The Multifaced Role of STAT3 in Cancer and Its Implication for Anticancer Therapy



Abstract
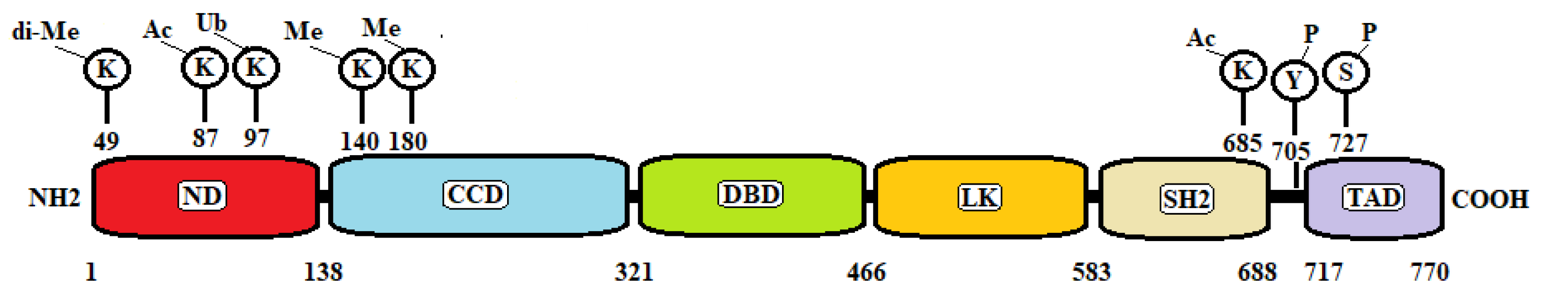
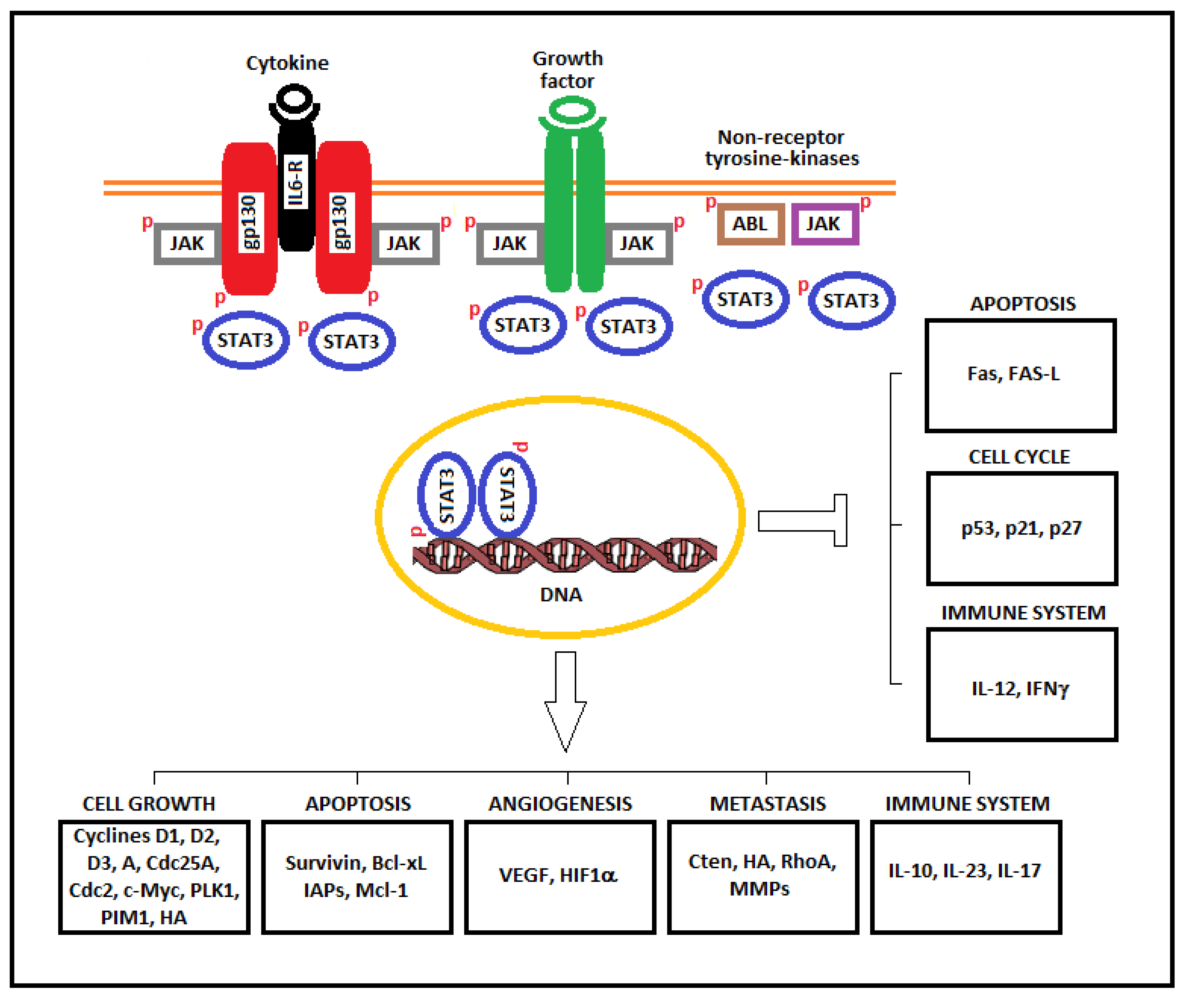
:1. Introduction
2. STAT3 and Tumorigenesis
3. Role of STAT3 in Oncogenic Transformation
4. Invasion, Metastasis, and Angiogenesis
5. Immune Escape
6. The Dual Role of STAT3 in Cancer
6.1. Glioblastomas
6.2. Prostate Cancer
6.3. Lung Cancer
6.4. Colorectal Cancer
6.5. Thyroid Cancer
6.6. Breast Cancer
6.7. Head and Neck Squamous Cell Cancers
6.8. STAT3 Isoforms
7. Treatment of Cancer with STAT3 Inhibitors
7.1. Inhibitors Targeting the SH2 Domain
7.2. Inhibitors Targeting the DBD
7.3. Inhibitors Targeting the ND and the CC Domain
7.4. Indirect Inhibitors
7.5. Clinical Trials
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Wegenka, U.M.; Buschmann, J.; Lütticken, C.; Heinrich, P.C.; Horn, F. Acute-phase response factor, a nuclear factor binding to acute-phase response elements, is rapidly activated by interleukin-6 at the posttranslational level. Mol. Cell. Biol. 1993, 13, 276–288. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Wen, Z.; Darnell, J.E. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Hemmann, U.; Gerhartz, C.; Heesel, B.; Sasse, J.; Kurapkat, G.; Grötzingerl, J.; Wollmert, A.; Zhong, Z.; Darnell, J.E.; Graeve, L.; et al. Differential activation of acute phase response factor Stat3 and Stat1 via the cytoplasmic domain of the interleukin 6 signal transducer gp130. II. Src homology SH2 domains define the specificity of stat factor activation. J. Biol. Chem. 1996, 271, 2999–3007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, C.P.; Cao, X. Structure, function, and regulation of STAT proteins. Mol. BioSyst. 2006, 2, 536–550. [Google Scholar] [CrossRef]
- Quesnelle, K.M.; Boehm, A.L.; Grandis, J.R. STAT-mediated EGFR signaling in cancer. J. Cell. Biochem. 2007, 102, 311–319. [Google Scholar] [CrossRef]
- Decker, T.; Kovarik, P. Serine phosphorylation of STATs. Oncogene 2000, 19, 2628–2637. [Google Scholar] [CrossRef] [Green Version]
- Ihle, J.N. The janus kinase family and signaling through members of the cytokine receptor superfamily. Proc. Soc. Exp. Biol. Med. 1994, 206, 268–272. [Google Scholar] [CrossRef]
- Darnell, J.E.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lupardus, P.; LaPorte, S.L.; Garcia, K.C. Structural biology of shared cytokine receptors. Ann. Rev. Immunol. 2009, 27, 29–60. [Google Scholar] [CrossRef] [Green Version]
- Seidel, H.M.; Milocco, L.H.; Lamb, P.; Darnell, J.E.; Stein, R.B.; Rosen, J. Spacing of palindromic half sites as a determinant of selective STAT (signal transducers and activators of transcription) DNA binding and transcriptional activity. Proc. Natl. Acad. Sci. USA 1995, 92, 3041–3045. [Google Scholar] [CrossRef] [Green Version]
- Garbers, C.; Hermanns, H.M.; Schaper, F.; Müller-Newen, G.; Grötzinger, J.; Rose-John, S.; Scheller, J. Plasticity and cross-talk of interleukin 6-type cytokines. Cytokine Growth Factor Rev. 2012, 23, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Jenkins, B.J. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 2018, 18, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Betz, U.A.K.; Bloch, W.; Van Den Broek, M.; Yoshida, K.; Taga, T.; Kishimoto, T.; Addicks, K.; Rajewsky, K.; Müller, W. Postnatally induced inactivation of gp130 in mice results in neurological, cardiac, hematopoietic, immunological, hepatic, and pulmonary defects. J. Exp. Med. 1998, 188, 1955–1965. [Google Scholar] [CrossRef] [PubMed]
- Turkson, J.; Jove, R. STAT proteins: Novel molecular targets for cancer drug discovery. Oncogene 2000, 19, 6613–6626. [Google Scholar] [CrossRef] [Green Version]
- Levy, D.E.; Inghirami, G. STAT3: STAT3: A multifaceted oncogene. Proc. Natl. Acad. Sci. USA 2006, 103, 10151–10152. [Google Scholar] [CrossRef] [Green Version]
- Pensa, S.; Regis, G.; Boselli, D.; Novelli, F.; Poli, V. STAT1 and STAT3 in tumorigenesis: Two sides of the same coin? In JAK-STAT Pathway in Disease; Landes Bioscience Books: Austin, TX, USA, 2009; pp. 100–121. [Google Scholar]
- Yu, C.L.; Meyer, D.J.; Campbell, G.S.; Larner, A.C.; Carter-Su, C.; Schwartz, J.; Jove, R. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science 1995, 269, 81–83. [Google Scholar] [CrossRef]
- Cao, X.; Tay, A.; Guy, G.R.; Tan, Y.H. Activation and association of Stat3 with Src in v-Src-transformed cell lines. Mol. Cell. Biol. 1996, 16, 1595–1603. [Google Scholar] [CrossRef] [Green Version]
- Rane, S.G.; Reddy, E.P. JAKs, STATs and Src kinases in hematopoiesis. Oncogene 2002, 21, 3334–3358. [Google Scholar] [CrossRef] [Green Version]
- Danial, N.N.; Rothman, P. Jak-STAT signaling induced by the v-abl oncogene. Oncogene 2000, 269, 1875–1877. [Google Scholar] [CrossRef]
- Besser, D.; Bromberg, J.F.; Darnell, J.E.; Hanafusa, H. A single amino acid substitution in the v-Eyk intracellular domain results in activation of Stat3 and enhances cellular transformation. Mol. Cell. Biol. 1999, 19, 1401–1409. [Google Scholar] [CrossRef] [Green Version]
- Zong, C.S.; Zeng, L.; Jiang, Y.; Sadowski, H.B.; Wang, L.H. Stat3 plays an important role in oncogenic Ros- and insulin-like growth factor I receptor-induced anchorage-independent growth. J. Biol. Chem. 1998, 273, 28065–28072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, R.; Yu, C.L.; Hudnall, A.; Catlett, R.; Nelson, K.L.; Smithgall, T.; Fujita, D.J.; Ethier, S.P.; Jove, R. Constitutive activation of Stat3 in fibroblasts transformed by diverse oncoproteins and in breast carcinoma cells. Cell Growth Differ. 1997, 8, 1267–1276. [Google Scholar] [PubMed]
- Wen, X.; Lin, H.H.; Shih, H.M.; Kung, H.J.; Ann, D.K. Kinase activation of the non-receptor tyrosine kinase Etk/BMX alone is sufficient to transactivate STAT-mediated gene expression in salivary and lung epithelial cells. J. Biol. Chem. 1999, 274, 38204–38210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, T.C.; Prator, P.C.; Medveczky, M.M.; Medveczky, P.G. The Lck binding domain of herpesvirus saimiri tip-484 constitutively activates Lck and STAT3 in T cells. J. Virol. 1999, 73, 1689–1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migone, T.S.; Lin, J.X.; Cereseto, A.; Mulloy, J.C.; O’Shea, J.J.; Franchini, G.; Leonard, W.J. Constitutively activated Jak-STAT pathway in T cells transformed with HTLV-I. Science 1995, 269, 79–81. [Google Scholar] [CrossRef] [PubMed]
