Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions


Abstract
:1. Mitochondria
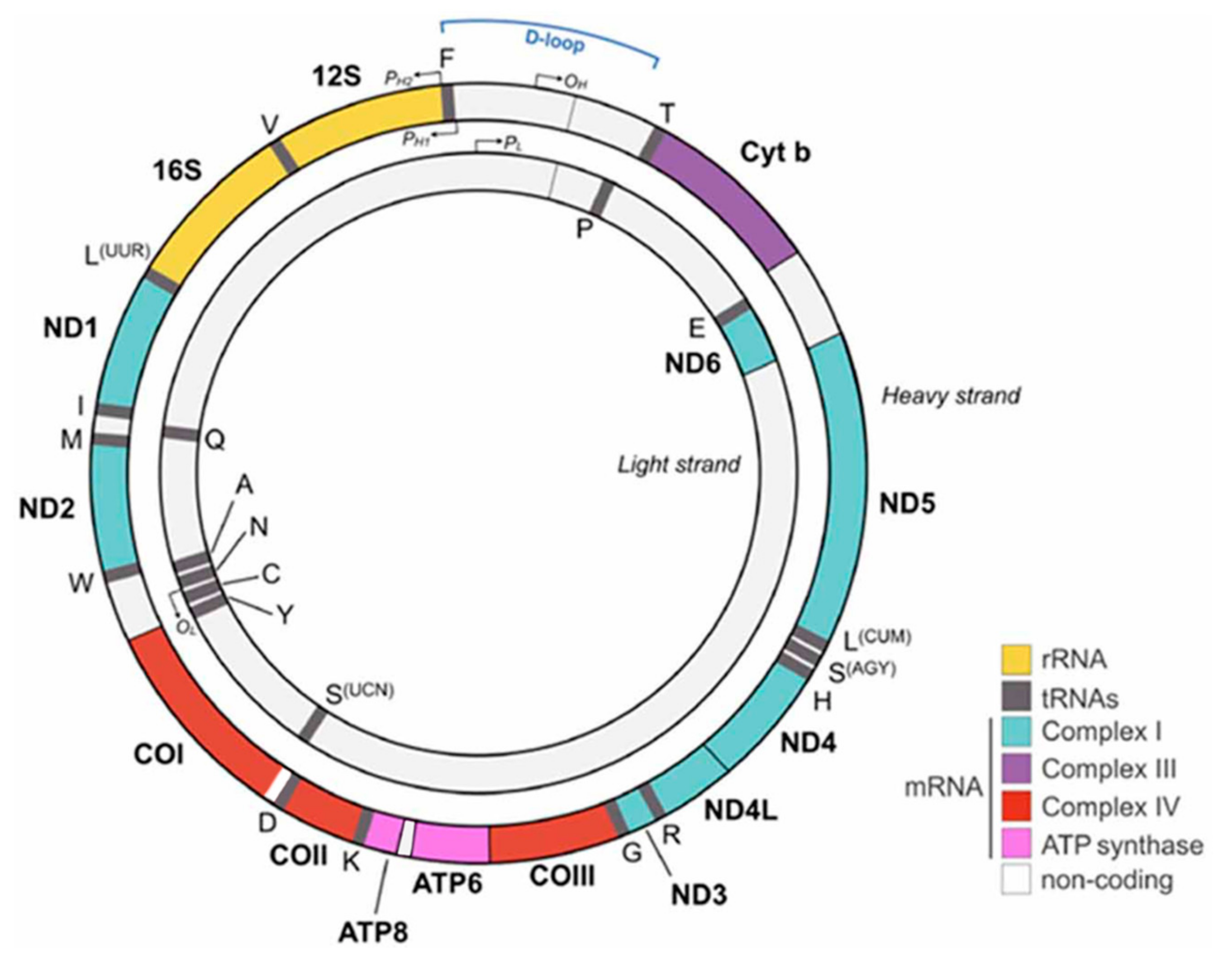
1.1. Origin of Mitochondria and Mitochondrial Genome
1.2. Mitochondrial DNA Mutations

1.3. Mitochondrial Membranes
1.4. Mitochondrial Cristae
1.5. Protein Transport through Mitochondrial Membranes
1.6. Mitochondrial Dynamics
1.7. Mitochondrial Functions
1.7.1. Energy Production
1.7.2. Apoptosis
1.7.3. Calcium Homeostasis
1.7.4. Heme Synthesis
1.7.5. Fe/S Clusters Synthesis
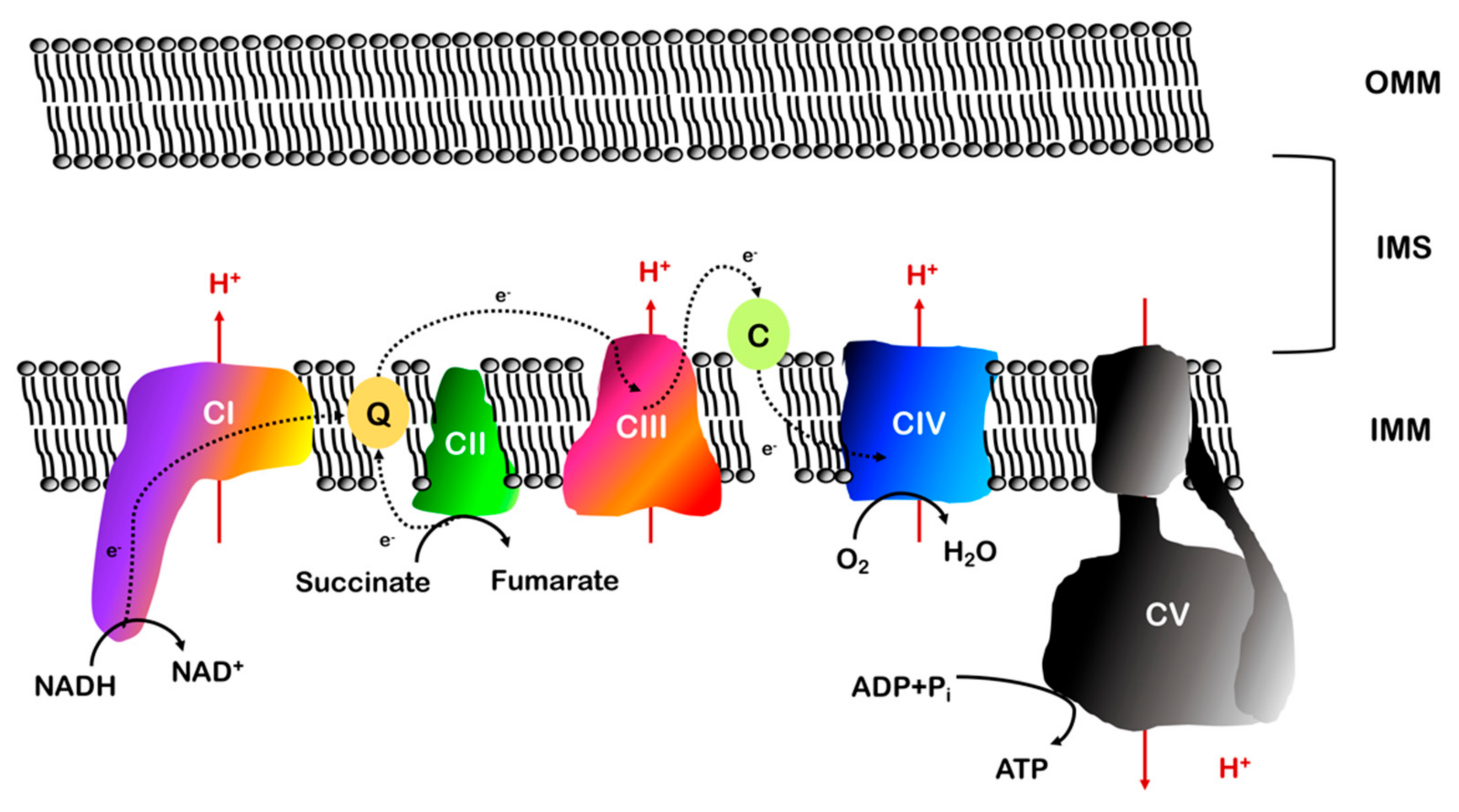
2. The Electron Transport Chain
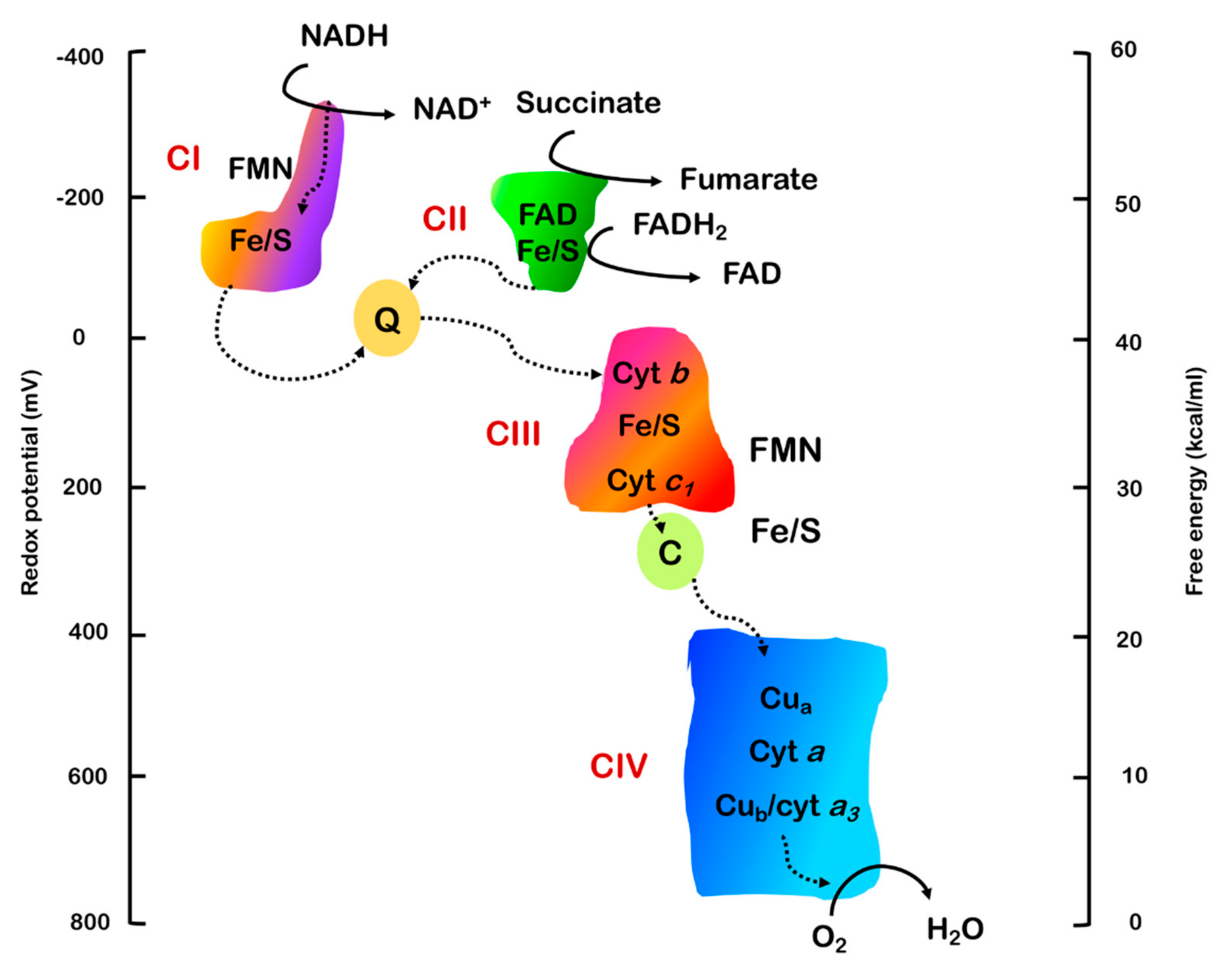
2.1. Proton Gradient and Proton Motive Force
2.2. Electron Transport and Oxidative Phosphorylation
2.3. Complex I
2.3.1. Structure and Assembly

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assembly Factor | Function | CI Interacting Module | References |
|---|---|---|---|
| ACAD9 | Binding of ND2 module | ND2/PP-b module | [113,114] |
| ECSIT | Insertion of ND2 | ND2/PP-b module | [115] |
| FOXRED1 | In a complex with AIFM1 and ACAD9 | ND4/PD module | [116,117] |
| ATP5SL | Binding of ND4 module | ND4/PD module | [118] |
| TMEM70 | Binding of ND4 module | ND4/PD module | [119,120] |
| NDUFAF1 | Insertion of ND2 module | N module, ND1 | [121] |
| NDUFAF2 | Binding of N module | N module | [122] |
| NDUFAF3 | Binding of Q with PP-a | Q module | [123] |
| NDUFAF4 | Binding of Q with PP-a | Q module | [124] |
| NDUFAF5 | Methyltransferase activity | Not known | [125,126] |
| NDUFAF6 | Squalene/phytoene synthase activity | Not known | [127] |
| NDUFAF7 | Methyltransferase activity | Not known | [128,129] |
| NUBPL | 4Fe/4S clusters insertion. Necessary for the entire enzyme stability | Supposed to interact with the developing N module and possibly Q module | [106,107,130] |
| TIMMDC1 | Translocase of inner mitochondrial membrane domain-containing protein 1 | ND1/PP-a | [118,131] |
| TMEM126B | Required for formation of the ND2 module | ND2/PP-b module | [132] |
| TMEM186 | Not known | ND2/PP-b module | [109] |
| DMAC1/TMEM261 | Stabilization and/or assembly of ND5 | ND5/PD-b | [111] |
| COA1 | CIV assembly factor, found bound to CI assembly intermediates | ND2/PP-b module | [109] |
2.3.2. Pathologies Associated with Complex I Deficiency
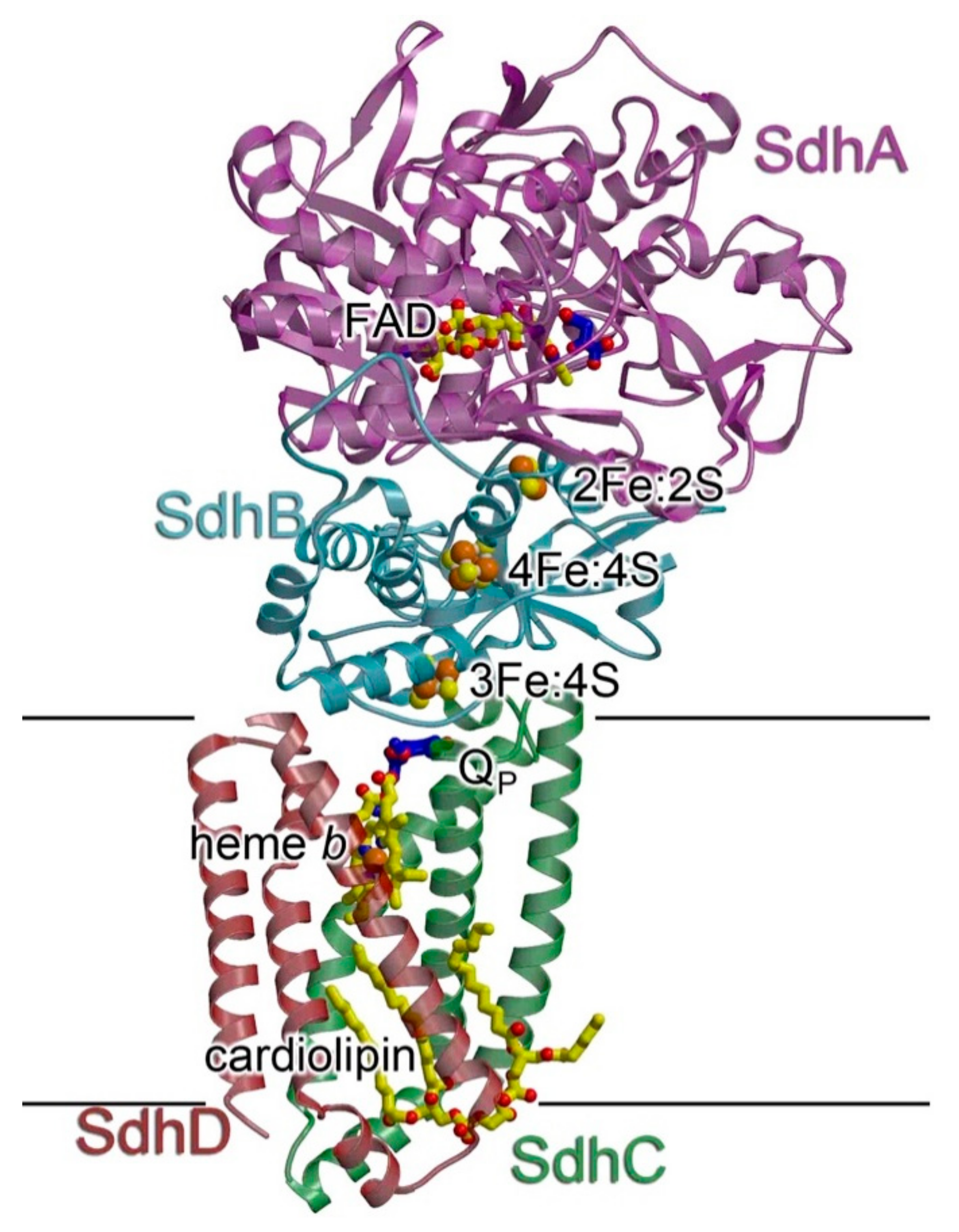
2.4. Complex II
2.4.1. Structure and Assembly
2.4.2. Pathologies Associated with Complex II Deficiency
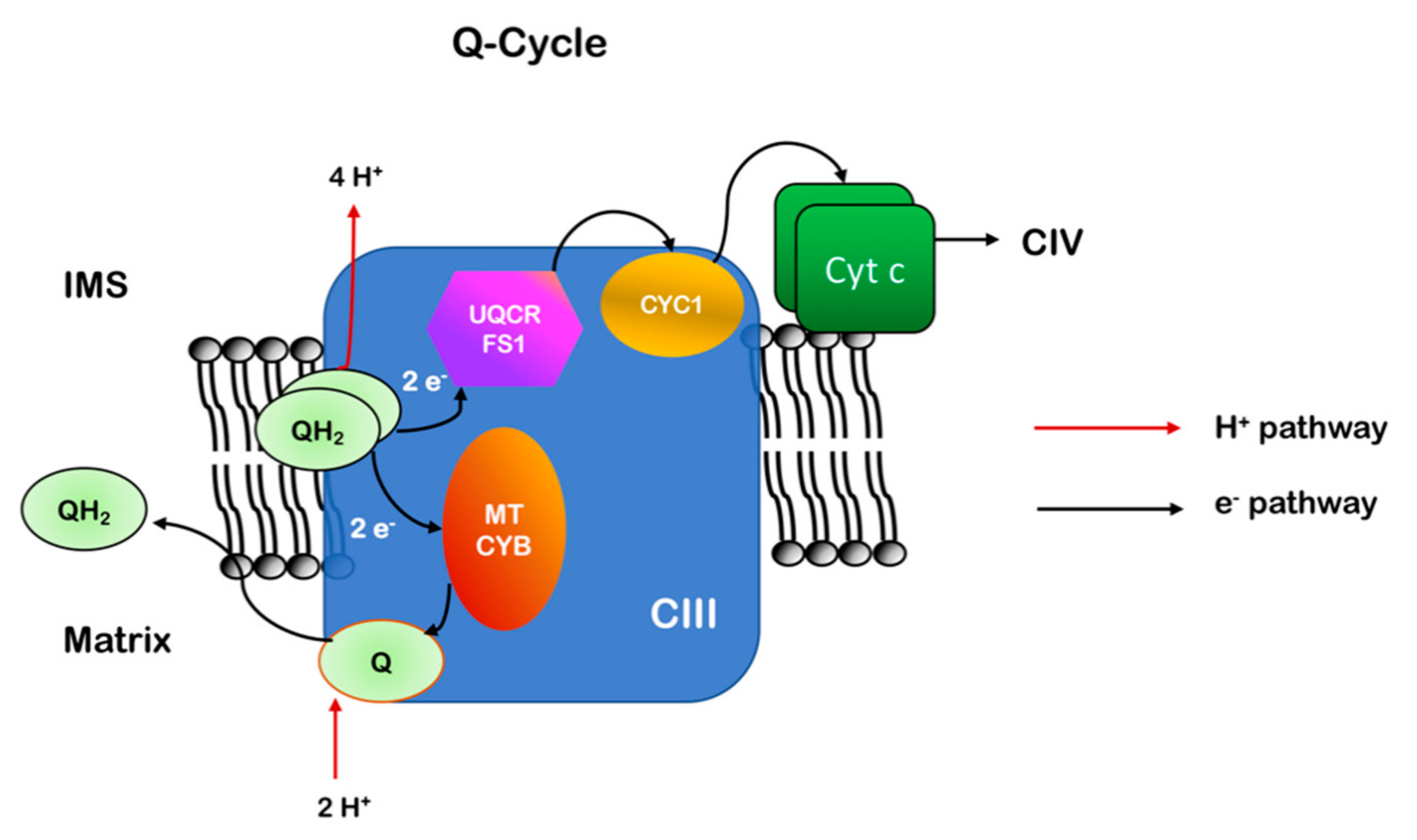
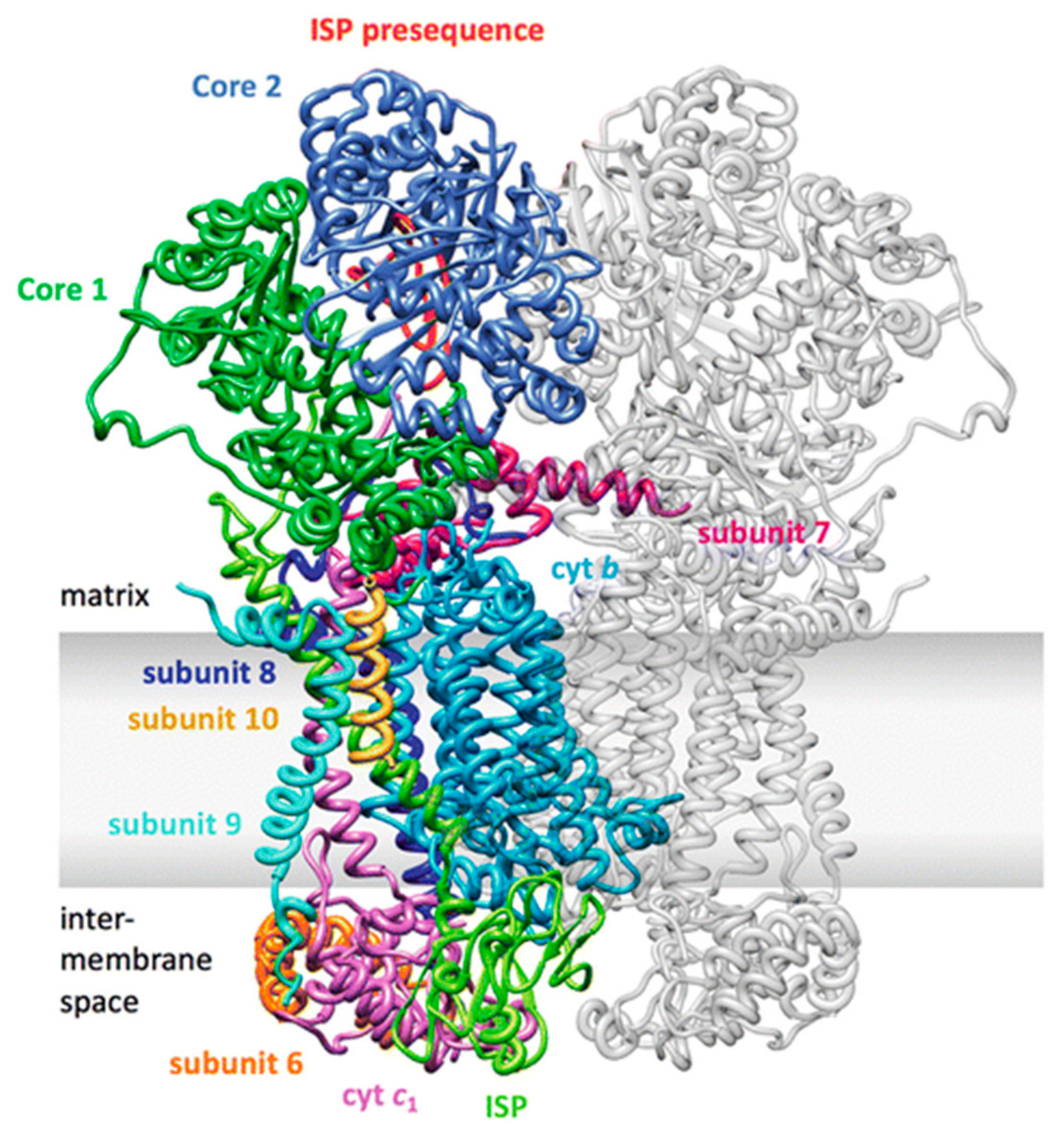
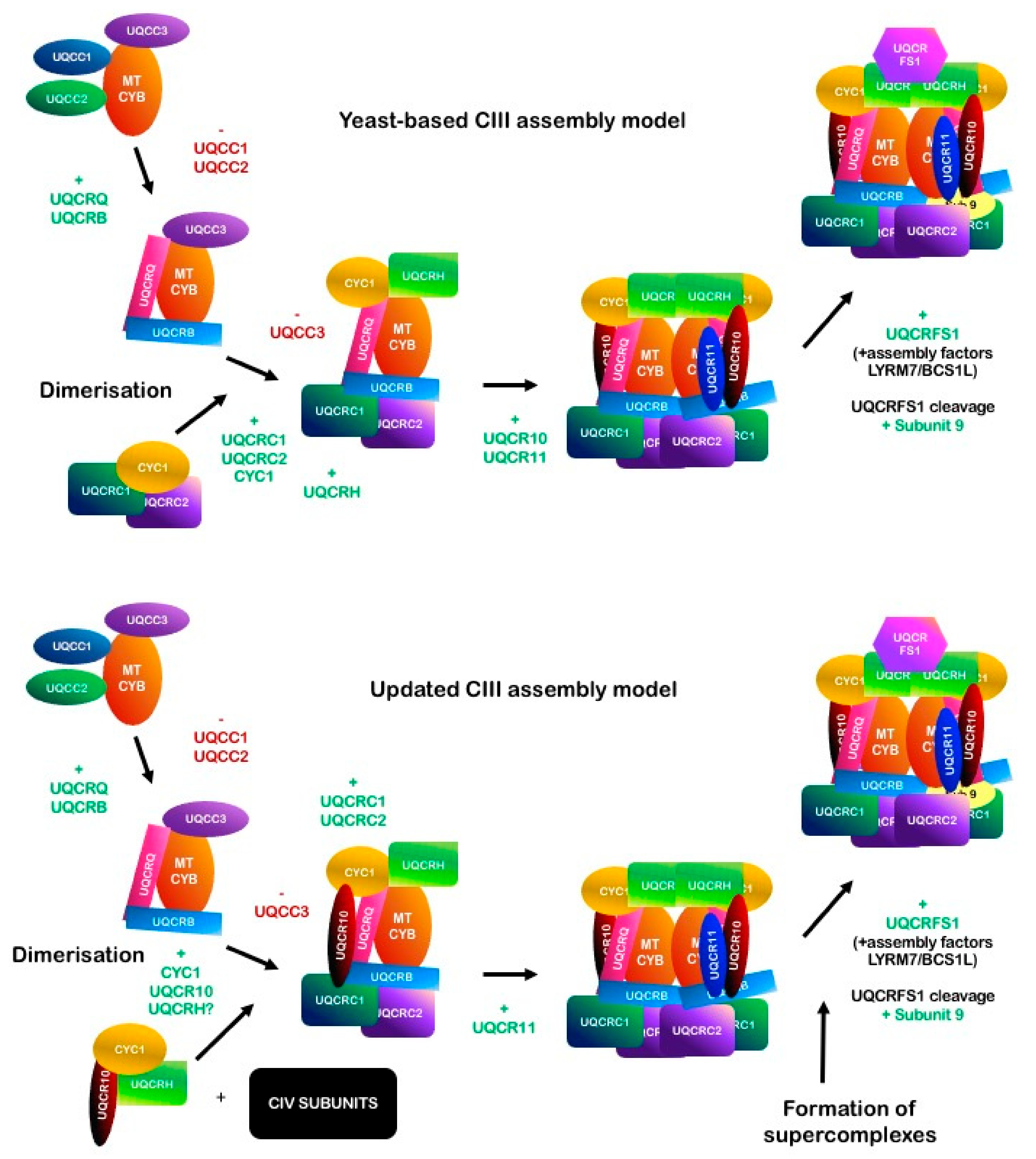
2.5. Complex III
2.5.1. Structure and Subunits
2.5.2. Assembly

| S. cerevisiae | Homo sapiens | ||||
|---|---|---|---|---|---|
| Gene | Protein | Gene | Protein | Reference (ID Yeast) | |
| Complex III subunits | |||||
| COR1 | Cor1 | UQCRC1 | UQCRC1 | [258] | |
| COR2 | Cor2 | UQCRC2 | UQCRC2 | [259] | |
| COB | Cytb | MT-CYB | Cytochrome b | [260] | |
| CYT1 | Cytc1 | CYC1 | CYC1 | [238] | |
| RIP1 | Rip1 | UQCRFS1 | UQCRFS1 | [261] | |
| QCR6 | Qcr6 | UQCRH | UQCRH | [262] | |
| QCR7 | Qcr7 | UQCRB | UQCRB | [263] | |
| QCR8 | Qcr8 | UQCRQ | UQCRQ | [264] | |
| QCR9 | Qcr9 | UQCR10 | UQCR10 | [242] | |
| QCR10 | Qcr10 | UQCR11 | UQCR11 | [244] | |
| - | - | UQCRFS1 | UQCRFS1 | - | |
| Translation factors | Function | Reference | |||
| CBP1 | Cbp1 | - | - | Translational activator of COB mRNA | [265] |
| CBS1 | Cbs1 | - | - | Translational activator of COB mRNA | [266] |
| CBS2 | Cbs2 | - | - | Translational activator of COB mRNA | [266] |
| CBP3 | Cbp3 | UQCC1 | UQCC1 | Translational activator of COB | [267] |
| CBP6 | Cbp6 | UQCC2 | UQCC2 | Translational activator of COB | [268] |
| Assembly factors | |||||
| CBP3 | Cbp3 | UQCC1 | UQCC1 | Cytochrome b assembly factor | [267] |
| CBP6 | Cbp6 | UQCC2 | UQCC2 | Cytochrome b assembly factor | [268] |
| CBP4 | Cbp4 | UQCC3 | UQCC3 | Cytochrome b assembly factor | [269,270] |
| FMP25 | Bca1 | - | - | Early/intermediate stages assembly factor in fungi | [237] |
| CYT2 | Cyt2 | VPS53 | HCCS1 | Heme lyase (Cytochrome c1) | [240] |
| CYC2 | Cyc2 | - | - | Cytochrome c1 and cytochrome c assembly factor | [271] |
| BCS1 | Bcs1 | BCS1L | BCS1L | AAA-ATPase involved in Rieske protein incorporation | [248,249,250,272] |
| MZM1 | Mzm1 | LYRM7 | LYRM7 | Matrix protein involved in Rieske protein incorporation | [253,255,257] |
| - | - | TTC19 | TTC19 | Rieske protein metabolism | [256] |
2.5.3. Pathologies Associated with Complex III Deficiency
2.6. Complex IV
2.6.1. Structure and Subunits

2.6.2. Assembly
2.6.3. Pathologies Associated with Complex IV Deficiency
2.7. Complex V
2.7.1. Structure and Assembly
2.7.2. Pathologies Associated with Complex V Deficiency
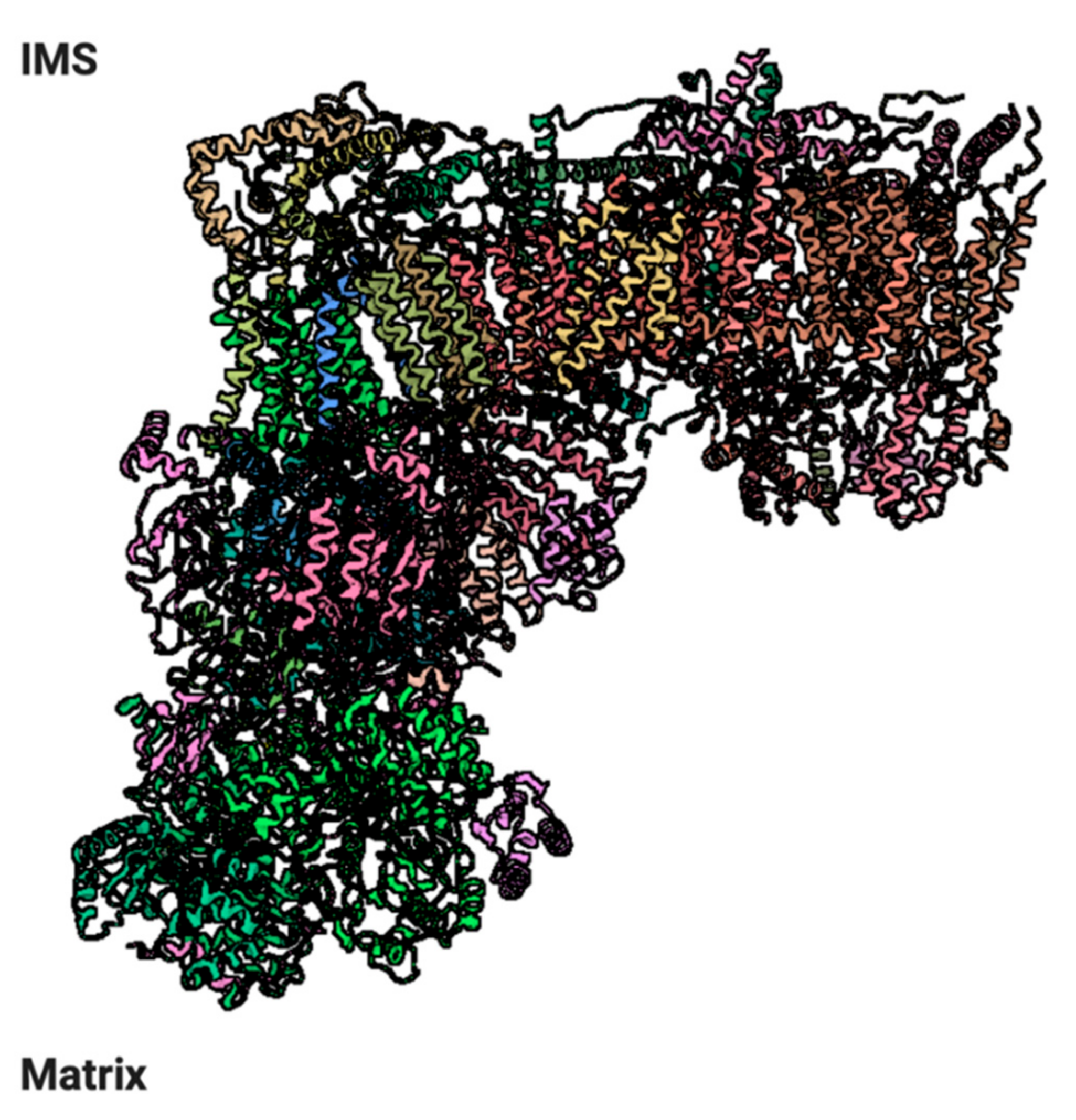
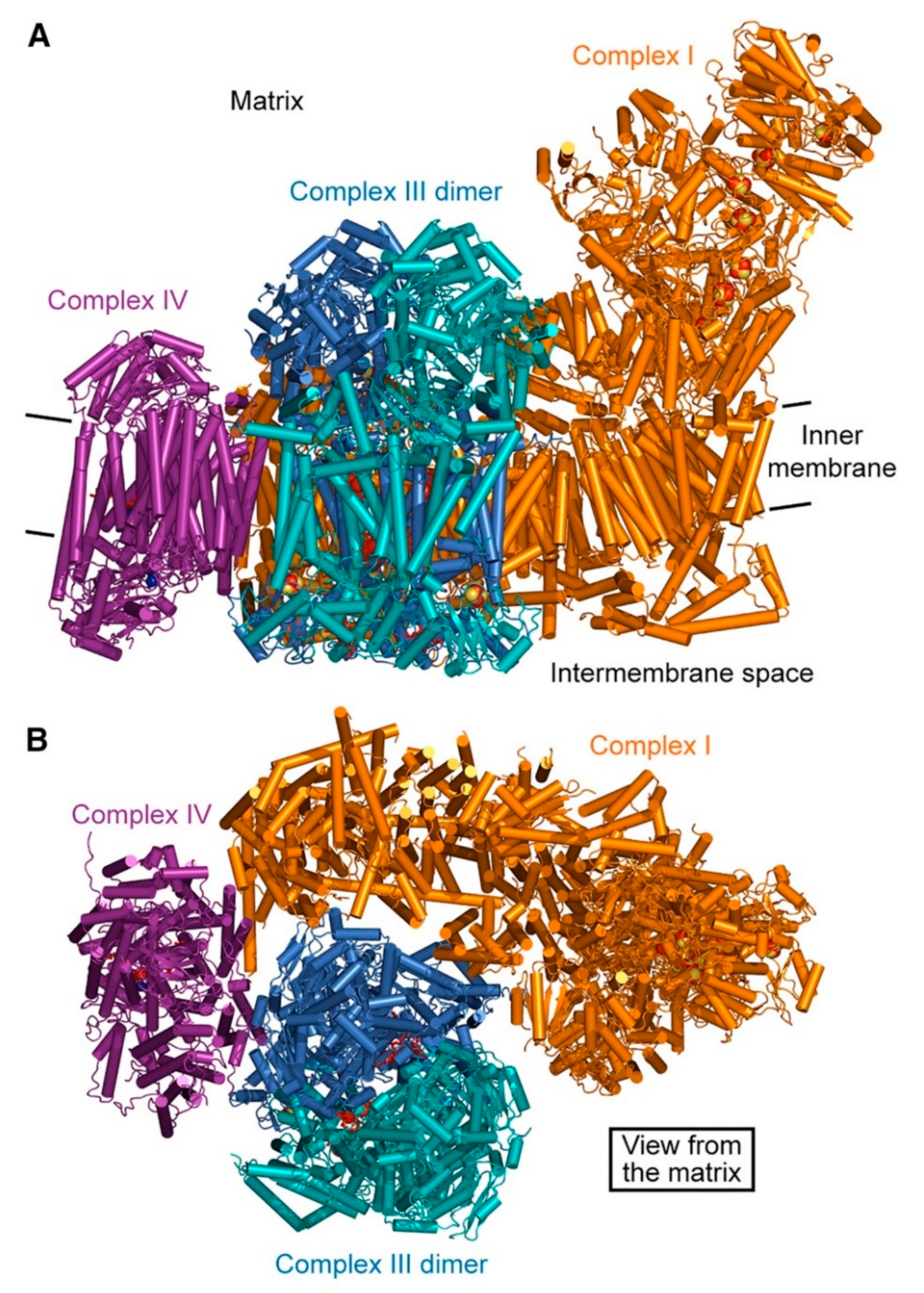
2.8. Localization of the OXPHOS Machinery in the IMM
2.9. Supercomplexes
2.9.1. Existing Models
2.9.2. Species of Supercomplexes and Complex–Complex Interactions

2.9.3. Possible Functional Roles of Supercomplexes
2.9.4. Assembly of Supercomplexes
2.9.5. Conclusive Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Margulis, L. Origin of Eukaryotic Cells: Evidence and Research Implications for a Theory of the Origin and Evolution of Microbial, Plant, and Animal Cells on the Precambrian Earth; Yale University Press: London, UK, 1970. [Google Scholar]
- Cavalier-Smith, T. Origin of mitochondria by intracellular enslavement of a photosynthetic purple bacterium. Proc. Biol. Sci. 2006, 273, 1943–1952. [Google Scholar] [CrossRef] [Green Version]
- Chinnery, P.F.; Hudson, G. Mitochondrial genetics. Br. Med. Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, C.M.; Falkenberg, M.; Larsson, N.G. Maintenance and Expression of Mammalian Mitochondrial DNA. Annu. Rev. Biochem. 2016, 85, 133–160. [Google Scholar] [CrossRef]
- Giles, R.E.; Blanc, H.; Cann, H.M.; Wallace, D.C. Maternal inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1980, 77, 6715–6719. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.; Spengler, D. The Mitochondrion as Potential Interface in Early-Life Stress Brain Programming. Front. Behav. Neurosci. 2018, 12, 306. [Google Scholar] [CrossRef] [Green Version]
- Kuhlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef] [Green Version]
- Ernster, L.; Schatz, G. Mitochondria: A historical review. J. Cell Biol. 1981, 91, 227–255. [Google Scholar]
- Palade, G.E. An electron microscope study of the mitochondrial structure. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1953, 1, 188–211. [Google Scholar] [CrossRef]
- Colombini, M. Voltage gating in the mitochondrial channel, VDAC. J. Membr. Biol. 1989, 111, 103–111. [Google Scholar]
- Nicholls, D.G. The influence of respiration and ATP hydrolysis on the proton-electrochemical gradient across the inner membrane of rat-liver mitochondria as determined by ion distribution. Eur. J. Biochem. 1974, 50, 305–315. [Google Scholar] [CrossRef]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Cogliati, S.; Calvo, E.; Loureiro, M.; Guaras, A.M.; Nieto-Arellano, R.; Garcia-Poyatos, C.; Ezkurdia, I.; Mercader, N.; Vazquez, J.; Enriquez, J.A. Mechanism of super-assembly of respiratory complexes III and IV. Nature 2016, 539, 579–582. [Google Scholar] [CrossRef]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Pfanner, N.; van der Laan, M.; Amati, P.; Capaldi, R.A.; Caudy, A.A.; Chacinska, A.; Darshi, M.; Deckers, M.; Hoppins, S.; Icho, T.; et al. Uniform nomenclature for the mitochondrial contact site and cristae organizing system. J. Cell Biol. 2014, 204, 1083–1086. [Google Scholar] [CrossRef] [Green Version]
- An, J.; Shi, J.; He, Q.; Lui, K.; Liu, Y.; Huang, Y.; Sheikh, M.S. CHCM1/CHCHD6, novel mitochondrial protein linked to regulation of mitofilin and mitochondrial cristae morphology. J. Biol. Chem. 2012, 287, 7411–7426. [Google Scholar] [CrossRef] [Green Version]
- Guarani, V.; McNeill, E.M.; Paulo, J.A.; Huttlin, E.L.; Frohlich, F.; Gygi, S.P.; Van Vactor, D.; Harper, J.W. QIL1 is a novel mitochondrial protein required for MICOS complex stability and cristae morphology. Elife 2015, 4. [Google Scholar] [CrossRef]
- Rampelt, H.; Zerbes, R.M.; van der Laan, M.; Pfanner, N. Role of the mitochondrial contact site and cristae organizing system in membrane architecture and dynamics. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 737–746. [Google Scholar]
- Barbot, M.J.D.; Schulz, C.; Denkert, N.; Kroppen, B.; Hoppert, M.; Jakobs, S.; Meinecke, M. Mic10 Oligomerizes to Bend Mitochondrial Inner Membranes at Cristae Junctions. Cell Metab. 2015, 21, 756–763. [Google Scholar]
- Darshi, M.; Mendiola, V.L.; Mackey, M.R.; Murphy, A.N.; Koller, A.; Perkins, G.A.; Ellisman, M.H.; Taylor, S.S. ChChd3, an inner mitochondrial membrane protein, is essential for maintaining crista integrity and mitochondrial function. J. Biol. Chem. 2011, 286, 2918–2932. [Google Scholar] [CrossRef] [Green Version]
- Huynen, M.A.; Mühlmeister, M.; Gotthardt, K.; Guerrero-Castillo, S.; Brandt, U. Evolution and structural organization of the mitochondrial contact site (MICOS) complex and the mitochondrial intermembrane space bridging (MIB) complex. Biochim. Biophys. Acta 2016, 1863, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Scharwey, M.; Tatsuta, T.; Langer, T. Mitochondrial lipid transport at a glance. J. Cell Sci. 2013, 126, 5317–5323. [Google Scholar] [CrossRef] [Green Version]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [Green Version]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Glytsou, C.; Calvo, E.; Cogliati, S.; Mehrotra, A.; Anastasia, I.; Rigoni, G.; Raimondi, A.; Shintani, N.; Loureiro, M.; Vazquez, J.; et al. Optic Atrophy 1 Is Epistatic to the Core MICOS Component MIC60 in Mitochondrial Cristae Shape Control. Cell Rep. 2016, 17, 3024–3034. [Google Scholar] [CrossRef] [Green Version]
- Paumard, P.; Vaillier, J.; Coulary, B.; Schaeffer, J.; Soubannier, V.; Mueller, D.M.; Brethes, D.; di Rago, J.P.; Velours, J. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 2002, 21, 221–230. [Google Scholar] [CrossRef]
- Palmieri, F.P.C. Mitochondrial metabolite transport. Essays Biochem. 2010, 47, 37–52. [Google Scholar]
- Sickmann, A.; Reinders, J.; Wagner, Y.; Joppich, C.; Zahedi, R.; Meyer, H.E.; Schonfisch, B.; Perschil, I.; Chacinska, A.; Guiard, B.; et al. The proteome of Saccharomyces cerevisiae mitochondria. Proc. Natl. Acad. Sci. USA 2003, 100, 13207–13212. [Google Scholar] [CrossRef] [Green Version]
- Neupert, W. Protein import into mitochondria. Annu. Rev. Biochem. 1997, 66, 863–917. [Google Scholar]
- Mori, M.; Terada, K. Mitochondrial protein import in animals. Biochim. Biophys. Acta 1998, 1403, 12–27. [Google Scholar]
- Wiedemann, N.; Kozjak, V.; Chacinska, A.; Schönfisch, B.; Rospert, S.; Ryan, M.T.; Pfanner, N.; Meisinger, C. Machinery for protein sorting and assembly in the mitochondrial outer membrane. Nature 2003, 424, 565–571. [Google Scholar]
- Stroud, D.; Becker, T.; Qiu, J.; Stojanovski, D.; Pfannschmidt, S.; Wirth, C.; Hunte, C.; Guiard, B.; Meisinger, C.; Pfanner, N.; et al. Biogenesis of mitochondrial β-barrel proteins: The POTRA domain is involved in precursor release from the SAM complex. Mol. Biol. Cell. 2011, 22, 2823–2833. [Google Scholar]
- Mordas, A.; Tokatlidis, K. The MIA pathway: A key regulator of mitochondrial oxidative protein folding and biogenesis. Acc. Chem. Res. 2015, 48, 2191–2199. [Google Scholar] [CrossRef]
- Fox, T. Mitochondrial Protein Synthesis, Import, and Assembly. Genetics 2012, 192, 1203–1234. [Google Scholar]
- Sirrenberg, C.; Bauer, M.F.; Guiard, B.; Neupert, W.; Brunner, M. Import of carrier proteins into the mitochondrial inner membrane mediated by Tim22. Nature 1996, 384, 582–585. [Google Scholar]
- Rehling, P.; Model, K.; Brandner, K.; Kovermann, P.; Sickmann, A.; Meyer, H.E.; Kühlbrandt, W.; Wagner, R.; Truscott, K.N.; Pfanner, N. Protein insertion into the mitochondrial inner membrane by a twin-pore translocase. Science (N. Y.) 2003, 299, 1747–1751. [Google Scholar]
- Krüger, V.; Deckers, M.; Hildenbeutel, M.; van der Laan, M.; Hellmers, M.; Dreker, C.; Preuss, M.; Herrmann, J.M.; Rehling, P.; Wagner, R.; et al. The mitochondrial oxidase assembly protein1 (Oxa1) insertase forms a membrane pore in lipid bilayers. J. Biol. Chem. 2012, 287, 33314–33326. [Google Scholar] [CrossRef] [Green Version]
- Demishtein-Zohary, K.A.A. The TIM23 mitochondrial protein import complex: Function and dysfunction. Cell Tissue Res. 2017, 367, 33–41. [Google Scholar]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [Green Version]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. CB 2018, 28, R170–R185. [Google Scholar] [CrossRef] [Green Version]
- Kraus, F.; Ryan, M.T. The constriction and scission machineries involved in mitochondrial fission. J. Cell Sci. 2017, 130, 2953–2960. [Google Scholar] [CrossRef] [Green Version]
- Waterham, H.R.; Koster, J.; van Roermund, C.W.; Mooyer, P.A.; Wanders, R.J.; Leonard, J.V. A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 2007, 356, 1736–1741. [Google Scholar] [CrossRef]
- Gerber, S.; Charif, M.; Chevrollier, A.; Chaumette, T.; Angebault, C.; Kane, M.S.; Paris, A.; Alban, J.; Quiles, M.; Delettre, C.; et al. Mutations in DNM1L, as in OPA1, result in dominant optic atrophy despite opposite effects on mitochondrial fusion and fission. Brain J. Neurol. 2017, 140, 2586–2596. [Google Scholar] [CrossRef]
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef]
- Pesch, U.E.; Leo-Kottler, B.; Mayer, S.; Jurklies, B.; Kellner, U.; Apfelstedt-Sylla, E.; Zrenner, E.; Alexander, C.; Wissinger, B. OPA1 mutations in patients with autosomal dominant optic atrophy and evidence for semi-dominant inheritance. Hum. Mol. Genet. 2001, 10, 1359–1368. [Google Scholar] [CrossRef] [Green Version]
- Toomes, C.; Marchbank, N.J.; Mackey, D.A.; Craig, J.E.; Newbury-Ecob, R.A.; Bennett, C.P.; Vize, C.J.; Desai, S.P.; Black, G.C.; Patel, N.; et al. Spectrum, frequency and penetrance of OPA1 mutations in dominant optic atrophy. Hum. Mol. Genet. 2001, 10, 1369–1378. [Google Scholar] [CrossRef] [Green Version]
- Delettre, C.; Griffoin, J.M.; Kaplan, J.; Dollfus, H.; Lorenz, B.; Faivre, L.; Lenaers, G.; Belenguer, P.; Hamel, C.P. Mutation spectrum and splicing variants in the OPA1 gene. Hum. Genet. 2001, 109, 584–591. [Google Scholar] [CrossRef]
- Barboni, P.; Carbonelli, M.; Savini, G.; Foscarini, B.; Parisi, V.; Valentino, M.L.; Carta, A.; De Negri, A.; Sadun, F.; Zeviani, M.; et al. OPA1 mutations associated with dominant optic atrophy influence optic nerve head size. Ophthalmology 2010, 117, 1547–1553. [Google Scholar] [CrossRef]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef] [Green Version]
- Civiletto, G.; Varanita, T.; Cerutti, R.; Gorletta, T.; Barbaro, S.; Marchet, S.; Lamperti, C.; Viscomi, C.; Scorrano, L.; Zeviani, M. Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab. 2015, 21, 845–854. [Google Scholar] [CrossRef] [Green Version]
- Frederick, R.L.; Shaw, J.M. Moving mitochondria: Establishing distribution of an essential organelle. Traffic (Cph. Den.) 2007, 8, 1668–1675. [Google Scholar] [CrossRef] [Green Version]
- Hollenbeck, P.J.; Saxton, W.M. The axonal transport of mitochondria. J. Cell Sci. 2005, 118, 5411–5419. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.R.; Zhang, X.N.; Chen, Z. Mitochondrial transport serves as a mitochondrial quality control strategy in axons: Implications for central nervous system disorders. CNS Neurosci. Ther. 2019, 25, 876–886. [Google Scholar] [CrossRef] [Green Version]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Herzig, S.; Raemy, E.; Montessuit, S.; Veuthey, J.L.; Zamboni, N.; Westermann, B.; Kunji, E.R.; Martinou, J.C. Identification and functional expression of the mitochondrial pyruvate carrier. Science (N. Y.) 2012, 337, 93–96. [Google Scholar] [CrossRef]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry, 5th ed.; W. H. Freeman: New York, NY, USA, 2002. [Google Scholar]
- Adeva-Andany, M.M.; Carneiro-Freire, N.; Seco-Filgueira, M.; Fernández-Fernández, C.; Mouriño-Bayolo, D. Mitochondrial β-oxidation of saturated fatty acids in humans. Mitochondrion 2019, 46, 73–90. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef]
- Große, L.; Wurm, C.A.; Brüser, C.; Neumann, D.; Jans, D.C.; Jakobs, S. Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J. 2016, 35, 402–413. [Google Scholar] [CrossRef]
- Salvador-Gallego, R.; Mund, M.; Cosentino, K.; Schneider, J.; Unsay, J.; Schraermeyer, U.; Engelhardt, J.; Ries, J.; García-Sáez, A.J. Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J. 2016, 35, 389–401. [Google Scholar] [CrossRef] [Green Version]
- Tait, S.W.; Green, D.R. Mitochondrial regulation of cell death. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [Green Version]
- Chinnaiyan, A.M. The apoptosome: Heart and soul of the cell death machine. Neoplasia (N. Y.) 1999, 1, 5–15. [Google Scholar] [CrossRef] [Green Version]
- Hill, M.M.; Adrain, C.; Duriez, P.J.; Creagh, E.M.; Martin, S.J. Analysis of the composition, assembly kinetics and activity of native Apaf-1 apoptosomes. EMBO J. 2004, 23, 2134–2145. [Google Scholar] [CrossRef] [Green Version]
