The Bile Salt Export Pump: Molecular Structure, Study Models and Small-Molecule Drugs for the Treatment of Inherited BSEP Deficiencies

 ,
,  and
and
Abstract
:1. Introduction
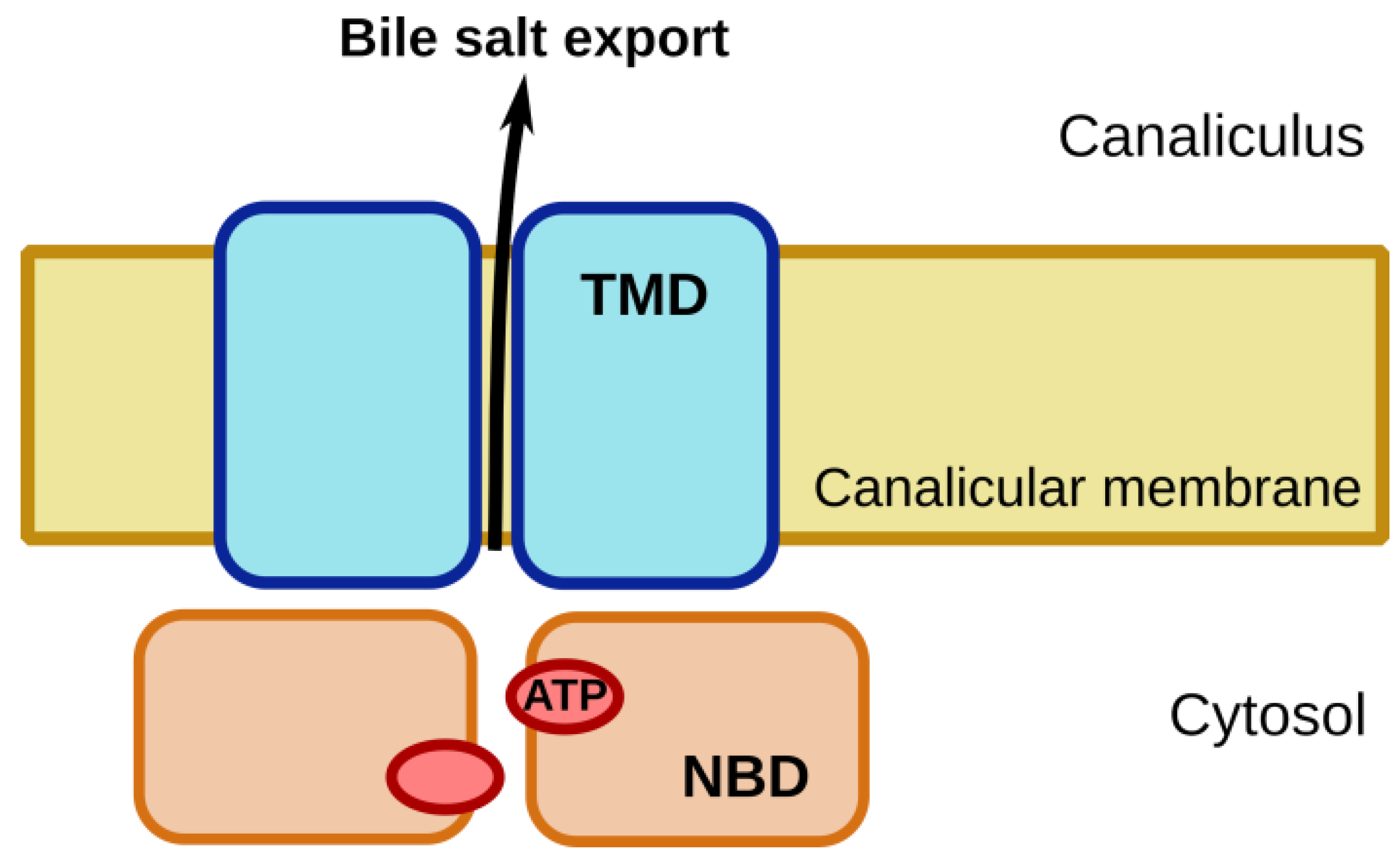
2. BSEP/ABCB11: Physiological Role
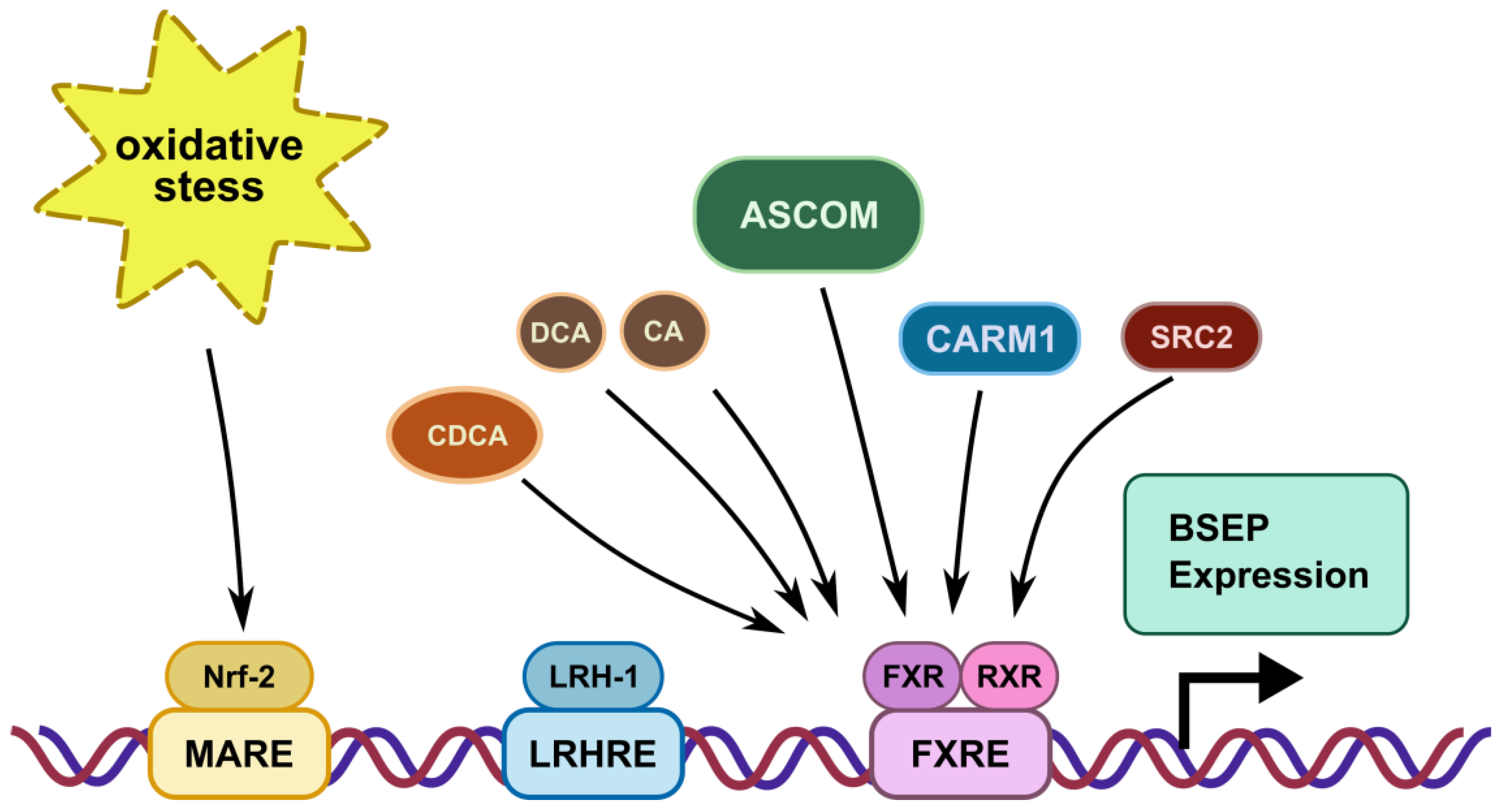
3. Transcriptional Regulation
4. Processing and Trafficking of BSEP
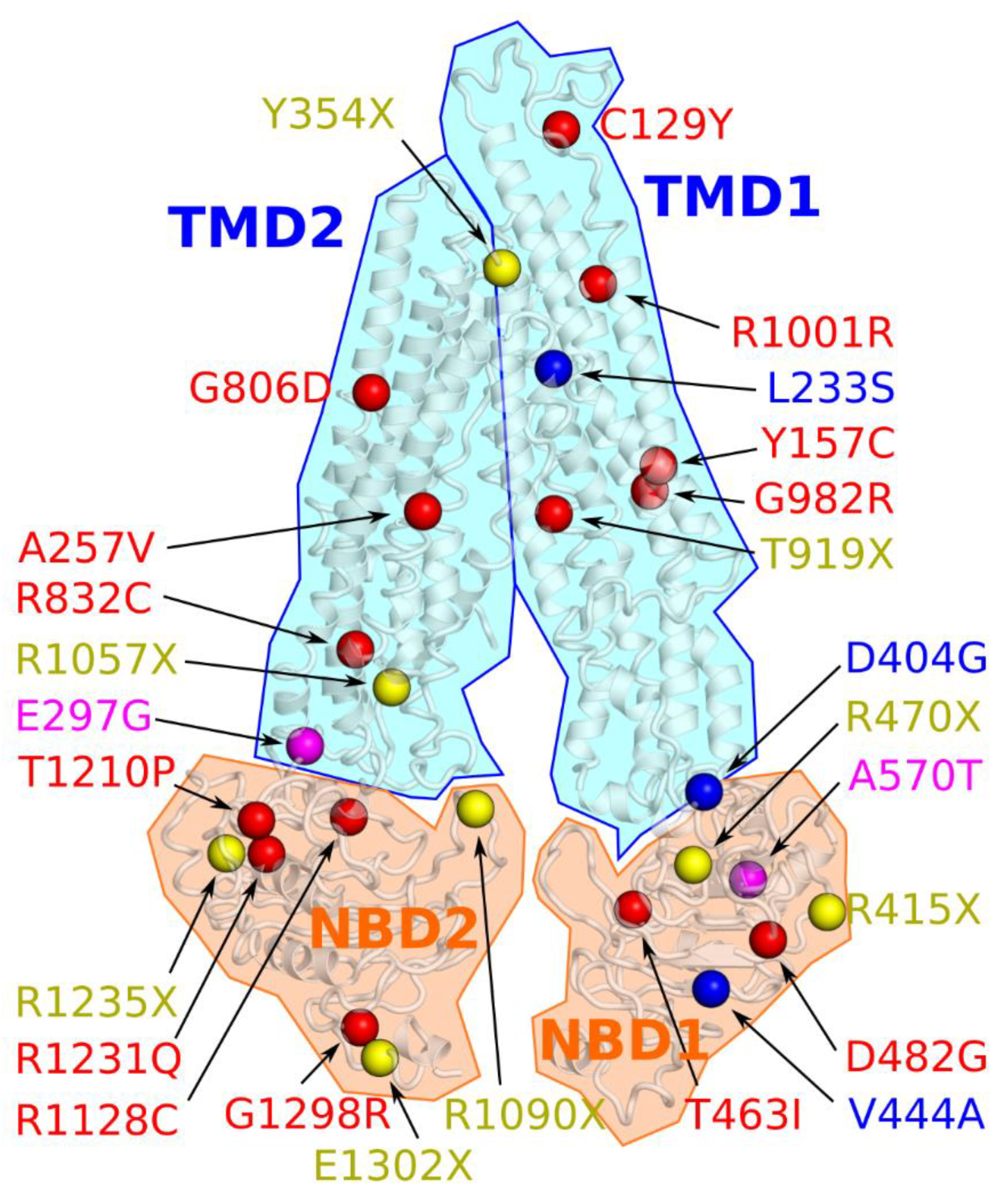
5. Structural Models of BSEP
6. Experimental Model Systems
6.1. In Vitro Models
6.1.1. Membrane Vesicles
6.1.2. Polarized Cell Lines Expressing BSEP
6.1.3. Primary Hepatocyte Cultures
6.2. BSEP Knockout Animals
6.2.1. Rodents
6.2.2. Zebrafish
7. Treatment Options for BSEP-Related Diseases
7.1. Transcriptional Modulators
7.2. Ursodeoxycholic Acid (UDCA)
7.3. Chemical Correction with 4-PB
7.4. Potentiation with Ivacaftor
7.5. Readthrough Therapy with Gentamicin
8. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| AMPK | AMP-activated protein kinase |
| AP-2 | adaptor protein 2 |
| ASCOM | activating signal cointegrator-2-containing complex |
| BSEP (ABCB11) | bile salt export pump |
| BRIC2 | benign recurrent intrahepatic cholestasis type 2 |
| CA | cholic acid |
| cAMP | cyclic adenosine monophosphate |
| CARM1 | co-activator-associated arginine methyltransferase 1 |
| CFTR (ABCC7) | cystic fibrosis transmembrane conductance regulator |
| CDCA | chenodeoxycholic acid |
| CM | canalicular membrane |
| cPKC | classical (Ca2+-dependent) protein kinase C |
| cryo-EM | cryo electron microscopy |
| DCA | deoxycholic acid |
| DILI | drug-induced liver injury |
| ECL | extracellular loop |
| Epac | exchange protein directly activated by cAMP |
| ICL | intracellular loop |
| ICP | intrahepatic cholestasis of pregnancy |
| ERAD | Endoplasmic-reticulum-associated degradation |
| E17G | estradiol 17 β-D-glucuronide |
| FXR (NR1H4) | farnesoid X receptor |
| FXRE | FXR response element |
| LKB1 | liver kinase B1 |
| LRH-1 (NR5A2) | liver receptor homolog-1 |
| MAPK | Mitogen-activated protein kinase |
| MRP3 (ABCC3) | Multidrug-resistance-associated protein 3 |
| MRP4 (ABCC4) | Multidrug-resistance-associated protein 4 |
| NASH | non-alcoholic steatohepatitis |
| NBD | nucleotide-binding domain |
| NBS | nucleotide-binding site |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| NTCP (SLC10A1) | sodium taurocholate co-transporting polypeptide |
| OCA | 6α-ethyl-CDCA (obeticholic acid) |
| 4-PB | 4-phenylbutyrate |
| PBC | primary biliary cholangitis |
| PFIC2 | progressive familial intrahepatic cholestasis type 2 |
| PI3K | phosphoinositide 3-kinase |
| PM | plasma membrane |
| RXR | retinoid X receptor |
| SRC2 | steroid receptor co-activator 2 |
| TCA | taurocholic acid |
| TMD | transmembrane domain |
| TUDCA | tauroursodeoxycholic acid |
| UDCA | ursodeoxycholic acid |
References
- Dean, M.; Rzhetsky, A.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001, 11, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, L. Structure and mechanism of ABC transporters. Curr. Opin. Struct. Biol. 2002, 12, 754–760. [Google Scholar] [CrossRef]
- Shintre, C.A.; Pike, A.C.W.; Li, Q.; Kim, J.-I.; Barr, A.J.; Goubin, S.; Shrestha, L.; Yang, J.; Berridge, G.; Ross, J.; et al. Structures of ABCB10, a human ATP-binding cassette transporter in apo- and nucleotide-bound states. Proc. Natl. Acad. Sci. USA 2013, 110, 9710–9715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Procko, E.; O’Mara, M.L.; Bennett, W.F.D.; Tieleman, D.P.; Gaudet, R. The mechanism of ABC transporters: General lessons from structural and functional studies of an antigenic peptide transporter. FASEB J. 2009, 23, 1287–1302. [Google Scholar] [CrossRef]
- Langmann, T.; Mauerer, R.; Zahn, A.; Moehle, C.; Probst, M.; Stremmel, W.; Schmitz, G. Real-Time Reverse Transcription-PCR Expression Profiling of the Complete Human ATP-Binding Cassette Transporter Superfamily in Various Tissues. Clin. Chem. 2003, 49, 230–238. [Google Scholar] [CrossRef]
- Stieger, B. The Role of the Sodium-Taurocholate Cotransporting Polypeptide (NTCP) and of the Bile Salt Export Pump (BSEP) in Physiology and Pathophysiology of Bile Formation. In Drug Transporters; Springer: Berlin/Heidelberg, Germany, 2011; pp. 205–259. [Google Scholar]
- Lagana, S.M.; Salomao, M.; Remotti, H.E.; Knisely, A.S.; Moreira, R.K. Bile salt export pump: A sensitive and specific immunohistochemical marker of hepatocellular carcinoma. Histopathology 2015, 66, 598–602. [Google Scholar] [CrossRef]
- Hilgendorf, C.; Ahlin, G.; Seithel, A.; Artursson, P.; Ungell, A.-L.; Karlsson, J. Expression of Thirty-six Drug Transporter Genes in Human Intestine, Liver, Kidney, and Organotypic Cell Lines. Drug Metab. Dispos. 2007, 35, 1333–1340. [Google Scholar] [CrossRef] [Green Version]
- Stieger, B.; Beuers, U. The Canalicular Bile Salt Export Pump BSEP (ABCB11) as a Potential Therapeutic Target. Curr. Drug Targets 2011, 12, 661–670. [Google Scholar] [CrossRef]
- Trauner, M.; Fuchs, C.D.; Halilbasic, E.; Paumgartner, G. New therapeutic concepts in bile acid transport and signaling for management of cholestasis. Hepatology 2017, 65, 1393–1404. [Google Scholar] [CrossRef] [Green Version]
- Trauner, M.; Boyer, J.L. Bile Salt Transporters: Molecular Characterization, Function, and Regulation. Physiol. Rev. 2003, 83, 633–671. [Google Scholar] [CrossRef] [Green Version]
- Lecureur, V.; Sun, D.; Hargrove, P.; Schuetz, E.G.; Kim, R.B.; Lan, L.B.; Schuetz, J.D. Cloning and expression of murine sister of P-glycoprotein reveals a more discriminating transporter than MDR1/P-glycoprotein. Mol. Pharmacol. 2000, 57, 24–35. [Google Scholar] [PubMed]