- Weber-Nordt, R.; Egen, C.; Wehinger, J.; Ludwig, W.; Gouilleux-Gruart, V.; Mertelsmann, R.; Finke, J. Constitutive activation of STAT proteins in primary lymphoid and myeloid leukemia cells and in Epstein-Barr virus (EBV)-related lymphoma cell lines. Blood 1996, 88, 809–816. [Google Scholar] [CrossRef] [Green Version]
- Lund, T.C.; Garcia, R.; Medveczky, M.M.; Jove, R.; Medveczky, P.G. Activation of STAT transcription factors by herpesvirus Saimiri Tip-484 requires p56lck. J. Virol. 1997, 71, 6677–6682. [Google Scholar] [CrossRef] [Green Version]
- Nepomuceno, R.R.; Snow, A.L.; Beatty, P.R.; Krams, S.M.; Martinez, O.M. Constitutive activation of Jak/STAT proteins in Epstein-Barr virus-infected B-cell lines from patients with post-transplant lymphoproliferative disorder. Transplantation 2002, 74, 396–402. [Google Scholar] [CrossRef]
- Samaan, A.; Mahana, W. Constitutive and induced activation of JAK/Stat pathway in leukemogenic and asymptomatic human T-cell lymphoptropic virus type 1 (HTLV-1) transformed rabbit cell lines. Immunol. Lett. 2007, 109, 113–119. [Google Scholar] [CrossRef]
- Tomita, M.; Kawakami, H.; Uchihara, J.N.; Okudaira, T.; Masuda, M.; Matsuda, T.; Tanaka, Y.; Ohshiro, K.; Mori, N. Inhibition of constitutively active Jak-Stat pathway suppresses cell growth of human T-cell leukemia virus type 1-infected T-cell lines and primary adult T-cell leukemia Cells. Retrovirology 2006, 3, 22. [Google Scholar] [CrossRef] [Green Version]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Catlett-Falcone, R.; Landowski, T.H.; Oshiro, M.M.; Turkson, J.; Levitzki, A.; Savino, R.; Ciliberto, G.; Moscinski, L.; Fernández-Luna, J.L.; Nuñez, G.; et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 1999, 10, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Lou, W.; Ni, Z.; Dyer, K.; Tweardy, D.J.; Gao, A.C. Interleukin-6 induces prostate cancer cell growth accompanied by activation of stat3 signaling pathway. Prostate 2000, 42, 239–242. [Google Scholar] [CrossRef]
- Niu, G.; Bowman, T.; Huang, M.; Shivers, S.; Reintgen, D.; Daud, A.; Chang, A.; Kraker, A.; Jove, R.; Yu, H. Roles of activated Src and Stat3 signaling in melanoma tumor cell growth. Oncogene 2002, 21, 7001–7007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.S.; Carbajal, S.; Kiguchi, K.; Clifford, J.; Sano, S.; DiGiovanni, J. Epidermal growth factor receptor-mediated activation of Stat3 during multistage skin carcinogenesis. Cancer Res. 2004, 64, 2382–2389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.I.; Grandis, J.R. STAT signaling in head and neck cancer. Oncogene 2000, 19, 2489–2495. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Turkson, J.; Karras, J.G.; Jove, R.; Haura, E.B. Activation of Stat3 by receptor tyrosine kinases and cytokines regulates survival in human non-small cell carcinoma cells. Oncogene 2003, 22, 4150–4165. [Google Scholar] [CrossRef] [Green Version]
- Grandis, J.R.; Zeng, Q.; Drenning, S.D. Epidermal growth factor receptor—Mediated stat3 signaling blocks apoptosis in head and neck cancer. Laryngoscope 2000, 119, 868–874. [Google Scholar] [CrossRef]
- Trevino, J.G.; Gray, M.J.; Nawrocki, S.T.; Summy, J.M.; Lesslie, D.P.; Evans, D.B.; Sawyer, T.K.; Shakespeare, W.C.; Watowich, S.S.; Chiao, P.J.; et al. Src activation of Stat3 is an independent requirement from NF-kappaB activation for constitutive IL-8 expression in human pancreatic adenocarcinoma cells. Angiogenesis 2006, 9, 101–110. [Google Scholar] [CrossRef]
- Pilati, C.; Amessou, M.; Bihl, M.P.; Balabaud, C.; Van Nhieu, J.T.; Paradis, V.; Nault, J.C.; Izard, T.; Bioulac-Sage, P.; Couchy, G.; et al. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. J. Exp. Med. 2011, 206, 1359–1366. [Google Scholar] [CrossRef]
- Koskela, H.L.M.; Eldfors, S.; Ellonen, P.; van Adrichem, A.J.; Kuusanmäki, H.; Andersson, E.I.; Lagström, S.; Clemente, M.J.; Olson, T.; Jalkanen, S.E.; et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N. Engl. J. Med. 2012, 366, 1905–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraud, S.; Hurlstone, A.; Avril, S.; Coqueret, O. Implication of BRG1 and cdk9 in the STAT3-mediated activation of the p21waf1. Oncogene 2004, 23, 7391–7398. [Google Scholar] [CrossRef] [PubMed]
- Giraud, S.; Bienvenu, F.; Avril, S.; Gascan, H.; Heery, D.M.; Coqueret, O. Functional interaction of STAT3 transcription factor with the coactivator NcoA/SRC1a. J. Biol. Chem. 2002, 277, 8004–8011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazan-Halevy, I.; Harris, D.; Liu, Z.; Liu, J.; Li, P.; Chen, X.; Shanker, S.; Ferrajoli, A.; Keating, M.J.; Estrov, Z. STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood 2010, 115, 2852–2863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef] [Green Version]
- Sellier, H.; Rébillard, A.; Guette, C.; Barré, B.; Coqueret, O. How should we define STAT3 as an oncogene and as a potential target for therapy? JAK-STAT 2013, 2, e24716. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Zhang, P.; Herrmann, A.; Yang, C.; Xin, H.; Wang, Z.; Hoon, D.S.B.; Forman, S.J.; Jove, R.; Riggs, A.D.; et al. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc. Natl. Acad. Sci. USA 2012, 109, 7765–7769. [Google Scholar] [CrossRef] [Green Version]
- Ray, S.; Boldogh, I.; Brasier, A.R. STAT3 NH2-terminal acetylation is activated by the hepatic acute-phase response and required for IL-6 induction of angiotensinogen. Gastroenterology 2005, 129, 1616–1632. [Google Scholar] [CrossRef]
- Dasgupta, M.; Dermawan, J.K.; Willard, B.; Stark, G.R. STAT3-driven transcription depends upon the dimethylation of K49 by EZH2. Proc. Natl. Acad. Sci USA 2015, 112, 3985–3990. [Google Scholar] [CrossRef] [Green Version]
- Ray, S.; Zhao, Y.; Jamaluddin, M.; Edeh, C.B.; Lee, C.; Brasier, A.R. Inducible STAT3 NH2 terminal mono-ubiquitination promotes BRD4 complex formation to regulate apoptosis. Cell Signal. 2014, 26, 1445–1455. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Huang, J.; Dasgupta, M.; Sears, N.; Miyagi, M.; Wang, B.; Chance, M.R.; Chen, X.; Du, Y.; Wang, Y.; et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 21499–21504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Kim, M.; Woo, D.H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell. 2013, 23, 839–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Du, Y.; Song, Q.; Nan, J.; Guan, P.; Guo, J.; Wang, X.; Yang, J.; Zhao, C. E2F is required for STAT3-mediated upregulation of cyclin B1 and Cdc2 expressions and contributes to G2-M phase transition. Acta Biochim. Biophys. Sin. 2019, 51, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Leslie, K.; Lang, C.; Devgan, G.; Azare, J.; Berishaj, M.; Gerald, W.; Kim, Y.B.; Paz, K.; Darnell, J.E.; Albanese, C.; et al. Cyclin D1 is transcriptionally regulated by and required for transformation by activated signal transducer and activator of transcription 3. Cancer Res. 2006, 66, 2544–2552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alle, K.M.; Henshall, S.M.; Field, A.S.; Sutherland, R.L. Cyclin D1 protein is overexpressed in hyperplasia and intraductal carcinoma of the breast. Clin. Cancer Res. 1998, 4, 847–854. [Google Scholar] [PubMed]