- Modjtahedi, N.; Giordanetto, F.; Madeo, F.; Kroemer, G. Apoptosis-inducing factor: Vital and lethal. Trends Cell Biol. 2006, 16, 264–272. [Google Scholar] [CrossRef]
- Ghezzi, D.; Sevrioukova, I.; Invernizzi, F.; Lamperti, C.; Mora, M.; D’Adamo, P.; Novara, F.; Zuffardi, O.; Uziel, G.; Zeviani, M. Severe X-linked mitochondrial encephalomyopathy associated with a mutation in apoptosis-inducing factor. Am. J. Hum. Genet. 2010, 86, 639–649. [Google Scholar] [CrossRef] [Green Version]
- Pozzan, T.; Rizzuto, R. The renaissance of mitochondrial calcium transport. Eur. J. Biochem. 2000, 267, 5269–5273. [Google Scholar] [CrossRef] [Green Version]
- Colombini, M. Structure and mode of action of a voltage dependent anion-selective channel (VDAC) located in the outer mitochondrial membrane. Ann. N. Y. Acad. Sci. 1980, 341, 552–563. [Google Scholar] [CrossRef]
- Giorgi, C.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Poletti, F.; Rimessi, A.; et al. Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion 2012, 12, 77–85. [Google Scholar] [CrossRef]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014, 5, 61. [Google Scholar] [CrossRef] [Green Version]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Molecular Cell Biology, 4th ed.; W. H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Ogun, A.S.V.M. Biochemistry, Heme Synthesis; StatPearls Publishing: Petersbur, FL, USA, 2019. [Google Scholar]
- Ajioka, R.S.; Phillips, J.D.; Kushner, J.P. Biosynthesis of heme in mammals. Biochim. Biophys. Acta 2006, 1763, 723–736. [Google Scholar] [CrossRef] [Green Version]
- Maio, N.; Rouault, T.A. Iron–sulfur cluster biogenesis in mammalian cells: New insights into the molecular mechanisms of cluster delivery. Biochim. Biophys. Acta (BBA) Bioenerg. 2015, 6, 1493–1512. [Google Scholar]
- Imlay, J. Iron-sulphur clusters and the problem with oxygen. Mol. Microbiol. 2006, 59, 1073–1082. [Google Scholar]
- Beinert, H.H.R.; Münck, E. Iron-sulfur clusters: Nature’s modular, multipurpose structures. Science (N. Y.) 1997, 277, 653–659. [Google Scholar]
- Ye, H.R.T. Erythropoiesis and iron sulfur cluster biogenesis. Adv. Hematol. 2010. [Google Scholar] [CrossRef] [Green Version]
- Roche, B.; Aussel, L.; Ezraty, B.; Mandin, P.; Py, B.; Barras, F. Iron/sulfur proteins biogenesis in prokaryotes: Formation, regulation and diversity. Biochim. Biophys. Acta 2013, 1827, 455–469. [Google Scholar] [CrossRef] [Green Version]
- Dean, D.R.; Dos Santos, P.C. Trading Places-Switching Frataxin Function by a Single Amino Acid Substitution within the [Fe-S] Cluster Assembly Scaffold. PLoS Genet. 2015, 11, e1005192. [Google Scholar] [CrossRef]
- Reid, R.A.; Moyle, J.; Mitchell, P. Synthesis of adenosine triphosphate by a protonmotive force in rat liver mitochondria. Nature 1966, 212, 257–258. [Google Scholar] [CrossRef]
- Capaldi, R.A.; Aggeler, R. Mechanism of the F(1)F(0)-type ATP synthase, a biological rotary motor. Trends Biochem. Sci. 2002, 27, 154–160. [Google Scholar] [CrossRef]
- Hunte, C.; Zickermann, V.; Brandt, U. Functional modules and structural basis of conformational coupling in mitochondrial complex I. Science (N. Y.) 2010, 329, 448–451. [Google Scholar] [CrossRef]
- Efremov, R.G.; Sazanov, L.A. Structure of the membrane domain of respiratory complex I. Nature 2011, 476, 414–420. [Google Scholar] [CrossRef]
- Kaila, V.R.; Wikström, M.; Hummer, G. Electrostatics, hydration, and proton transfer dynamics in the membrane domain of respiratory complex I. Proc. Natl. Acad. Sci. USA 2014, 111, 6988–6993. [Google Scholar] [CrossRef] [Green Version]
- Verkhovsky, M.; Bloch, D.A.; Verkhovskaya, M. Tightly-bound ubiquinone in the Escherichia coli respiratory complex I. Biochim. Biophys. Acta 2012, 1817, 1550–1556. [Google Scholar] [CrossRef] [Green Version]
- Brandt, U. A two-state stabilization-change mechanism for proton-pumping complex I. Biochim. Biophys. Acta 2011, 1807, 1364–1369. [Google Scholar] [CrossRef] [Green Version]
- Hirst, J.; Roessler, M.M. Energy conversion, redox catalysis and generation of reactive oxygen species by respiratory complex I. Biochim. Biophys. Acta 2016, 1857, 872–883. [Google Scholar] [CrossRef] [Green Version]
- Cabrera-Orefice, A.; Yoga, E.G.; Wirth, C.; Siegmund, K.; Zwicker, K.; Guerrero-Castillo, S.; Zickermann, V.; Hunte, C.; Brandt, U. Locking loop movement in the ubiquinone pocket of complex I disengages the proton pumps. Nat. Commun. 2018, 9, 4500. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P. Possible molecular mechanisms of the protonmotive function of cytochrome systems. J. Theor. Biol. 1976, 62, 327–367. [Google Scholar] [CrossRef]
- Trumpower, B.L. The protonmotive Q cycle. Energy transduction by coupling of proton translocation to electron transfer by the cytochrome bc1 complex. J. Biol. Chem. 1990, 265, 11409–11412. [Google Scholar]
- Xia, D.; Yu, C.A.; Kim, H.; Xia, J.Z.; Kachurin, A.M.; Zhang, L.; Yu, L.; Deisenhofer, J. Crystal structure of the cytochrome bc1 complex from bovine heart mitochondria. Science (N. Y.) 1997, 277, 60–66. [Google Scholar]
- Iwata, S.; Lee, J.W.; Okada, K.; Lee, J.K.; Iwata, M.; Rasmussen, B.; Link, T.A.; Ramaswamy, S.; Jap, B.K. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science (N. Y.) 1998, 281, 64–71. [Google Scholar]
- Zhang, Z.H.L.; Shulmeister, V.M.; Chi, Y.; Kim, K.K.; Hung, L.; Crofts, A.R.; Berry, E.A.; Kim, S. Electron transfer by domain movement in cytochrome bc1. Nature 1998, 392, 677–684. [Google Scholar]
- Urban, P.F.; Klingenberg, M. On the redox potentials of ubiquinone and cytochrome b in the respiratory chain. Eur. J. Biochem. 1969, 9, 519–525. [Google Scholar] [CrossRef]
- Peters, J.W.; Beratan, D.N.; Bothner, B.; Dyer, R.B.; Harwood, C.S.; Heiden, Z.M.; Hille, R.; Jones, A.K.; King, P.W.; Lu, Y.; et al. A new era for electron bifurcation. Curr. Opin. Chem. Biol. 2018, 47, 32–38. [Google Scholar] [CrossRef]
- Saraste, M. Oxidative phosphorylation at the fin de siecle. Science (N. Y.) 1999, 283, 1488–1493. [Google Scholar] [CrossRef]
- Michel, H. Proton pumping by cytochrome c oxidase. Nature 1999, 402, 602–603. [Google Scholar] [CrossRef]
- Wikstrom, M. Cytochrome c oxidase: 25 years of the elusive proton pump. Biochim. Biophys. Acta 2004, 1655, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Pilet, E.; Jasaitis, A.; Liebl, U.; Vos, M.H. Electron transfer between hemes in mammalian cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2004, 101, 16198–16203. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, S.; Shimada, A. Reaction mechanism of cytochrome c oxidase. Chem. Rev. 2015, 115, 1936–1989. [Google Scholar] [CrossRef]
- Vinothkumar, K.R.; Zhu, J.; Hirst, J. Architecture of mammalian respiratory complex I. Nature 2014, 515, 80–84. [Google Scholar] [CrossRef] [Green Version]
- Vartak, R.S.; Semwal, M.K.; Bai, Y. An update on complex I assembly: The assembly of players. J. Bioenerg. Biomembr. 2014, 46, 323–328. [Google Scholar] [CrossRef] [Green Version]
- Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Post-translational modifications near the quinone binding site of mammalian complex I. J. Biol. Chem. 2013, 288, 24799–24808. [Google Scholar] [CrossRef] [Green Version]
- Claros, M.G.; Vincens, P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 1996, 241, 779–786. [Google Scholar] [CrossRef]
- Sanchez-Caballero, L.; Guerrero-Castillo, S.; Nijtmans, L. Unraveling the complexity of mitochondrial complex I assembly: A dynamic process. Biochim. Biophys. Acta 2016, 1857, 980–990. [Google Scholar] [CrossRef]
- Sheftel, A.D.; Stehling, O.; Pierik, A.J.; Netz, D.J.; Kerscher, S.; Elsasser, H.P.; Wittig, I.; Balk, J.; Brandt, U.; Lill, R. Human ind1, an iron-sulfur cluster assembly factor for respiratory complex I. Mol. Cell Biol. 2009, 29, 6059–6073. [Google Scholar] [CrossRef] [Green Version]
- Bych, K.; Kerscher, S.; Netz, D.J.; Pierik, A.J.; Zwicker, K.; Huynen, M.A.; Lill, R.; Brandt, U.; Balk, J. The iron-sulphur protein Ind1 is required for effective complex I assembly. EMBO J. 2008, 27, 1736–1746. [Google Scholar] [CrossRef] [Green Version]
- Signes, A.; Fernandez-Vizarra, E. Assembly of mammalian oxidative phosphorylation complexes I-V and supercomplexes. Essays Biochem. 2018, 62, 255–270. [Google Scholar] [CrossRef]
- Guerrero-Castillo, S.; Baertling, F.; Kownatzki, D.; Wessels, H.J.; Arnold, S.; Brandt, U.; Nijtmans, L. The Assembly Pathway of Mitochondrial Respiratory Chain Complex I. Cell Metab. 2017, 25, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Formosa, L.E.; Muellner-Wong, L.; Reljic, B.; Sharpe, A.J.; Jackson, T.D.; Beilharz, T.H.; Stojanovski, D.; Lazarou, M.; Stroud, D.A.; Ryan, M.T. Dissecting the Roles of Mitochondrial Complex I Intermediate Assembly Complex Factors in the Biogenesis of Complex I. Cell Rep. 2020, 31, 107541. [Google Scholar] [CrossRef]
- Stroud, D.A.; Surgenor, E.E.; Formosa, L.E.; Reljic, B.; Frazier, A.E.; Dibley, M.G.; Osellame, L.D.; Stait, T.; Beilharz, T.H.; Thorburn, D.R.; et al. Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature 2016, 538, 123–126. [Google Scholar] [CrossRef] [Green Version]
- Giachin, G.; Bouverot, R.; Acajjaoui, S.; Pantalone, S.; Soler-López, M. Dynamics of Human Mitochondrial Complex I Assembly: Implications for Neurodegenerative Diseases. Front. Mol. Biosci. 2016, 3, 43. [Google Scholar]
- Nouws, J.; Nijtmans, L.; Houten, S.M.; van den Brand, M.; Huynen, M.; Venselaar, H.; Hoefs, S.; Gloerich, J.; Kronick, J.; Hutchin, T.; et al. Acyl-CoA dehydrogenase 9 is required for the biogenesis of oxidative phosphorylation complex I. Cell Metab. 2010, 12, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Haack, T.B.; Danhauser, K.; Haberberger, B.; Hoser, J.; Strecker, V.; Boehm, D.; Uziel, G.; Lamantea, E.; Invernizzi, F.; Poulton, J.; et al. Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat. Genet. 2010, 42, 1131–1134. [Google Scholar] [CrossRef]
- Vogel, R.O.; Janssen, R.J.; van den Brand, M.A.; Dieteren, C.E.; Verkaart, S.; Koopman, W.J.; Willems, P.H.; Pluk, W.; van den Heuvel, L.P.; Smeitink, J.A.; et al. Cytosolic signaling protein Ecsit also localizes to mitochondria where it interacts with chaperone NDUFAF1 and functions in complex I assembly. Genes Dev. 2007, 21, 615–624. [Google Scholar] [CrossRef] [Green Version]
- Formosa, L.E.; Mimaki, M.; Frazier, A.E.; McKenzie, M.; Stait, T.L.; Thorburn, D.R.; Stroud, D.A.; Ryan, M.T. Characterization of mitochondrial FOXRED1 in the assembly of respiratory chain complex I. Hum. Mol. Genet. 2015, 24, 2952–2965. [Google Scholar] [CrossRef] [Green Version]
- Rendón, O.; Antonicka, H.; Horvath, R.; Shoubridge, E.A. A mutation in the FAD-dependent oxidoreductase FOXRED1 results in cell-type specific assembly defects in oxidative phosphorylation complexes I and II. Mol. Cell. Biol. 2016, 36, 2132–2140. [Google Scholar]
- Andrews, B.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Assembly factors for the membrane arm of human complex I. Proc. Natl. Acad. Sci. USA 2013, 110, 18934–18939. [Google Scholar] [CrossRef] [Green Version]
- Cizkova, A.; Stranecky, V.; Mayr, J.A.; Tesarova, M.; Havlickova, V.; Paul, J.; Ivanek, R.; Kuss, A.W.; Hansikova, H.; Kaplanova, V.; et al. TMEM70 mutations cause isolated ATP synthase deficiency and neonatal mitochondrial encephalocardiomyopathy. Nat. Genet. 2008, 40, 1288–1290. [Google Scholar] [CrossRef]
- Sánchez-Caballero, L.; Elurbe, D.M.; Baertling, F.; Guerrero-Castillo, S.; van den Brand, M.; van Strien, J.; van Dam, T.J.P.; Rodenburg, R.; Brandt, U.; Huynen, M.A.; et al. TMEM70 functions in the assembly of complexes I and V. Biochim. Biophys. Acta. Bioenerg. 2020, 1861, 148202. [Google Scholar] [CrossRef]
- Vogel, R.O.; Janssen, R.J.; Ugalde, C.; Grovenstein, M.; Huijbens, R.J.; Visch, H.J.; van den Heuvel, L.P.; Willems, P.H.; Zeviani, M.; Smeitink, J.A.; et al. Human mitochondrial complex I assembly is mediated by NDUFAF1. FEBS J. 2005, 272, 5317–5326. [Google Scholar] [CrossRef]
- Ogilvie, I.; Kennaway, N.G.; Shoubridge, E.A. A molecular chaperone for mitochondrial complex I assembly is mutated in a progressive encephalopathy. J. Clin. Investig. 2005, 115, 2784–2792. [Google Scholar] [CrossRef]
- Saada, A.; Vogel, R.O.; Hoefs, S.J.; van den Brand, M.A.; Wessels, H.J.; Willems, P.H.; Venselaar, H.; Shaag, A.; Barghuti, F.; Reish, O.; et al. Mutations in NDUFAF3 (C3ORF60), encoding an NDUFAF4 (C6ORF66)-interacting complex I assembly protein, cause fatal neonatal mitochondrial disease. Am. J. Hum. Genet. 2009, 84, 718–727. [Google Scholar] [CrossRef] [Green Version]
- Saada, A.; Edvardson, S.; Rapoport, M.; Shaag, A.; Amry, K.; Miller, C.; Lorberboum-Galski, H.; Elpeleg, O. C6ORF66 is an assembly factor of mitochondrial complex I. Am. J. Hum. Genet. 2008, 82, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Sugiana, C.; Pagliarini, D.J.; McKenzie, M.; Kirby, D.M.; Salemi, R.; Abu-Amero, K.K.; Dahl, H.H.; Hutchison, W.M.; Vascotto, K.A.; Smith, S.M.; et al. Mutation of C20orf7 disrupts complex I assembly and causes lethal neonatal mitochondrial disease. Am. J. Hum. Genet. 2008, 83, 468–478. [Google Scholar] [CrossRef] [Green Version]
- Rhein, V.F.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. NDUFAF5 Hydroxylates NDUFS7 at an Early Stage in the Assembly of Human Complex I. J. Biol. Chem. 2016, 291, 14851–14860. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, M.; Tucker, E.J.; Compton, A.G.; Lazarou, M.; George, C.; Thorburn, D.R.; Ryan, M.T. Mutations in the gene encoding C8orf38 block complex I assembly by inhibiting production of the mitochondria-encoded subunit ND1. J. Mol. Biol. 2011, 414, 413–426. [Google Scholar] [CrossRef]
- Carilla-Latorre, S.; Gallardo, M.E.; Annesley, S.J.; Calvo-Garrido, J.; Grana, O.; Accari, S.L.; Smith, P.K.; Valencia, A.; Garesse, R.; Fisher, P.R.; et al. MidA is a putative methyltransferase that is required for mitochondrial complex I function. J. Cell Sci. 2010, 123, 1674–1683. [Google Scholar] [CrossRef] [Green Version]
- Rhein, V.F.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. NDUFAF7 methylates arginine 85 in the NDUFS2 subunit of human complex I. J. Biol. Chem. 2013, 288, 33016–33026. [Google Scholar] [CrossRef] [Green Version]
- Protasoni, M.; Bruno, C.; Donati, M.A.; Mohamoud, K.; Severino, M.; Allegri, A.; Robinson, A.J.; Reyes, A.; Zeviani, M.; Garone, C. Novel compound heterozygous pathogenic variants in nucleotide-binding protein like protein (NUBPL) cause leukoencephalopathy with multi-systemic involvement. Mol. Genet. Metab. 2020, 129, 26–34. [Google Scholar] [CrossRef]
- Guarani, V.; Paulo, J.; Zhai, B.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. TIMMDC1/C3orf1 functions as a membrane-embedded mitochondrial complex I assembly factor through association with the MCIA complex. Mol. Cell Biol. 2014, 34, 847–861. [Google Scholar] [CrossRef] [Green Version]
- Heide, H.; Bleier, L.; Steger, M.; Ackermann, J.; Drose, S.; Schwamb, B.; Zornig, M.; Reichert, A.S.; Koch, I.; Wittig, I.; et al. Complexome profiling identifies TMEM126B as a component of the mitochondrial complex I assembly complex. Cell Metab. 2012, 16, 538–549. [Google Scholar] [CrossRef] [Green Version]
- Janssen, R.J.; Nijtmans, L.G.; van den Heuvel, L.P.; Smeitink, J.A. Mitochondrial complex I: Structure, function and pathology. J. Inherit. Metab. Dis. 2006, 29, 499–515. [Google Scholar] [CrossRef]
- Scheffler, I.E. Mitochondrial disease associated with complex I (NADH-CoQ oxidoreductase) deficiency. J. Inherit. Metab. Dis. 2015, 38, 405–415. [Google Scholar] [CrossRef]
- Simon, D.K.; Friedman, J.; Breakefield, X.O.; Jankovic, J.; Brin, M.F.; Provias, J.; Bressman, S.B.; Charness, M.E.; Tarsy, D.; Johns, D.R.; et al. A heteroplasmic mitochondrial complex I gene mutation in adult-onset dystonia. Neurogenetics 2003, 4, 199–205. [Google Scholar] [CrossRef]
- Kirby, D.M.; McFarland, R.; Ohtake, A.; Dunning, C.; Ryan, M.T.; Wilson, C.; Ketteridge, D.; Turnbull, D.M.; Thorburn, D.R.; Taylor, R.W. Mutations of the mitochondrial ND1 gene as a cause of MELAS. J. Med. Genet. 2004, 41, 784–789. [Google Scholar] [CrossRef] [Green Version]
- Howell, N.; Bindoff, L.A.; McCullough, D.A.; Kubacka, I.; Poulton, J.; Mackey, D.; Taylor, L.; Turnbull, D.M. Leber hereditary optic neuropathy: Identification of the same mitochondrial ND1 mutation in six pedigrees. Am. J. Hum. Genet. 1991, 49, 939–950. [Google Scholar]
- Johns, D.R.; Berman, J. Alternative, simultaneous complex I mitochondrial DNA mutations in Leber’s hereditary optic neuropathy. Biochem. Biophys. Res. Commun. 1991, 174, 1324–1330. [Google Scholar] [CrossRef]
- McFarland, R.; Kirby, D.M.; Fowler, K.J.; Ohtake, A.; Ryan, M.T.; Amor, D.J.; Fletcher, J.M.; Dixon, J.W.; Collins, F.A.; Turnbull, D.M.; et al. De novo mutations in the mitochondrial ND3 gene as a cause of infantile mitochondrial encephalopathy and complex I deficiency. Ann. Neurol. 2004, 55, 58–64. [Google Scholar] [CrossRef]