- Hirano, M.; Maeda, K.; Hayashi, H.; Kusuhara, H.; Sugiyama, Y. Bile Salt Export Pump (BSEP/ABCB11) Can Transport a Nonbile Acid Substrate, Pravastatin. J. Pharmacol. Exp. Ther. 2005, 314, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Maeda, K.; Hayashi, H.; Debori, Y.; Schinkel, A.H.; Schuetz, J.D.; Kusuhara, H.; Sugiyama, Y. Involvement of Multiple Efflux Transporters in Hepatic Disposition of Fexofenadine. Mol. Pharmacol. 2008, 73, 1474–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, R.H.; Leake, B.F.; Kilkenny, D.M.; Meyer zu Schwabedissen, H.E.; Glaeser, H.; Kroetz, D.L.; Kim, R.B. Polymorphic variants in the human bile salt export pump (BSEP; ABCB11): Functional characterization and interindividual variability. Pharmacogenet. Genom. 2010, 20, 45–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makishima, M. Identification of a Nuclear Receptor for Bile Acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous Bile Acids Are Ligands for the Nuclear Receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. [Google Scholar] [CrossRef]
- Parks, D.J. Bile Acids: Natural Ligands for an Orphan Nuclear Receptor. Science 1999, 284, 1365–1368. [Google Scholar] [CrossRef]
- Baghdasaryan, A.; Chiba, P.; Trauner, M. Clinical application of transcriptional activators of bile salt transporters. Mol. Aspects Med. 2014, 37, 57–76. [Google Scholar] [CrossRef]
- Plass, J.R.M.; Mol, O.; Heegsma, J.; Geuken, M.; Faber, K.N.; Jansen, P.L.M.; Müller, M. Farnesoid X receptor and bile salts are involved in transcriptional regulation of the gene encoding the human bile salt export pump. Hepatology 2002, 35, 589–596. [Google Scholar] [CrossRef]
- Ananthanarayanan, M.; Li, Y.; Surapureddi, S.; Balasubramaniyan, N.; Ahn, J.; Goldstein, J.A.; Suchy, F.J. Histone H3K4 trimethylation by MLL3 as part of ASCOM complex is critical for NR activation of bile acid transporter genes and is downregulated in cholestasis. Am. J. Physiol. Liver Physiol. 2011, 300, G771–G781. [Google Scholar] [CrossRef] [Green Version]
- Ananthanarayanan, M.; Li, S.; Balasubramaniyan, N.; Suchy, F.J.; Walsh, M.J. Ligand-dependent Activation of the Farnesoid X-receptor Directs Arginine Methylation of Histone H3 by CARM1. J. Biol. Chem. 2004, 279, 54348–54357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chopra, A.R.; Kommagani, R.; Saha, P.; Louet, J.-F.; Salazar, C.; Song, J.; Jeong, J.; Finegold, M.; Viollet, B.; DeMayo, F.; et al. Cellular Energy Depletion Resets Whole-Body Energy by Promoting Coactivator-Mediated Dietary Fuel Absorption. Cell Metab. 2011, 13, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Kaimal, R.; Yan, B.; Deng, R. Liver receptor homolog 1 transcriptionally regulates human bile salt export pump expression. J. Lipid Res. 2008, 49, 973–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mataki, C.; Magnier, B.C.; Houten, S.M.; Annicotte, J.-S.; Argmann, C.; Thomas, C.; Overmars, H.; Kulik, W.; Metzger, D.; Auwerx, J.; et al. Compromised Intestinal Lipid Absorption in Mice with a Liver-Specific Deficiency of Liver Receptor Homolog 1. Mol. Cell. Biol. 2007, 27, 8330–8339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weerachayaphorn, J.; Cai, S.-Y.; Soroka, C.J.; Boyer, J.L. Nuclear factor erythroid 2-related factor 2 is a positive regulator of human bile salt export pump expression. Hepatology 2009, 50, 1588–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Dong, H.; Soroka, C.J.; Wei, N.; Boyer, J.L.; Hochstrasser, M. Degradation of the bile salt export pump at endoplasmic reticulum in progressive familial intrahepatic cholestasis type II. Hepatology 2008, 48, 1558–1569. [Google Scholar] [CrossRef] [Green Version]
- Kleizen, B.; van Vlijmen, T.; de Jonge, H.R.; Braakman, I. Folding of CFTR Is Predominantly Cotranslational. Mol. Cell 2005, 20, 277–287. [Google Scholar] [CrossRef]
- Rudashevskaya, E.L.; Stockner, T.; Trauner, M.; Freissmuth, M.; Chiba, P. Pharmacological correction of misfolding of ABC proteins. Drug Discov. Today Technol. 2014, 12, e87–e94. [Google Scholar] [CrossRef] [Green Version]
- Chiba, P.; Freissmuth, M.; Stockner, T. Defining the blanks—Pharmacochaperoning of SLC6 transporters and ABC transporters. Pharmacol. Res. 2014, 83, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Guna, A.; Hegde, R.S. Transmembrane Domain Recognition during Membrane Protein Biogenesis and Quality Control. Curr. Biol. 2018, 28, R498–R511. [Google Scholar] [CrossRef] [Green Version]
- Printsev, I.; Curiel, D.; Carraway, K.L. Membrane Protein Quantity Control at the Endoplasmic Reticulum. J. Membr. Biol. 2017, 250, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Needham, P.G.; Guerriero, C.J.; Brodsky, J.L. Chaperoning Endoplasmic Reticulum–Associated Degradation (ERAD) and Protein Conformational Diseases. Cold Spring Harb. Perspect. Biol. 2019, 11, a033928. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.D.; Novak, P.; Lake, A.D.; Hardwick, R.N.; Cherrington, N.J. Impaired N-linked glycosylation of uptake and efflux transporters in human non-alcoholic fatty liver disease. Liver Int. 2017, 37, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, K.; Kagawa, T.; Numari, A.; Harris, M.J.; Itoh, J.; Watanabe, N.; Mine, T.; Arias, I.M. Two N -linked glycans are required to maintain the transport activity of the bile salt export pump (ABCB11) in MDCK II cells. Am. J. Physiol. Liver Physiol. 2007, 292, G818–G828. [Google Scholar] [CrossRef] [Green Version]
- Plass, J.R.; Mol, O.; Heegsma, J.; Geuken, M.; de Bruin, J.; Elling, G.; Müller, M.; Faber, K.N.; Jansen, P.L. A progressive familial intrahepatic cholestasis type 2 mutation causes an unstable, temperature-sensitive bile salt export pump. J. Hepatol. 2004, 40, 24–30. [Google Scholar] [CrossRef]