- Wang, T.C.; Cardiff, R.D.; Zukerberg, L.; Lees, E.; Arnold, A.; Schmidt, E.V. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature 1994, 369, 669–671. [Google Scholar] [CrossRef]
- Robles, A.I.; Rodriguez-Puebla, M.L.; Glick, A.B.; Trempus, C.; Hansen, L.; Sicinski, P.; Tennant, R.W.; Weinberg, R.A.; Yuspa, S.H.; Conti, C.J. Reduced skin tumor development in cyclin D1-deficient mice highlights the oncogenic ras pathway in vivo. Genes Dev. 1998, 12, 2469–2474. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Geng, Y.; Sicinski, P. Specific protection against breast cancers by cyclin D1 ablation. Nature 2001, 411, 1017–1021. [Google Scholar] [CrossRef]
- Bowman, T.; Broome, M.A.; Sinibaldi, D.; Wharton, W.; Pledger, W.J.; Sedivy, J.M.; Irby, R.; Yeatman, T.; Courtneidge, S.A.; Jove, R. Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 7319–7324. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Du, X.; Wang, C.; Lin, D.; Ruan, X.; Feng, Y.; Huo, Y.; Peng, H.; Cui, J.; Zhang, T.; et al. Reciprocal activation between PLK1 and Stat3 contributes to survival and proliferation of esophageal cancer cells. Gastroenterology 2012, 142, 521–530. [Google Scholar] [CrossRef]
- Vogt, P.K. Fortuitous convergences: The beginnings of JUN. Nat. Rev. Cancer 2002, 2, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Parri, E.; Kuusanmäki, H.; van Adrichem, A.J.; Kaustio, M.; Wennerberg, K. Identification of novel regulators of STAT3 activity. PLoS ONE 2020, 142, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Schuringa, J.J.; Timmer, H.; Luttickhuizen, D.; Vellenga, E.; Kruijer, W. c-Jun and c-Fos cooperate with STAT3 in IL-6-induced transactivation of the IL-6 respone element (IRE). Cytokine 2001, 14, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Erikson, R.L. Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 5789–5794. [Google Scholar] [CrossRef] [Green Version]
- Shirogane, T.; Fukada, T.; Muller, J.M.M.; Shima, D.T.; Hibi, M.; Hirano, T. Synergistic roles for Pim-1 and c-Myc in STAT3-mediated cell cycle progression and antiapoptosis. Immunity 1999, 11, 709–719. [Google Scholar] [CrossRef] [Green Version]
- Albasri, A.; Seth, R.; Jackson, D.; Benhasouna, A.; Crook, S.; Nateri, A.S.; Chapman, R.; Ilyas, M. C-terminal Tensin-like (CTEN) is an oncogene which alters cell motility possibly through repression of E-cadherin in colorectal cancer. J. Pathol. 2009, 218, 57–65. [Google Scholar] [CrossRef]
- Liao, Y.C.; Chen, N.T.; Shih, Y.P.; Dong, Y.; Lo, S.H. Up-regulation of C-terminal tensin-like molecule promotes the tumorigenicity of colon cancer through β-catenin. Cancer Res. 2009, 69, 4563–4566. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, H.; Yukiue, H.; Kobayashi, Y.; Fukai, I.; Fujii, Y. Cten mRNA expression is correlated with tumor progression in thymoma. Tumor Biol. 2003, 24, 271–274. [Google Scholar] [CrossRef]
- Sakashita, K.; Mimori, K.; Tanaka, F.; Kamohara, Y.; Inoue, H.; Sawada, T.; Hirakawa, K.; Mori, M. Prognostic relevance of Tensin4 expression in human gastric cancer. Ann. Surg. Oncol. 2008, 15, 2606–2613. [Google Scholar] [CrossRef]
- Sasaki, H.; Moriyama, S.; Mizuno, K.; Yukiue, H.; Konishi, A.; Yano, M.; Kaji, M.; Fukai, I.; Kiriyama, M.; Yamakawa, Y.; et al. Cten mRNA expression was correlated with tumor progression in lung cancers. Lung Cancer 2003, 40, 151–155. [Google Scholar] [CrossRef]
- Bohrer, L.R.; Chuntova, P.; Bade, L.K.; Beadnell, T.C.; Leon, R.P.; Brady, N.J.; Ryu, Y.; Goldberg, J.E.; Schmechel, S.C.; Koopmeiners, J.S.; et al. Activation of the FGFR-STAT3 pathway in breast cancer cells induces a hyaluronan-rich microenvironment that licenses tumor formation. Cancer Res. 2014, 74, 374–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.M.; Su, F.; Kalyana-Sundaram, S.; Khazanov, N.; Ateeq, B.; Cao, X.; Lonigro, R.J.; Vats, P.; Wang, R.; Lin, S.F.; et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013, 3, 636–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gritsko, T.; Williams, A.; Turkson, J.; Kaneko, S.; Bowman, T.; Huang, M.; Nam, S.; Eweis, I.; Diaz, N.; Sullivan, D.; et al. Persistent activation of Stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin. Cancer Res. 2006, 12, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.-L.; Yeh, J.; Van Waes, C.; Chen, Z. Bcl-xL is regulated by NF-KappaB and STAT3 through p53 -dependent control in head and neck squamous cell carcinoma. Cancer Res. 2004, 64 (Suppl. 7), 1115. [Google Scholar]
- Kunigal, S.; Laka, S.S.; Sodadasu, P.K.; Estes, N.; Rao, J.S. Stat3-siRNA induces Fas-mediated apoptosis in vitro and in vivo in breast cancer. Int. J. Oncol. 2009, 34, 1209–1220. [Google Scholar]
- Bhattacharya, S.; Ray, R.M.; Johnson, L.R. STAT3-mediated transcription of Bcl-2, Mcl-1 and C-IAP2 prevents apoptosis in polyamine-depleted cells. Biochem. J. 2005, 392, 335–344. [Google Scholar] [CrossRef] [Green Version]
- Xiang, W.; Yang, C.Y.; Bai, L. MCL-1 inhibition in cancer treatment. Onco. Targets. Ther. 2018, 11, 7301–7314. [Google Scholar] [CrossRef] [Green Version]
- Niu, G.; Wright, K.L.; Ma, Y.; Wright, G.M.; Huang, M.; Irby, R.; Briggs, J.; Karras, J.; Cress, W.D.; Pardoll, D.; et al. Role of Stat3 in Regulating p53 Expression and Function. Mol. Cell. Biol. 2005, 25, 7432–7440. [Google Scholar] [CrossRef] [Green Version]
- Ranger, J.J.; Levy, D.E.; Shahalizadeh, S.; Hallett, M.; Muller, W.J. Identification of a Stat3-dependent transcription regulatory network involved in metastatic progression. Cancer Res. 2009, 69, 6823–6830. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, I.; Quaglino, E.; Maritano, D.; Pannellini, T.; Riera, L.; Cavallo, F.; Forni, G.; Musiani, P.; Chiarle, R.; Poli, V. Stat3 is required for anchorage-independent growth and metastasis but not for mammary tumor development downstream of the ErbB-2 oncogene. Mol. Carcinog. 2010, 49, 114–120. [Google Scholar] [CrossRef]