- Torroni, A.; Petrozzi, M.; D’Urbano, L.; Sellitto, D.; Zeviani, M.; Carrara, F.; Carducci, C.; Leuzzi, V.; Carelli, V.; Barboni, P.; et al. Haplotype and phylogenetic analyses suggest that one European-specific mtDNA background plays a role in the expression of Leber hereditary optic neuropathy by increasing the penetrance of the primary mutations 11778 and 14484. Am. J. Hum. Genet. 1997, 60, 1107–1121. [Google Scholar]
- Lertrit, P.; Noer, A.S.; Jean-Francois, M.J.; Kapsa, R.; Dennett, X.; Thyagarajan, D.; Lethlean, K.; Byrne, E.; Marzuki, S. A new disease-related mutation for mitochondrial encephalopathy lactic acidosis and strokelike episodes (MELAS) syndrome affects the ND4 subunit of the respiratory complex I. Am. J. Hum. Genet. 1992, 51, 457–468. [Google Scholar]
- Brown, M.D.; Starikovskaya, E.; Derbeneva, O.; Hosseini, S.; Allen, J.C.; Mikhailovskaya, I.E.; Sukernik, R.I.; Wallace, D.C. The role of mtDNA background in disease expression: A new primary LHON mutation associated with Western Eurasian haplogroup J. Hum. Genet. 2002, 110, 130–138. [Google Scholar] [CrossRef]
- Brown, M.D.; Voljavec, A.S.; Lott, M.T.; MacDonald, I.; Wallace, D.C. Leber’s hereditary optic neuropathy: A model for mitochondrial neurodegenerative diseases. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1992, 6, 2791–2799. [Google Scholar] [CrossRef]
- Liolitsa, D.; Rahman, S.; Benton, S.; Carr, L.J.; Hanna, M.G. Is the mitochondrial complex I ND5 gene a hot-spot for MELAS causing mutations? Ann. Neurol. 2003, 53, 128–132. [Google Scholar] [CrossRef]
- Ravn, K.; Wibrand, F.; Hansen, F.J.; Horn, N.; Rosenberg, T.; Schwartz, M. An mtDNA mutation, 14453G−>A, in the NADH dehydrogenase subunit 6 associated with severe MELAS syndrome. Eur. J. Hum. Genet. EJHG 2001, 9, 805–809. [Google Scholar] [CrossRef] [Green Version]
- Schuelke, M.; Smeitink, J.; Mariman, E.; Loeffen, J.; Plecko, B.; Trijbels, F.; Stöckler-Ipsiroglu, S.; van den Heuvel, L. Mutant NDUFV1 subunit of mitochondrial complex I causes leukodystrophy and myoclonic epilepsy. Nat. Genet. 1999, 21, 260–261. [Google Scholar] [CrossRef]
- Bénit, P.; Chretien, D.; Kadhom, N.; de Lonlay-Debeney, P.; Cormier-Daire, V.; Cabral, A.; Peudenier, S.; Rustin, P.; Munnich, A.; Rötig, A. Large-scale deletion and point mutations of the nuclear NDUFV1 and NDUFS1 genes in mitochondrial complex I deficiency. Am. J. Hum. Genet. 2001, 68, 1344–1352. [Google Scholar] [CrossRef] [Green Version]
- Bénit, P.; Beugnot, R.; Chretien, D.; Giurgea, I.; De Lonlay-Debeney, P.; Issartel, J.P.; Corral-Debrinski, M.; Kerscher, S.; Rustin, P.; Rötig, A.; et al. Mutant NDUFV2 subunit of mitochondrial complex I causes early onset hypertrophic cardiomyopathy and encephalopathy. Hum. Mutat. 2003, 21, 582–586. [Google Scholar] [CrossRef]
- Loeffen, J.; Elpeleg, O.; Smeitink, J.; Smeets, R.; Stöckler-Ipsiroglu, S.; Mandel, H.; Sengers, R.; Trijbels, F.; van den Heuvel, L. Mutations in the complex I NDUFS2 gene of patients with cardiomyopathy and encephalomyopathy. Ann. Neurol. 2001, 49, 195–201. [Google Scholar] [CrossRef]
- Bénit, P.; Slama, A.; Cartault, F.; Giurgea, I.; Chretien, D.; Lebon, S.; Marsac, C.; Munnich, A.; Rötig, A.; Rustin, P. Mutant NDUFS3 subunit of mitochondrial complex I causes Leigh syndrome. J. Med. Genet. 2004, 41, 14–17. [Google Scholar] [CrossRef]
- Budde, S.M.; van den Heuvel, L.P.; Janssen, A.J.; Smeets, R.J.; Buskens, C.A.; DeMeirleir, L.; Van Coster, R.; Baethmann, M.; Voit, T.; Trijbels, J.M.; et al. Combined enzymatic complex I and III deficiency associated with mutations in the nuclear encoded NDUFS4 gene. Biochem. Biophys. Res. Commun. 2000, 275, 63–68. [Google Scholar] [CrossRef]
- Spiegel, R.; Shaag, A.; Mandel, H.; Reich, D.; Penyakov, M.; Hujeirat, Y.; Saada, A.; Elpeleg, O.; Shalev, S.A. Mutated NDUFS6 is the cause of fatal neonatal lactic acidemia in Caucasus Jews. Eur. J. Hum. Genet. EJHG 2009, 17, 1200–1203. [Google Scholar] [CrossRef] [Green Version]
- Smeitink, J.; van den Heuvel, L. Human mitochondrial complex I in health and disease. Am. J. Hum. Genet. 1999, 64, 1505–1510. [Google Scholar] [CrossRef] [Green Version]
- Loeffen, J.; Smeitink, J.; Triepels, R.; Smeets, R.; Schuelke, M.; Sengers, R.; Trijbels, F.; Hamel, B.; Mullaart, R.; van den Heuvel, L. The first nuclear-encoded complex I mutation in a patient with Leigh syndrome. Am. J. Hum. Genet. 1998, 63, 1598–1608. [Google Scholar] [CrossRef] [Green Version]
- Berger, I.; Hershkovitz, E.; Shaag, A.; Edvardson, S.; Saada, A.; Elpeleg, O. Mitochondrial complex I deficiency caused by a deleterious NDUFA11 mutation. Ann. Neurol. 2008, 63, 405–408. [Google Scholar] [CrossRef]
- Fernandez-Moreira, D.; Ugalde, C.; Smeets, R.; Rodenburg, R.J.; Lopez-Laso, E.; Ruiz-Falco, M.L.; Briones, P.; Martin, M.A.; Smeitink, J.A.; Arenas, J. X-linked NDUFA1 gene mutations associated with mitochondrial encephalomyopathy. Ann. Neurol. 2007, 61, 73–83. [Google Scholar] [CrossRef]
- Hoefs, S.J.; Dieteren, C.E.; Distelmaier, F.; Janssen, R.J.; Epplen, A.; Swarts, H.G.; Forkink, M.; Rodenburg, R.J.; Nijtmans, L.G.; Willems, P.H.; et al. NDUFA2 complex I mutation leads to Leigh disease. Am. J. Hum. Genet. 2008, 82, 1306–1315. [Google Scholar] [CrossRef] [Green Version]
- Alston, C.L.; Heidler, J.; Dibley, M.G.; Kremer, L.S.; Taylor, L.S.; Fratter, C.; French, C.E.; Glasgow, R.I.C.; Feichtinger, R.G.; Delon, I.; et al. Bi-allelic Mutations in NDUFA6 Establish Its Role in Early-Onset Isolated Mitochondrial Complex I Deficiency. Am. J. Hum. Genet. 2018, 103, 592–601. [Google Scholar] [CrossRef] [Green Version]
- Bugiani, M.; Invernizzi, F.; Alberio, S.; Briem, E.; Lamantea, E.; Carrara, F.; Moroni, I.; Farina, L.; Spada, M.; Donati, M.A.; et al. Clinical and molecular findings in children with complex I deficiency. Biochim. Biophys. Acta 2004, 1659, 136–147. [Google Scholar] [CrossRef] [Green Version]
- van den Bosch, B.J.; Gerards, M.; Sluiter, W.; Stegmann, A.P.; Jongen, E.L.; Hellebrekers, D.M.; Oegema, R.; Lambrichs, E.H.; Prokisch, H.; Danhauser, K.; et al. Defective NDUFA9 as a novel cause of neonatally fatal complex I disease. J. Med. Genet. 2012, 49, 10–15. [Google Scholar] [CrossRef]
- Hoefs, S.J.; van Spronsen, F.J.; Lenssen, E.W.; Nijtmans, L.G.; Rodenburg, R.J.; Smeitink, J.A.; van den Heuvel, L.P. NDUFA10 mutations cause complex I deficiency in a patient with Leigh disease. Eur. J. Hum. Genet. EJHG 2011, 19, 270–274. [Google Scholar] [CrossRef] [Green Version]
- Ostergaard, E.; Rodenburg, R.J.; van den Brand, M.; Thomsen, L.L.; Duno, M.; Batbayli, M.; Wibrand, F.; Nijtmans, L. Respiratory chain complex I deficiency due to NDUFA12 mutations as a new cause of Leigh syndrome. J. Med. Genet. 2011, 48, 737–740. [Google Scholar] [CrossRef]
- Angebault, C.; Charif, M.; Guegen, N.; Piro-Megy, C.; Mousson de Camaret, B.; Procaccio, V.; Guichet, P.O.; Hebrard, M.; Manes, G.; Leboucq, N.; et al. Mutation in NDUFA13/GRIM19 leads to early onset hypotonia, dyskinesia and sensorial deficiencies, and mitochondrial complex I instability. Hum. Mol. Genet. 2015, 24, 3948–3955. [Google Scholar] [CrossRef] [Green Version]
- Haack, T.B.; Madignier, F.; Herzer, M.; Lamantea, E.; Danhauser, K.; Invernizzi, F.; Koch, J.; Freitag, M.; Drost, R.; Hillier, I.; et al. Mutation screening of 75 candidate genes in 152 complex I deficiency cases identifies pathogenic variants in 16 genes including NDUFB9. J. Med. Genet. 2012, 49, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Piekutowska-Abramczuk, D.; Assouline, Z.; Mataković, L.; Feichtinger, R.G.; Koňařiková, E.; Jurkiewicz, E.; Stawiński, P.; Gusic, M.; Koller, A.; Pollak, A.; et al. NDUFB8 Mutations Cause Mitochondrial Complex I Deficiency in Individuals with Leigh-like Encephalomyopathy. Am. J. Hum. Genet. 2018, 102, 460–467. [Google Scholar] [CrossRef] [Green Version]
- Friederich, M.W.; Erdogan, A.J.; Coughlin, C.R., 2nd; Elos, M.T.; Jiang, H.; O’Rourke, C.P.; Lovell, M.A.; Wartchow, E.; Gowan, K.; Chatfield, K.C.; et al. Mutations in the accessory subunit NDUFB10 result in isolated complex I deficiency and illustrate the critical role of intermembrane space import for complex I holoenzyme assembly. Hum. Mol. Genet. 2017, 26, 702–716. [Google Scholar] [CrossRef] [Green Version]
- van Rahden, V.A.; Fernandez-Vizarra, E.; Alawi, M.; Brand, K.; Fellmann, F.; Horn, D.; Zeviani, M.; Kutsche, K. Mutations in NDUFB11, encoding a complex I component of the mitochondrial respiratory chain, cause microphthalmia with linear skin defects syndrome. Am. J. Hum. Genet. 2015, 96, 640–650. [Google Scholar] [CrossRef] [Green Version]
- Reinson, K.; Kovacs-Nagy, R.; Õiglane-Shlik, E.; Pajusalu, S.; Nõukas, M.; Wintjes, L.T.; van den Brandt, F.C.A.; Brink, M.; Acker, T.; Ahting, U.; et al. Diverse phenotype in patients with complex I deficiency due to mutations in NDUFB11. Eur. J. Med. Genet. 2019, 62, 103572. [Google Scholar] [CrossRef]
- Alahmad, A.; Nasca, A.; Heidler, J.; Thompson, K.; Oláhová, M.; Legati, A.; Lamantea, E.; Meisterknecht, J.; Spagnolo, M.; He, L.; et al. Bi-allelic pathogenic variants in NDUFC2 cause early-onset Leigh syndrome and stalled biogenesis of complex I. EMBO Mol. Med. 2020, 12, e12619. [Google Scholar] [CrossRef]
- Calvo, S.E.; Tucker, E.J.; Compton, A.G.; Kirby, D.M.; Crawford, G.; Burtt, N.P.; Rivas, M.; Guiducci, C.; Bruno, D.L.; Goldberger, O.A.; et al. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat. Genet. 2010, 42, 851–858. [Google Scholar] [CrossRef]
- Dunning, C.J.; McKenzie, M.; Sugiana, C.; Lazarou, M.; Silke, J.; Connelly, A.; Fletcher, J.M.; Kirby, D.M.; Thorburn, D.R.; Ryan, M.T. Human CIA30 is involved in the early assembly of mitochondrial complex I and mutations in its gene cause disease. EMBO J. 2007, 26, 3227–3237. [Google Scholar] [CrossRef] [Green Version]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Alston, C.L.; Veling, M.T.; Heidler, J.; Taylor, L.S.; Alaimo, J.T.; Sung, A.Y.; He, L.; Hopton, S.; Broomfield, A.; Pavaine, J.; et al. Pathogenic Bi-allelic Mutations in NDUFAF8 Cause Leigh Syndrome with an Isolated Complex I Deficiency. Am. J. Hum. Genet. 2020, 106, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Kremer, L.S.; Bader, D.M.; Mertes, C.; Kopajtich, R.; Pichler, G.; Iuso, A.; Haack, T.B.; Graf, E.; Schwarzmayr, T.; Terrile, C.; et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat. Commun. 2017, 8, 15824. [Google Scholar] [CrossRef]
- Sánchez-Caballero, L.; Ruzzenente, B.; Bianchi, L.; Assouline, Z.; Barcia, G.; Metodiev, M.D.; Rio, M.; Funalot, B.; van den Brand, M.A.; Guerrero-Castillo, S.; et al. Mutations in Complex I Assembly Factor TMEM126B Result in Muscle Weakness and Isolated Complex I Deficiency. Am. J. Hum. Genet. 2016, 99, 208–216. [Google Scholar] [CrossRef] [Green Version]
- Alston, C.L.; Compton, A.G.; Formosa, L.E.; Strecker, V.; Oláhová, M.; Haack, T.B.; Smet, J.; Stouffs, K.; Diakumis, P.; Ciara, E.; et al. Biallelic Mutations in TMEM126B Cause Severe Complex I Deficiency with a Variable Clinical Phenotype. Am. J. Hum. Genet. 2016, 99, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Martinez Lyons, A.; Reyes, A.; Robinson, A.J.; Moroni, I.; Ghezzi, D.; Fernandez-Vizarra, E.; Zeviani, M. COA7 (C1orf163/RESA1) mutations associated with mitochondrial leukoencephalopathy and cytochrome c oxidase deficiency. J. Med. Genet. 2016, 53, 846–849. [Google Scholar]
- Rutter, J.W.D.; Schiffman, J.D. Succinate dehydrogenase—Assembly, regulation and role in human disease. Mitochondrion 2010, 10, 393–401. [Google Scholar]
- Cecchini, G. Function and structure of complex II of the respiratory chain. Annu. Rev. Biochem. 2003, 72, 77–109. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.; Huo, X.; Zhai, Y.; Wang, A.; Xu, J.; Su, D.; Bartlam, M.; Rao, Z. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell 2005, 121, 1043–1057. [Google Scholar]
- Iverson, T.M. Catalytic mechanisms of complex II enzymes: A structural perspective. Biochim. Biophys. Acta 2013, 1827, 648–657. [Google Scholar] [CrossRef] [Green Version]
- Van Vranken, J.; Na, U.; Winge, D.R.; Rutter, J. Protein-mediated assembly of succinate dehydrogenase and its cofactors. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 168–180. [Google Scholar]
- Hao, H.-X.; Khalimonchuk, O.; Schraders, M.; Dephoure, N.; Bayley, J.-P.; Kunst, H.; Devilee, P.; Cremers CW, R.J.; Schiffman, J.D.; Bentz, B.G.; et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science (N. Y.) 2009, 325, 1139–1142. [Google Scholar]
- Ghezzi, D.; Goffrini, P.; Uziel, G.; Horvath, R.; Klopstock, T.; Lochmüller, H.; D’Adamo, P.; Gasparini, P.; Strom, T.M.; Prokisch, H.; et al. SDHAF1, encoding a LYR complex-II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nat. Genet. 2009, 41, 654–656. [Google Scholar]
- Maio, N.; Ghezzi, D.; Verrigni, D.; Rizza, T.; Bertini, E.; Martinelli, D.; Zeviani, M.; Singh, A.; Carrozzo, R.; Rouault, T.A. Disease- Causing SDHAF1 Mutations Impair Transfer of Fe-S Clusters to SDHB. Cell Metab. 2016, 23, 292–302. [Google Scholar]
- Dwight, T.; Na, U.; Kim, E.; Zhu, Y.; Richardson, A.L.; Robinson, B.G.; Tucker, K.M.; Gill, A.J.; Benn, D.E.; Clifton-Bligh, R.J.; et al. Analysis of SDHAF3 in familial and sporadic pheochromocytoma and paraganglioma. BMC Cancer 2017, 17, 497. [Google Scholar]
- Munnich, A.; Rustin, P. Clinical spectrum and diagnosis of mitochondrial disorders. Am. J. Med. Genet. 2001, 106, 4–17. [Google Scholar]
- Alston, C.L.; Davison, J.E.; Meloni, F.; van der Westhuizen, F.H.; He, L.; Hornig-Do, H.-T.; Peet, A.C.; Gissen, P.; Goffrini, P.; Ferrero, I.; et al. Recessive germline SDHA and SDHB mutations causing leukodystrophy and isolated mitochondrial complex II deficiency. J. Med. Genet. 2012, 49, 569–577. [Google Scholar]
- Skoldberg, F.; Grimelius, L.; Woodward, E.R.; Rorsman, F.; Van Schothorst, E.W.; Winqvist, O.; Karlsson, F.A.; Akerstrom, G.; Kampe, O.; Husebye, E.S. A family with hereditary extra-adrenal paragangliomas without evidence for mutations in the von Hippel-Lindau disease or ret genes. Clin. Endocr. 1998, 48, 11–16. [Google Scholar]
- Baysal, B.E.; Willett-Brozick, J.E.; Filho, P.A.A.; Lawrence, E.C.; Myers, E.N.; Ferrell, R.E. An Alu-mediated partial SDHC deletion causes familial and sporadic paraganglioma. J. Med. Genet. 2004, 41, 703–709. [Google Scholar]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; van der May, A.; Taschner, P.E.M.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science (N. Y.) 2000, 287, 848–851. [Google Scholar]
- Lussey-Lepoutre, C.; Buffet, A.; Gimenez-Roqueplo, A.P.; Favier, J. Mitochondrial Deficiencies in the Predisposition to Paraganglioma. Metabolites 2017, 7, 17. [Google Scholar] [CrossRef]
- Ghezzi, D.; Zeviani, M. Human diseases associated with defects in assembly of OXPHOS complexes. Essays Biochem. 2018, 62, 271–286. [Google Scholar]
- Parfait, B.; Chretien, D.; Rötig, A.; Marsac, C.; Munnich, A.; Rustin, P. Compound heterozygous mutations in the flavoprotein gene of the respiratory chain complex II in a patient with Leigh syndrome. Hum. Genet. 2000, 106, 236–243. [Google Scholar] [CrossRef]
- Levitas, A.; Muhammad, E.; Harel, G.; Saada, A.; Caspi, V.C.; Manor, E.; Beck, J.C.; Sheffield, V.; Parvari, R. Familial neonatal isolated cardiomyopathy caused by a mutation in the flavoprotein subunit of succinate dehydrogenase. Eur. J. Hum. Genet. EJHG 2010, 18, 1160–1165. [Google Scholar] [CrossRef]
- Burnichon, N.; Brière, J.J.; Libé, R.; Vescovo, L.; Rivière, J.; Tissier, F.; Jouanno, E.; Jeunemaitre, X.; Bénit, P.; Tzagoloff, A.; et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum. Mol. Genet. 2010, 19, 3011–3020. [Google Scholar] [CrossRef] [Green Version]
- Astuti, D.; Latif, F.; Dallol, A.; Dahia, P.L.; Douglas, F.; George, E.; Sköldberg, F.; Husebye, E.S.; Eng, C.; Maher, E.R. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am. J. Hum. Genet. 2001, 69, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Janeway, K.A.; Kim, S.Y.; Lodish, M.; Nosé, V.; Rustin, P.; Gaal, J.; Dahia, P.L.; Liegl, B.; Ball, E.R.; Raygada, M.; et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc. Natl. Acad. Sci. USA 2011, 108, 314–318. [Google Scholar] [CrossRef] [Green Version]
- McWhinney, S.R.; Pasini, B.; Stratakis, C.A. Familial gastrointestinal stromal tumors and germ-line mutations. N. Engl. J. Med. 2007, 357, 1054–1056. [Google Scholar] [CrossRef]
- Brandt, U.; Yu, L.; Yu, C.A.; Trumpower, B.L. The mitochondrial targeting presequence of the Rieske iron-sulfur protein is processed in a single step after insertion into the cytochrome bc1 complex in mammals and retained as a subunit in the complex. J. Biol. Chem. 1993, 268, 8387–8390. [Google Scholar]
- Bottani, E.; Cerutti, R.; Harbour, M.E.; Ravaglia, S.; Dogan, S.A.; Giordano, C.; Fearnley, I.M.; D’Amati, G.; Viscomi, C.; Fernandez-Vizarra, E.; et al. TTC19 Plays a Husbandry Role on UQCRFS1 Turnover in the Biogenesis of Mitochondrial Respiratory Complex III. Mol. Cell 2017, 67, 96–105.e104. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Vizarra, E.; Zeviani, M. Mitochondrial complex III Rieske Fe-S protein processing and assembly. Cell Cycle (Georget. Tex.) 2018, 17, 681–687. [Google Scholar] [CrossRef] [Green Version]