- Kipp, H.; Pichetshote, N.; Arias, I.M. Transporters on Demand. J. Biol. Chem. 2001, 276, 7218–7224. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, Y.; Lippincott-Schwartz, J.; Arias, I.M. Intracellular Trafficking of Bile Salt Export Pump (ABCB11) in Polarized Hepatic Cells: Constitutive Cycling between the Canalicular Membrane and rab11-positive Endosomes. Mol. Biol. Cell 2004, 15, 3485–3496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, P.; Soroka, C.; Boyer, J. The Bile Salt Export Pump: Clinical and Experimental Aspects of Genetic and Acquired Cholestatic Liver Disease. Semin. Liver Dis. 2010, 30, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Lam, P.; Xu, S.; Soroka, C.J.; Boyer, J.L. A C-terminal tyrosine-based motif in the bile salt export pump directs clathrin-dependent endocytosis. Hepatology 2012, 55, 1901–1911. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, H.; Inamura, K.; Aida, K.; Naoi, S.; Horikawa, R.; Nagasaka, H.; Takatani, T.; Fukushima, T.; Hattori, A.; Yabuki, T.; et al. AP2 adaptor complex mediates bile salt export pump internalization and modulates its hepatocanalicular expression and transport function. Hepatology 2012, 55, 1889–1900. [Google Scholar] [CrossRef]
- Chan, W.; Calderon, G.; Swift, A.L.; Moseley, J.; Li, S.; Hosoya, H.; Arias, I.M.; Ortiz, D.F. Myosin II Regulatory Light Chain Is Required for Trafficking of Bile Salt Export Protein to the Apical Membrane in Madin-Darby Canine Kidney Cells. J. Biol. Chem. 2005, 280, 23741–23747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, Y.; Dutt, P.; Lippincott-Schwartz, J.; Arias, I.M. Rab11a and myosin Vb are required for bile canalicular formation in WIF-B9 cells. Proc. Natl. Acad. Sci. USA 2005, 102, 15087–15092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crocenzi, F.A. Localization status of hepatocellular transporters in cholestasis. Front. Biosci. 2012, 17, 1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crocenzi, F.A.; Mottino, A.D.; Cao, J.; Veggi, L.M.; Pozzi, E.J.S.; Vore, M.; Coleman, R.; Roma, M.G. Estradiol-17β-D-glucuronide induces endocytic internalization of Bsep in rats. Am. J. Physiol. Liver Physiol. 2003, 285, G449–G459. [Google Scholar] [CrossRef]
- Miszczuk, G.S.; Barosso, I.R.; Larocca, M.C.; Marrone, J.; Marinelli, R.A.; Boaglio, A.C.; Sánchez Pozzi, E.J.; Roma, M.G.; Crocenzi, F.A. Mechanisms of canalicular transporter endocytosis in the cholestatic rat liver. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1072–1085. [Google Scholar] [CrossRef]
- Crocenzi, F.A.; Sánchez Pozzi, E.J.; Ruiz, M.L.; Zucchetti, A.E.; Roma, M.G.; Mottino, A.D.; Vore, M. Ca2+-dependent protein kinase C isoforms are critical to estradiol 17β-D-glucuronide-induced cholestasis in the rat. Hepatology 2008, 48, 1885–1895. [Google Scholar] [CrossRef] [Green Version]
- Boaglio, A.C.; Zucchetti, A.E.; Sánchez Pozzi, E.J.; Pellegrino, J.M.; Ochoa, J.E.; Mottino, A.D.; Vore, M.; Crocenzi, F.A.; Roma, M.G. Phosphoinositide 3-kinase/protein kinase B signaling pathway is involved in estradiol 17β-d-glucuronide-induced cholestasis: Complementarity with classical protein kinase c. Hepatology 2010, 52, 1465–1476. [Google Scholar] [CrossRef]
- Boaglio, A.C.; Zucchetti, A.E.; Toledo, F.D.; Barosso, I.R.; Sánchez Pozzi, E.J.; Crocenzi, F.A.; Roma, M.G. ERK1/2 and p38 MAPKs Are Complementarily Involved in Estradiol 17ß-d-Glucuronide-Induced Cholestasis: Crosstalk with cPKC and PI3K. PLoS ONE 2012, 7, e49255. [Google Scholar] [CrossRef] [Green Version]
- Fu, D.; Wakabayashi, Y.; Lippincott-Schwartz, J.; Arias, I.M. Bile acid stimulates hepatocyte polarization through a cAMP-Epac-MEK-LKB1-AMPK pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 1403–1408. [Google Scholar] [CrossRef] [Green Version]
- Homolya, L.; Fu, D.; Sengupta, P.; Jarnik, M.; Gillet, J.-P.; Vitale-Cross, L.; Gutkind, J.S.; Lippincott-Schwartz, J.; Arias, I.M. LKB1/AMPK and PKA Control ABCB11 Trafficking and Polarization in Hepatocytes. PLoS ONE 2014, 9, e91921. [Google Scholar] [CrossRef] [Green Version]
- Misra, S.; Varticovski, L.; Arias, I.M. Mechanisms by which cAMP increases bile acid secretion in rat liver and canalicular membrane vesicles. Am. J. Physiol. Liver Physiol. 2003, 285, G316–G324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurz, A.K.; Graf, D.; Schmitt, M.; Vom Dahl, S.; Häussinger, D. Tauroursodesoxycholate-induced choleresis involves p38MAPK activation and translocation of the bile salt export pump in rats. Gastroenterology 2001, 121, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Kubitz, R.; Sütfels, G.; Kühlkamp, T.; Kölling, R.; Häussinger, D. Trafficking of the bile salt export pump from the Golgi to the canalicular membrane is regulated by the p38 MAP kinase. Gastroenterology 2004, 126, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Noe, J. Characterization of the mouse bile salt export pump overexpressed in the baculovirus system. Hepatology 2001, 33, 1223–1231. [Google Scholar] [CrossRef]
- Hayashi, H.; Sugiyama, Y. Short-Chain Ubiquitination Is Associated with the Degradation Rate of a Cell-Surface-Resident Bile Salt Export Pump (BSEP/ABCB11). Mol. Pharmacol. 2009, 75, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Aida, K.; Hayashi, H.; Inamura, K.; Mizuno, T.; Sugiyama, Y. Differential Roles of Ubiquitination in the Degradation Mechanism of Cell Surface–Resident Bile Salt Export Pump and Multidrug Resistance–Associated Protein 2. Mol. Pharmacol. 2014, 85, 482–491. [Google Scholar] [CrossRef] [Green Version]
