Structure, Activation and Regulation of NLRP3 and AIM2 Inflammasomes


Abstract
:1. Introduction
2. NLRP3
2.1. General Information on NLRP3
2.2. Role of the NLRP3 Inflammasome in COVID-19
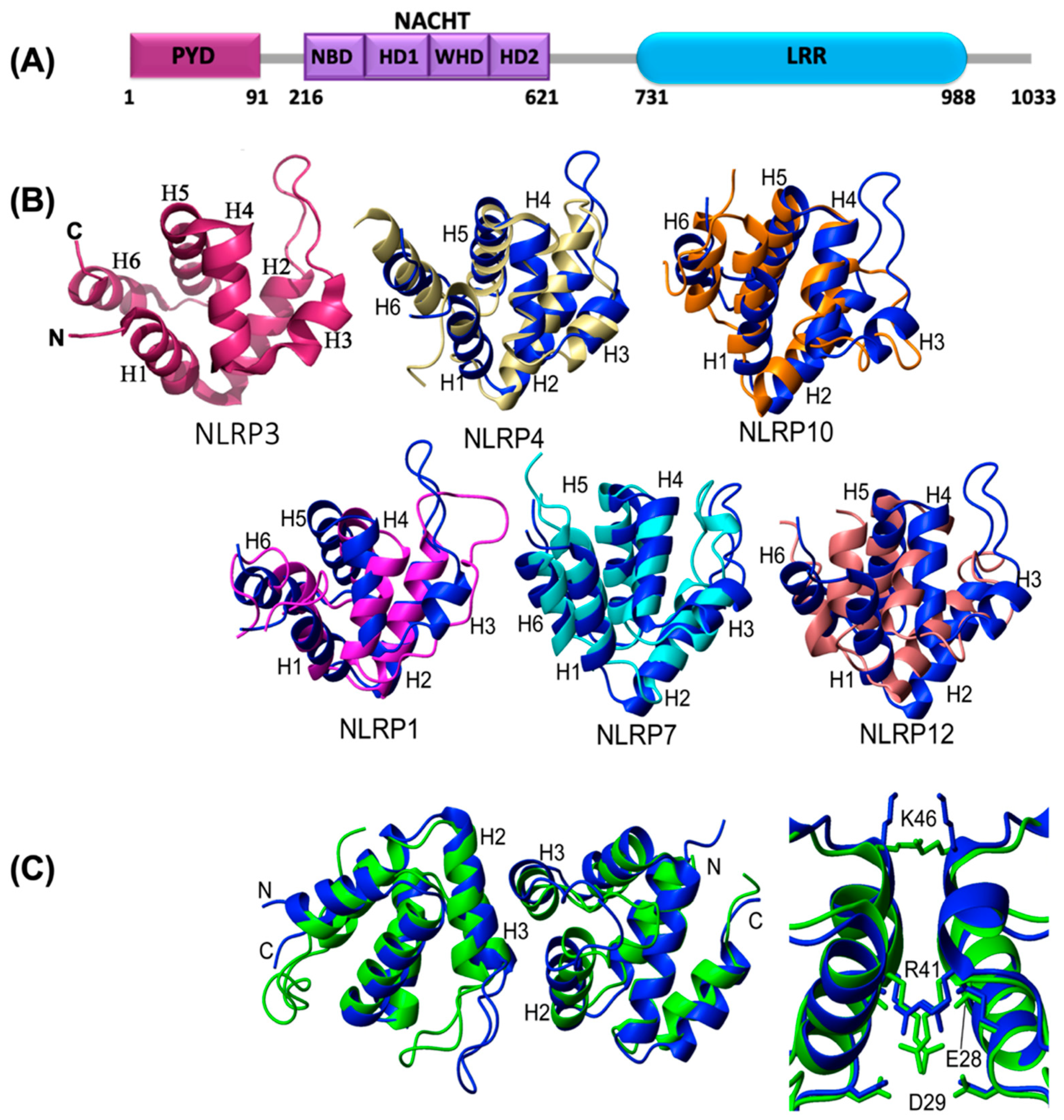