- Schagger, H.; Link, T.A.; Engel, W.D.; von Jagow, G. Isolation of the eleven protein subunits of the bc1 complex from beef heart. Methods Enzymol. 1986, 126, 224–237. [Google Scholar]
- Yu, C.; Xia, J.Z.; Kachurin, A.M.; Yu, L.; Xia, D.; Kim, H.; Deisenhofer, J. Crystallization and preliminary structure of beef heart mitochondrial cytochrome-bc1 complex. Biochim. Biophys. Acta 1996, 1275, 47–53. [Google Scholar]
- Berry, E.; Huang, L.S.; Zhang, Z.; Kim, S.H. Structure of the avian mitochondrial cytochrome bc1 complex. J. Bioenerg. Biomembr. 1999, 31, 177–190. [Google Scholar]
- Lange, C.H.C. Crystal structure of the yeast cytochrome bc1 complex with its bound substrate cytochrome c. Proc. Natl. Acad. Sci. USA 2002, 99, 2800–2805. [Google Scholar]
- Sousa, J.S.; Vonck, J. Mitochondrial Respiratory Chain Complexes; Springer: Singapore, 2018; Volume 87. [Google Scholar]
- Kim, H.J.; Khalimonchuk, O.; Smith, P.M.; Winge, D.R. Structure, function, and assembly of heme centers in mitochondrial respiratory complexes. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2012, 1823, 1604–1616. [Google Scholar]
- Yun, C.H.; Gennis, R.B. Assignment of the histidine axial ligands to the cytochrome bH and cytochrome bL components of the bc1 complex from Rhodobacter sphaeroides by site-directed mutagenesis. Biochemistry 1991, 30, 6747–6754. [Google Scholar]
- Iwata, M.; Bjorkman, J.; Iwata, S. Conformational change of the Rieske [2Fe-2S] protein in cytochrome bc1 complex. J. Bioenerg. Biomembr. 1999, 31, 169–175. [Google Scholar] [CrossRef]
- Graham, L.A.; Brandt, U.; Trumpower, B.L. Protease maturation of the Rieske iron-sulphur protein after its insertion into the mitochondrial cytochrome bc1 complex of Saccharomyces cerevisiae. Biochem. Soc. Trans. 1994, 22, 188–191. [Google Scholar] [CrossRef] [Green Version]
- Deng, K.; Shenoy, S.K.; Tso, S.C.; Yu, L.; Yu, C.A. Reconstitution of mitochondrial processing peptidase from the core proteins (subunits I and II) of bovine heart mitochondrial cytochrome bc(1) complex. J. Biol. Chem. 2001, 276, 6499–6505. [Google Scholar] [CrossRef] [Green Version]
- Deng, K.; Zhang, L.; Kachurin, A.M.; Yu, L.; Xia, D.; Kim, H.; Deisenhofer, J.; Yu, C.A. Activation of a matrix processing peptidase from the crystalline cytochrome bc1 complex of bovine heart mitochondria. J. Biol. Chem. 1998, 273, 20752–20757. [Google Scholar] [CrossRef] [Green Version]
- Berry, E.A.; De Bari, H.; Huang, L.S. Unanswered questions about the structure of cytochrome bc1 complexes. Biochim. Biophys. Acta 2013, 1827, 1258–1277. [Google Scholar] [CrossRef] [Green Version]
- Barel, O.; Shorer, Z.; Flusser, H.; Ofir, R.; Narkis, G.; Finer, G.; Shalev, H.; Nasasra, A.; Saada, A.; Birk, O.S. Mitochondrial complex III deficiency associated with a homozygous mutation in UQCRQ. Am. J. Hum. Genet. 2008, 82, 1211–1216. [Google Scholar]
- Haut, S.; Brivet, M.; Touati, G.; Rustin, P.; Lebon, S.; Garcia-Cazorla, A.; Saudubray, J.M.; Boutron, A.; Legrand, A.; Slama, A. A deletion in the human QP-C gene causes a complex III deficiency resulting in hypoglycaemia and lactic acidosis. Hum. Genet. 2003, 113, 118–122. [Google Scholar]
- Zara, V.; Conte, L.; Trumpower, B.L. Identification and characterization of cytochrome bc(1) subcomplexes in mitochondria from yeast with single and double deletions of genes encoding cytochrome bc(1) subunits. FEBS J. 2007, 274, 4526–4539. [Google Scholar] [CrossRef]
- Zara, V.; Conte, L.; Trumpower, B.L. Biogenesis of the yeast cytochrome bc1 complex. Biochim. Biophys. Acta 2009, 1793, 89–96. [Google Scholar] [CrossRef] [Green Version]
- Zara, V.; Conte, L.; Trumpower, B.L. Evidence that the assembly of the yeast cytochrome bc1 complex involves the formation of a large core structure in the inner mitochondrial membrane. FEBS J. 2009, 276, 1900–1914. [Google Scholar] [CrossRef] [Green Version]
- Ndi, M.; Marin-Buera, L.; Salvatori, R.; Singh, A.P.; Ott, M. Biogenesis of the bc1 Complex of the Mitochondrial Respiratory Chain. J. Mol. Biol. 2018, 430, 3892–3905. [Google Scholar] [CrossRef]
- Smith, P.M.; Fox, J.L.; Winge, D.R. Biogenesis of the cytochrome bc(1) complex and role of assembly factors. Biochim. Biophys. Acta 2012, 1817, 276–286. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Vizarra, E.; Zeviani, M. Nuclear gene mutations as the cause of mitochondrial complex III deficiency. Front. Genet. 2015, 6, 134. [Google Scholar] [CrossRef]
- Protasoni, M.; Pérez-Pérez, R.; Lobo-Jarne, T.; Harbour, M.E.; Ding, S.; Peñas, A.; Diaz, F.; Moraes, C.T.; Fearnley, I.M.; Zeviani, M.; et al. Respiratory supercomplexes act as a platform for complex III-mediated maturation of human mitochondrial complexes I and IV. EMBO J. 2020, 39, e102817. [Google Scholar] [CrossRef]
- Christianson, T.; Edwards, J.C.; Mueller, D.M.; Rabinowitz, M. Identification of a single transcriptional initiation site for the glutamic tRNA and COB genes in yeast mitochondria. Proc. Natl. Acad. Sci. USA 1983, 80, 5564–5568. [Google Scholar]
- Hallberg, B.; Larsson, N.G. Making proteins in the powerhouse. Cell Metab. 2014, 20, 226–240. [Google Scholar]
- Xu, F.; Ackerley, C.; Maj, M.C.; Addis, J.B.; Levandovskiy, V.; Lee, J.; Mackay, N.; Cameron, J.M.; Robinson, B.H. Disruption of a mitochondrial RNA-binding protein gene results in decreased cytochrome b expression and a marked reduction in ubiquinol-cytochrome c reductase activity in mouse heart mitochondria. Biochem. J. 2008, 416, 15–26. [Google Scholar]
- Kehrein, K.; Schilling, R.; Möller-Hergt, B.V.; Wurm, C.A.; Jakobs, S.; Lamkemeyer, T.; Langer, T.; Ott, M. Organization of mitochondrial gene expression in two distinct ribosome-containing assemblies. Cell Rep. 2015, 10, 843–853. [Google Scholar]
- Islas-Osuna, M.A.E.T.; Mittelmeier, T.M.; Dieckmann, C.L. Suppressor mutations define two regions in the Cbp1 protein important for mitochondrial cytochrome b mRNA stability in Saccharomyces cerevisiae. Curr. Genet. 2003, 43, 327–336. [Google Scholar]
- Krause-Buchholz, U.; Lauffer, S.; Rödel, G. Saccharomyces cerevisiae translational activator Cbs1p is associated with translationally active mitochondrial ribosomes. Biol. Chem. 2005, 386, 407–415. [Google Scholar]
- Naithani, S.; Saracco, S.A.; Butler, C.A.; Fox, T.D. Interactions among COX1, COX2, and COX3 mRNA-specific translational activator proteins on the inner surface of the mitochondrial inner membrane of Saccharomyces cerevisiae. Mol. Biol. Cell 2003, 14, 324–333. [Google Scholar]
- Salvatori, R.; Kehrein, K.; Singh, A.P.; Aftab, W.; Möller-Hergt, B.V.; Forne, I.; Imhof, A.; Ott, M. Molecular Wiring of a Mitochondrial Translational Feedback Loop. Mol. Cell 2020, 77, 887–900.e885. [Google Scholar] [CrossRef]
- Gruschke, S.; Kehrein, K.; Römpler, K.; Gröne, K.; Israel, L.; Imhof, A.; Herrmann, J.M.; Ott, M. Cbp3–Cbp6 interacts with the yeast mitochondrial ribosomal tunnel exit and promotes cytochrome b synthesis and assembly. J. Cell Biol. 2011, 193, 101–114. [Google Scholar]
- Hildenbeutel, M.; Hegg, E.L.; Stephan, K.; Gruschke, S.; Meunier, B.; Ott, M. Assembly factors monitor sequential hemylation of cytochrome b to regulate mitochondrial translation. J. Cell Biol. 2014, 205, 511–524. [Google Scholar]
- Tucker, E.J.; Wanschers, B.F.; Szklarczyk, R.; Mountford, H.S.; Wijeyeratne, X.W.; van den Brand, M.A.; Leenders, A.M.; Rodenburg, R.J.; Reljic, B.; Compton, A.G.; et al. Mutations in the UQCC1-interacting protein, UQCC2, cause human complex III deficiency associated with perturbed cytochrome b protein expression. PLoS Genet. 2013, 9, e1004034. [Google Scholar] [CrossRef] [Green Version]
- Zara, V.P.I.; Conte, L.; Trumpower, B.L. Further insights into the assembly of the yeast cytochrome bc1 complex based on analysis of single and double deletion mutants lacking supernumerary subunits and cytochrome b. Eur. J. Biochem. 2004, 271, 1209–1218. [Google Scholar]
- Stephan, K.; Ott, M. Timing of dimerization of the bc(1) complex during mitochondrial respiratory chain assembly. Biochim. Biophys. Acta. Bioenerg. 2020, 1861, 148177. [Google Scholar] [CrossRef]
- Mathieu, L.; Saint-Georges, Y.; Jacq, C.; Dujardin, G. A transcriptome screen in yeast identifies a novel assembly factor for the mitochondrial complex III. Mitochondrion 2011, 11, 391–396. [Google Scholar]
- Sadler, I.; Suda, K.; Schatz, G.; Kaudewitz, F.; Haid, A. Sequencing of the nuclear gene for the yeast cytochrome c1 precursor reveals an unusually complex amino-terminal presequence. EMBO J. 1984, 3, 2137–2143. [Google Scholar]
- Arnold, I.; Folsch, H.; Neupert, W.; Stuart, R.A. Two distinct and independent mitochondrial targeting signals function in the sorting of an inner membrane protein, cytochrome c1. J. Biol. Chem. 1998, 273, 1469–1476. [Google Scholar]
- Zollner, A.; Haid, A. Molecular cloning and characterization of the Saccharomyces cerevisiae CYT2 gene encoding cytochrome-c1– heme lyase. FEBS J. 1992, 207, 1093–1100. [Google Scholar]
- van Loon, A.; Brandli, A.W.; Pesold-Hurt, B.; Blank, D.; Schatz, G. Transport of proteins to the mitochondrial intermembrane space: The ‘matrix-targeting’ and the ‘sorting’ domains in the cytochrome c1 presequence. EMBO J. 1987, 6, 2433–2439. [Google Scholar]
- Phillips, J.; Schmitt, M.E.; Brown, T.A.; Beckmann, J.D.; Trumpower, B.L. Isolation and characterization of QCR9, a nuclear gene encoding the 7.3-kDa subunit 9 of the Saccharomyces cerevisiae ubiquinol-cytochrome c oxidoreductase complex. An intron-containing gene with a conserved sequence occurring in the intron of COX4. J. Biol. Chem. 1990, 265, 20813–20821. [Google Scholar]
- Phillips, J.; Graham, L.A.; Trumpower, B.L. Subunit 9 of the Saccharomyces cerevisiae cytochrome bc1 complex is required for insertion of EPR-detectable iron-sulfur cluster into the Rieske iron-sulfur protein. J. Biol. Chem. 1993, 268, 11727–11736. [Google Scholar]
- Brandt, U.; Uribe, S.; Schägger, H.; Trumpower, B.L. Isolation and characterization of QCR10, the nuclear gene encoding the 8.5-kDa subunit 10 of the Saccharomyces cerevisiae cytochrome bc1 complex. J. Biol. Chem. 1994, 269, 12947–12953. [Google Scholar]
- Fu, W.; Japa, S.; Beattie, D.S. Import of the iron-sulfur protein of the cytochrome b.c1 complex into yeast mitochondria. J. Biol. Chem. 1990, 265, 16541–16547. [Google Scholar]
- Fölsch, H.; Neupert, W.; Stuart, R.A. Internal targeting signal of the BCS1 protein: A novel mechanism of import into mitochondria. EMBO J. 1996, 15, 479–487. [Google Scholar]
- Nouet, C.; Mathieu, L.; Dujardin, G. Functional analysis of yeast bcs1 mutants highlights the role of Bcs1p-specific amino acids in the AAA domain. J. Mol. Biol. 2009, 388, 252–261. [Google Scholar]
- Nobrega, F.; Nobrega, M.P.; Tzagoloff, A. BCS1, a novel gene required for the expression of functional Rieske iron-sulfur protein in Saccharomyces cerevisiae. EMBO J. 1992, 11, 3821–3829. [Google Scholar]
- Cruciat, C.M.; Fölsch, H.; Neupert, W.; Stuart, R.A. Bcs1p, an AAA-family member, is a chaperone for the assembly of the cytochrome bc1 complex. EMBO J. 1999, 18, 5226–5233. [Google Scholar]
- Wagener, N.; Funes, S.; Neupert, W. A pathway of protein translocation in mitochondria mediated by the AAA-ATPase Bcs1. Mol. Cell 2011, 44, 191–202. [Google Scholar]
- Kater, L.; Wagener, N.; Berninghausen, O.; Becker, T.; Neupert, W.; Beckmann, R. Structure of the Bcs1 AAA-ATPase suggests an airlock-like translocation mechanism for folded proteins. Nat. Struct. Mol. Biol. 2020, 27, 142–149. [Google Scholar] [CrossRef]
- Tang, W.K.; Borgnia, M.J.; Hsu, A.L.; Esser, L.; Fox, T.; de Val, N.; Xia, D. Structures of AAA protein translocase Bcs1 suggest translocation mechanism of a folded protein. Nat. Struct. Mol. Biol. 2020, 27, 202–209. [Google Scholar] [CrossRef]
- Atkinson, A.; Khalimonchuk, O.; Smith, P.; Sabic, H.; Eide, D.; Winge, D.R. Mzm1 influences a labile pool of mitochondrial zinc important for respiratory function. J. Biol. Chem. 2010, 285, 19450–19459. [Google Scholar]
- Atkinson, A.; Smith, P.; Fox, J.L.; Cui, T.Z.; Khalimonchuk, O.; Winge, D.R. The LYR protein Mzm1 functions in the insertion of the Rieske Fe/S protein in yeast mitochondria. Mol. Cell Biol. 2011, 31, 3988–3996. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, E.; Lobo, T.; Fox, J.L.; Zeviani, M.; Winge, D.R.; Fernandez-Vizarra, E. LYRM7/MZM1L is a UQCRFS1 chaperone involved in the last steps of mitochondrial Complex III assembly in human cells. Biochim. Biophys. Acta 2013, 1827, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, D.; Arzuffi, P.; Zordan, M.; Da Re, C.; Lamperti, C.; Benna, C.; D’Adamo, P.; Diodato, D.; Costa, R.; Mariotti, C.; et al. Mutations in TTC19 cause mitochondrial complex III deficiency and neurological impairment in humans and flies. Nat. Genet. 2011, 43, 259–263. [Google Scholar]
- Cui, T.Z.; Smith, P.M.; Fox, J.L.; Khalimonchuk, O.; Winge, D.R. Late-stage maturation of the Rieske Fe/S protein: Mzm1 stabilizes Rip1 but does not facilitate its translocation by the AAA ATPase Bcs1. Mol. Cell Biol. 2012, 32, 4400–4409. [Google Scholar] [CrossRef] [Green Version]
- Tzagoloff, A.; Wu, M.A.; Crivellone, M. Assembly of the mitochondrial membrane system. Characterization of COR1, the structural gene for the 44-kilodalton core protein of yeast coenzyme QH2-cytochrome c reductase. J. Biol. Chem. 1986, 261, 17163–17169. [Google Scholar]
- Oudshoorn, P.; Van Steeg, H.; Swinkels, B.W.; Schoppink, P.; Grivell, L.A. Subunit II of yeast QH2:cytochrome-c oxidoreductase. Nucleotide sequence of the gene and features of the protein. Eur. J. Biochem. 1987, 163, 97–103. [Google Scholar] [CrossRef]
- Nobrega, F.G.; Tzagoloff, A. Assembly of the mitochondrial membrane system. DNA sequence and organization of the cytochrome b gene in Saccharomyces cerevisiae D273-10B. J. Biol. Chem. 1980, 255, 9828–9837. [Google Scholar]
- Beckmann, J.D.; Ljungdahl, P.O.; Lopez, J.L.; Trumpower, B.L. Isolation and characterization of the nuclear gene encoding the Rieske iron-sulfur protein (RIP1) from Saccharomyces cerevisiae. J. Biol. Chem. 1987, 262, 8901–8909. [Google Scholar]
- Van Loon, A.P.; De Groot, R.J.; De Haan, M.; Dekker, A.; Grivell, L.A. The DNA sequence of the nuclear gene coding for the 17-kd subunit VI of the yeast ubiquinol-cytochrome c reductase: A protein with an extremely high content of acidic amino acids. EMBO J. 1984, 3, 1039–1043. [Google Scholar]
- De Haan, M.; van Loon, A.P.; Kreike, J.; Vaessen, R.T.; Grivell, L.A. The biosynthesis of the ubiquinol-cytochrome c reductase complex in yeast. DNA sequence analysis of the nuclear gene coding for the 14-kDa subunit. Eur. J. Biochem. 1984, 138, 169–177. [Google Scholar] [CrossRef]
- Maarse, A.C.; Grivell, L.A. Nucleotide sequence of the gene encoding the 11-kDa subunit of the ubiquinol-cytochrome-c oxidoreductase in Saccharomyces cerevisiae. Eur. J. Biochem. 1987, 165, 419–425. [Google Scholar] [CrossRef]
- Dieckmann, C.; Pape, L.K.; Tzagoloff, A. Identification and cloning of a yeast nuclear gene (CBP1) involved in expression of mitochondrial cytochrome b. Proc. Natl. Acad. Sci. USA 1982, 79, 1805–1809. [Google Scholar]
- Rödel, G.; Michaelis, U.; Forsbach, V.; Kreike, J.; Kaudewitz, F. Molecular cloning of the yeast nuclear genes CBS1 and CBS2. Curr. Genet. 1986, 11, 47–53. [Google Scholar]
- Wu, M.T.A. Identification and characterization of a new gene (CBP3) required for the expression of yeast coenzyme QH2-cytochrome c reductase. J. Biol. Chem. 1989, 264, 11122–11130. [Google Scholar]
- Dieckmann, C.; Tzagoloff, A. Assembly of the mitochondrial membrane system. CBP6, a yeast nuclear gene necessary for synthesis of cytochrome b. J. Biol. Chem. 1985, 260, 1513–1520. [Google Scholar]
- Crivellone, M. Characterization of CBP4, a new gene essential for the expression of ubiquinol-cytochrome c reductase in Saccharomyces cerevisiae. J. Biol. Chem. 1994, 269, 21284–21292. [Google Scholar]
- Wanschers, B.F.J.; Szklarczyk, R.; van den Brand, M.A.M.; Jonckheere, A.; Suijskens, J.; Smeets, R.; Rodenburg, R.J.; Stephan, K.; Helland, I.B.; Elkamil, A. A mutation in the human CBP4 ortholog UQCC3 impairs complex III assembly, activity and cytochrome b stability. Hum. Mol. Genet. 2014, 23, 6356–6365. [Google Scholar]
- Dumont, M.E.; Schlichter, J.B.; Cardillo, T.S.; Hayes, M.K.; Bethlendy, G.; Sherman, F. CYC2 encodes a factor involved in mitochondrial import of yeast cytochrome c. Mol. Cell Biol. 1993, 13, 6442–6451. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Vizarra, E.; Bugiani, M.; Goffrini, P.; Carrara, F.; Farina, L.; Procopio, E.; Donati, A.; Uziel, G.; Ferrero, I.; Zeviani, M. Impaired complex III assembly associated with BCS1L gene mutations in isolated mitochondrial encephalopathy. Hum. Mol. Genet. 2007, 16, 1241–1252. [Google Scholar] [CrossRef] [Green Version]
- Baker, R.A.; Priestley, J.R.C.; Wilstermann, A.M.; Reese, K.J.; Mark, P.R. Clinical spectrum of BCS1L Mitopathies and their underlying structural relationships. Am. J. Med. Genetics. Part A 2019, 179, 373–380. [Google Scholar] [CrossRef]