- Czuba, L.C.; Hillgren, K.M.; Swaan, P.W. Post-translational modifications of transporters. Pharmacol. Ther. 2018, 192, 88–99. [Google Scholar] [CrossRef]
- Wang, L.; Hou, W.-T.; Chen, L.; Jiang, Y.-L.; Xu, D.; Sun, L.; Zhou, C.-Z.; Chen, Y. Cryo-EM structure of human bile salts exporter ABCB11. Cell Res. 2020, 30, 623–625. [Google Scholar] [CrossRef]
- Keitel, V.; Burdelski, M.; Vojnisek, Z.; Schmitt, L.; Häussinger, D.; Kubitz, R. De novo bile salt transporter antibodies as a possible cause of recurrent graft failure after liver transplantation: A novel mechanism of cholestasis. Hepatology 2009, 50, 510–517. [Google Scholar] [CrossRef]
- Kubitz, R.; Dröge, C.; Stindt, J.; Weissenberger, K.; Häussinger, D. The bile salt export pump (BSEP) in health and disease. Clin. Res. Hepatol. Gastroenterol. 2012, 36, 536–553. [Google Scholar] [CrossRef]
- Dawson, R.J.P.; Locher, K.P. Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 2007, 581, 935–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovannoni, I.; Callea, F.; Bellacchio, E.; Torre, G.; De Ville De Goyet, J.; Francalanci, P. Genetics and Molecular Modeling of New Mutations of Familial Intrahepatic Cholestasis in a Single Italian Center. PLoS ONE 2015, 10, e0145021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Jaimes, K.F.; Aller, S.G. Refined structures of mouse P-glycoprotein. Protein Sci. 2014, 23, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Dröge, C.; Bonus, M.; Baumann, U.; Klindt, C.; Lainka, E.; Kathemann, S.; Brinkert, F.; Grabhorn, E.; Pfister, E.-D.; Wenning, D.; et al. Sequencing of FIC1, BSEP and MDR3 in a large cohort of patients with cholestasis revealed a high number of different genetic variants. J. Hepatol. 2017, 67, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Grandits, M.; Richter, L.; Ecker, G.F. Structure based classification for bile salt export pump (BSEP) inhibitors using comparative structural modeling of human BSEP. J. Comput. Aided. Mol. Des. 2017, 31, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Sohail, M.I.; Schmid, D.; Wlcek, K.; Spork, M.; Szakács, G.; Trauner, M.; Stockner, T.; Chiba, P. Molecular Mechanism of Taurocholate Transport by the Bile Salt Export Pump, an ABC Transporter Associated with Intrahepatic Cholestasis. Mol. Pharmacol. 2017, 92, 401–413. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Thonghin, N.; Collins, R.F.; Barbieri, A.; Shafi, T.; Siebert, A.; Ford, R.C. Novel features in the structure of P-glycoprotein (ABCB1) in the post-hydrolytic state as determined at 7.9 Å resolution. BMC Struct. Biol. 2018, 18, 17. [Google Scholar] [CrossRef]
- Kagawa, T.; Watanabe, N.; Mochizuki, K.; Numari, A.; Ikeno, Y.; Itoh, J.; Tanaka, H.; Arias, I.M.; Mine, T. Phenotypic differences in PFIC2 and BRIC2 correlate with protein stability of mutant Bsep and impaired taurocholate secretion in MDCK II cells. Am. J. Physiol. Liver Physiol. 2008, 294, G58–G67. [Google Scholar] [CrossRef] [Green Version]
- Goda, K.; Dönmez-Cakil, Y.; Tarapcsák, S.; Szalóki, G.; Szöllősi, D.; Parveen, Z.; Türk, D.; Szakács, G.; Chiba, P.; Stockner, T. Human ABCB1 with an ABCB11-like degenerate nucleotide binding site maintains transport activity by avoiding nucleotide occlusion. PLoS Genet. 2020, 16, e1009016. [Google Scholar] [CrossRef]
- Shukla, S.; Schwartz, C.; Kapoor, K.; Kouanda, A.; Ambudkar, S.V. Use of Baculovirus BacMam Vectors for Expression of ABC Drug Transporters in Mammalian Cells. Drug Metab. Dispos. 2012, 40, 304–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stieger, B.; Fattinger, K.; Madon, J.; Kullak-Ublick, G.A.; Meier, P.J. Drug- and estrogen-induced cholestasis through inhibition of the hepatocellular bile salt export pump (Bsep) of rat liver. Gastroenterology 2000, 118, 422–430. [Google Scholar] [CrossRef]
- Kondo, T.; Dale, G.L.; Beutler, E. Simple and rapid purification of inside-out vesicles from human erythrocytes. Biochim. Biophys. Acta Biomembr. 1980, 602, 127–130. [Google Scholar] [CrossRef]
- Guyot, C.; Stieger, B. Interaction of bile salts with rat canalicular membrane vesicles: Evidence for bile salt resistant microdomains. J. Hepatol. 2011, 55, 1368–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horikawa, M.; Kato, Y.; Tyson, C.A.; Sugiyama, Y. Potential Cholestatic Activity of Various Therapeutic Agents Assessed by Bile Canalicular Membrane Vesicles Isolated from Rats and Humans. Drug Metab. Pharmacokinet. 2003, 18, 16–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stieger, B.; Mahdi, Z.M. Model Systems for Studying the Role of Canalicular Efflux Transporters in Drug-Induced Cholestatic Liver Disease. J. Pharm. Sci. 2017, 106, 2295–2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mita, S.; Suzuki, H.; Akita, H.; Hayashi, H.; Onuki, R.; Hofmann, A.F.; Sugiyama, Y. Vectorial transport of unconjugated and conjugated bile salts by monolayers of LLC-PK1 cells doubly transfected with human NTCP and BSEP or with rat Ntcp and Bsep. Am. J. Physiol. Liver Physiol. 2006, 290, G550–G556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montanari, F.; Pinto, M.; Khunweeraphong, N.; Wlcek, K.; Sohail, M.I.; Noeske, T.; Boyer, S.; Chiba, P.; Stieger, B.; Kuchler, K.; et al. Flagging Drugs That Inhibit the Bile Salt Export Pump. Mol. Pharm. 2016, 13, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Kenna, J.G.; Taskar, K.S.; Battista, C.; Bourdet, D.L.; Brouwer, K.L.R.; Brouwer, K.R.; Dai, D.; Funk, C.; Hafey, M.J.; Lai, Y.; et al. Can Bile Salt Export Pump Inhibition Testing in Drug Discovery and Development Reduce Liver Injury Risk? An International Transporter Consortium Perspective. Clin. Pharmacol. Ther. 2018, 104, 916–932. [Google Scholar] [CrossRef] [Green Version]
