 | |||
| DNA-binding transcription factor activity | >3 | >3 | >3 | >3 | >3 | ||||
| Transcription regulator activity | >3 | >3 | >3 | >3 | >3 | ||||
| Antioxidant activity | >3 | 2.7 | >3 | >3 | |||||
| Cofactor binding | 1.4 | 1.6 | >3 | 3.0 | |||||
| DNA binding | 2.5 | 2.7 | >3 | >3 | |||||
| Endopeptidase inhibitor activity | 3.0 | 2.7 | 3.0 | 2.3 | |||||
| Endopeptidase regulator activity | 3.0 | 2.7 | 3.0 | 2.3 | |||||
| Oxidoreductase activity, acting on peroxide as acceptor | >3 | 3.0 | >3 | >3 | |||||
| Peptidase inhibitor activity | 3.0 | 2.7 | 3.0 | 2.3 | |||||
| Peptidase regulator activity | 3.0 | 2.4 | 2.4 | 1.9 | |||||
| Peroxidase activity | >3 | 3.0 | >3 | >3 | |||||
| Serine-type endopeptidase inhibitor activity | 3.0 | 3.0 | 1.4 | 1.3 | |||||
| Catalytic activity | >3 | 1.7 | >3 | ||||||
| Dioxygenase activity | >3 | 1.7 | 2.7 | ||||||
| Electron transfer activity | 1.6 | 3.0 | 3.0 | ||||||
| Gene | 75A:258 | Wonga | Emir | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 1 | 5 | 9 | 16 | 23 | 1 | 5 | 9 | 1 | 5 | 9 | |
| TanjilG_24849 | 5.8 2 | 9.3 | 7.7 | 11.9 | 8.9 | 5.2 | 6.4 | 10.3 | 5.9 | 6.9 | 10.2 |
| TanjilG_24253 | 5.2 3 | 7.4 | 4.0 | 18.9 | 23.5 | 6.2 | 5.9 | 9.5 | 8.1 | 20.0 | 9.5 |
| TanjilG_05213 | 2.9 | 6.5 | 3.7 | 4.1 | 5.0 | 4.1 | 4.7 | 5.0 | 1.5 | 3.0 | 7.8 |
| TanjilG_08982 | −2.0 | 20.7 | 18.4 | - 4 | 17.3 | 21.0 | 21.1 | 7.2 | 17.6 | - | 23.1 |
| TanjilG_19904 | 2.6 | 4.5 | 3.9 | 3.5 | 4.4 | 3.0 | 3.3 | 9.0 | 2.3 | 3.3 | 7.7 |
| TanjilG_10317 | 2.0 | 4.5 | 3.2 | 3.2 | 3.8 | 2.5 | 2.4 | 6.3 | 3.2 | 2.9 | 6.3 |
| TanjilG_13015 | 2.4 | 4.2 | 2.8 | 3.5 | 3.7 | 2.6 | 3.5 | 5.5 | 2.9 | 3.9 | 8.1 |
| TanjilG_27897 | 4.7 | 24.8 | 24.0 | 19.4 | 23.5 | 4.9 | 25.0 | 27.5 | 22.1 | 21.3 | 11.1 |
| TanjilG_02482 | 18.0 | 23.8 | 23.8 | - | 25.9 | - | 22.7 | 27.7 | 21.4 | 22.8 | 28.4 |
| TanjilG_23505 | 3.5 | 4.7 | 3.9 | 6.4 | 6.8 | 3.7 | 3.2 | 8.1 | 1.7 | 4.7 | 6.8 |
| TanjilG_15237 | 3.9 | 6.8 | 24.7 | 22.5 | 7.0 | 7.6 | 8.0 | 28.1 | 2.6 | 3.5 | 25.8 |
| TanjilG_10302 | 15.4 | 42.7 | 3.1 | 38.3 | 40.8 | 36.8 | 5.4 | 8.0 | 2.7 | 38.4 | 9.4 |
| Gene | 75A:258 | Wonga | Emir | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 1 | 5 | 9 | 16 | 23 | 1 | 5 | 9 | 16 | 23 | 1 | 5 | 9 | 16 | 23 | |
| TanjilG_24849 | 10.1 2 | 11.3 | 8.4 | 8.6 | 8.4 | 4.2 | 9.9 | 10.9 | 9.2 | 8.2 | 4.6 | 9.1 | 10.2 | 12.5 | 11.4 |
| TanjilG_24253 | 8.1 | 9.3 | 5.2 | 5.7 | 5.6 | −0.7 | 6.0 | 11.1 | 8.8 | 7.7 | 1.9 | 5.4 | 11.8 | 12.6 | 11.6 |