- Miyake, N.; Yano, S.; Sakai, C.; Hatakeyama, H.; Matsushima, Y.; Shiina, M.; Watanabe, Y.; Bartley, J.; Abdenur, J.E.; Wang, R.Y.; et al. Mitochondrial Complex III Deficiency Caused by a Homozygous UQCRC2 Mutation Presenting with Neonatal-Onset Recurrent Metabolic Decompensation. Hum. Mutat. 2013, 34, 446–452. [Google Scholar]
- Brown, M.D.; Voljavec, A.S.; Lott, M.T.; Torroni, A.; Yang, C.-C.; Wallace, D.C. Mitochondrial DNA complex I and III mutations associated with Leber’s hereditary optic neuropathy. Genetics 1992, 130, 163–173. [Google Scholar]
- Bouzidi, M.F.; Schagger, H.; Collombet, J.M.; Carrier, H.; Flocard, F.; Quard, S.; Mousson, B.; Godinot, C. Decreased expression of ubiquinol-cytochrome c reductase subunits in patients exhibiting mitochondrial myopathy with progressive exercise intolerance. Neuromusc. Disord. 1993, 3, 599–604. [Google Scholar]
- Keightley, J.A.; Anitori, R.; Burton, M.D.; Quan, F.; Buist, N.R.M.; Kennaway, N.G. Mitochondrial encephalomyopathy and complex III deficiency associated with a stop-codon mutation in the cytochrome b gene. Am. J. Hum. Genet. 2000, 67, 1400–1410. [Google Scholar]
- Andreu, A.L.; Checcarelli, N.; Iwata, S.; Shanske, S.; DiMauro, S. A missense mutation in the mitochondrial cytochrome b gene in a revisited case with histiocytoid cardiomyopathy. Pediat. Res. 2000, 48, 311–314. [Google Scholar]
- Wibrand, F.; Ravn, K.; Schwartz, M.; Rosenberg, T.; Horn, N.; Vissing, J. Multisystem disorder associated with a missense mutation in the mitochondrial cytochrome b gene. Ann. Neurol. 2001, 50, 540–543. [Google Scholar]
- Gaignard, P.; Menezes, M.; Schiff, M.; Bayot, A.; Rak, M.; Ogier de Baulny, H.; Su, C.H.; Gilleron, M.; Lombes, A.; Abida, H.; et al. Mutations in CYC1, encoding cytochrome c1 subunit of respiratory chain complex III, cause insulin-responsive hyperglycemia. Am. J. Hum. Genet. 2013, 93, 384–389. [Google Scholar]
- Gusic, M.; Schottmann, G.; Feichtinger, R.G.; Du, C.; Scholz, C.; Wagner, M.; Mayr, J.A.; Lee, C.Y.; Yépez, V.A.; Lorenz, N.; et al. Bi-Allelic UQCRFS1 Variants Are Associated with Mitochondrial Complex III Deficiency, Cardiomyopathy, and Alopecia Totalis. Am. J. Hum. Genet. 2020, 106, 102–111. [Google Scholar] [CrossRef]
- Visapaa, I.; Fellman, V.; Varilo, T.; Palotie, A.; Raivio, K.O.; Peltonen, L. Assignment of the locus for a new lethal neonatal metabolic syndrome to 2q33-37. Am. J. Hum. Genet. 1998, 63, 1396–1403. [Google Scholar]
- Siddiqi, S.; Siddiq, S.; Mansoor, A.; Oostrik, J.; Ahmad, N.; Kazmi, S.A.; Kremer, H.; Qamar, R.; Schraders, M. Novel mutation in AAA domain of BCS1L causing Bjornstad syndrome. J. Hum. Genet. 2013, 58, 819–821. [Google Scholar]
- de Lonlay, P.; Valnot, I.; Barrientos, A.; Gorbatyuk, M.; Tzagoloff, A.; Taanman, J.W.; Benayoun, E.; Chretien, D.; Kadhom, N.; Lombes, A.; et al. A mutant mitochondrial respiratory chain assembly protein causes complex III deficiency in patients with tubulopathy, encephalopathy and liver failure. Nat. Genet. 2001, 29, 57–60. [Google Scholar] [CrossRef]
- Hinson, J.T.; Fantin, V.R.; Schonberger, J.; Breivik, N.; Siem, G.; McDonough, B.; Sharma, P.; Keogh, I.; Godinho, R.; Santos, F.; et al. Missense mutations in the BCS1L gene as a cause of the Bjornstad syndrome. N. Engl. J. Med. 2007, 356, 809–819. [Google Scholar] [CrossRef]
- Gil-Borlado, M.C.; Gonzalez-Hoyuela, M.; Blazquez, A.; Garcia-Silva, M.T.; Gabaldon, T.; Manzanares, J.; Vara, J.; Martin, M.A.; Seneca, S.; Arenas, J.; et al. Pathogenic mutations in the 5’ untranslated region of BCS1L mRNA in mitochondrial complex III deficiency. Mitochondrion 2009, 9, 299–305. [Google Scholar] [CrossRef]
- Oláhová, M.; Ceccatelli Berti, C.; Collier, J.J.; Alston, C.L.; Jameson, E.; Jones, S.A.; Edwards, N.; He, L.; Chinnery, P.F.; Horvath, R.; et al. Molecular genetic investigations identify new clinical phenotypes associated with BCS1L-related mitochondrial disease. Hum. Mol. Genet. 2019, 28, 3766–3776. [Google Scholar] [CrossRef]
- Blázquez, A.; Gil-Borlado, M.C.; Morán, M.; Verdú, A.; Cazorla-Calleja, M.R.; Martín, M.A.; Arenas, J.; Ugalde, C. Infantile mitochondrial encephalomyopathy with unusual phenotype caused by a novel BCS1L mutation in an isolated complex III-deficient patient. Neuromuscul. Disord. NMD 2009, 19, 143–146. [Google Scholar] [CrossRef]
- Morino, H.; Miyamoto, R.; Ohnishi, S.; Maruyama, H.; Kawakami, H. Exome sequencing reveals a novel TTC19 mutation in an autosomal recessive spinocerebellar ataxia patient. BMC Neurol. 2014, 14, 1–5. [Google Scholar]
- Nogueira, C.; Barros, J.; Sa, M.J.; Azevedo, L.; Taipa, R.; Torraco, A.; Meschini, M.C.; Verrigni, D.; Nesti, C.; Rizza, T.; et al. Novel TTC19 mutation in a family with severe psychiatric manifestations and complex III deficiency. Neurogenetics 2013, 14, 153–160. [Google Scholar]
- Habibzadeh, P.; Inaloo, S.; Silawi, M.; Dastsooz, H.; Farazi Fard, M.A.; Sadeghipour, F.; Faghihi, Z.; Rezaeian, M.; Yavarian, M.; Böhm, J.; et al. A Novel TTC19 Mutation in a Patient With Neurological, Psychological, and Gastrointestinal Impairment. Front. Neurol. 2019, 10, 944. [Google Scholar] [CrossRef]
- Mordaunt, D.A.; Jolley, A.; Balasubramaniam, S.; Thorburn, D.R.; Mountford, H.S.; Compton, A.G.; Nicholl, J.; Manton, N.; Clark, D.; Bratkovic, D.; et al. Phenotypic variation of TTC19-deficient mitochondrial complex III deficiency: A case report and literature review. Am. J. Med. Genetics. Part A 2015, 167, 1330–1336. [Google Scholar] [CrossRef]
- Invernizzi, F.; Tigano, M.; Dallabona, C.; Donnini, C.; Ferrero, I.; Cremonte, M.; Ghezzi, D.; Lamperti, C.; Zeviani, M. A homozygous mutation in LYRM7/MZM1L associated with early onset encephalopathy, lactic acidosis, and severe reduction of mitochondrial complex III activity. Hum. Mutat. 2013, 34, 1619–1622. [Google Scholar] [CrossRef] [Green Version]
- Dallabona, C.; Abbink, T.E.; Carrozzo, R.; Torraco, A.; Legati, A.; van Berkel, C.G.; Niceta, M.; Langella, T.; Verrigni, D.; Rizza, T.; et al. LYRM7 mutations cause a multifocal cavitating leukoencephalopathy with distinct MRI appearance. Brain J. Neurol. 2016, 139, 782–794. [Google Scholar] [CrossRef] [Green Version]
- Feichtinger, R.G.; Brunner-Krainz, M.; Alhaddad, B.; Wortmann, S.B.; Kovacs-Nagy, R.; Stojakovic, T.; Erwa, W.; Resch, B.; Windischhofer, W.; Verheyen, S.; et al. Combined Respiratory Chain Deficiency and UQCC2 Mutations in Neonatal Encephalomyopathy: Defective Supercomplex Assembly in Complex III Deficiencies. Oxidative Med. Cell. Longev. 2017, 2017, 7202589. [Google Scholar] [CrossRef] [Green Version]
- Capaldi, R.A. Structure and function of cytochrome c oxidase. Annu. Rev. Biochem. 1990, 59, 569–596. [Google Scholar]
- Balsa, E.M.R.; Perales-Clemente, E.; Szklarczyk, R.; Calvo, E.; Landázuri, M.O.; Enríquez, J.A. NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell Metab. 2012, 16, 378–386. [Google Scholar]
- Pitceathly, R.; Rahman, S.; Wedatilake, Y.; Polke, J.M.; Cirak, S.; Foley, A.R.; Sailer, A.; Hurles, M.E.; Stalker, J.; Hargreaves, I.; et al. NDUFA4 mutations underlie dysfunction of a cytochrome c oxidase subunit linked to human neurological disease. Cell Rep. 2013, 3, 1795–1805. [Google Scholar]
- Zong, S.; Wu, M.; Gu, J.; Liu, T.; Guo, R.; Yang, M. Structure of the intact 14-subunit human cytochrome c oxidase. Cell Res. 2018, 28, 1026–1034. [Google Scholar] [CrossRef] [Green Version]
- Hill, B. The sequence of electron carriers in the reaction of cytochrome c oxidase with oxygen. J. Bioenerg. Biomembr. 1993, 25, 115–120. [Google Scholar]
- Sinkler, C.A.; Kalpage, H.; Shay, J.; Lee, I.; Malek, M.H.; Grossman, L.I.; Hüttemann, M. Tissue- and Condition-Specific Isoforms of Mammalian Cytochrome c Oxidase Subunits: From Function to Human Disease. Oxidative Med. Cell. Longev. 2017, 2017, 1534056. [Google Scholar] [CrossRef] [Green Version]
- Kadenbach, B.; Huttemann, M. The subunit composition and function of mammalian cytochrome c oxidase. Mitochondrion 2015, 24, 64–76. [Google Scholar]
- Ishigami, I.; Zatsepin, N.A.; Hikita, M.; Conrad, C.E.; Nelson, G.; Coe, J.D.; Basu, S.; Grant, T.D.; Seaberg, M.H.; Sierra, R.G.; et al. Crystal structure of CO-bound cytochrome c oxidase determined by serial femtosecond X-ray crystallography at room temperature. Proc. Natl. Acad. Sci. USA 2017, 114, 8011–8016. [Google Scholar] [CrossRef] [Green Version]
- Nijtmans, L.G.; Muijsers, A.O.; Speijer, D.; Van den Bogert, C. Assembly of cytochrome-c oxidase in cultured human cells. Eur. J. Biochem. 1998, 254, 389–394. [Google Scholar]
- Timón-Gómez, A.; Abriata, L.A.; Vila, A.J.; Hosler, J.; Barrientos, A. Mitochondrial cytochrome c oxidase biogenesis: Recent developments. Semin. Cell Dev. Biol. 2018, 76, 163–178. [Google Scholar]
- Vidoni, S.H.; Guerrero-Castillo, S.; Signes, A.; Ding, S.; Fearnley, I.M.; Taylor, R.W.; Tiranti, V.; Arnold, S.; Fernandez-Vizarra, E.; Zeviani, M. MR-1S Interacts with PET100 and PET117 in Module-Based Assembly of Human Cytochrome c Oxidase. Cell Rep. 2017, 18, 1727–1738. [Google Scholar]
- Hayashi, T.; Asano, Y.; Shintani, Y.; Aoyama, H.; Kioka, H.; Tsukamoto, O.; Hikita, M.; Shinzawa-Itoh, K.; Takafuji, K.; Higo, S.; et al. Higd1a is a positive regulator of cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2015, 112, 1553–1558. [Google Scholar] [CrossRef] [Green Version]
- Mick, D.; Dennerlein, S.; Wiese, H.; Reinhold, R.; Pacheu-Grau, D.; Lorenzi, I.; Sasarman, F.; Weraarpachai, W.; Shoubridge, E.A.; Warscheid, B.; et al. MITRAC links mitochondrial protein translocation to respiratory-chain assembly and translational regulation. Cell 2012, 15, 1528–1541. [Google Scholar]
- Dennerlein, S.O.S.; Jans, D.; Hellwig, C.; Bareth, B.; Jakobs, S.; Deckers, M.; Warscheid, B.; Rehling, P. MITRAC7 Acts as a COX1-Specific Chaperone and Reveals a Checkpoint during Cytochrome c Oxidase Assembly. Cell Rep. 2015, 12, 1644–1655. [Google Scholar]
- Xu, F.; Morin, C.; Mitchell, G.; Ackerley, C.; Robinson, B.H. The role of the LRPPRC (leucine-rich pentatricopeptide repeat cassette) gene in cytochrome oxidase assembly: Mutation causes lowered levels of COX (cytochrome c oxidase) I and COX III mRNA. Biochem. J. 2004, 382, 331–336. [Google Scholar]
- Weraarpachai, W.; Antonicka, H.; Sasarman, F.; Seeger, J.; Schrank, B.; Kolesar, J.E.; Lochmüller, H.; Chevrette, M.; Kaufman, B.A.; Horvath, R.; et al. Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset Leigh syndrome. Nat. Genet. 2009, 41, 833–837. [Google Scholar]
- Szklarczyk, R.W.B.; Cuypers, T.D.; Esseling, J.J.; Riemersma, M.; van den Brand, M.A.; Gloerich, J.; Lasonder, E.; van den Heuvel, L.P.; Nijtmans, L.G.; Huynen, M.A. Iterative orthology prediction uncovers new mitochondrial proteins and identifies C12orf62 as the human ortholog of COX14, a protein involved in the assembly of cytochrome c oxidase. Genome Biol. 2012, 13, R12. [Google Scholar]
- Clemente, P.; Peralta, S.; Cruz-Bermudez, A.; Echevarría, L.; Fontanesi, F.; Barrientos, A.; Fernandez-Moreno, M.A.; Garesse, R. hCOA3 stabilizes cytochrome c oxidase 1 (COX1) and promotes cytochrome c oxidase assembly in human mitochondria. J. Biol. Chem. 2013, 288, 8321–8331. [Google Scholar]
- Mick, D.; Vukotic, M.; Piechura, H.; Meyer, H.E.; Warscheid, B.; Deckers, M.; Rehling, P. Coa3 and Cox14 are essential for negative feedback regulation of COX1 translation in mitochondria. J. Cell Biol. 2010, 191, 141–154. [Google Scholar]
- Bourens, M.; Barrientos, A. A CMC1-knockout reveals translation-independent control of human mitochondrial complex IV biogenesis. EMBO Rep. 2017, 18, 477–494. [Google Scholar]
- Antonicka, H.; Leary, S.C.; Guercin, G.H.; Agar, J.N.; Horvath, R.; Kennaway, N.G.; Harding, C.O.; Jaksch, M.; Shoubridge, E.A. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum. Mol. Genet. 2003, 12, 2693–2702. [Google Scholar]
- Diaz, F.; Thomas, C.K.; Garcia, S.; Hernandez, D.; Moraes, C.T. Mice lacking COX10 in skeletal muscle recapitulate the phenotype of progressive mitochondrial myopathies associated with cytochrome c oxidase deficiency. Hum. Mol. Genet. 2015, 14, 2737–2748. [Google Scholar]
- Taylor, N.G.S.S.; Harris, N.J.; Germany, E.M.; Fox, J.L.; Khalimonchuk, O. The Assembly Factor Pet117 Couples Heme a Synthase Activity to Cytochrome Oxidase Assembly. J. Biol. Chem. 2017, 292, 1815–1825. [Google Scholar]
- Hiser, L.D.V.M.; Hamer, A.G.; Hosler, J.P. Cox11p is required for stable formation of the Cu(B) and magnesium centers of cytochrome c oxidase. J. Biol. Chem. 2000, 275, 619–623. [Google Scholar]
- Bode, M.; Woellhaf, M.W.; Bohnert, M.; van der Laan, M.; Sommer, F.; Jung, M.; Zimmermann, R.; Schroda, M.; Herrmann, J.M. Redox-regulated dynamic interplay between Cox19 and the copper-binding protein Cox11 in the intermembrane space of mitochondria facilitates biogenesis of cytochrome c oxidase. Mol. Biol. Cell 2015, 26, 2385–2401. [Google Scholar]
- Glerum, D.; Shtanko, A.; Tzagoloff, A. Characterization of COX17, a yeast gene involved in copper metabolism and assembly of cytochrome oxidase. J. Biol. Chem. 1996, 271, 14504–14509. [Google Scholar]
- Mansilla, N.; Racca, S.; Gras, D.E.; Gonzalez, D.H.; Welchen, E. The Complexity of Mitochondrial Complex IV: An Update of Cytochrome c Oxidase Biogenesis in Plants. Int. J. Mol. Sci. 2018, 19, 662. [Google Scholar]
- Bourens, M.; Boulet, A.; Leary, S.C.; Barrientos, A. Human COX20 cooperates with SCO1 and SCO2 to mature COX2 and promote the assembly of cytochrome c oxidase. Hum. Mol. Genet. 2014, 23, 2901–2913. [Google Scholar]
- Lorenzi, I.; Oeljeklaus, S.; Aich, A.; Ronsör, C.; Callegari, S.; Dudek, J.; Warscheid, B.; Dennerlein, S.; Rehling, P. The mitochondrial TMEM177 associates with COX20 during COX2 biogenesis. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 323–333. [Google Scholar]
- Leary, S.C.; Sasarman, F.; Nishimura, T.; Shoubridge, E.A. Human SCO2 is required for the synthesis of CO II and as a thiol-disulphide oxidoreductase for SCO1. Hum. Mol. Genet. 2009, 18, 2230–2240. [Google Scholar]
- Leary, S.; Cobine, P.A.; Kaufman, B.A.; Guercin, G.H.; Mattman, A.; Palaty, J.; Lockitch, G.; Winge, D.R.; Rustin, P.; Horvath, R.; et al. The human cytochrome c oxidase assembly factors SCO1 and SCO2 have regulatory roles in the maintenance of cellular copper homeostasis. Cell Metab. 2007, 5, 9–20. [Google Scholar]
- Leary, S.; Kaufman, B.A.; Pellecchia, G.; Guercin, G.H.; Mattman, A.; Jaksch, M.; Shoubridge, E.A. Human SCO1 and SCO2 have independent, cooperative functions in copper delivery to cytochrome c oxidase. Hum. Mol. Genet. 2004, 13, 1839–1848. [Google Scholar]
- Stroud, D.; Maher, M.J.; Lindau, C.; Vögtle, F.N.; Frazier, A.E.; Surgenor, E.; Mountford, H.; Singh, A.P.; Bonas, M.; Oeljeklaus, S.; et al. COA6 is a mitochondrial complex IV assembly factor critical for biogenesis of mtDNA-encoded COX2. Hum. Mol. Genet. 2015, 24, 5404–5415. [Google Scholar]
- Pacheu-Grau, D.; Bareth, B.; Dudek, J.; Juris, L.; Vögtle, F.N.; Wissel, M.; Leary, S.C.; Dennerlein, S.; Rehling, P.; Deckers, M. Cooperation between COA6 and SCO2 in COX2 maturation during cytochrome c oxidase assembly links two mitochondrial cardiomyopathies. Cell Metab. 2015, 21, 823–833. [Google Scholar]
- Aich, A.; Wang, C.; Chowdhury, A.; Ronsör, C.; Pacheu-Grau, D.; Richter-Dennerlein, R.; Dennerlein, S.; Rehling, P. COX16 promotes COX2 metallation and assembly during respiratory complex IV biogenesis. Elife 2018, 7, e32572. [Google Scholar]
- Cerqua, C.; Morbidoni, V.; Desbats, M.A.; Doimo, M.; Frasson, C.; Sacconi, S.; Baldoin, M.C.; Sartori, G.; Basso, G.; Salviati, L.; et al. COX16 is required for assembly of cytochrome c oxidase in human cells and is involved in copper delivery to COX2. Biochim. Biophys. Acta 2018, 1859, 244–252. [Google Scholar]
- Church, C.G.B.; Forsha, D.; Wazny, P.; Poyton, R.O. A role for Pet100p in the assembly of yeast cytochrome c oxidase: Interaction with a subassembly that accumulates in a pet100 mutant. J. Biol. Chem. 2005, 280, 1854–1863. [Google Scholar]
- Antonicka, H.; Shoubridge, E.A. Mitochondrial RNA Granules Are Centers for Posttranscriptional RNA Processing and Ribosome Biogenesis. Cell Rep. 2015, 10, 920–932. [Google Scholar] [CrossRef] [Green Version]
- Nobrega, M.; Nobrega, F.G.; Tzagoloff, A. COX10 codes for a protein homologous to the ORF1 product of Paracoccus denitrificans and is required for the synthesis of yeast cytochrome oxidase. J. Biol. Chem. 1990, 265, 14220–14226. [Google Scholar]
- Glerum, D.M.; Muroff, I.; Jin, C.; Tzagoloff, A. COX15 codes for a mitochondrial protein essential for the assembly of yeast cytochrome oxidase. J. Biol. Chem. 1997, 272, 19088–19094. [Google Scholar] [CrossRef] [Green Version]
- Antonicka, H.; Mattman, A.; Carlson, C.G.; Glerum, D.M.; Hoffbuhr, K.C.; Leary, S.C.; Kennaway, N.G.; Shoubridge, E.A. Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am. J. Hum. Genet. 2003, 72, 101–114. [Google Scholar]
- Smith, D.G.J.; Mitchell, L.; Antholine, W.E.; Hosler, J.P. Assembly of Cytochrome-c Oxidase in the Absence of Assembly Protein Surf1p Leads to Loss of the Active Site Heme. J. Biol. Chem. 2005, 280, 17652–17656. [Google Scholar]