- Brouwer, K.L.R.; Keppler, D.; Hoffmaster, K.A.; Bow, D.A.J.; Cheng, Y.; Lai, Y.; Palm, J.E.; Stieger, B.; Evers, R. In Vitro Methods to Support Transporter Evaluation in Drug Discovery and Development. Clin. Pharmacol. Ther. 2013, 94, 95–112. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Guo, C.; Woodhead, J.L.; St. Claire, R.L.; Watkins, P.B.; Siler, S.Q.; Howell, B.A.; Brouwer, K.L.R. Sandwich-Cultured Hepatocytes as a Tool to Study Drug Disposition and Drug-Induced Liver Injury. J. Pharm. Sci. 2016, 105, 443–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bruyn, T.; Chatterjee, S.; Fattah, S.; Keemink, J.; Nicolaï, J.; Augustijns, P.; Annaert, P. Sandwich-cultured hepatocytes: Utility for in vitro exploration of hepatobiliary drug disposition and drug-induced hepatotoxicity. Expert Opin. Drug Metab. Toxicol. 2013, 9, 589–616. [Google Scholar] [CrossRef] [PubMed]
- Swift*, B.; Pfeifer*, N.D.; Brouwer, K.L.R. Sandwich-cultured hepatocytes: An in vitro model to evaluate hepatobiliary transporter-based drug interactions and hepatotoxicity. Drug Metab. Rev. 2010, 42, 446–471. [Google Scholar] [CrossRef] [Green Version]
- Li, A.P.; Gorycki, P.D.; Hengstler, J.G.; Kedderis, G.L.; Koebe, H.G.; Rahmani, R.; de Sousas, G.; Silva, J.M.; Skett, P. Present status of the application of cryopreserved hepatocytes in the evaluation of xenobiotics: Consensus of an international expert panel. Chem. Biol. Interact. 1999, 121, 117–123. [Google Scholar] [CrossRef]
- Lundquist, P.; Englund, G.; Skogastierna, C.; Lööf, J.; Johansson, J.; Hoogstraate, J.; Afzelius, L.; Andersson, T.B. Functional ATP-Binding Cassette Drug Efflux Transporters in Isolated Human and Rat Hepatocytes Significantly Affect Assessment of Drug Disposition. Drug Metab. Dispos. 2014, 42, 448–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yucha, R.W.; He, K.; Shi, Q.; Cai, L.; Nakashita, Y.; Xia, C.Q.; Liao, M. In Vitro Drug-Induced Liver Injury Prediction: Criteria Optimization of Efflux Transporter IC50 and Physicochemical Properties. Toxicol. Sci. 2017, 157, 487–499. [Google Scholar] [CrossRef]
- Cheng, Y.; Woolf, T.F.; Gan, J.; He, K. In vitro model systems to investigate bile salt export pump (BSEP) activity and drug interactions: A review. Chem. Biol. Interact. 2016, 255, 23–30. [Google Scholar] [CrossRef]
- Cheng, Y.; Freeden, C.; Zhang, Y.; Abraham, P.; Shen, H.; Wescott, D.; Humphreys, W.G.; Gan, J.; Lai, Y. Biliary excretion of pravastatin and taurocholate in rats with bile salt export pump (Bsep) impairment. Biopharm. Drug Dispos. 2016, 37, 276–286. [Google Scholar] [CrossRef]
- Wang, R. Targeted inactivation of sister of P-glycoprotein gene (spgp) in mice results in nonprogressive but persistent intrahepatic cholestasis. Proc. Natl. Acad. Sci. USA 2001, 98, 2011–2016. [Google Scholar] [CrossRef]
- Pankowicz, F.P.; Barzi, M.; Kim, K.H.; Legras, X.; Martins, C.S.; Wooton-Kee, C.R.; Lagor, W.R.; Marini, J.C.; Elsea, S.H.; Bissig-Choisat, B.; et al. Rapid Disruption of Genes Specifically in Livers of Mice Using Multiplex CRISPR/Cas9 Editing. Gastroenterology 2018, 155, 1967–1970.e6. [Google Scholar] [CrossRef]
- Alves-Bezerra, M.; Furey, N.; Johnson, C.G.; Bissig, K.-D. Using CRISPR/Cas9 to model human liver disease. JHEP Rep. 2019, 1, 392–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, C.D.; Paumgartner, G.; Wahlström, A.; Schwabl, P.; Reiberger, T.; Leditznig, N.; Stojakovic, T.; Rohr-Udilova, N.; Chiba, P.; Marschall, H.-U.; et al. Metabolic preconditioning protects BSEP/ABCB11−/− mice against cholestatic liver injury. J. Hepatol. 2017, 66, 95–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Chen, H.-L.; Liu, L.; Sheps, J.A.; Phillips, M.J.; Ling, V. Compensatory role of P-glycoproteins in knockout mice lacking the bile salt export pump. Hepatology 2009, 50, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.L.; Bove, K.E.; Schuetz, E.G.; Leino, D.; Valencia, C.A.; Schuetz, J.D.; Miethke, A.; Yin, C. Zebrafish abcb11b mutant reveals strategies to restore bile excretion impaired by bile salt export pump deficiency. Hepatology 2018, 67, 1531–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, P.; Wang, R.; Ling, V. Bile Acid Transport in Sister of P-Glycoprotein (ABCB11) Knockout Mice. Biochemistry 2005, 44, 12598–12605. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, E.; Grosse, B.; Schuller, B.; Davit-Spraul, A.; Conti, F.; Guettier, C.; Cassio, D.; Jacquemin, E. Targeted pharmacotherapy in progressive familial intrahepatic cholestasis type 2: Evidence for improvement of cholestasis with 4-phenylbutyrate. Hepatology 2015, 62, 558–566. [Google Scholar] [CrossRef]
- Gonzales, E.; Grosse, B.; Cassio, D.; Davit-Spraul, A.; Fabre, M.; Jacquemin, E. Successful mutation-specific chaperone therapy with 4-phenylbutyrate in a child with progressive familial intrahepatic cholestasis type 2. J. Hepatol. 2012, 57, 695–698. [Google Scholar] [CrossRef]
- The Human Gene Mutation Database. Available online: http://www.hgmd.cf.ac.uk/ac/gene.php?gene=ABCB11 (accessed on 12 November 2020).
- Imagawa, K.; Hayashi, H.; Sabu, Y.; Tanikawa, K.; Fujishiro, J.; Kajikawa, D.; Wada, H.; Kudo, T.; Kage, M.; Kusuhara, H.; et al. Clinical phenotype and molecular analysis of a homozygous ABCB11 mutation responsible for progressive infantile cholestasis. J. Hum. Genet. 2018, 63, 569–577. [Google Scholar] [CrossRef]