2.3. Structural Details of NLRP3
2.4. Oligomerization of NLRP3PYD
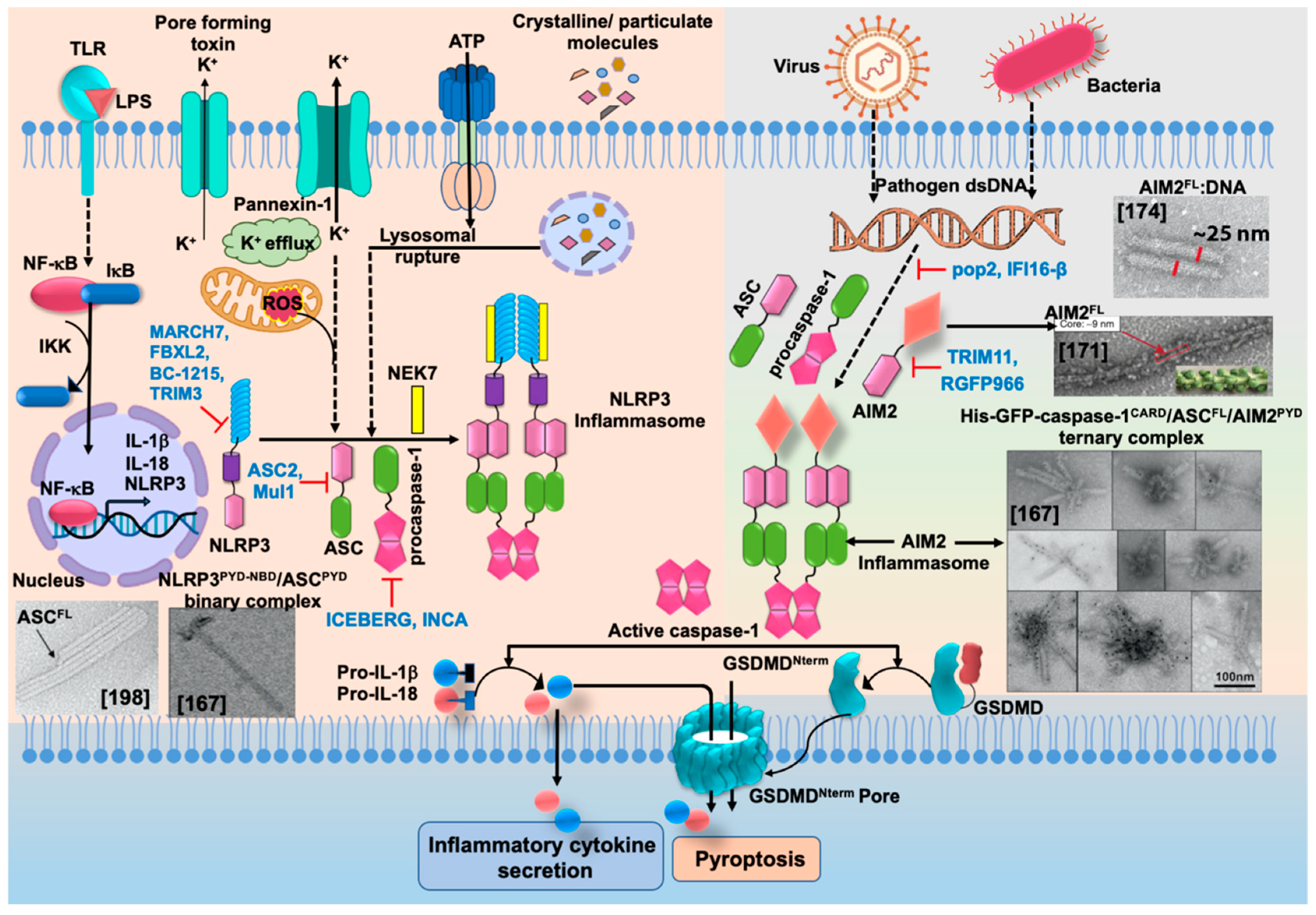
2.5. Activation and Regulation of NLRP3
2.5.1. Post-Translational Modifications