- Vögtle, F.N.B.J.; Rao, S.; Gerbeth, C.; Hinrichs, J.; Martinou, J.C.; Chacinska, A.; Sickmann, A.; Zahedi, R.P.; Meisinger, C. Intermembrane space proteome of yeast mitochondria. Mol. Cell Proteomics. 2012, 11, 1840–1852. [Google Scholar]
- Schulze, M.R.G. SCO1, a yeast nuclear gene essential for accumulation of mitochondrial cytochrome c oxidase subunit II. Mol. Gen. Genet. 1988, 211, 492–498. [Google Scholar]
- Papadopoulou, L.S.C.M.; Davidson, M.M.; Tanji, K.; Nishino, I.; Sadlock, J.E.; Krishna, S.; Walker, W.; Selby, J.; Glerum, D.M.; Coster, R.V.; et al. Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat. Genet. 1999, 23, 333–337. [Google Scholar]
- Tzagoloff, A.; Capitanio, N.; Nobrega, M.P.; Gatti, D. Cytochrome oxidase assembly in yeast requires the product of COX11, a homolog of the P. denitrificans protein encoded by ORF3. EMBO J. 1990, 9, 2759–2764. [Google Scholar]
- Carlson, C.G.B.A.; Tzagoloff, A.; Glerum, D.M. COX16 encodes a novel protein required for the assembly of cytochrome oxidase in Saccharomyces cerevisiae. J. Biol. Chem. 2003, 278, 3770–3775. [Google Scholar]
- Nobrega, M.; Bandeira, S.C.; Beers, J.; Tzagoloff, A. Characterization of COX19, a widely distributed gene required for expression of mitochondrial cytochrome oxidase. J. Biol. Chem. 2002, 277, 40206–40211. [Google Scholar]
- Weraarpachai, W.; Sasarman, F.; Nishimura, T.; Antonicka, H.; Aure, K.; Rotig, A.; Lombes, A.; Shoubridge, E.A. Mutations in C12orf62, a factor that couples COX I synthesis with cytochrome c oxidase assembly, cause fatal neonatal lactic acidosis. Am. J. Hum. Genet. 2012, 90, 142–151. [Google Scholar]
- Hell, K.; Tzagoloff, A.; Neupert, W.; Stuart, R.A. Identification of Cox20p, a novel protein involved in the maturation and assembly of cytochrome oxidase subunit 2. J. Biol. Chem. 2000, 275, 4571–4578. [Google Scholar]
- Church, C.C.C.; Poyton, R.O. Cloning and characterization of PET100, a gene required for the assembly of yeast cytochrome c oxidase. J. Biol. Chem. 1996, 271, 18499–18507. [Google Scholar]
- Lim, S.C.S.K.; Stroud, D.A.; Compton, A.G.; Tucker, E.J.; Dasvarma, A.; Gandolfo, L.C.; Marum, J.E.; McKenzie, M.; Peters, H.L.; Mowat, D.; et al. A founder mutation in PET100 causes isolated complex IV deficiency in Lebanese individuals with Leigh syndrome. Am. J. Hum. Genet. 2014, 94, 209–222. [Google Scholar]
- Oláhová, M.; Haack, T.B.; Alston, C.L.; Houghton, J.A.; He, L.; Morris, A.A.; Brown, G.K.; McFarland, R.; Chrzanowska-Lightowlers, Z.M.; Lightowlers, R.N.; et al. A truncating PET100 variant causing fatal infantile lactic acidosis and isolated cytochrome c oxidase deficiency. Eur. J. Hum. Genet. EJHG 2015, 23, 935–939. [Google Scholar] [CrossRef] [Green Version]
- McEwen, J.; Hong, K.H.; Park, S.; Preciado, G.T. Sequence and chromosomal localization of two PET genes required for cytochrome c oxidase assembly in Saccharomyces cerevisiae. Curr. Genet. 1993, 23, 9–14. [Google Scholar]
- Renkema, G.H.V.G.; Baertling, F.; Wintjes, L.T.; Wolters, V.M.; van Montfrans, J.; de Kort, G.A.P.; Nikkels, P.G.J.; van Hasselt, P.M.; van der Crabben, S.N.; Rodenburg, R.J.T. Mutated PET117 causes complex IV deficiency and is associated with neurodevelopmental regression and medulla oblongata lesions. Hum. Genet. 2017, 136, 759–769. [Google Scholar]
- Signes, A.C.R.; Dickson, A.S.; Benincá, C.; Hinchy, E.C.; Ghezzi, D.; Carrozzo, R.; Bertini, E.; Murphy, M.P.; Nathan, J.A.; Viscomi, C.; et al. APOPT1/COA8 assists COX assembly and is oppositely regulated by UPS and ROS. EMBO Mol. Med. 2019, 11, e9582. [Google Scholar]
- Souza, R.L.G.-W.N.; Fox, T.D.; Tzagoloff, A.; Nobrega, F.G. Cloning and characterization of COX18, a Saccharomyces cerevisiae PET gene required for the assembly of cytochrome oxidase. J. Biol. Chem. 2000, 275, 14898–14902. [Google Scholar]
- Bourens, M.; Barrientos, A. Human mitochondrial cytochrome c oxidase assembly factor COX18 acts transiently as a membrane insertase within the subunit 2 maturation module. J. Biol. Chem. 2017, 292, 7774–7783. [Google Scholar] [CrossRef] [Green Version]
- Brischigliaro, M.; Zeviani, M. Cytochrome c oxidase deficiency. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148335. [Google Scholar] [CrossRef]
- Lamperti, C.; Diodato, D.; Lamantea, E.; Carrara, F.; Ghezzi, D.; Mereghetti, P.; Rizzi, R.; Zeviani, M. MELAS-like encephalomyopathy caused by a new pathogenic mutation in the mitochondrial DNA encoded cytochrome c oxidase subunit I. Neuromuscul. Disord. 2012, 22, 990–994. [Google Scholar]
- Valente, L.P.D.; Lamantea, E.; Carrara, F.; Uziel, G.; Cudia, P.; Zani, A.; Farina, L.; Morandi, L.; Mora, M. Identification of novel mutations in five patients with mitochondrial encephalomyopathy. Biochim. Biophys. Acta 2009, 1787, 491–501. [Google Scholar]
- Comi, G.P.B.A.; Salani, S.; Franceschina, L.; Sciacco, M.; Prelle, A.; Fortunato, F.; Zeviani, M.; Napoli, L.; Bresolin, N. Cytochrome c oxidase subunit I microdeletion in a patient with motor neuron disease. Ann. Neurol. 1998, 43, 110–116. [Google Scholar]
- D’Aurelio, M.P.F.; Barrientos, A.; Gajewski, C.D.; Kwong, J.Q.; Bruno, C.; Beal, M.F.; Manfredi, G. In vivo regulation of oxidative phosphorylation in cells harboring a stop-codon mutation in mitochondrial DNA-encoded cytochrome c oxidase subunit I. J. Biol. Chem. 2001, 276, 46925–46932. [Google Scholar]
- Nishigaki, Y.U.H.; Coku, J.; Koga, Y.; Fujii, T.; Sahashi, K.; Nakano, K.; Yoneda, M.; Nonaka, M.; Tang, L. Extensive screening system using suspension array technology to detect mitochondrial DNA point mutations. Mitochondrion 2010, 10, 300–308. [Google Scholar]
- Clark, K.M.; Taylor, R.W.; Johnson, M.A.; Chinnery, P.F.; Chrzanowska-Lightowlers, Z.M.; Andrews, R.M.; Nelson, I.P.; Wood, N.W.; Lamont, P.J.; Hanna, M.G. An mtDNA mutation in the initiation codon of the cytochrome C oxidase subunit II gene results in lower levels of the protein and a mitochondrial encephalomyopathy. Am. J. Hum. Genet. 1999, 64, 1330–1339. [Google Scholar]
- Abu-Amero, K.K.; Bosley, T.M. Mitochondrial abnormalities in patients with LHON-like optic neuropathies. Invest. Ophthalmol. Vis. Sci. 2006, 47, 4211–4220. [Google Scholar]
- Rahman, S.; Taanman, J.W.; Cooper, J.M.; Nelson, I.; Hargreaves, I.; Meunier, B.; Hanna, M.G.; Garcia, J.J.; Capaldi, R.A.; Lake, B.D. A missense mutation of cytochrome oxidase subunit II causes defective assembly and myopathy. Am. J. Hum. Genet. 1999, 65, 1030–1039. [Google Scholar]
- Wei, Y.L.; Yu, C.A.; Yang, P.; Li, A.L.; Wen, J.Y.; Zhao, S.M.; Liu, H.X.; Ke, Y.N.; Campbell, W.; Zhang, Y.G. Novel mitochondrial DNA mutations associated with Chinese familial hypertrophic cardiomyopathy. Clin. Exp. Pharmacol. Physiol. 2006, 36, 933–939. [Google Scholar]
- Tabebi, M.; Mkaouar-Rebai, E.; Mnif, M.; Kallabi, F.; Ben Mahmoud, A.; Ben Saad, W.; Charfi, N.; Keskes-Ammar, L.; Kamoun, H.; Abid, M. A novel mutation MT-COIII m.9267G>C and MT-COI m.5913G>A mutation in mitochondrial genes in a Tunisian family with maternally inherited diabetes and deafness (MIDD) associated with severe nephropathy. Biochem. Biophys. Res. Commun. 2015, 459, 353–360. [Google Scholar]
- Horvath, R.S.C.; Hoeltzenbein, M.; Do, B.H.; Schroder, C.; Warzok, R.; Vogelgesang, S.; Lochmuller, H.; Muller-Hocker, J.; Gerbitz, K.D. Childhood onset mitochondrial myopathy and lactic acidosis caused by a stop mutation in the mitochondrial cytochrome c oxidase III gene. J. Med. Genet. 2002, 39, 812–816. [Google Scholar]
- Mkaouar-Rebai, E.E.E.; Chamkha, I.; Kammoun, F.; Triki, C.; Fakhfakh, F. Molecular-clinical correlation in a family with a novel heteroplasmic Leigh syndrome missense mutation in the mitochondrial cytochrome c oxidase III gene. J. Child Neurol. 2011, 26, 12–20. [Google Scholar]
- Bosley, T.M.B.M.C.; Glasier, C.M.; Abu-Amero, K.K. Sporadic bilateral optic neuropathy in children: The role of mitochondrial abnormalities. Invest. Ophthalmol. Vis. Sci. 2008, 49, 5250–5256. [Google Scholar]
- Marotta, R.C.J.; Kirby, D.M.; Chiotis, M.; Cook, M.; Collins, S.J. Novel single base pair COX III subunit deletion of mitochondrial DNA associated with rhabdomyolysis. J. Clin. Neurosci. 2011, 18, 290–292. [Google Scholar]
- Hanna, M.G.; Nelson, I.P.; Rahman, S.; Lane, R.J.; Land, J.; Heales, S.; Cooper, M.J.; Schapira, A.H.; Morgan-Hughes, J.A.; Wood, N.W. Cytochrome c oxidase deficiency associated with the first stop-codon point mutation in human mtDNA. Am. J. Hum. Genet. 1998, 63, 29–36. [Google Scholar]
- Abu-Libdeh, B.; Douiev, L.; Amro, S.; Shahrour, M.; Ta-Shma, A.; Miller, C.; Elpeleg, O.; Saada, A. Mutation in the COX4I1 gene is associated with short stature, poor weight gain and increased chromosomal breaks, simulating Fanconi anemia. Eur. J. Hum. Genet. EJHG 2017, 25, 1142–1146. [Google Scholar] [CrossRef]
- Shteyer, E.; Saada, A.; Shaag, A.; Al-Hijawi, F.A.; Kidess, R.; Revel-Vilk, S.; Elpeleg, O. Exocrine pancreatic insufficiency, dyserythropoeitic anemia, and calvarial hyperostosis are caused by a mutation in the COX4I2 gene. Am. J. Hum. Genet. 2009, 84, 412–417. [Google Scholar] [CrossRef] [Green Version]
- Baertling, F.; Al-Murshedi, F.; Sánchez-Caballero, L.; Al-Senaidi, K.; Joshi, N.P.; Venselaar, H.; van den Brand, M.A.; Nijtmans, L.G.; Rodenburg, R.J. Mutation in mitochondrial complex IV subunit COX5A causes pulmonary arterial hypertension, lactic acidemia, and failure to thrive. Hum. Mutat. 2017, 38, 692–703. [Google Scholar] [CrossRef]
- Tamiya, G.M.S.; Hayashi, M.; Abe, A.; Numakura, C.; Ueki, M.; Tanaka, A.; Ito, C.; Toshimori, K.; Ogawa, N.; Terashima, T.; et al. A mutation of COX6A1 causes a recessive axonal or mixed form of Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 2014, 95, 294–300. [Google Scholar]
- Inoue, M.; Uchino, S.; Iida, A.; Noguchi, S.; Hayashi, S.; Takahashi, T.; Fujii, K.; Komaki, H.; Takeshita, E.; Nonaka, I.; et al. COX6A2 variants cause a muscle-specific cytochrome c oxidase deficiency. Ann. Neurol. 2019, 86, 193–202. [Google Scholar] [CrossRef]
- Massa, V.F.-V.E.; Alshahwan, S.; Bakhsh, E.; Goffrini, P.; Ferrero, I.; Mereghetti, P.; D’Adamo, P.; Gasparini, P.; Zeviani, M. Severe infantile encephalomyopathy caused by a mutation in COX6B1, a nucleus-encoded subunit of cytochrome c oxidase. Am. J. Hum. Genet. 2008, 82, 1281–1289. [Google Scholar]
- Vondrackova, A.; Vesela, K.; Hansikova, H.; Docekalova, D.Z.; Rozsypalova, E.; Zeman, J.; Tesarova, M. High-resolution melting analysis of 15 genes in 60 patients with cytochrome-c oxidase deficiency. J. Hum. Genet. 2012, 57, 442–448. [Google Scholar] [CrossRef] [Green Version]
- Indrieri, A.v.R.V.; Tiranti, V.; Morleo, M.; Iaconis, D.; Tammaro, R.; D’Amato, I.; Conte, I.; Maystadt, I.; Demuth, S.; Zvulunov, A.; et al. Mutations in COX7B cause microphthalmia with linear skin lesions, an unconventional mitochondrial disease. Am. J. Hum. Genet. 2012, 91, 942–949. [Google Scholar]
- Hallmann, K.; Kudin, A.P.; Zsurka, G.; Kornblum, C.; Reimann, J.; Stüve, B.; Waltz, S.; Hattingen, E.; Thiele, H.; Nürnberg, P.; et al. Loss of the smallest subunit of cytochrome c oxidase, COX8A, causes Leigh-like syndrome and epilepsy. Brain J. Neurol. 2016, 139, 338–345. [Google Scholar] [CrossRef]
- Tiranti, V.H.K.; Carrozzo, R. Mutations of SURF-1 in Leigh disease associated with cytochrome c oxidase deficiency. Am. J. Hum. Genet. 1998, 63, 1609–1621. [Google Scholar]
- Echaniz-Laguna, A.; Ghezzi, D.; Chassagne, M.; Mayençon, M.; Padet, S.; Melchionda, L.; Rouvet, I.; Lannes, B.; Bozon, D.; Latour, P.; et al. SURF1 deficiency causes demyelinating Charcot-Marie-Tooth disease. Neurology 2013, 81, 1523–1530. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, R.W.B.; Nijtmans, L.G.; Rodenburg, R.J.; Zschocke, J.; Dikow, N.; van den Brand, M.A.; Hendriks-Franssen, M.G.; Gilissen, C.; Veltman, J.A.; Nooteboom, M.; et al. A mutation in the FAM36A gene, the human ortholog of COX20, impairs cytochrome c oxidase assembly and is associated with ataxia and muscle hypotonia. Hum. Mol. Genet. 2013, 22, 656–667. [Google Scholar]
- Melchionda, L.H.T.B.; Hardy, S.; Abbink, T.E.; Fernandez-Vizarra, E.; Lamantea, E. Mutations in APOPT1, encoding a mitochondrial protein, cause cavitating leukoencephalopathy with cytochrome c oxidase deficiency. Am. J. Hum. Genet. 2014, 95, 315–325. [Google Scholar]
- Pronicki, M.; Kowalski, P.; Piekutowska-Abramczuk, D.; Taybert, J.; Karkucinska-Wieckowska, A.; Szymanska-Debinska, T.; Karczmarewicz, E.; Pajdowska, M.; Migdal, M.; Milewska-Bobula, B.; et al. A homozygous mutation in the SCO2 gene causes a spinal muscular atrophy like presentation with stridor and respiratory insufficiency. Eur. J. Paediatr. Neurol. 2010, 14, 253–260. [Google Scholar]
- Rebelo, A.; Saade, D.; Pereira, C.V.; Farooq, A.; Huff, T.C.; Abreu, L.; Moraes, C.T.; Mnatsakanova, D.; Mathews, K.; Yang, H.; et al. SCO2 mutations cause early-onset axonal Charcot-Marie-Tooth disease associated with cellular copper deficiency. Brain J. Neurol. 2018, 141, 662–672. [Google Scholar]
- Baertling, F.A.M.; van den Brand, M.; Hertecant, J.L.; Al-Shamsi, A.; van den Heuvel, L.P.; Distelmaier, F.; Mayatepek, E.; Smeitink, J.A.; Nijtmans, L.G.; Rodenburg, R.J. Mutations in COA6 cause cytochrome c oxidase deficiency and neonatal hypertrophic cardiomyopathy. Hum. Mutat. 2015, 36, 34–38. [Google Scholar]
- Huigsloot, M.; Nijtmans, L.G.; Szklarczyk, R.; Baars, M.J.; van den Brand, M.A.; Hendriksfranssen, M.G.; van den Heuvel, L.P.; Smeitink, J.A.; Huynen, M.A.; Rodenburg, R.J. A mutation in C2orf64 causes impaired cytochrome c oxidase assembly and mitochondrial cardiomyopathy. Am. J. Hum. Genet. 2011, 88, 488–493. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, D.; Saada, A.; D’Adamo, P.; Fernandez-Vizarra, E.; Gasparini, P.; Tiranti, V.; Elpeleg, O.; Zeviani, M. FASTKD2 nonsense mutation in an infantile mitochondrial encephalomyopathy associated with cytochrome c oxidase deficiency. Am. J. Hum. Genet. 2008, 83, 415–423. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Du, M.; Li, D.; Wen, S.; Xie, J.; Li, Y.; Chen, A.; Zhang, K.; Xu, P.; Jia, M.; et al. Mutations in FASTKD2 are associated with mitochondrial disease with multi-OXPHOS deficiency. Hum. Mutat. 2020, 41, 961–972. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lepage, P.; Miller, K.; Bunkenborg, J.; Reich, M.; Hjerrild, M.; Delmonte, T.; Villeneuve, A.; Sladek, R.; Xu, F.; et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc. Natl. Acad. Sci. USA 2003, 100, 605–610. [Google Scholar] [CrossRef] [Green Version]
- Jonckheere, A.I.; Smeitink, J.A.; Rodenburg, R.J. Mitochondrial ATP synthase: Architecture, function and pathology. J. Inherit. Metab. Dis. 2012, 35, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Abrahams, J.P.; Leslie, A.G.; Lutter, R.; Walker, J.E. Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria. Nature 1994, 370, 621–628. [Google Scholar] [CrossRef]
- He, J.; Ford, H.C.; Carroll, J.; Douglas, C.; Gonzales, E.; Ding, S.; Fearnley, I.M.; Walker, J.E. Assembly of the membrane domain of ATP synthase in human mitochondria. Proc. Natl. Acad. Sci. USA 2018, 115, 2988–2993. [Google Scholar] [CrossRef] [Green Version]
- Watt, I.N.; Montgomery, M.G.; Runswick, M.J.; Leslie, A.G.; Walker, J.E. Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc. Natl. Acad. Sci. USA 2010, 107, 16823–16827. [Google Scholar] [CrossRef] [Green Version]
- Kuhlbrandt, W. Structure and Mechanisms of F-Type ATP Synthases. Annu. Rev. Biochem. 2019, 88, 515–549. [Google Scholar] [CrossRef]
- Wittig, I.; Schagger, H. Structural organization of mitochondrial ATP synthase. Biochim. Biophys. Acta 2008, 1777, 592–598. [Google Scholar] [CrossRef] [Green Version]
- Ackerman, S.H.; Tzagoloff, A. Identification of two nuclear genes (ATP11, ATP12) required for assembly of the yeast F1-ATPase. Proc. Natl. Acad. Sci. USA 1990, 87, 4986–4990. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.E. The ATP synthase: The understood, the uncertain and the unknown. Biochem. Soc. Trans. 2013, 41, 1–16. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Ford, H.C.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc. Natl. Acad. Sci. USA 2017, 114, 3409–3414. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Barros, M.H.; Shulman, T.; Tzagoloff, A. ATP25, a new nuclear gene of Saccharomyces cerevisiae required for expression and assembly of the Atp9p subunit of mitochondrial ATPase. Mol. Biol. Cell 2008, 19, 1366–1377. [Google Scholar] [CrossRef] [Green Version]
- Rak, M.; Gokova, S.; Tzagoloff, A. Modular assembly of yeast mitochondrial ATP synthase. EMBO J. 2011, 30, 920–930. [Google Scholar] [CrossRef] [Green Version]
- Lytovchenko, O.; Naumenko, N.; Oeljeklaus, S.; Schmidt, B.; von der Malsburg, K.; Deckers, M.; Warscheid, B.; van der Laan, M.; Rehling, P. The INA complex facilitates assembly of the peripheral stalk of the mitochondrial F1Fo-ATP synthase. EMBO J. 2014, 33, 1624–1638. [Google Scholar] [CrossRef]