- Sahai, E.; Marshall, C.J. Differing modes for tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 2003, 5, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Wyckoff, J.B.; Pinner, S.E.; Gschmeissner, S.; Condeelis, J.S.; Sahai, E. ROCK- and myosin-dependent Matrix deformation enables protease-independent tumor-cell invasion in vivo. Curr. Biol. 2006, 16, 1515–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.R.; Chen, C.C.; Chan, Y.T.; Wang, H.J.; Chien, F.T.; Chen, Y.L.; Liu, J.L.; Yang, M.H. STAT3-coordinated migration facilitates the dissemination of diffuse large B-cell lymphomas. Nat. Commun. 2018, 12, 3696. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.X.; Wei, D.; Liu, M.; Gao, A.C.; Ali-Osman, F.; Sawaya, R.; Huang, S. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene 2004, 23, 3550–3560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dechow, T.N.; Pedranzini, L.; Leitch, A.; Leslie, K.; Gerald, W.L.; Linkov, I.; Bromberg, J.F. Requirement of matrix metalloproteinase-9 for the transformation of human mammary epithelial cells by Stat3-C. Proc. Natl. Acad. Sci. USA 2004, 101, 10602–10607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, M.; Murata, T.; Suzuki, T.; Shindoh, M.; Nakajima, K.; Imai, K.; Yoshida, K. Requirement of STAT3 activation for maximal collagenase-1 (MMP-1) induction by epidermal growth factor and malignant characteristics in T24 bladder cancer cells. Oncogene 2006, 25, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Huang, C.; Huang, K.; Wu, W.; Jiang, T.; Cao, J.; Feng, Z.; Qiu, Z. STAT3 knockdown reduces pancreatic cancer cell invasiveness and matrix metalloproteinase-7 expression in nude mice. PLoS ONE 2011, 6, e25941. [Google Scholar] [CrossRef]
- Nguyen, D.X.; Bos, P.D.; Massagué, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef]
- Wang, T.; Niu, G.; Kortylewski, M.; Burdelya, L.; Shain, K.; Zhang, S.; Bhattacharya, R.; Gabrilovich, D.; Heller, R.; Coppola, D.; et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 2004, 10, 48–54. [Google Scholar] [CrossRef]
- Grunstein, J.; Roberts, W.G.; Mathieu-Costello, O.; Hanahan, D.; Johnson, R.S. Tumor-derived expression of vascular endothelial growth factor is a critical factor in tumor expansion and vascular function. Cancer Res. 1999, 59, 1592–1598. [Google Scholar] [PubMed]
- Chen, Z.; Zhong, C.H. STAT3: A critical transcription activator in angiogenesis. Med. Res. Rev. 2008, 28, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.K.; Park, H.J.; Kim, N.H.; Park, S.J.; Park, I.Y.; Kim, I.S. Hypoxia-inducible factor-1α enhances haptoglobin gene expression by improving binding of STAT3 to the promoter. J. Biol. Chem. 2011, 286, 8857–8865. [Google Scholar] [CrossRef] [Green Version]
- Trinchieri, G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef]
- Matsukawa, A.; Kudo, S.; Maeda, T.; Numata, K.; Watanabe, H.; Takeda, K.; Akira, S.; Ito, T. Stat3 in resident macrophages as a repressor protein of inflammatory response. J. Immunol. 2005, 175, 3354–3359. [Google Scholar] [CrossRef] [Green Version]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef]
- Cheng, F.; Wang, H.W.; Cuenca, A.; Huang, M.; Ghansah, T.; Brayer, J.; Kerr, W.G.; Takeda, K.; Akira, S.; Schoenberger, S.P.; et al. A critical role for Stat3 signaling in immune tolerance. Immunity 2003, 19, 425–436. [Google Scholar] [CrossRef] [Green Version]
- Jove, R. Preface: Stat signaling. Oncogene 2000, 19, 2466–2467. [Google Scholar] [CrossRef] [Green Version]
- Murphy, T.L.; Cleveland, M.G.; Kulesza, P.; Magram, J.; Murphy, K.M. Regulation of interleukin 12 p40 expression through an NF-kappa B half-site. Mol. Cell. Biol. 1995, 15, 5258–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortylewski, M.; Xin, H.; Kujawski, M.; Lee, H.; Liu, Y.; Harris, T.; Drake, C.; Pardoll, D.; Yu, H. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 2009, 15, 114–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, H.H.; Ivashkiv, L.B. Role of STAT3 in type I interferon responses: Negative regulation of STAT1-dependent inflammatory gene activation. J. Biol. Chem. 2006, 281, 14111–14118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, K.; Clausen, B.E.; Kaisho, T.; Tsujimura, T.; Terada, N.; Förster, I.; Akira, S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of stat3 in macrophages and neutrophils. Immunity 1999, 10, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Herbeuval, J.P.; Lelievre, E.; Lambert, C.; Dy, M.; Genin, C. Recruitment of STAT3 for production of IL-10 by colon carcinoma cells induced by macrophage-derived IL-6. J. Immunol. 2004, 172, 4630–4636. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Laurence, A.; Kanno, Y.; Pacher-Zavisin, M.; Zhu, B.M.; Tato, C.; Yoshimura, A.; Hennighausen, L.; O’Shea, J.J. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 8137–8142. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Rhee, K.J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Zorn, E.; Nelson, E.A.; Mohseni, M.; Porcheray, F.; Kim, H.; Litsa, D.; Bellucci, R.; Raderschall, E.; Canning, C.; Soiffer, R.J.; et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood 2006, 108, 1571–1579. [Google Scholar] [CrossRef] [Green Version]
- Cheng, P.; Corzo, C.A.; Luetteke, N.; Yu, B.; Nagaraj, S.; Bui, M.M.; Ortiz, M.; Nacken, W.; Sorg, C.; Vogl, T.; et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J. Exp. Med. 2008, 205, 2235–2249. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; O’Shea, J.J. Th17 cells: A new fate for differentiating helper T cells. Immunol. Res. 2008, 41, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Kortylewski, M.; Kujawski, M.; Wang, T.; Wei, S.; Zhang, S.; Pilon-Thomas, S.; Niu, G.; Kay, H.; Mulé, J.; Kerr, W.G.; et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 2005, 11, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Kobayashi, T.; Ichiyama, K.; Yoshida, R.; Hashimoto, M.; Takimoto, T.; Tanaka, K.; Chinen, T.; Shichita, T.; Wyss-Coray, T.; et al. Selective expansion of Foxp3-positive regulatory T cells and immunosuppression by suppressors of cytokine signaling 3-deficient dendritic cells. J. Immunol. 2007, 179, 2170–2179. [Google Scholar] [CrossRef] [Green Version]
- Zou, W. Regulatory T cells, tumour immunity and immunotherapy. Nat. Rev. Immunol. 2006, 6, 295–307. [Google Scholar] [CrossRef]
- Anderson, D.J. Stem cells and pattern formation in the nervous system: The possible versus the actual. Neuron 2001, 30, 19–35. [Google Scholar] [CrossRef] [Green Version]