- Malatack, J.J.; Doyle, D. A Drug Regimen for Progressive Familial Cholestasis Type 2. Pediatrics 2018, 141, e20163877. [Google Scholar] [CrossRef] [Green Version]
- Arthur Lorio, E.; Valadez, D.; Alkhouri, N.; Loo, N. Cholestasis in Benign Recurrent Intrahepatic Cholestasis 2. ACG Case Rep. J. 2020, 7, e00412. [Google Scholar] [CrossRef]
- Lam, P.; Pearson, C.L.; Soroka, C.J.; Xu, S.; Mennone, A.; Boyer, J.L. Levels of plasma membrane expression in progressive and benign mutations of the bile salt export pump (Bsep/Abcb11) correlate with severity of cholestatic diseases. Am. J. Physiol. Physiol. 2007, 293, C1709–C1716. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, H.; Sugiyama, Y. 4-phenylbutyrate enhances the cell surface expression and the transport capacity of wild-type and mutated bile salt export pumps. Hepatology 2007, 45, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.A.; Strautnieks, S.S.; Ihrke, G.; Pagani, F.; Knisely, A.S.; Linton, K.J.; Mieli-Vergani, G.; Thompson, R.J. Missense mutations and single nucleotide polymorphisms in ABCB11 impair bile salt export pump processing and function or disrupt pre-messenger RNA splicing. Hepatology 2009, 49, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Misawa, T.; Hayashi, H.; Sugiyama, Y.; Hashimoto, Y. Discovery and structural development of small molecules that enhance transport activity of bile salt export pump mutant associated with progressive familial intrahepatic cholestasis type 2. Bioorg. Med. Chem. 2012, 20, 2940–2949. [Google Scholar] [CrossRef] [PubMed]
- Misawa, T.; Hayashi, H.; Makishima, M.; Sugiyama, Y.; Hashimoto, Y. E297G mutated bile salt export pump (BSEP) function enhancers derived from GW4064: Structural development study and separation from farnesoid X receptor-agonistic activity. Bioorg. Med. Chem. Lett. 2012, 22, 3962–3966. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Hayashi, H.; Sugiyama, Y. Short- and medium-chain fatty acids enhance the cell surface expression and transport capacity of the bile salt export pump (BSEP/ABCB11). Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Naoi, S.; Hirose, Y.; Matsuzaka, Y.; Tanikawa, K.; Igarashi, K.; Nagasaka, H.; Kage, M.; Inui, A.; Kusuhara, H. Successful treatment with 4-phenylbutyrate in a patient with benign recurrent intrahepatic cholestasis type 2 refractory to biliary drainage and bilirubin absorption. Hepatol. Res. 2016, 46, 192–200. [Google Scholar] [CrossRef]
- Mareux, E.; Lapalus, M.; Amzal, R.; Almes, M.; Aït-Slimane, T.; Delaunay, J.; Adnot, P.; Collado-Hilly, M.; Davit-Spraul, A.; Falguières, T.; et al. Functional rescue of an ABCB11 mutant by ivacaftor: A new targeted pharmacotherapy approach in bile salt export pump deficiency. Liver Int. 2020, 40, 1917–1925. [Google Scholar] [CrossRef]
- Kagawa, T.; Orii, R.; Hirose, S.; Arase, Y.; Shiraishi, K.; Mizutani, A.; Tsukamoto, H.; Mine, T. Ursodeoxycholic acid stabilizes the bile salt export pump in the apical membrane in MDCK II cells. J. Gastroenterol. 2014, 49, 890–899. [Google Scholar] [CrossRef]
- Imagawa, K.; Takayama, K.; Isoyama, S.; Tanikawa, K.; Shinkai, M.; Harada, K.; Tachibana, M.; Sakurai, F.; Noguchi, E.; Hirata, K.; et al. Generation of a bile salt export pump deficiency model using patient-specific induced pluripotent stem cell-derived hepatocyte-like cells. Sci. Rep. 2017, 7, 41806. [Google Scholar] [CrossRef] [Green Version]
- Engelmann, G.; Wenning, D.; Herebian, D.; Sander, O.; Droge, C.; Kluge, S.; Kubitz, R. Two Case Reports of Successful Treatment of Cholestasis with Steroids in Patients with PFIC-2. Pediatrics 2015, 135, e1326–e1332. [Google Scholar] [CrossRef] [Green Version]
- Dröge, C.; Schaal, H.; Engelmann, G.; Wenning, D.; Häussinger, D.; Kubitz, R. Exon-skipping and mRNA decay in human liver tissue: Molecular consequences of pathogenic bile salt export pump mutations. Sci. Rep. 2016, 6, 24827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellinger, P.; Stindt, J.; Dröge, C.; Sattler, K.; Stross, C.; Kluge, S.; Herebian, D.; Smits, S.H.J.; Burdelski, M.; Schulz-Jürgensen, S.; et al. Partial external biliary diversion in bile salt export pump deficiency: Association between outcome and mutation. World J. Gastroenterol. 2017, 23, 5295. [Google Scholar] [CrossRef] [PubMed]
- Davit-Spraul, A.; Oliveira, C.; Gonzales, E.; Gaignard, P.; Thérond, P.; Jacquemin, E. Liver transcript analysis reveals aberrant splicing due to silent and intronic variations in the ABCB11 gene. Mol. Genet. Metab. 2014, 113, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Naoi, S.; Hayashi, H.; Inoue, T.; Tanikawa, K.; Igarashi, K.; Nagasaka, H.; Kage, M.; Takikawa, H.; Sugiyama, Y.; Inui, A.; et al. Improved Liver Function and Relieved Pruritus after 4-Phenylbutyrate Therapy in a Patient with Progressive Familial Intrahepatic Cholestasis Type 2. J. Pediatr. 2014, 164, 1219–1227.e3. [Google Scholar] [CrossRef] [PubMed]
- Amzal, R.; Thébaut, A.; Lapalus, M.; Almes, M.; Grosse, B.; Mareux, E.; Collado-Hilly, M.; Davit-Spraul, A.; Bidou, L.; Namy, O.; et al. Pharmacological premature termination codon readthrough of ABCB11 in bile salt export pump deficiency: An in vitro study. Hepatology 2020, hep.31476. [Google Scholar] [CrossRef]
- Strautnieks, S.S.; Byrne, J.A.; Pawlikowska, L.; Cebecauerová, D.; Rayner, A.; Dutton, L.; Meier, Y.; Antoniou, A.; Stieger, B.; Arnell, H.; et al. Severe Bile Salt Export Pump Deficiency: 82 Different ABCB11 Mutations in 109 Families. Gastroenterology 2008, 134, 1203–1214.e8. [Google Scholar] [CrossRef] [Green Version]
- Highlights of Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/207999s003lbl.pdf (accessed on 6 November 2020).