- López-Gallardo, E.; Solano, A.; Herrero-Martín, M.D.; Martínez-Romero, I.; Castaño-Pérez, M.D.; Andreu, A.L.; Herrera, A.; López-Pérez, M.J.; Ruiz-Pesini, E.; Montoya, J. NARP syndrome in a patient harbouring an insertion in the MT-ATP6 gene that results in a truncated protein. J. Med. Genet. 2009, 46, 64–67. [Google Scholar] [CrossRef]
- D’Aurelio, M.; Vives-Bauza, C.; Davidson, M.M.; Manfredi, G. Mitochondrial DNA background modifies the bioenergetics of NARP/MILS ATP6 mutant cells. Hum. Mol. Genet. 2010, 19, 374–386. [Google Scholar] [CrossRef] [Green Version]
- Jonckheere, A.I.; Hogeveen, M.; Nijtmans, L.; van den Brand, M.; Janssen, A.; Diepstra, H.; van den Brandt, F.; van den Heuvel, B.; Hol, F.; Hofste, T.; et al. A novel mitochondrial ATP8 gene mutation in a patient with apical hypertrophic cardiomyopathy and neuropathy. BMJ Case Rep. 2009, 2009. [Google Scholar] [CrossRef] [Green Version]
- Mayr, J.A.H.V.; Zimmermann, F.; Magler, I.; Kaplanová, V.; Jesina, P.; Pecinová, A.; Nusková, H.; Koch, J.; Sperl, W.; Houstek, J. Mitochondrial ATP synthase deficiency due to a mutation in the ATP5E gene for the F1 epsilon subunit. Hum. Mol. Genet. 2010, 19, 3430–3439. [Google Scholar]
- Tatuch, Y.; Robinson, B.H. The mitochondrial DNA mutation at 8993 associated with NARP slows the rate of ATP synthesis in isolated lymphoblast mitochondria. Biochem. Biophys. Res. Commun. 1993, 192, 124–128. [Google Scholar] [CrossRef]
- Jonckheere, A.I.H.M.; Nijtmans, L.G.; van den Brand, M.A.; Janssen, A.J.; Diepstra, J.H.; van den Brandt, F.C.; van den Heuvel, L.P.; Hol, F.A.; Hofste, T.G.; Kapusta, L.; et al. A novel mitochondrial ATP8 gene mutation in a patient with apical hypertrophic cardiomyopathy and neuropathy. J. Med. Genet. 2008, 45, 129–133. [Google Scholar]
- Jonckheere, A.I.; Renkema, G.H.; Bras, M.; van den Heuvel, L.P.; Hoischen, A.; Gilissen, C.; Nabuurs, S.B.; Huynen, M.A.; de Vries, M.C.; Smeitink, J.A.; et al. A complex V ATP5A1 defect causes fatal neonatal mitochondrial encephalopathy. Brain J. Neurol. 2013, 136, 1544–1554. [Google Scholar] [CrossRef] [Green Version]
- Hejzlarová, K.; Mráček, T.; Vrbacký, M.; Kaplanová, V.; Karbanová, V.; Nůsková, H.; Pecina, P.; Houštěk, J. Nuclear genetic defects of mitochondrial ATP synthase. Physiol. Res. 2014, 63 (Suppl. 1), S57–S71. [Google Scholar]
- de Coo, I.F.; Smeets, H.J.; Gabreëls, F.J.; Arts, N.; van Oost, B.A. Isolated case of mental retardation and ataxia due to a de novo mitochondrial T8993G mutation. Am. J. Hum. Genet. 1996, 58, 636–638. [Google Scholar]
- Ware, S.M.; El-Hassan, N.; Kahler, S.G.; Zhang, Q.; Ma, Y.W.; Miller, E.; Wong, B.; Spicer, R.L.; Craigen, W.J.; Kozel, B.A.; et al. Infantile cardiomyopathy caused by a mutation in the overlapping region of mitochondrial ATPase 6 and 8 genes. J. Med. Genet. 2009, 46, 308–314. [Google Scholar] [CrossRef]
- De Meirleir, L.; Seneca, S.; Lissens, W.; De Clercq, I.; Eyskens, F.; Gerlo, E.; Smet, J.; Van Coster, R. Respiratory chain complex V deficiency due to a mutation in the assembly gene ATP12. J. Med. Genet. 2004, 41, 120–124. [Google Scholar]
- Lieber, D.S.; Calvo, S.E.; Shanahan, K.; Slate, N.G.; Liu, S.; Hershman, S.G.; Gold, N.B.; Chapman, B.A.; Thorburn, D.R.; Berry, G.T.; et al. Targeted exome sequencing of suspected mitochondrial disorders. Neurology 2013, 80, 1762–1770. [Google Scholar] [CrossRef] [Green Version]
- Oláhová, M.; Yoon, W.H.; Thompson, K.; Jangam, S.; Fernandez, L.; Davidson, J.M.; Kyle, J.E.; Grove, M.E.; Fisk, D.G.; Kohler, J.N.; et al. Biallelic Mutations in ATP5F1D, which Encodes a Subunit of ATP Synthase, Cause a Metabolic Disorder. Am. J. Hum. Genet. 2018, 102, 494–504. [Google Scholar] [CrossRef] [Green Version]
- Scheffler, I.E. Mitochondria; Wiley-Liss: New York, NY, USA, 1999. [Google Scholar]
- Gilkerson, R.W.S.J.; Capaldi, R.A. The cristal membrane of mitochondria is the principal site of oxidative phosphorylation. FEBS Lett. 2003, 546, 355–358. [Google Scholar]
- Vogel, F.B.C.; Neupert, W.; Reichert, A.S. Dynamic subcompartmentalization of the mitochondrial inner membrane. J. Cell Biol. 2006, 175, 237–247. [Google Scholar]
- Stoldt, S.W.D.; Kehrein, K.; Riedel, D.; Ott, M.; Jakobs, S. Spatial orchestration of mitochondrial translation and OXPHOS complex assembly. Nat. Cell Biol. 2018, 20, 528–534. [Google Scholar]
- Schägger, H.; von Jagow, G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 1991, 199, 223–231. [Google Scholar]
- Hackenbrock, C.; Chazotte, B.; Gupte, S.S. The random collision model and a critical assessment of diffusion and collision in mitochondrial electron transport. J. Bioenerg. Biomembr. 1986, 18, 331–368. [Google Scholar]
- Höchli, M.H.C. Lateral translational diffusion of cytochrome c oxidase in the mitochondrial energy-transducing membrane. Proc. Natl. Acad. Sci. USA 1979, 76, 1236–1240. [Google Scholar]
- Sowers, A.E.H.C. Rate of lateral diffusion of intramembrane particles: Measurement by electrophoretic displacement and rerandomization. Proc. Natl. Acad. Sci. USA 1981, 78, 6246–6250. [Google Scholar]
- Fato, R.; Battino, M.; Degli Esposti, M.; Parenti Castelli, G.; Lenaz, G. Determination of partition and lateral diffusion coefficients of ubiquinones by fluorescence quenching of n-(9-anthroyloxy)stearic acids in phospholipid vesicles and mitochondrial membranes. Biochemistry 1986, 25, 3378–3390. [Google Scholar]
- Lenaz, G.F.R.; Di Bernardo, S.; Jarreta, D.; Costa, A.; Genova, M.L.; Parenti Castelli, G. Localization and mobility of coenzyme Q in lipid bilayers and membranes. Biofactors 1999, 9, 87–93. [Google Scholar]
- Schägger, H.P.K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000, 19, 1777–1783. [Google Scholar]
- Schägger, H.P.K. The ratio of oxidative phosphorylation complexes I-V in bovine heart mitochondria and the composition of respiratory chain supercomplexes. J. Biol. Chem. 2001, 276, 37861–37867. [Google Scholar]
- Stroh, A.A.O.; Pfeiffer, K.; Yagi, T.; Finel, M.; Ludwig, B.; Schägger, H. Assembly of respiratory complexes I, III, and IV into NADH oxidase supercomplex stabilizes complex I in Paracoccus denitrificans. J. Biol. Chem. 2004, 279, 5000–5007. [Google Scholar]
- Schäfer, E.; Seelert, H.; Reifschneider, N.H.; Krause, F.; Dencher, N.A.; Vonck, J. Architecture of active mammalian respiratory chain supercomplexes. J. Biol. Chem. 2006, 281, 15370–15375. [Google Scholar]
- Dudkina, N.; Eubel, H.; Keegstra, W.; Boekema, E.J.; Braun, H.P. Structure of a mitochondrial supercomplex formed by respiratory-chain complexes I and III. Proc. Natl. Acad. Sci. USA 2005, 102, 3225–3229. [Google Scholar]
- Keilin, D.; Hartree, E.F. Activity of the cytochrome system in heart muscle preparations. Biochem. J. 1947, 41, 500–502. [Google Scholar] [CrossRef] [Green Version]
- Chance, B.; Williams, G.R. A method for the localization of sites for oxidative phosphorylation. Nature 1955, 176, 250–254. [Google Scholar] [CrossRef]
- Hatefi, Y.; Haavik, A.G.; Fowler, L.R.; Griffiths, D.E. Studies on the electron transfer system. XLII. Reconstitution of the electron transfer system. J. Biol. Chem. 1962, 237, 2661–2669. [Google Scholar]
- Trouillard, M.; Meunier, B.; Rappaport, F. Questioning the functional relevance of mitochondrial supercomplexes by time-resolved analysis of the respiratory chain. Proc. Natl. Acad. Sci. USA 2011, 108, E1027–E1034. [Google Scholar] [CrossRef] [Green Version]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acin-Perez, R.; Latorre-Pellicer, A.; Colas, C.; Balsa, E.; Perales-Clemente, E.; Quiros, P.M.; Calvo, E.; Rodriguez-Hernandez, M.A.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science (N. Y.) 2013, 340, 1567–1570. [Google Scholar] [CrossRef]
- Blaza, J.N.; Serreli, R.; Jones, A.J.; Mohammed, K.; Hirst, J. Kinetic evidence against partitioning of the ubiquinone pool and the catalytic relevance of respiratory-chain supercomplexes. Proc. Natl. Acad. Sci. USA 2014, 111, 15735–15740. [Google Scholar] [CrossRef] [Green Version]
- Fedor, J.G.; Hirst, J. Mitochondrial Supercomplexes Do Not Enhance Catalysis by Quinone Channeling. Cell Metab. 2018, 28, 525–531.e524. [Google Scholar] [CrossRef] [Green Version]
- Acin-Perez, R.; Fernandez-Silva, P.; Peleato, M.L.; Perez-Martos, A.; Enriquez, J.A. Respiratory active mitochondrial supercomplexes. Mol. Cell 2008, 32, 529–539. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Enriquez, J.A. The function of the respiratory supercomplexes: The plasticity model. Biochim. Biophys. Acta 2014, 1837, 444–450. [Google Scholar] [CrossRef] [Green Version]
- Letts, J.A.F.K.; Sazanov, L.A. The architecture of respiratory supercomplexes. Nature 2016, 537, 644–648. [Google Scholar]
- Gu, J.; Wu, M.; Guo, R.; Yan, K.; Lei, J.; Gao, N.; Yang, M. The architecture of the mammalian respirasome. Nature 2016, 537, 639–643. [Google Scholar]
- Guo, R.; Zong, S.; Wu, M.; Gu, J.; Yang, M. Architecture of Human Mitochondrial Respiratory Megacomplex I2III2IV2. Cell 2017, 170, 1247–1257. [Google Scholar]
- Letts, J.A.; Fiedorczuk, K.; Degliesposti, G.; Skehel, M.; Sazanov, L.A. Structures of Respiratory Supercomplex I+III(2) Reveal Functional and Conformational Crosstalk. Mol. Cell 2019, 75, 1131–1146.e1136. [Google Scholar] [CrossRef] [Green Version]
- Dudkina, N.V.K.M.; Stahlberg, H.; Boekema, E.J. Interaction of complexes I, III, and IV within the bovine respirasome by single particle cryoelectron tomography. Proc. Natl. Acad. Sci. USA 2011, 108, 15196–15200. [Google Scholar]
- Davies, K.M.; Blum, T.B.; Kühlbrandt, W. Conserved in situ arrangement of complex I and III2 in mitochondrial respiratory chain supercomplexes of mammals, yeast, and plants. Proc. Natl. Acad. Sci. USA 2018, 115, 3024–3029. [Google Scholar]
- Hartley, A.M.; Lukoyanova, N.; Zhang, Y.; Cabrera-Orefice, A.; Arnold, S.; Meunier, B.; Pinotsis, N.; Maréchal, A. Structure of yeast cytochrome c oxidase in a supercomplex with cytochrome bc(1). Nat. Struct. Mol. Biol. 2019, 26, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Rathore, S.; Berndtsson, J.; Marin-Buera, L.; Conrad, J.; Carroni, M.; Brzezinski, P.; Ott, M. Cryo-EM structure of the yeast respiratory supercomplex. Nat. Struct. Mol. Biol. 2019, 26, 50–57. [Google Scholar] [CrossRef]
- Schagger, H. Respiratory chain supercomplexes of mitochondria and bacteria. Biochim. Biophys. Acta 2002, 1555, 154–159. [Google Scholar] [CrossRef] [Green Version]
- Vukotic, M.; Oeljeklaus, S.; Wiese, S.; Vogtle, F.N.; Meisinger, C.; Meyer, H.E.; Zieseniss, A.; Katschinski, D.M.; Jans, D.C.; Jakobs, S.; et al. Rcf1 mediates cytochrome oxidase assembly and respirasome formation, revealing heterogeneity of the enzyme complex. Cell Metab. 2012, 15, 336–347. [Google Scholar] [CrossRef] [Green Version]
- Marcet-Houben, M.M.G.; Gabaldón, T. Phylogenomics of the oxidative phosphorylation in fungi reveals extensive gene duplication followed by functional divergence. BMC Evol Biol. 2009, 9, 295. [Google Scholar]
- Friedrich, T.; Dekovic, D.K.; Burschel, S. Assembly of the Escherichia coli NADH:ubiquinone oxidoreductase (respiratory complex I). Biochim. Biophys. Acta 2016, 1857, 214–223. [Google Scholar] [CrossRef]
- Llorente-Garcia, I.; Lenn, T.; Erhardt, H.; Harriman, O.L.; Liu, L.N.; Robson, A.; Chiu, S.W.; Matthews, S.; Willis, N.J.; Bray, C.D.; et al. Single-molecule in vivo imaging of bacterial respiratory complexes indicates delocalized oxidative phosphorylation. Biochim. Biophys. Acta 2014, 1837, 811–824. [Google Scholar] [CrossRef] [Green Version]
- Milenkovic, D.; Blaza, J.N.; Larsson, N.G.; Hirst, J. The Enigma of the Respiratory Chain Supercomplex. Cell Metab. 2017, 25, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Lobo-Jarne, T.; Nyvltova, E.; Perez-Perez, R.; Timon-Gomez, A.; Molinie, T.; Choi, A.; Mourier, A.; Fontanesi, F.; Ugalde, C.; Barrientos, A. Human COX7A2L Regulates Complex III Biogenesis and Promotes Supercomplex Organization Remodeling without Affecting Mitochondrial Bioenergetics. Cell Rep. 2018, 25, 1786–1799.e1784. [Google Scholar] [CrossRef] [Green Version]
- Letts, J.A.; Sazanov, L.A. Clarifying the supercomplex: The higher-order organization of the mitochondrial electron transport chain. Nat. Struct. Mol. Biol. 2017, 24, 800–808. [Google Scholar] [CrossRef]
- Greggio, C.; Jha, P.; Kulkarni, S.S.; Lagarrigue, S.; Broskey, N.T.; Boutant, M.; Wang, X.; Conde Alonso, S.; Ofori, E.; Auwerx, J.; et al. Enhanced Respiratory Chain Supercomplex Formation in Response to Exercise in Human Skeletal Muscle. Cell Metab. 2017, 25, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Lobo-Jarne, T.; Ugalde, C. Respiratory chain supercomplexes: Structures, function and biogenesis. Semin. Cell Dev. Biol. 2018, 76, 179–190. [Google Scholar] [CrossRef]
- Mileykovskaya, E.; Penczek, P.A.; Fang, J.; Mallampalli, V.K.; Sparagna, G.C.; Dowhan, W. Arrangement of the respiratory chain complexes in Saccharomyces cerevisiae supercomplex III2IV2 revealed by single particle cryo-electron microscopy. J. Biol. Chem. 2012, 287, 23095–23103. [Google Scholar] [CrossRef] [Green Version]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Maranzana, E.; Barbero, G.; Falasca, A.I.; Lenaz, G.; Genova, M.L. Mitochondrial respiratory supercomplex association limits production of reactive oxygen species from complex I. Antioxid. Redox Signal. 2013, 19, 1469–1480. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Fabuel, I.; Le Douce, J.; Logan, A.; James, A.M.; Bonvento, G.; Murphy, M.P.; Almeida, A.; Bolanos, J.P. Complex I assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 13063–13068. [Google Scholar] [CrossRef] [Green Version]
- Blakely, E.L.; Mitchell, A.L.; Fisher, N.; Meunier, B.; Nijtmans, L.G.; Schaefer, A.M.; Jackson, M.J.; Turnbull, D.M.; Taylor, R.W. A mitochondrial cytochrome b mutation causing severe respiratory chain enzyme deficiency in humans and yeast. FEBS J. 2005, 272, 3583–3592. [Google Scholar] [CrossRef]
- Andreu, A.L.; Hanna, M.G.; Reichmann, H.; Bruno, C.; Penn, A.S.; Tanji, K.; Pallotti, F.; Iwata, S.; Bonilla, E.; Lach, B.; et al. Exercise intolerance due to mutations in the cytochrome b gene of mitochondrial DNA. N. Engl. J. Med. 1999, 341, 1037–1044. [Google Scholar] [CrossRef]
- Lamantea, E.; Carrara, F.; Mariotti, C.; Morandi, L.; Tiranti, V.; Zeviani, M. A novel nonsense mutation (Q352X) in the mitochondrial cytochrome b gene associated with a combined deficiency of complexes I and III. Neuromuscul. Disord. NMD 2002, 12, 49–52. [Google Scholar] [CrossRef]
- Moran, M.; Marin-Buera, L.; Gil-Borlado, M.C.; Rivera, H.; Blazquez, A.; Seneca, S.; Vazquez-Lopez, M.; Arenas, J.; Martin, M.A.; Ugalde, C. Cellular pathophysiological consequences of BCS1L mutations in mitochondrial complex III enzyme deficiency. Hum. Mutat. 2010, 31, 930–941. [Google Scholar] [CrossRef] [Green Version]
- Diaz, F.; Fukui, H.; Garcia, S.; Moraes, C.T. Cytochrome c oxidase is required for the assembly/stability of respiratory complex I in mouse fibroblasts. Mol. Cell Biol. 2006, 26, 4872–4881. [Google Scholar] [CrossRef] [Green Version]
- Acin-Perez, R.; Bayona-Bafaluy, M.P.; Fernandez-Silva, P.; Moreno-Loshuertos, R.; Perez-Martos, A.; Bruno, C.; Moraes, C.T.; Enriquez, J.A. Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol. Cell 2004, 13, 805–815. [Google Scholar] [CrossRef]
- Guaras, A.; Perales-Clemente, E.; Calvo, E.; Acin-Perez, R.; Loureiro-Lopez, M.; Pujol, C.; Martinez-Carrascoso, I.; Nunez, E.; Garcia-Marques, F.; Rodriguez-Hernandez, M.A.; et al. The CoQH2/CoQ Ratio Serves as a Sensor of Respiratory Chain Efficiency. Cell Rep. 2016, 15, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Diaz, F.; Enríquez, J.A.; Moraes, C.T. Cells lacking Rieske iron-sulfur protein have a reactive oxygen species-associated decrease in respiratory complexes I and IV. Mol. Cell Biol. 2012, 32, 415–429. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Lastres, D.; Fontanesi, F.; Garcia-Consuegra, I.; Martin, M.A.; Arenas, J.; Barrientos, A.; Ugalde, C. Mitochondrial complex I plays an essential role in human respirasome assembly. Cell Metab. 2012, 15, 324–335. [Google Scholar] [CrossRef] [Green Version]
- Habersetzer, J.; Ziani, W.; Larrieu, I.; Stines-Chaumeil, C.; Giraud, M.F.; Brèthes, D.; Dautant, A.; Paumard, P. ATP synthase oligomerization: From the enzyme models to the mitochondrial morphology. Int. J. Biochem. Cell Biol. 2013, 45, 99–105. [Google Scholar] [CrossRef]
- Strauss, M.; Hofhaus, G.; Schroder, R.R.; Kuhlbrandt, W. Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 2008, 27, 1154–1160. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Pérez, R.L.-J.T.; Milenkovic, D.; Mourier, A.; Bratic, A.; García-Bartolomé, A.; Fernández-Vizarra, E.; Cadenas, S.; Delmiro, A.; García-Consuegra, I.; Arenas, J.; et al. COX7A2L Is a Mitochondrial Complex III Binding Protein that Stabilizes the III2+IV Supercomplex without Affecting Respirasome Formation. Cell Rep. 2016, 16, 2387–2398. [Google Scholar]
- Williams, E.G.; Wu, Y.; Jha, P.; Dubuis, S.; Blattmann, P.; Argmann, C.A.; Houten, S.M.; Amariuta, T.; Wolski, W.; Zamboni, N.; et al. Systems proteomics of liver mitochondria function. Science (N. Y.) 2016, 352, aad0189. [Google Scholar] [CrossRef] [Green Version]