- Gage, F.H. Mammalian neural stem cells. Science 2000, 287, 1433–1438. [Google Scholar] [CrossRef]
- Qian, X.; Shen, Q.; Goderie, S.K.; He, W.; Capela, A.; Davis, A.A.; Temple, S. Timing of CNS cell generation: A programmed sequence of neuron and glial cell production from isolated murine cortical stem cells. Neuron 2000, 28, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Ware, C.B.; Horowitz, M.C.; Renshaw, B.R.; Hunt, J.S.; Liggitt, D.; Koblar, S.A.; Gliniak, B.C.; McKenna, H.J.; Papayannopoulou, T.; Thoma, B.; et al. Targeted disruption of the low-affinity leukemia inhibitory factor receptor gene causes placental, skeletal, neural and metabolic defects and results in perinatal death. Development 1995, 121, 1283–1299. [Google Scholar]
- Rajan, P.; McKay, R.D.G. Multiple routes to astrocytic differentiation in the CNS. J. Neurosci. 1998, 18, 3620–3629. [Google Scholar] [CrossRef]
- Yoshimatsu, T.; Kawaguchi, D.; Oishi, K.; Takeda, K.; Akira, S.; Masuyama, N.; Gotoh, Y. Non-cell-autonomous action of STAT3 in maintenance of neural precursor cells in the mouse neocortex. Development 2006, 133, 2553–2563. [Google Scholar] [CrossRef] [Green Version]
- Weissenberger, J.; Loeffler, S.; Kappeler, A.; Kopf, M.; Lukes, A.; Afanasieva, T.A.; Aguzzi, A.; Weis, J. IL-6 is required for glioma development in a mouse model. Oncogene 2004, 23, 3308–3316. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, L.K.; Ren, Z.; Fuller, G.N.; Schaefer, T.S. Constitutive of Stat3α in brain tumors: Localization to tumor endothelial cells and activation by the endothelial tyrosine kinase receptor (VEGFR-2). Oncogene 2002, 21, 2058–2065. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, H.; Zhang, W.; Huang, H.J.; Liao, W.S.L.; Fuller, G.N. Analysis of the activation status of Akt, NFκB, and Stat3 in human diffuse gliomas. Lab. Investig. 2004, 84, 941–951. [Google Scholar] [CrossRef] [Green Version]
- La Iglesia, N.; Puram, S.; Bonni, A. STAT3 Regulation of Glioblastoma Pathogenesis. Curr. Mol. Med. 2009, 9, 580–590. [Google Scholar] [CrossRef] [Green Version]
- Pencik, J.; Schlederer, M.; Gruber, W.; Unger, C.; Walker, S.M.; Chalaris, A.; Marié, I.J.; Hassler, M.R.; Javaheri, T.; Aksoy, O.; et al. STAT3 regulated ARF expression suppresses prostate cancer metastasis. Nat. Commun. 2015, 22, 7736. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.P.; Mark, K.G.; Leslie, K.; Pao, W.; Motoi, N.; Gerald, W.L.; Travis, W.D.; Bornmann, W.; Veach, D.; Clarkson, B.; et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J. Clin. Investig. 2007, 117, 3846–3856. [Google Scholar] [CrossRef] [Green Version]
- Haura, E.B.; Zheng, Z.; Song, L.; Cantor, A.; Bepler, G. Activated epidermal growth factor receptor-stat-3 signaling promotes tumor survival in vivo in non-small cell lung cancer. Clin. Cancer Res. 2005, 11, 8288–8294. [Google Scholar] [CrossRef] [Green Version]
- Grabner, B.; Schramek, D.; Mueller, K.M.; Moll, H.P.; Svinka, J.; Hoffmann, T.; Bauer, E.; Blaas, L.; Hruschka, N.; Zboray, K.; et al. Disruption of STAT3 signalling promotes KRAS-induced lung tumorigenesis. Nat. Commun. 2015, 6, 6285. [Google Scholar] [CrossRef] [Green Version]
- Takaku, K.; Oshima, M.; Miyoshi, H.; Matsui, M.; Seldin, M.F.; Taketo, M.M. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell 1998, 6, 645–656. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Bacani, J.; Begthel, H.; Jonkeer, S.; Gregorieff, A.; Van De Born, M.; Malats, N.; Sancho, E.; Boon, E.; Pawson, T.; et al. EphB receptor activity suppresses colorectal cancer progression. Nature 2005, 435, 1126–1130. [Google Scholar] [CrossRef]
- Lee, J.; Kim, J.C.K.; Lee, S.E.; Quinley, C.; Kim, H.R.; Herdman, S.; Corr, M.; Raz, E. Signal transducer and activator of transcription 3 (STAT3) protein suppresses adenoma-to-carcinoma transition in Apcmin/+ mice via regulation of Snail-1 (SNAI) protein stability. J. Biol. Chem. 2012, 287, 18182–18189. [Google Scholar] [CrossRef] [Green Version]
- Demaria, M.; Misale, S.; Giorgi, C.; Miano, V.; Camporeale, A.; Campisi, J.; Pinton, P.; Poli, V. STAT3 can serve as a hit in the process of malignant transformation of primary cells. Cell Death Differ. 2012, 19, 1390–1397. [Google Scholar] [CrossRef] [Green Version]
- Couto, J.P.; Daly, L.; Almeida, A.; Knauf, J.A.; Fagin, J.A.; Sobrinho-Simões, M.; Lima, J.; Máximo, V.; Soares, P.; Lyden, D.; et al. STAT3 negatively regulates thyroid tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 2361–2370. [Google Scholar] [CrossRef] [Green Version]
- Kritikou, E.A.; Sharkey, A.; Abell, K.; Came, P.J.; Anderson, E.; Clarkson, R.W.E.; Watson, C.J. A dual, non-redundant, role for LIF as a regulator of development and STAT3-mediated cell death in mammary gland. Development 2003, 130, 3459–3468. [Google Scholar] [CrossRef] [Green Version]
- Kreuzaler, P.A.; Staniszewska, A.D.; Li, W.; Omidvar, N.; Kedjouar, B.; Turkson, J.; Poli, V.; Flavell, R.A.; Clarkson, R.W.E.; Watson, C.J. Stat3 controls lysosomal-mediated cell death in vivo. Nat. Cell Biol. 2011, 13, 303–309. [Google Scholar] [CrossRef]
- Lloyd-Lewis, B.; Krueger, C.C.; Sargeant, T.J.; D’Angelo, M.E.; Deery, M.J.; Feret, R.; Howard, J.A.; Lilley, K.S.; Watson, C.J. Stat3-mediated alterations in lysosomal membrane protein composition. J. Biol. Chem. 2018, 293, 4244–4261. [Google Scholar] [CrossRef] [Green Version]
- Resemann, H.K.; Watson, C.J.; Lloyd-Lewis, B. The stat3 paradox: A killer and an oncogene. Mol. Cell. Endocrinol. 2014, 382, 603–611. [Google Scholar] [CrossRef]
- Wake, M.S.; Watson, C.J. STAT3 the oncogene—Still eluding therapy? FEBS J. 2015, 282, 2600–2611. [Google Scholar] [CrossRef] [Green Version]
- Watson, C.J.; Miller, W.R. Elevated levels of members of the STAT family of transcription factors in breast carcinoma nuclear extracts. Br. J. Cancer 1995, 71, 840–844. [Google Scholar] [CrossRef]
- Alvarez, J.V.; Febbo, P.G.; Ramaswamy, S.; Loda, M.; Richardson, A.; Frank, D.A. Identification of a genetic signature of activated signal transducer and activator of transcription 3 in human tumors. Cancer Res. 2005, 65, 5054–5062. [Google Scholar] [CrossRef] [Green Version]
- Dolled-Filhart, M.; Camp, R.L.; Kowalski, D.P.; Smith, B.L.; Rimm, D.L. Tissue microarray analysis of signal transducers and activators of transcription 3 (Stat3) and phospho-Stat3 (Tyr705) in node-negative breast cancer shows nuclear localization is associated with a better prognosis. Clin. Cancer Res. 2003, 9, 594–600. [Google Scholar]