| TanjilG_05213 | 3.7 | 8.4 | 5.2 | 5.8 | 4.9 | 4.4 | 5.2 | 8.2 | 3.5 | 3.7 | 2.4 | 4.3 | 8.7 | 8.7 | 8.2 |
| TanjilG_08982 | 0.7 | 0.6 3 | 0.2 | −0.4 | 0.0 | 0.3 | 2.2 | 3.0 | 1.4 | 1.4 | 0.4 | 2.1 | 3.6 | 3.9 | 2.1 |
| TanjilG_19904 | 2.3 | 2.8 | 2.2 | 3.4 | 3.0 | 5.3 | 4.6 | 7.6 | 5.8 | 6.4 | 1.1 | 2.6 | 5.7 | 7.5 | 6.5 |
| TanjilG_10317 | 2.1 | 4.3 | 2.8 | 1.4 | 2.4 | 1.1 | 1.7 | 6.2 | 5.6 | 5.8 | 1.7 | 1.5 | 4.1 | 5.7 | 6.4 |
| TanjilG_13015 | 3.5 | 5.1 | 3.5 | 1.7 | 3.1 | 3.1 | 4.8 | 7.6 | 3.6 | 4.6 | 1.1 | 3.4 | 6.7 | 6.7 | 6.9 |
| TanjilG_27897 | 7.1 | 7.8 | 7.8 | 4.0 | 6.1 | 6.8 | 10.0 | 10.5 | 10.2 | 10.6 | 2.0 | 3.4 | 10.6 | 11.7 | 12.4 |
| TanjilG_02482 | 1.7 | 1.4 | 0.1 | 9.4 | 3.3 | −0.8 | 1.7 | 4.2 | 1.6 | 0.6 | −1.0 | 0.6 | 7.2 | 3.1 | 1.9 |
| TanjilG_23505 | 6.5 | 4.3 | 4.6 | 3.8 | 5.1 | 4.2 | 4.1 | 8.2 | 6.1 | 5.4 | 2.9 | 4.6 | 6.9 | 6.6 | 8.4 |
| TanjilG_15237 | 8.2 | 9.0 | 8.9 | 3.6 | 5.3 | 7.8 | 9.7 | 14.4 | 11.8 | 8.3 | 4.8 | 4.3 | 9.5 | 15.5 | 11.9 |
| TanjilG_10302 | 6.3 | 7.9 | 6.1 | 8.7 | 4.7 | 3.8 | 7.8 | 8.5 | 5.2 | 6.1 | 2.9 | 5.6 | 10.2 | 10.7 | 8.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Książkiewicz, M.; Rychel-Bielska, S.; Plewiński, P.; Nuc, M.; Irzykowski, W.; Jędryczka, M.; Krajewski, P. The Resistance of Narrow-Leafed Lupin to Diaporthe toxica Is Based on the Rapid Activation of Defense Response Genes. Int. J. Mol. Sci. 2021, 22, 574. https://doi.org/10.3390/ijms22020574
Książkiewicz M, Rychel-Bielska S, Plewiński P, Nuc M, Irzykowski W, Jędryczka M, Krajewski P. The Resistance of Narrow-Leafed Lupin to Diaporthe toxica Is Based on the Rapid Activation of Defense Response Genes. International Journal of Molecular Sciences. 2021; 22(2):574. https://doi.org/10.3390/ijms22020574
Chicago/Turabian StyleKsiążkiewicz, Michał, Sandra Rychel-Bielska, Piotr Plewiński, Maria Nuc, Witold Irzykowski, Małgorzata Jędryczka, and Paweł Krajewski. 2021. "The Resistance of Narrow-Leafed Lupin to Diaporthe toxica Is Based on the Rapid Activation of Defense Response Genes" International Journal of Molecular Sciences 22, no. 2: 574. https://doi.org/10.3390/ijms22020574
APA StyleKsiążkiewicz, M., Rychel-Bielska, S., Plewiński, P., Nuc, M., Irzykowski, W., Jędryczka, M., & Krajewski, P. (2021). The Resistance of Narrow-Leafed Lupin to Diaporthe toxica Is Based on the Rapid Activation of Defense Response Genes. International Journal of Molecular Sciences, 22(2), 574. https://doi.org/10.3390/ijms22020574