- Mourier, A.; Matic, S.; Ruzzenente, B.; Larsson, N.G.; Milenkovic, D. The respiratory chain supercomplex organization is independent of COX7a2l isoforms. Cell Metab. 2014, 20, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Dawitz, H.; Schäfer, J.; Schaart, J.M.; Magits, W.; Brzezinski, P.; Ott, M. Rcf1 Modulates Cytochrome c Oxidase Activity Especially Under Energy-Demanding Conditions. Front. Physiol. 2019, 10, 1555. [Google Scholar] [CrossRef]
- Strogolova, V.; Hoang, N.H.; Hosler, J.; Stuart, R.A. The yeast mitochondrial proteins Rcf1 and Rcf2 support the enzymology of the cytochrome c oxidase complex and generation of the proton motive force. J. Biol. Chem. 2019, 294, 4867–4877. [Google Scholar] [CrossRef] [Green Version]
- Hartley, A.M.; Meunier, B.; Pinotsis, N.; Maréchal, A. Rcf2 revealed in cryo-EM structures of hypoxic isoforms of mature mitochondrial III-IV supercomplexes. Proc. Natl. Acad. Sci. USA 2020, 117, 9329–9337. [Google Scholar] [CrossRef] [Green Version]
- Timón-Gómez, A.; Garlich, J.; Stuart, R.A.; Ugalde, C.; Barrientos, A. Distinct Roles of Mitochondrial HIGD1A and HIGD2A in Respiratory Complex and Supercomplex Biogenesis. Cell Rep. 2020, 31, 107607. [Google Scholar] [CrossRef]
- Chen, Y.C.; Taylor, E.B.; Dephoure, N.; Heo, J.M.; Tonhato, A.; Papandreou, I.; Nath, N.; Denko, N.C.; Gygi, S.P.; Rutter, J. Identification of a protein mediating respiratory supercomplex stability. Cell Metab. 2012, 15, 348–360. [Google Scholar] [CrossRef] [Green Version]
- Rieger, B.; Shalaeva, D.N.; Sohnel, A.C.; Kohl, W.; Duwe, P.; Mulkidjanian, A.Y.; Busch, K.B. Lifetime imaging of GFP at CoxVIIIa reports respiratory supercomplex assembly in live cells. Sci. Rep. 2017, 7, 46055. [Google Scholar] [CrossRef]
- Hock, D.H.; Reljic, B.; Ang, C.S.; Muellner-Wong, L.; Mountford, H.S.; Compton, A.G.; Ryan, M.T.; Thorburn, D.R.; Stroud, D.A. HIGD2A is required for assembly of the COX3 module of human mitochondrial complex IV. Mol. Cell. Proteom. MCP 2020. [Google Scholar] [CrossRef] [Green Version]
- Reinders, J.; Wagner, K.; Zahedi, R.P.; Stojanovski, D.; Eyrich, B.; van der Laan, M.; Rehling, P.; Sickmann, A.; Pfanner, N.; Meisinger, C. Profiling phosphoproteins of yeast mitochondria reveals a role of phosphorylation in assembly of the ATP synthase. Mol. Cell. Proteom. MCP 2007, 6, 1896–1906. [Google Scholar] [CrossRef] [Green Version]
- Brandner, K.; Mick, D.U.; Frazier, A.E.; Taylor, R.D.; Meisinger, C.; Rehling, P. Taz1, an outer mitochondrial membrane protein, affects stability and assembly of inner membrane protein complexes: Implications for Barth Syndrome. Mol. Biol. Cell 2005, 16, 5202–5214. [Google Scholar] [CrossRef]
- McKenzie, M.; Lazarou, M.; Thorburn, D.R.; Ryan, M.T. Mitochondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. J. Mol. Biol. 2006, 361, 462–469. [Google Scholar] [CrossRef]










| Gene/Protein | OMIM | Associated Phenotype | Reference |
|---|---|---|---|
| Complex I subunits | |||
| MTND1 | 516000 | Leber optic atrophy, MELAS syndrome, dystonia, spasticity and myopathy. | [135,136,137] |
| MTND2 | 516001 | Leber optic atrophy. | [138] |
| MTND3 | 516002 | Infantile encephalopathy and Leigh syndrome. | [139] |
| MTND4 | 516003 | Leber optic atrophy and MELAS syndrome. | [140,141] |
| MTND4L | 516004 | Leber optic atrophy. | [142] |
| MTND5 | 516005 | Leber optic atrophy and MELAS syndrome. | [143,144] |
| MTND6 | 516006 | Leber optic atrophy and MELAS syndrome. | [143,145] |
| NDUFV1 | 161015 | Severe encephalopathy and neurologic abnormalities. | [146,147] |
| NDUFV2 | 600532 | Hypertrophic cardiomyopathy, truncal hypotonia and encephalopathy. | [148] |
| NDUFS1 | 157655 | Growth retardation, axial hypotonia, hepatomegaly, dystonia and persistent hyperlactatemia. | [147] |
| NDUFS2 | 602985 | Neonatal lactic acidosis and hypertrophic cardiomyopathy. | [149] |
| NDUFS3 | 603846 | Leigh syndrome, severe axial dystonia with oral and pharyngeal motor dysfunction, dysphagia and a tetraparetic syndrome. | [150] |
| NDUFS4 | 602694 | Muscular hypotonia, absence of visual and auditive attention and cardiac defects. | [151] |
| NDUFS6 | 603848 | Fatal infantile lactic acidosis. | [152] |
| NDUFS7 | 601825 | Leigh syndrome, feeding problems, dysarthria and ataxia. | [153] |
| NDUFS8 | 602141 | Leigh syndrome, poor feeding and episodes of apnea and cyanosis. | [154] |
| NDUFA11 | 612638 | Fatal infantile metabolic acidosis, brain atrophy, no motor development and hypertrophic cardiomyopathy. | [155] |
| NDUFA1 | 300078 | Leigh syndrome, hypotonia, nystagmus, generalized choreoathetosis and decreased reflexes. | [156] |
| NDUFA2 | 602137 | Leigh syndrome, hypertrophic cardiomyopathy and developmental delay. | [157] |
| NDUFA6 | 602138 | Intrauterine growth retardation, respiratory insufficiency, lactic acidosis and hypoglycemia. | [158] |
| NDUFA8 | 603359 | Severe neonatal hypotonia, dysmorphic features, epilepsy and signs of brainstem involvement. | [159] |
| NDUFA9 | 603834 | Respiratory and metabolic acidosis, hearing loss, apneas and retinitis pigmentosa. | [160] |
| NDUFA10 | 603835 | Leigh syndrome and delayed psychomotor development. | [161] |
| NDUFA12 | 614530 | Leigh syndrome, progressive loss of motor abilities, scoliosis and dystonia. | [162] |
| NDUFA13 | 609435 | Delayed development, hypotonia, poor eye contact, abnormal eye movements, poor feeding, encephalopathy and hearing loss. | [163] |
| NDUFB3 | 603839 | Encephalopathy, myopathy, hypotonia, developmental delay and lactic acidosis. | [164] |
| NDUFB8 | 602140 | Leigh syndrome, respiratory failure, seizures, hypotonia, cardiac hypertrophy, failure to thrive and severely delayed psychomotor development. | [165] |
| NDUFB9 | 601445 | Progressive hypotonia associated with increased serum lactate. | [164] |
| NDUFB10 | 603843 | Lethal complex I deficiency. | [166] |
| NDUFB11 | 300403 | X-linked microphthalmia with linear skin defects (MLS) syndrome, cardiomyopathy and other congenital anomalies. | [167,168] |
| NDUFC2 | 603845 | Leigh syndrome. | [169] |
| Complex I assembly factors | |||
| ACAD9 | 611103 | Cardiorespiratory depression, hypertrophic cardiomyopathy, encephalopathy and severe lactic acidosis. | [114] |
| FOXRED1 | 613622 | Leigh syndrome, congenital lactic acidosis, athetoid movements of the limbs in early childhood, hypotonia and cerebellar atrophy. | [170] |
| NDUFAF1 | 606934 | Hypertrophic cardiomyopathy, developmental delay, lactic acidosis, hypotonia and Wolff–Parkinson–White syndrome. | [171] |
| NDUFAF2 | 609653 | Ataxia, lethargy, nystagmus, hypotonia, optic atrophy and episodic respiratory insufficiency. | [122] |
| NDUFAF3 | 612911 | Macrocephaly, weak cry, no eye contact, wide anterior fontanel and axial hypotonia. | [123] |
| NDUFAF4 | 611776 | Severe encephalopathy and antenatal cardiomyopathy. | [124] |
| NDUFAF5 | 612360 | Facial dysmorphism, progressive lactic acidosis and neurological defects. | [125] |
| NDUFAF6 | 612392 | Focal seizures, decreased movement and strength, ataxia, lactic acidosis and Leigh syndrome. | [172] |
| NDUFAF8 | 618461 | Leigh syndrome. | [173] |
| NUBPL | 613621 | Infantile-onset hepatopathy, renal tubular acidosis, developmental delay, short stature, leukoencephalopathy, myopathy, nystagmus and ataxia. | [106,107,130] |
| TIMMDC1 | 615534 | Infantile-onset hypotonia, failure to thrive, delayed or minimal psychomotor development, sensorineural deafness, dysmetria, dyskinetic movements, peripheral neuropathy, nystagmus and Leigh syndrome. | [174] |
| TMEM126B | 615533 | Exercise intolerance, muscle weakness, myalgia, early-onset renal tubular acidosis and hypertrophic cardiomyopathy. | [175,176] |
| COA7 | 615623 | Autosomal recessive spinocerebellar ataxia with axonal neuropathy-3. | [177] |
| Gene/Protein | OMIM | Associated Phenotype | Reference |
|---|---|---|---|
| SDHA | 600857 | Leigh syndrome, neonatal dilated cardiomyopathy, catecholamine-secreting extra-adrenal paraganglioma. | [194,195,196] |
| SDHB | 185470 | Paraganglioma, pheochromocytoma, gastrointestinal stromal tumors. | [197,198] |
| SDHC | 602413 | Paraganglioma, gastric stromal sarcoma. | [190,199] |
| SDHD | 602690 | Paraganglioma, pheochromocytoma, gastric stromal sarcoma. | [191,199] |
| SDHAF1 | 612848 | Leukoencephalopathy, spastic quadriplegia, psychomotor regression. | [184] |
| SDHAF2 | 613019 | Paraganglioma. | [183] |
| Gene/Protein | OMIM | Associated Phenotype | Reference |
|---|---|---|---|
| Complex III subunits | |||
| UQCRC2 | 191329 | Hypoglycemia, lactic acidosis, ketosis and hyperammonemia. | [274] |
| MTCYB | 516020 | Leber optic atrophy, exercise intolerance, encephalomyopathy, cardiomyopathy and multisystemic disorder. | [275,276,277,278,279] |
| CYC1 | 123980 | Neurologic deterioration, insulin-responsive hyperglycemia, ketoacidosis with increased serum lactate, liver failure and hyperammonemia. | [280] |
| UQCRB | 191330 | Gastroenteritis, liver enlargement, hypoglycemia and metabolic acidosis but normal psychomotor development at age 4. | [216] |
| UQCRQ | 612080 | Severe neurologic phenotype. | [215] |
| UQCRFS1 | 191327 | Cardiomyopathy and alopecia totalis. | [281] |
| Complex III assembly factors | |||
| BCS1L | 603647 | GRACILE Syndrome, Bjornstad Syndrome, myopathy, encephalopathy, proximal tubulopathy and liver failure. | [272,273,282,283,284,285,286,287,288] |
| TTC19 | 613814 | Progressive encephalopathy, ataxia, spastic paraparesis and psychiatric phenotype. | [256,289,290,291,292] |
| LYRM7 | 615831 | Neurological decompensation and regression, leukoencephalopathy and liver failure. | [293,294] |
| UQCC2 | 614461 | Intrauterine growth retardation, neonatal lactic acidosis and renal tubular dysfunction. | [234,295] |
| UQCC3 | 616097 | Lactic acidosis, hypoglycemia, hypotonia and delayed development. | [270] |
| Assembly Factor (Yeast) | Assembly Factor (Mammals) | Function | CIV Interacting Module | References |
|---|---|---|---|---|
| RNA stability and translation | ||||
| - | TACO1 | Translational activator of mitochondria-encoded MTCO1. | MTCO1-translation | [311] |
| - | LRPPRC | Mitochondrial mRNA stability. | - | [310] |
| - | FASTKD2 | Involved in post-transcriptional RNA maturation, ribosome biogenesis and translation. | - | [333] |
| Heme a biosynthesis and insertion | ||||
| Cox10 | COX10 | Heme a synthesis (conversion of heme b into heme o). | MTCO1 module | [316,334] |
| Cox15 | COX15 | Heme a synthesis (conversion of heme o into heme a). | MTCO1 module | [335,336] |
| Shy1 | SURF1 | Involved in the insertion or stabilization of heme a3. | Early MTCO1 subcomplexes | [337] |
| Copper metabolism and insertion | ||||
| Coa6 | COA6 | Copper homeostasis and transport to CIV. | MTCO2 module | [328,338] |
| Sco1 | SCO1 | Incorporation of copper atoms. | MTCO2 module | [327,339] |
| - | SCO2 | Incorporation of copper atoms. | MTCO2 module | [340] |
| Cox11 | COX11 | Copper chaperone. | MTCO1 module | [319,341] |
| Cox16 | COX16 | MTCO2 maturation. | MTCO2 module | [331,342] |
| Cox17 | COX17 | Copper transfer. | MTCO1 module | [321] |
| Cox19 | COX19 | Stabilization of COX11. | MTCO1 module | [320,343] |
| Assembly | ||||
| Coa3 | COA3/MITRAC12 | Required for MTCO1 stability and assembly. Involved in translational regulation of MTCO1 and prevention of MTCO1 aggregation before assembly. | MTCO1 module | [313,314] |
| - | COA7 | Unknown. | Unknown | [177] |
| Cox14 | COX14/c12orf62 | MTCO1 stability and assembly; avoids MTCO1 aggregation. | MTCO1 module | [312,344] |
| Cmc1 | CMC1 | Stabilizes the interaction between MTCO1, COX14 and COA3. | MTCO1 module | [315] |
| - | COX20/FAM36A | MTCO2 chaperone for copper metalation. | MTCO2 module | [345] |
| Pet100 | PET100 | Assembly factor. | S3 intermediary | [346,347,348] |
| Pet117 | PET117 | Assembly factor; possible role in Cox15 oligomerization and function. | S3 intermediary | [318,349,350] |
| - | MR-1S | Interacts with PET117 and PET100. | S3 intermediary | [306] |
| - | APOPT1/COA8 | Intermediate assembly steps. Putative role in CIV protection from ROS damage. | Unknown | [351] |
| Cox18 | COX18 | Promotes the translocation of MTCO2 globular domain through the IMM. | MTCO2 | [352,353] |
| Gene/Protein | OMIM | Associated Phenotype | Reference |
|---|---|---|---|
| Complex IV subunits | |||
| MTCO1 | 516030 | MELAS syndrome, myopathy, myoglobinuria, motor neurone disease, exercise intolerance, epilepsy, multisystem disorders, deafness, LHON or mitochondrial sensorineural hearing loss. | [355,356,357,358,359] |
| MTCO2 | 516040 | Encephalomyopathy, LHON, myopathy, hypertrophic cardiomyopathy. | [360,361,362,363] |
| MTCO3 | 516050 | MIDD, LHON, myopathy, Leigh disease, myoglobinuria, sporadic bilateral optic neuropathy, rhabdomyolysis, encephalopathy. | [364,365,366,367,368,369] |
| COX4I1 | 123864 | Short stature, poor weight gain, mild dysmorphic features, Fanconi anemia. | [370] |
| COX4I2 | 607976 | Exocrine pancreatic insufficiency, dyserythropoietic anemia, calvarial hyperostosis. | [371] |
| COX5A | 603773 | Early-onset pulmonary arterial hypertension, lactic acidemia, failure to thrive. | [372] |
| COX6A1 | 602072 | Charcot–Marie–Tooth disease. | [373] |
| COX6A2 | 602009 | Muscle weakness and hypotonia, cardiomyopathy. | [374] |
| COX6B1 | 124089 | Severe infantile encephalomyopathy. | [375] |
| COX7A1 | 123995 | Failure to thrive, encephalopathy, hypotonia. | [376] |
| COX7B | 300885 | Microphthalmia with linear skin lesions. | [377] |
| COX8A | 123870 | Leigh-like syndrome presenting with leukodystrophy and severe epilepsy. | [378] |
| NDUFA4 | 603833 | Leigh syndrome. | [298] |
| Complex IV assembly factors | |||
| SURF1 | 185620 | Leigh syndrome, Charcot–Marie–Tooth disease. | [379,380] |
| COA3/MITRAC12 | 614775 | Mild phenotype, exercise intolerance, peripheral neuropathy, obesity and short stature. | [308] |
| COA7 | 615623 | Ataxia and peripheral neuropathy, cognitive impairments, leukodystrophy. | [177] |
| COX14/c12orf62 | 614478 | Severe lactic acidosis and dysmorphic features. | [344] |
| COX20/FAM36A | 614698 | Growth delay, hypotonia, cerebellar ataxia. | [381] |
| PET100 | 614770 | Early-onset psychomotor delay, seizures, hypotonia, Leigh syndrome. | [347,348] |
| PET117 | 614771 | Neurodevelopmental regression. | [350] |
| APOPT1/COA8 | 616003 | Leukodystrophy, neurological signs. | [382] |
| SCO1/SCO2 | 603644/604272 | Cardioencephalomyopathy, Leigh syndrome-like symptoms, spinal muscular atrophy-like presentations, Charcot–Marie–Tooth disease type 4. | [383,384] |
| COX10/COX15 | 602125/603646 | Leigh syndrome, encephalopathy, cardiomyopathy, sensorineural deafness and metabolic acidosis. | [316,336] |
| COA6/C1orf31 | 614772 | Fatal infantile cardioencephalopathy. | [385] |
| TACO1 | 612958 | Leigh syndrome. | [311] |
| COA5 | 613920 | Fatal infantile cardioencephalomyopathy. | [386] |
| FASTKD2 | 612322 | Brain atrophy, epilepsy, delayed psychomotor development, bilateral optic atrophy, spastic hemiparesis, cardiomyopathy. | [387,388] |
| LRPPRC | 607544 | French Canadian type of Leigh syndrome. | [389] |
| Gene/Protein | OMIM | Associated Phenotype | Reference |
|---|---|---|---|
| MT-ATP6 | 516060 | Neuropathy, ataxia and retinitis pigmentosa (NARP), maternally inherited Leigh’s syndrome (MILS), mental retardation, ataxia, cardiomyopathy. | [402,403,410,411] |
| MT-ATP8 | 516070 | Hypertrophic cardiomyopathy and neuropathy. | [404] |
| ATP5E | 614053 | Neonatal-onset lactic acidosis, 3-methylglutaconic aciduria, mental retardation, hypertrophic cardiomyopathy and peripheral neuropathy. | [405] |
| ATP5A1 | 615228 | Fatal infantile encephalopathy. | [408] |
| ATPAF2 | 608918 | Degenerative encephalopathy, elevated lactate levels, developmental delay. | [412] |
| ATP5F1A | 164360 | Fatal infantile mitochondrial encephalopathy | [408,413] |
| ATP5F1D | 603150 | Metabolic decompensation with lactic acidosis, hypoglycemia, hyperammonemia, 3-methylglutaconic aciduria, encephalopathy. | [414] |
| ATP5F1E | 606153 | Neonatal-onset lactic acidosis, 3-methylglutaconic aciduria, mild mental retardation, hypertrophic cardiomyopathy, peripheral neuropathy. | [405] |
| TMEM70 | 612418 | Neonatal mitochondrial encephalocardiomyopathy. | [119] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586. https://doi.org/10.3390/ijms22020586
Protasoni M, Zeviani M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. International Journal of Molecular Sciences. 2021; 22(2):586. https://doi.org/10.3390/ijms22020586
Chicago/Turabian StyleProtasoni, Margherita, and Massimo Zeviani. 2021. "Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions" International Journal of Molecular Sciences 22, no. 2: 586. https://doi.org/10.3390/ijms22020586
APA StyleProtasoni, M., & Zeviani, M. (2021). Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. International Journal of Molecular Sciences, 22(2), 586. https://doi.org/10.3390/ijms22020586