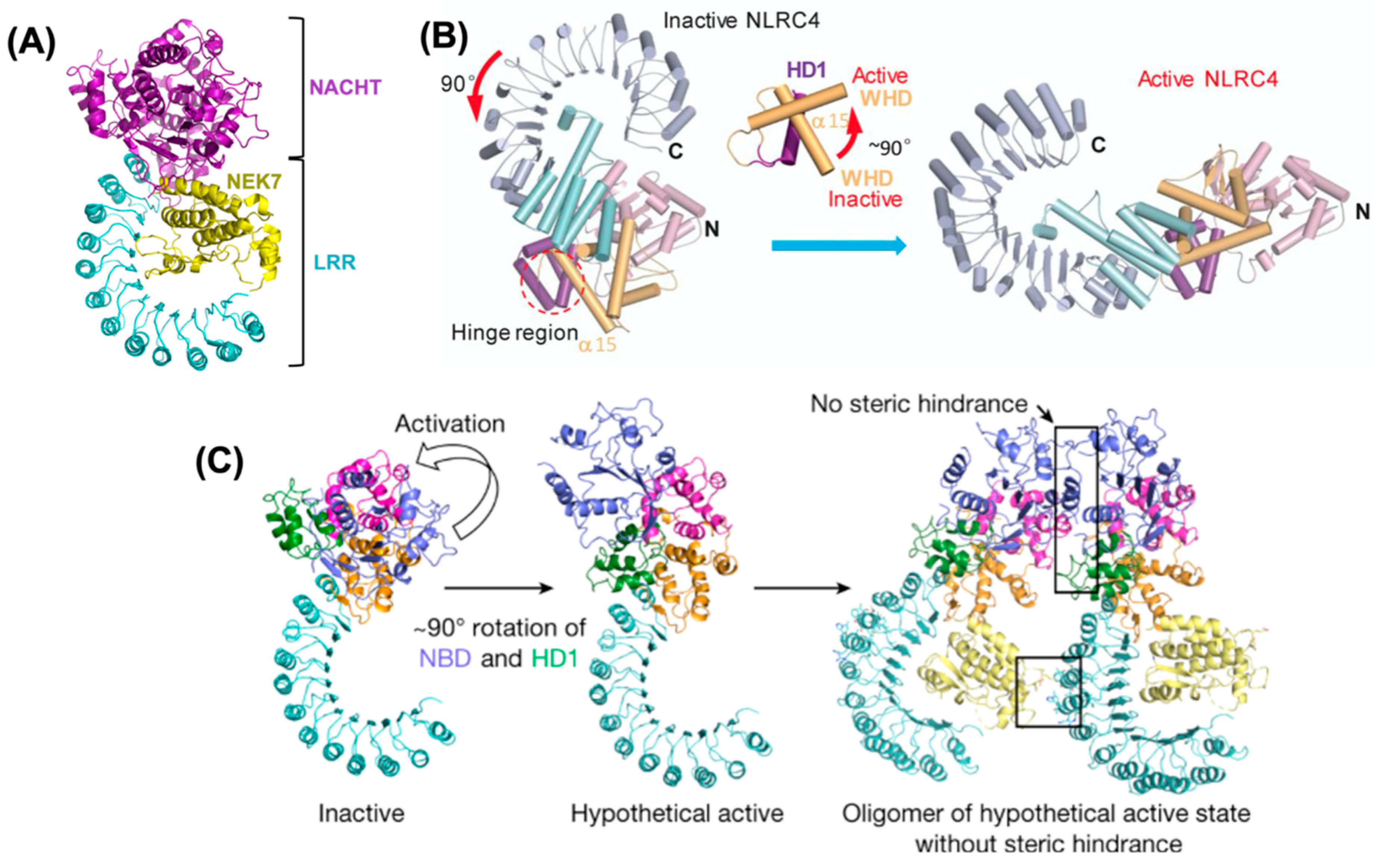
2.5.2. NEK7 Mediated Activation of NLRP3
2.5.3. Role of Caspase-8 in Inflammasome Activation
3. AIM2
3.1. AIM2PYD Domain Structure
3.2. AIM2PYD Self-Association
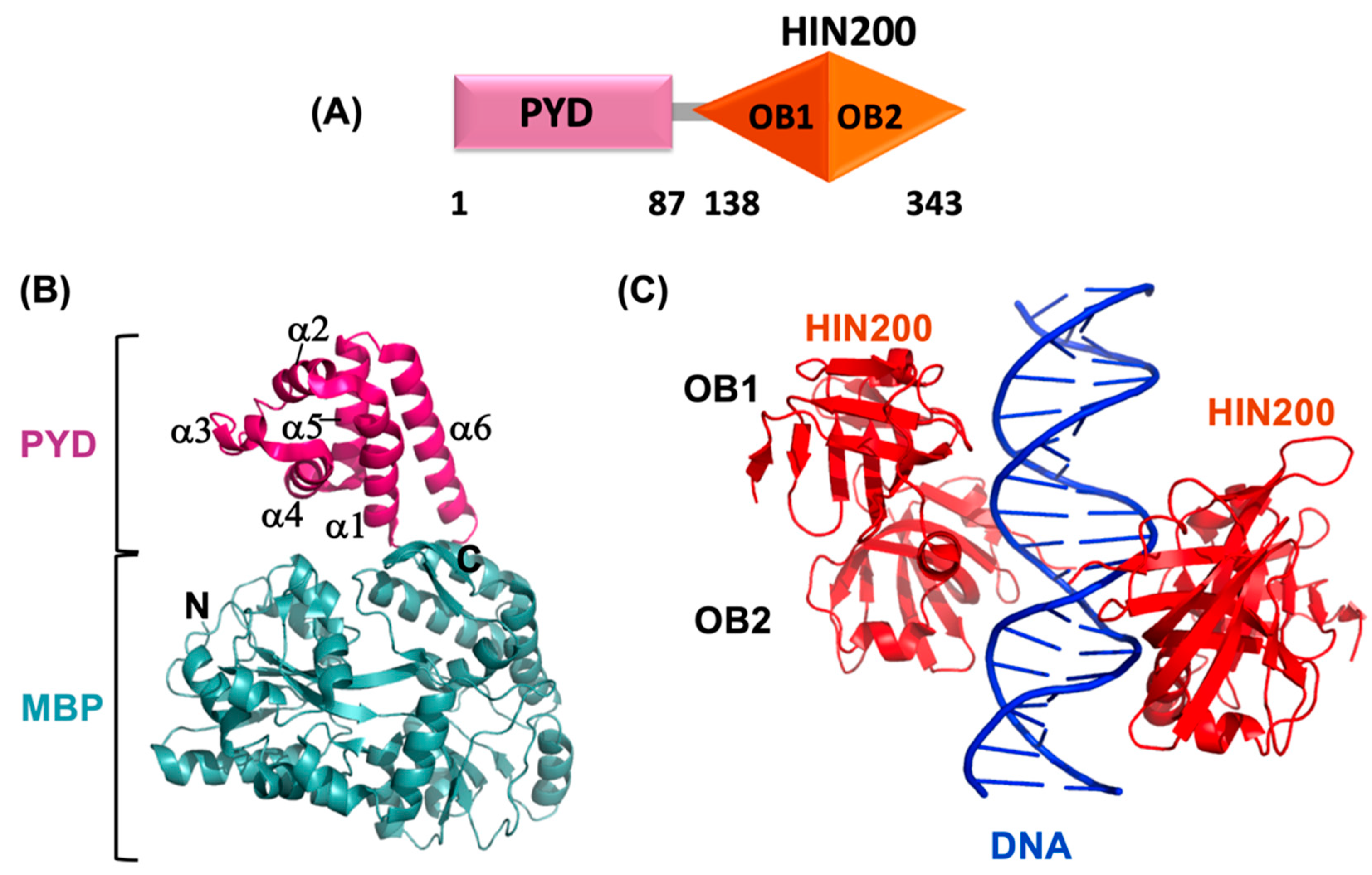