- Yates, L.R.; Knappskog, S.; Wedge, D.; Farmery, J.H.R.; Gonzalez, S.; Martincorena, I.; Alexandrov, L.B.; Van Loo, P.; Haugland, H.K.; Lilleng, P.K.; et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 2017, 32, 169–184. [Google Scholar] [CrossRef] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Johnson, R.W.; Finger, E.C.; Olcina, M.M.; Vilalta, M.; Aguilera, T.; Miao, Y.; Merkel, A.R.; Johnson, J.R.; Sterling, J.A.; Wu, J.Y.; et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat. Cell Biol. 2016, 18, 1078–1089. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.S.; Avivar-Valderas, A.; Estrada, Y.; Bragado, P.; Sosa, M.S.; Aguirre-Ghiso, J.A.; Segall, J.E. Dormancy signatures and metastasis in estrogen receptor positive and negative breast cancer. PLoS ONE 2012, 7, e35569. [Google Scholar] [CrossRef]
- Pectasides, E.; Egloff, A.M.; Sasaki, C.; Kountourakis, P.; Burtness, B.; Fountzilas, G.; Dafni, U.; Zaramboukas, T.; Rampias, T.; Rimm, D.; et al. Nuclear localization of signal transducer and activator of transcription 3 in head and neck squamous cell carcinoma is associated with a better prognosis. Clin. Cancer Res. 2010, 16, 2427–2434. [Google Scholar] [CrossRef] [Green Version]
- Grandis, J.R.; Drenning, S.D.; Zeng, Q.; Watkins, S.C.; Melhem, M.F.; Endo, S.; Johnson, D.E.; Huang, L.; He, Y.; Kim, J.D. Constitutive activation of Stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 4227–4232. [Google Scholar] [CrossRef] [Green Version]
- Grandis, J.R.; Drenning, S.D.; Chakraborty, A.; Zhou, M.Y.; Zeng, Q.; Pitt, A.S.; Tweardy, D.J. Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor-mediated cell growth in vitro. J. Clin. Investig. 1998, 102, 1385–1392. [Google Scholar] [CrossRef] [Green Version]
- Shao, H.; Quintero, A.J.; Tweardy, D.J. Identification and characterization of cis elements in the STAT3 gene regulating STAT3α and STAT3β messenger RNA splicing. Blood 2001, 98, 3853–3856. [Google Scholar] [CrossRef]
- Tang, J.Z.; Kong, X.J.; Banerjee, A.; Muniraj, N.; Pandey, V.; Steiner, M.; Perry, J.K.; Zhu, T.; Liu, D.X.; Lobie, P.E. STAT3α is oncogenic for endometrial carcinoma cells and mediates the oncogenic effects of autocrine human growth hormone. Endocrinology 2010, 151, 4133–4145. [Google Scholar] [CrossRef] [Green Version]
- Dewilde, S.; Vercelli, A.; Chiarle, R.; Poli, V. Of alphas and betas: Distinct and overlapping functions of STAT3 isoforms. Front. Biosci. 2008, 13, 6501–6514. [Google Scholar] [CrossRef] [Green Version]
- Karni, R.; Jove, R.; Levitzki, A. Inhibition of pp60(c-Src) reduces Bcl-X(L) expression and reverses the transformed phenotype of cells overexpressing EGF and HER-2 receptors. Oncogene 1999, 18, 4654–4662. [Google Scholar] [CrossRef] [Green Version]
- Sinibaldi, D.; Wharton, W.; Turkson, J.; Bowman, T.; Pledger, W.J.; Jove, R. Induction of p21(WAF1/CIP1) and cyclin D1 expression by the Src oncoprotein in mouse fibroblasts: Role of activated STAT3 signaling. Oncogene 2000, 19, 5419–5427. [Google Scholar] [CrossRef] [Green Version]
- Turkson, J.; Bowman, T.; Garcia, R.; Caldenhoven, E.; De Groot, R.P.; Jove, R. Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol. Cell. Biol. 1998, 18, 2545–2552. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, V.N.; Zhou, H.; Partridge, M.A.; Hei, T.K. Inhibition of ataxia telangiectasia mutated kinase activity enhances TRAIL-mediated apoptosis in human melanoma cells. Cancer Res. 2009, 69, 3510–3519. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, V.N.; Krasilnikov, M.; Ronai, Z. Regulation of Fas expression by STAT3 and c-Jun is mediated by phosphatidylinositol 3-kinase-AKT signaling. J. Biol. Chem. 2002, 277, 4932–4944. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.S.; Pal, A.D.; Banerjee, S. Epstein-Barr virus-encoded small non-coding RNAs induce cancer cell chemoresistance and migration. Virology 2013, 443, 294–305. [Google Scholar] [CrossRef] [Green Version]
- Benekli, M.; Baer, M.R.; Baumann, H.; Wetzler, M. Signal transducer and activator of transcription proteins in leukemias. Blood 2003, 101, 2940–2954. [Google Scholar] [CrossRef] [Green Version]
- Niu, G.; Shain, K.H.; Huang, M.; Ravi, R.; Bedi, A.; Dalton, W.S.; Jove, R.; Yu, H. Overexpression of a dominant-negative signal transducer and activator of transcription 3 variant in tumor cells leads to production of soluble factors that induce apoptosis and cell cycle arrest. Cancer Res. 2001, 61, 3276–3280. [Google Scholar]
- Niu, G.; Heller, R.; Catlett-Falcone, R.; Coppola, D.; Jaroszeski, M.; Dalton, W.; Jove, R.; Yu, H. Gene therapy with dominant-negative Stat3 suppresses growth of the murine melanoma B16 tumor in vivo. Cancer Res. 1999, 59, 5059–5063. [Google Scholar]
- Burke, W.M.; Jin, X.; Lin, H.J.; Huang, M.; Liu, R.; Reynolds, R.K.; Lin, J. Inhibition of constitutively active Stat3 suppresses growth of human ovarian and breast cancer cells. Oncogene 2001, 20, 7925–7934. [Google Scholar] [CrossRef] [Green Version]
- Christine, R.; Sylvie, R.; Erik, B.; Geneviève, P.; Amélie, R.; Gérard, R.; Marc, B.; Christian, G.; Samir, A. Implication of STAT3 signaling in human colonic cancer cells during intestinal trefoil factor 3 (TFF3)—And vascular endothelial growth factor-mediated cellular invasion and tumor growth. Cancer Res. 2005, 65, 195–202. [Google Scholar]
- Zhang, H.F.; Chen, Y.; Wu, C.; Wu, Z.Y.; Tweardy, D.J.; Alshareef, A.; Liao, L.D.; Xue, Y.J.; Wu, J.Y.; Chen, B.; et al. The opposing function of STAT3 as an oncoprotein and tumor suppressor is dictated by the expression status of STAT3β in esophageal squamous cell carcinoma. Clin. Cancer Res. 2016, 22, 691–703. [Google Scholar] [CrossRef] [Green Version]
- Haura, E.B.; Turkson, J.; Jove, R. Mechanisms of disease: Insights into the emerging role of signal transducers and activators of transcription in cancer. Nat. Clin. Pract. Oncol. 2005, 2, 315–324. [Google Scholar] [CrossRef]
- Debnath, B.; Xu, S.; Neamati, N. Small molecule inhibitors of signal transducer and activator of transcription 3 (stat3) protein. J. Med. Chem. 2012, 55, 6645–6668. [Google Scholar] [CrossRef]
- Deng, J.; Grande, F.; Neamati, N. Small molecule inhibitors of Stat3 signaling pathway. Curr. Cancer Drug Targets 2007, 7, 91–107. [Google Scholar] [CrossRef]