- Fiorucci, S.; Antonelli, E.; Rizzo, G.; Renga, B.; Mencarelli, A.; Riccardi, L.; Orlandi, S.; Pellicciari, R.; Morelli, A. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology 2004, 127, 1497–1512. [Google Scholar] [CrossRef]
- Trauner, M.; Nevens, F.; Shiffman, M.L.; Drenth, J.P.H.; Bowlus, C.L.; Vargas, V.; Andreone, P.; Hirschfield, G.M.; Pencek, R.; Malecha, E.S.; et al. Long-term efficacy and safety of obeticholic acid for patients with primary biliary cholangitis: 3-year results of an international open-label extension study. Lancet Gastroenterol. Hepatol. 2019, 4, 445–453. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef] [Green Version]
- Fiorucci, S.; Biagioli, M.; Sepe, V.; Zampella, A.; Distrutti, E. Bile acid modulators for the treatment of nonalcoholic steatohepatitis (NASH). Expert Opin. Investig. Drugs 2020, 29, 623–632. [Google Scholar] [CrossRef]
- Garzel, B.; Yang, H.; Zhang, L.; Huang, S.-M.; Polli, J.E.; Wang, H. The Role of Bile Salt Export Pump Gene Repression in Drug-Induced Cholestatic Liver Toxicity. Drug Metab. Dispos. 2014, 42, 318–322. [Google Scholar] [CrossRef] [Green Version]
- Hiebl, V.; Ladurner, A.; Latkolik, S.; Dirsch, V.M. Natural products as modulators of the nuclear receptors and metabolic sensors LXR, FXR and RXR. Biotechnol. Adv. 2018, 36, 1657–1698. [Google Scholar] [CrossRef]
- Wu, G.; Wen, M.; Sun, L.; Li, H.; Liu, Y.; Li, R.; Wu, F.; Yang, R.; Lin, Y. Mechanistic insights into geniposide regulation of bile salt export pump (BSEP) expression. RSC Adv. 2018, 8, 37117–37128. [Google Scholar] [CrossRef] [Green Version]
- Hoeke, M.O.; Plass, J.R.M.; Heegsma, J.; Geuken, M.; van Rijsbergen, D.; Baller, J.F.W.; Kuipers, F.; Moshage, H.; Jansen, P.L.M.; Faber, K.N. Low retinol levels differentially modulate bile salt-induced expression of human and mouse hepatic bile salt transporters. Hepatology 2009, 49, 151–159. [Google Scholar] [CrossRef]
- Telbisz, Á.; Homolya, L. Recent advances in the exploration of the bile salt export pump (BSEP/ABCB11) function. Expert Opin. Ther. Targets 2016, 20, 501–514. [Google Scholar] [CrossRef] [Green Version]
- Halilbasic, E.; Steinacher, D.; Trauner, M. Nor-Ursodeoxycholic Acid as a Novel Therapeutic Approach for Cholestatic and Metabolic Liver Diseases. Dig. Dis. 2017, 35, 288–292. [Google Scholar] [CrossRef]
- Kim, D.J.; Yoon, S.; Ji, S.C.; Yang, J.; Kim, Y.-K.; Lee, S.; Yu, K.-S.; Jang, I.-J.; Chung, J.-Y.; Cho, J.-Y. Ursodeoxycholic acid improves liver function via phenylalanine/tyrosine pathway and microbiome remodelling in patients with liver dysfunction. Sci. Rep. 2018, 8, 11874. [Google Scholar] [CrossRef]
- Halilbasic, E.; Fiorotto, R.; Fickert, P.; Marschall, H.-U.; Moustafa, T.; Spirli, C.; Fuchsbichler, A.; Gumhold, J.; Silbert, D.; Zatloukal, K.; et al. Side chain structure determines unique physiologic and therapeutic properties of norursodeoxycholic acid in Mdr2−/− mice. Hepatology 2009, 49, 1972–1981. [Google Scholar] [CrossRef] [Green Version]
- Moustafa, T.; Fickert, P.; Magnes, C.; Guelly, C.; Thueringer, A.; Frank, S.; Kratky, D.; Sattler, W.; Reicher, H.; Sinner, F.; et al. Alterations in Lipid Metabolism Mediate Inflammation, Fibrosis, and Proliferation in a Mouse Model of Chronic Cholestatic Liver Injury. Gastroenterology 2012, 142, 140–151.e12. [Google Scholar] [CrossRef]
- Ito, S.; Hayashi, H.; Sugiura, T.; Ito, K.; Ueda, H.; Togawa, T.; Endo, T.; Tanikawa, K.; Kage, M.; Kusuhara, H.; et al. Effects of 4-phenylbutyrate therapy in a preterm infant with cholestasis and liver fibrosis. Pediatr. Int. 2016, 58, 506–509. [Google Scholar] [CrossRef]
- Vitale, G.; Simonetti, G.; Pirillo, M.; Taruschio, G.; Pietro, A. Bipolar and Related Disorders Induced by Sodium 4-Phenylbutyrate in a Male Adolescent with Bile Salt Export Pump Deficiency Disease. Psychiatry Investig. 2016, 13, 580. [Google Scholar] [CrossRef] [Green Version]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830. [Google Scholar] [CrossRef] [Green Version]
- De Boeck, K.; Munck, A.; Walker, S.; Faro, A.; Hiatt, P.; Gilmartin, G.; Higgins, M. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J. Cyst. Fibros. 2014, 13, 674–680. [Google Scholar] [CrossRef] [Green Version]
- Delaunay, J.; Bruneau, A.; Hoffmann, B.; Durand-Schneider, A.; Barbu, V.; Jacquemin, E.; Maurice, M.; Housset, C.; Callebaut, I.; Aït-Slimane, T. Functional defect of variants in the adenosine triphosphate–binding sites of ABCB4 and their rescue by the cystic fibrosis transmembrane conductance regulator potentiator, ivacaftor (VX-770). Hepatology 2017, 65, 560–570. [Google Scholar] [CrossRef] [Green Version]
- Van Wessel, D.B.E.; Thompson, R.J.; Gonzales, E.; Jankowska, I.; Sokal, E.; Grammatikopoulos, T.; Kadaristiana, A.; Jacquemin, E.; Spraul, A.; Lipiński, P.; et al. Genotype correlates with the natural history of severe bile salt export pump deficiency. J. Hepatol. 2020, 73, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Dabrowski, M.; Bukowy-Bieryllo, Z.; Zietkiewicz, E. Advances in therapeutic use of a drug-stimulated translational readthrough of premature termination codons. Mol. Med. 2018, 24, 25. [Google Scholar] [CrossRef] [Green Version]
- Cuyx, S.; De Boeck, K. Treating the Underlying Cystic Fibrosis Transmembrane Conductance Regulator Defect in Patients with Cystic Fibrosis. Semin. Respir. Crit. Care Med. 2019, 40, 762–774. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Nucleotide Change | Type of Mutation | Amino Acid | Defect | Potential Corrective Therapy | Cell Line/Organism | Disease | References |
|---|---|---|---|---|---|---|---|
| c.386GA | Missense | C129Y 1 | Impaired membrane trafficking, reduced level of mature protein | 4-PB | HEK293T | PFIC2 | [100] |
| c.470AG c.3892GA | Missense | Y157C G1298R | Reduced/absent BSEP activity | 4-PB in combination with oxcarbazepine and maralixibat | Patient with 2 heterozygous missense mutations | PFIC2 | [101] |
| c.698TC | Missense | L233S | - | Methylprednisolone | Patient with heterozygosity in ABCB11, as well as in CFTR, NPHP4 and A1ATD | BRIC2 | [102] |