- Siddiquee, K.A.Z.; Gunning, P.T.; Glen, M.; Katt, W.P.; Zhang, S.; Schroeck, C.; Sebti, S.M.; Jove, R.; Hamilton, A.D.; Turkson, J. An oxazole-based small-molecule stat3 inhibitor modulates stat3 stability and processing and induces antitumor cell effects. ACS Chem. Biol. 2007, 2, 787–798. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, Y.; Pireddu, R.; Yang, H.; Urlam, M.K.; Lawrence, H.R.; Guida, W.C.; Lawrence, N.J.; Sebti, S.M. A novel inhibitor of STAT3 homodimerization selectively suppresses STAT3 activity and malignant transformation. Cancer Res. 2013, 73, 1922–1933. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, S.; Page, B.D.G.; Zhang, X.; Yue, P.; Li, Z.H.; Sharmeen, S.; Singh, J.; Zhao, W.; Schimmer, A.D.; Trudel, S.; et al. Antagonism of the Stat3-Stat3 protein dimer with salicylic acid based small molecules. ChemMedChem 2011, 6, 1459–1470. [Google Scholar] [CrossRef] [Green Version]
- Turkson, J.; Ryan, D.; Kim, J.S.; Zhang, Y.; Chen, Z.; Haura, E.; Laudano, A.; Sebti, S.; Hamilton, A.D.; Jove, R. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J. Biol. Chem. 2001, 276, 45443–45455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Z.; Cabell, L.A.; Schaefer, T.S.; McMurray, J.S. Identification of a high-affinity phosphopeptide inhibitor of Stat3. Bioorgan. Med. Chem. Lett. 2003, 13, 633–636. [Google Scholar] [CrossRef]
- Chen, J.; Nikolovska-Coleska, Z.; Yang, C.Y.; Gomez, C.; Gao, W.; Krajewski, K.; Jiang, S.; Roller, P.; Wang, S. Design and synthesis of a new, conformationally constrained, macrocyclic small-molecule inhibitor of STAT3 via “click chemistry. ” Bioorgan. Med. Chem. Lett. 2007, 17, 3939–3942. [Google Scholar] [CrossRef]
- Song, H.; Wang, R.; Wang, S.; Lin, J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 4700–4705. [Google Scholar] [CrossRef] [Green Version]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: A small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, F.; Sugimoto, K.; Harada, Y.; Hashimoto, N.; Ohi, N.; Kurahashi, S.; Naoe, T. A novel STAT inhibitor, OPB-31121, has a significant antitumor effect on leukemia with STAT-addictive oncokinases. Blood Cancer J. 2013, 3, e166. [Google Scholar] [CrossRef]
- Kim, M.J.; Nam, H.J.; Kim, H.P.; Han, S.W.; Im, S.A.; Kim, T.Y.; Oh, D.Y.; Bang, Y.J. OPB-31121, a novel small molecular inhibitor, disrupts the JAK2/STAT3 pathway and exhibits an antitumor activity in gastric cancer cells. Cancer Lett. 2013, 335, 145–152. [Google Scholar] [CrossRef]
- Okusaka, T.; Ueno, H.; Ikeda, M.; Mitsunaga, S.; Ozaka, M.; Ishii, H.; Yokosuka, O.; Ooka, Y.; Yoshimoto, R.; Yanagihara, Y.; et al. Phase 1 and pharmacological trial of OPB-31121, a signal transducer and activator of transcription-3 inhibitor, in patients with advanced hepatocellular carcinoma. Hepatol. Res. 2015, 45, 1283–1291. [Google Scholar] [CrossRef]
- Alexandrow, M.G.; Song, L.J.; Altiok, S.; Gray, J.; Haura, E.B.; Kumar, N.B. Curcumin: A novel Stat3 pathway inhibitor for chemoprevention of lung cancer. Eur. J. Cancer Prev. 2012, 21, 407–412. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, X.; Zeng, S.; Zhang, X.; Zhao, J.; Zhang, X.; Chen, X.; Yang, W.; Yang, Y.; Dong, Z.; et al. The natural polyphenol curcumin induces apoptosis by suppressing STAT3 signaling in esophageal squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 303. [Google Scholar] [CrossRef]
- Kumar, A.; Bora, U. Molecular docking studies on inhibition of Stat3 dimerization by curcumin natural derivatives and its conjugates with amino acids. Bioinformation 2012, 8, 988–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Wang, L.; Li, F.; Hu, R.; Ma, J.; Zhang, K.; Cheng, X. Resveratrol downregulates STAT3 expression and astrocyte activation in primary astrocyte cultures of rat. Neurochem. Res. 2020, 45, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Kim, H.N.; Shin, K.D.; Yoon, Y.J.; Kim, S.J.; Han, D.C.; Kwon, B.M. Cryptotanshinone inhibits constitutive signal transducer and activator of transcription 3 function through blocking the dimerization in DU145 prostate cancer cells. Cancer Res. 2009, 69, 193–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Lu, B.; Zhang, X.; Zhang, J.; Lai, L.; Li, D.; Wu, Y.; Song, Y.; Luo, J.; Pang, X.; et al. Cucurbitacin E, a tetracyclic triterpenes compound from Chinese medicine, inhibits tumor angiogenesis through VEGFR2-mediated Jak2-STAT3 signaling pathway. Carcinogenesis 2010, 31, 2097–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Dong, Z.; Chen, Y.; Wang, F.; Wang, C.J.; Peng, H.; He, Y.; Hangoc, G.; Pollok, K.; Sandusky, G.; et al. Small-molecule inhibitors targeting the DNA-binding domain of STAT3 suppress tumor growth, metastasis and STAT3 target gene expression in vivo. Oncogene 2016, 35, 783–792. [Google Scholar] [CrossRef]
- Bharadwaj, U.; Eckols, T.K.; Kolosov, M.; Kasembeli, M.M.; Adam, A.; Torres, D.; Zhang, X.; Dobrolecki, L.E.; Wei, W.; Lewis, M.T.; et al. Drug-repositioning screening identified piperlongumine as a direct STAT3 inhibitor with potent activity against breast cancer. Oncogene 2015, 34, 1341–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosch-Barrera, J.; Menendez, J.A. Silibinin and STAT3: A natural way of targeting transcription factors for cancer therapy. Cancer Treat. Rev. 2015, 41, 540–546. [Google Scholar] [CrossRef]
- Buettner, R.; Corzano, R.; Rashid, R.; Lin, J.; Senthil, M.; Hedvat, M.; Schroeder, A.; Mao, A.; Herrmann, A.; Yim, J.; et al. Alkylation of cysteine 468 in stat3 defines a novel site for therapeutic development. Proc. ACS Chem. Biol. 2011, 6, 432–443. [Google Scholar] [CrossRef]
- Huang, W.; Dong, Z.; Wang, F.; Peng, H.; Liu, J.Y.; Zhang, J.T. A small molecule compound targeting STAT3 DNA-binding domain inhibits cancer cell proliferation, migration, and invasion. ACS Chem. Biol. 2014, 9, 1188–1196. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.M.; Wang, C.M.; Jeng, J.C.; Leprince, D.; Shih, H.M. HIC1 interacts with and modulates the activity of STAT3. Cell Cycle 2013, 12, 2266–2276. [Google Scholar] [CrossRef] [Green Version]
- Nagel-Wolfrum, K.; Buerger, C.; Wittig, I.; Butz, K.; Hoppe-Seyler, F.; Groner, B. The interaction of specific peptide aptamers with the DNA binding domain and the dimerization domain of the transcription factor Stat3 inhibits transactivation and induces apoptosis in tumor cells. Mol. Cancer Res. 2004, 2, 170–182. [Google Scholar] [PubMed]
- Turkson, J.; Zhang, S.; Palmer, J.; Kay, H.; Stanko, J.; Mora, L.B.; Sebti, S.; Yu, H.; Jove, R. Inhibition of constitutive signal transducer and activator of transcription 3 activation by novel platinum complexes with potent antitumor activity. Mol. Cancer Ther. 2004, 3, 1533–1542. [Google Scholar] [PubMed]
- Turkson, J.; Zhang, S.; Mora, L.B.; Burns, A.; Sebti, S.; Jove, R. A novel platinum compound inhibits constitutive Stat3 signaling and induces cell cycle arrest and apoptosis of malignant cells. J. Biol. Chem. 2005, 280, 32979–32988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timofeeva, O.A.; Tarasova, N.I.; Zhang, X.; Chasovskikh, S.; Cheema, A.K.; Wang, H.; Brown, M.L.; Dritschilo, A. STAT3 suppresses transcription of proapoptotic genes in cancer cells with the involvement of its N-terminal domain. Proc. Natl. Acad. Sci. USA 2013, 110, 1267–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Sala, G.; Michiels, C.; Kükenshöner, T.; Brandstoetter, T.; Maurer, B.; Koide, A.; Lau, K.; Pojer, F.; Koide, S.; Sexl, V.; et al. Selective inhibition of STAT3 signaling using monobodies targeting the coiled-coil and N-terminal domains. Nat. Commun. 2020, 28, 253–262. [Google Scholar] [CrossRef]
- Buerger, C.; Nagel-Wolfrum, K.; Kunz, C.; Wittig, I.; Butz, K.; Hoppe-Seyler, F.; Groner, B. Sequence-specific peptide aptamers, interacting with the intracellular domain of the epidermal growth factor receptor, interfere with Stat3 activation and inhibit the growth of tumor cells. J. Biol. Chem. 2003, 278, 37610–37621. [Google Scholar] [CrossRef] [Green Version]
- Ge, H.; Liu, H.; Fu, Z.; Sun, Z. Therapeutic and preventive effects of an epidermal growth factor receptor inhibitor on oral squamous cell carcinoma. J. Int. Med. Res. 2012, 40, 455–466. [Google Scholar] [CrossRef] [Green Version]
- Aittomäki, S.; Pesu, M. Therapeutic targeting of the JAK/STAT pathway. Basic Clin. Pharmacol. Toxicol. 2014, 114, 18–23. [Google Scholar] [CrossRef]
- Buchert, M.; Burns, C.J.; Ernst, M. Targeting JAK kinase in solid tumors: Emerging opportunities and challenges. Oncogene 2016, 35, 939. [Google Scholar] [CrossRef]
- Plimack, E.R.; LoRusso, P.M.; McCoon, P.; Tang, W.; Krebs, A.D.; Curt, G.; Eckhardt, S.G. AZD1480: A Phase I study of a novel JAK2 inhibitor in solid tumors. Oncologist 2013, 18, 819–820. [Google Scholar] [CrossRef] [Green Version]
- Furtek, S.L.; Backos, D.S.; Matheson, C.J.; Reigan, P. Strategies and approaches of targeting STAT3 for cancer treatment. ACS Chem. Biol. 2016, 11, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Puls, L.N.; Eadens, M.; Messersmith, W. Current status of Src inhibitors in solid tumor malignancies. Oncologist 2011, 16, 566–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, S.; Wen, W.; Schroeder, A.; Herrmann, A.; Yu, H.; Cheng, X.; Merz, K.H.; Eisenbrand, G.; Li, H.; Yuan, Y.C.; et al. Dual inhibition of Janus and Src family kinases by novel indirubin derivative blocks constitutively-activated Stat3 signaling associated with apoptosis of human pancreatic cancer cells. Mol. Oncol. 2013, 7, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Gaboriaud, N.; Vougogianopoulou, K.; Tian, Y.; Wu, J.; Wen, W.; Skaltsounis, L.; Jove, R. MLS-2384, a new 6-bromoindirubin derivative with dual JAK/Src kinase inhibitory activity, suppresses growth of diverse cancer cells. Cancer Biol. Ther. 2014, 15, 178–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, T.; Takigawa, N.; Ninomiya, T.; Ochi, N.; Yasugi, M.; Honda, Y.; Kubo, T.; Ichihara, E.; Hotta, K.; Tanimoto, M.; et al. Effect of AZD1480 in an epidermal growth factor receptor-driven lung cancer model. Lung Cancer 2014, 83, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Lau, Y.T.K.; Ramaiyer, M.; Johnson, D.E.; Grandis, J.R. Targeting STAT3 in cancer with nucleotide therapeutics. Cancers 2019, 11, 1681. [Google Scholar] [CrossRef] [Green Version]
- Guha, P.; Gardell, J.; Darpolor, J.; Cunetta, M.; Lima, M.; Miller, G.; Espat, N.J.; Junghans, R.P.; Katz, S.C. STAT3 inhibition induces Bax-dependent apoptosis in liver tumor myeloid-derived suppressor cells. Oncogene 2019, 38, 533–548. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Dong, Y.; Chen, Z.; Eckols, T.K.; Kasembeli, M.M.; Tweardy, D.J.; Mitch, W.E. Pharmacokinetics and pharmacodynamics of TTI-101, a STAT3 inhibitor that blocks muscle proteolysis in rats with chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2020, 319, F84–F92. [Google Scholar] [CrossRef]
- Verstovsek, S.; Manshouri, T.; Quintás-Cardama, A.; Harris, D.; Cortes, J.; Giles, F.J.; Kantarjian, H.; Priebe, W.; Estrov, Z. WP1066, a novel JAK2 inhibitor, suppresses proliferation and induces apoptosis in erythroid human cells carrying the JAK2 V617F mutation. Clin. Cancer Res. 2008, 14, 788–796. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Tumor | Tumor Background | Tumor Suppressor Activity | Tumor Promoter Activity |
|---|---|---|---|
| Glioma | Normal PTEN expression | LIFRβ activation of STAT3 that inhibits IL-8 expression. | |
| EGFRvIII expression | Formation of oncogenic EGFRvIII-STAT3 complex. | ||
| Prostate cancer | STAT3 activation of ARF-p53 pathway. | ||
| Lung Adenocarcinomas | KRAS mutations | STAT3 dependent sequestration of NF-kB in the cytoplasm and inhibition of IL-8 expression. | |
| EGFR mutations | STAT3 tumor promoter activity. | ||
| Colorectal cancer | STAT3 mediated degradation of Snail-1 and inhibition of epithelial-mesenchymal transition. | STAT3 mutations associated with APC mutations. | |
| STAT3β expression. | |||
| Thyroid cancer | STAT3 dependent suppression of HIF1α and aerobic glycolysis under hypoxic stress. | ||
| STAT3 dependent activation of IGFBP7. | |||
| Breast cancer | Primary tumor | STAT3 tumor promoter activity. | |
| Metastasis | STAT3 activation of dormancy-associated genes under hypoxic condition. | ||
| Esophageal squamous cell carcinoma | STAT3α expression. | ||
| Esophageal squamous cell carcinoma with longer overall survival | STAT3β expression. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolomeo, M.; Cascio, A. The Multifaced Role of STAT3 in Cancer and Its Implication for Anticancer Therapy. Int. J. Mol. Sci. 2021, 22, 603. https://doi.org/10.3390/ijms22020603
Tolomeo M, Cascio A. The Multifaced Role of STAT3 in Cancer and Its Implication for Anticancer Therapy. International Journal of Molecular Sciences. 2021; 22(2):603. https://doi.org/10.3390/ijms22020603
Chicago/Turabian StyleTolomeo, Manlio, and Antonio Cascio. 2021. "The Multifaced Role of STAT3 in Cancer and Its Implication for Anticancer Therapy" International Journal of Molecular Sciences 22, no. 2: 603. https://doi.org/10.3390/ijms22020603
APA StyleTolomeo, M., & Cascio, A. (2021). The Multifaced Role of STAT3 in Cancer and Its Implication for Anticancer Therapy. International Journal of Molecular Sciences, 22(2), 603. https://doi.org/10.3390/ijms22020603