| c.890AG | Missense | E297G 2 | Protein instability, ubiquitin-dependent degradation [103], impaired membrane trafficking, reduced level of mature protein | 4-PB | Madin-Darby canine kidney (MDCK) II cells and Sprague–Dawley rats | BRIC2, PFIC2 | [104] |
| Glycerol, glycerol at 28 °C | CHO-K1 cells | [105] | |||||
| CA, CDCA, DCA, UDCA, GW4064 (FXR agonist) | MDCK II cells | [106,107] | |||||
| Butyrate and octanoic acid | MDCK II cells | [108] | |||||
| c.1211AG | Missense | D404G | Reduced level of mature protein, ER-like distribution | 4-PB | HEK293T cells | BRIC2 | [109] |
| c.1211AG c.1331TC | Missense | D404G V444A | Reduced level of mature protein | 4-PB | Patient compound heterozygous for D404G and homozygous for V444A mutations | BRIC2 | [109] |
| c.1388CT | Missense | T463I | Impaired ATP-binding, BSEP dysfunction | Ivacaftor | MDCK II cells | PFIC2 | [110] |
| c.1445AG | Missense | D482G 2 | Protein instability, ubiquitin-dependent [103], impaired membrane trafficking, reduced level of mature protein, severe differential splicing [105] | 4-PB | MDCK II cells and Sprague– Dawley rats | PFIC2 | [104] |
| Sodium butyrate and 4-PB | HEK293T cells | [103] | |||||
| Butyrate and octanoic acid | MDCK II cells | [108] | |||||
| c.1708GA | Missense | A570T | Reduced level of mature protein, reduced BSEP activity [105] | UDCA | MDCK II cells | BRIC2 PFIC2 | [111] |
| Glycerol at 28 °C | CHO-K1 cells | [105] | |||||
| c.2417GA | Missense | G806D | Reduced level of mature protein, aberrant splicing | 4-PB | BSEP-deficient hepatocyte-like cells | PFIC2 | [112] |
| c.-24CA | 5′-UTR (five prime untranslated region) | ||||||
| c.2494CT | Missense | R832C | Differential splice products [105] | Steroid | Patient with compound heterozygosity | PFIC2 | [113] |
| c.150+3AC | Splice-site mutation | Partial exon skipping [114] | |||||
| c.2756_2758delCCA | Deletion | T919del | Reduced BSEP activity [115] | Steroid | Patient with compound heterozygosity | PFIC2 | [113] |
| c.3703CT | Nonsense | R1235X | Truncated, non-functional transporter [115] | ||||
| c.2944GA | Missense | G982R | Retention in ER, reduced level of mature protein | UDCA, 4-PB single agents or in combination | Can 10 cells | PFIC2 | [97] |
| c.2944GA | Missense | G982R | Retention in ER, reduced level of mature protein | 4-PB | Patient with compound heterozygosity | PFIC2 | [97] |
| c.770CT | Missense | A257V | Normal canalicular expression of BSEP | ||||
| c.2944GA | Missense | G982R | Retention in ER, reduced level of mature protein | 4-PB | Patient with compound heterozygosity | PFIC2 | [97] |
| c.3003AG | Silent | R1001R | Abnormal splicing [116] | ||||
| c.3382CT | Missense | R1128C | Retention in ER, reduced level of mature protein, Mild exon skipping [105] | UDCA, 4-PB single agents or in combination | Can 10 cells | PFIC2 | [97] |
| 4-PB | Patient homozygous for R1128C | ||||||
| c.3628AC | Missense | T1210P | Retention in ER, reduced level of mature protein | UDCA, 4-PB single agents or in combination | Can 10 cells | PFIC2 | [97] |
| 4-PB | Can 10 cells | [98] | |||||
| 4-PB | Patient with homozygous mutation | [97,98] | |||||
| c.3692GA | Missense | R1231Q | Retention in ER [117], no splicing, immature protein [105] | 4-PB | HEK293T cells, McA-RH7777 cells, patient with homozygous mutation | PFIC2 | [117] |
| c.1062TA | Nonsense | Y354X | Premature termination codon | G418, gentamicin | NIH3T3 cells (increased readthrough) | PFIC2 | [118] |
| c.1243CT | Nonsense | R415X | Premature termination codon | G418, gentamicin | NIH3T3 cells (increased readthrough) | PFIC2 | [118] |
| Gentamicin | HEK293 cells (production of a full-length BSEP protein) | ||||||
| c.1408CT | Nonsense | R470X | Premature termination codon | G418, gentamicin | NIH3T3 cells (increased readthrough) | PFIC2 | [118] |
| Gentamicin | HEK293 cells (production of a full-length BSEP protein) | ||||||
| c.3169CT | Nonsense | R1057X | Premature termination codon | G418, gentamicin | NIH3T3 cells (increased readthrough) | PFIC2 | [118] |
| Gentamicin | HEK293 cells (production of a full-length BSEP protein) | ||||||
| c.3268CT | Nonsense | R1090X | Premature termination codon | G418, gentamicin | NIH3T3 cells (increased readthrough) | PFIC2 | [118] |
| Gentamicin | HEK293, Can10 and HepG2 cells (production of a full-length BSEP protein and localization at the PM of HEK293 and at the CM of Can 10 and HepG2 cells) | ||||||
| Gentamicin treatment with UDCA, 4-PB and UDCA + 4-PB, gentamicin at 27 °C | Can10 cells (increased canalicular expression) | ||||||
| Gentamicin, gentamicin with 4-PB, gentamicin at 27 °C | NTCP expressing MDCK cells (significantly increased transport of [3H]TC) | ||||||
| c.3904GT | Nonsense | E1302X | Premature termination codon | G418, gentamicin, PTC124 | NIH3T3 cells (increased readthrough) | PFIC2 | [118] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sohail, M.I.; Dönmez-Cakil, Y.; Szöllősi, D.; Stockner, T.; Chiba, P. The Bile Salt Export Pump: Molecular Structure, Study Models and Small-Molecule Drugs for the Treatment of Inherited BSEP Deficiencies. Int. J. Mol. Sci. 2021, 22, 784. https://doi.org/10.3390/ijms22020784
Sohail MI, Dönmez-Cakil Y, Szöllősi D, Stockner T, Chiba P. The Bile Salt Export Pump: Molecular Structure, Study Models and Small-Molecule Drugs for the Treatment of Inherited BSEP Deficiencies. International Journal of Molecular Sciences. 2021; 22(2):784. https://doi.org/10.3390/ijms22020784
Chicago/Turabian StyleSohail, Muhammad Imran, Yaprak Dönmez-Cakil, Dániel Szöllősi, Thomas Stockner, and Peter Chiba. 2021. "The Bile Salt Export Pump: Molecular Structure, Study Models and Small-Molecule Drugs for the Treatment of Inherited BSEP Deficiencies" International Journal of Molecular Sciences 22, no. 2: 784. https://doi.org/10.3390/ijms22020784
APA StyleSohail, M. I., Dönmez-Cakil, Y., Szöllősi, D., Stockner, T., & Chiba, P. (2021). The Bile Salt Export Pump: Molecular Structure, Study Models and Small-Molecule Drugs for the Treatment of Inherited BSEP Deficiencies. International Journal of Molecular Sciences, 22(2), 784. https://doi.org/10.3390/ijms22020784