3.3. AIM HIN Domain Structure
3.4. AIM2 HIN:dsDNA Interaction
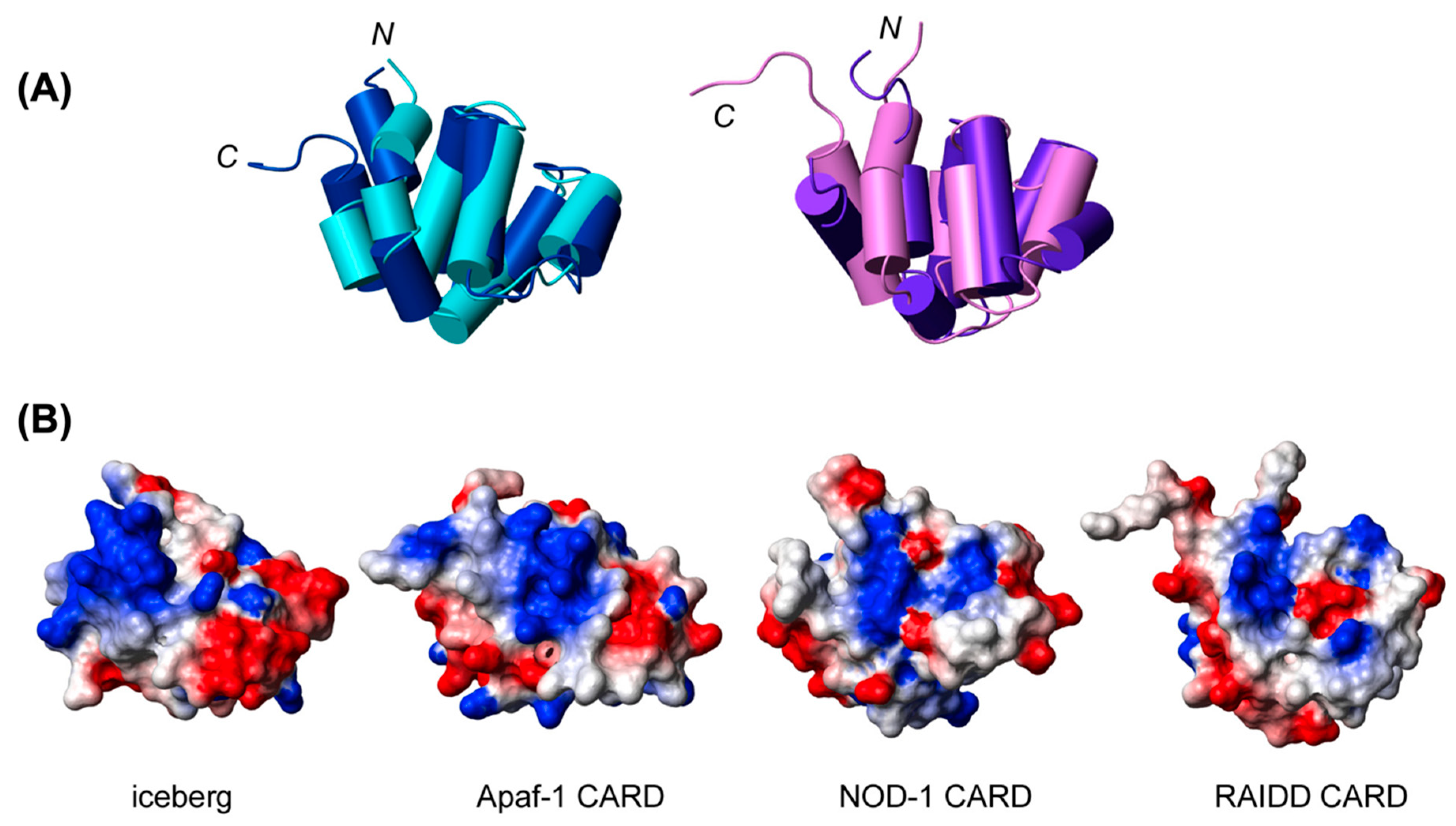
3.5. AIM2 PYD:HIN Interaction
3.6. Importance of AIM2PYD in dsDNA Interaction and Oligomerization
3.7. Regulation of AIM2
3.7.1. Negative Regulators of AIM2 Inflammasome Activation
3.7.2. IFI16-β Mediated Regulation of AIM2 Inflammasome Activation
3.7.3. Post-Translational Modifications of AIM2
4. ASC
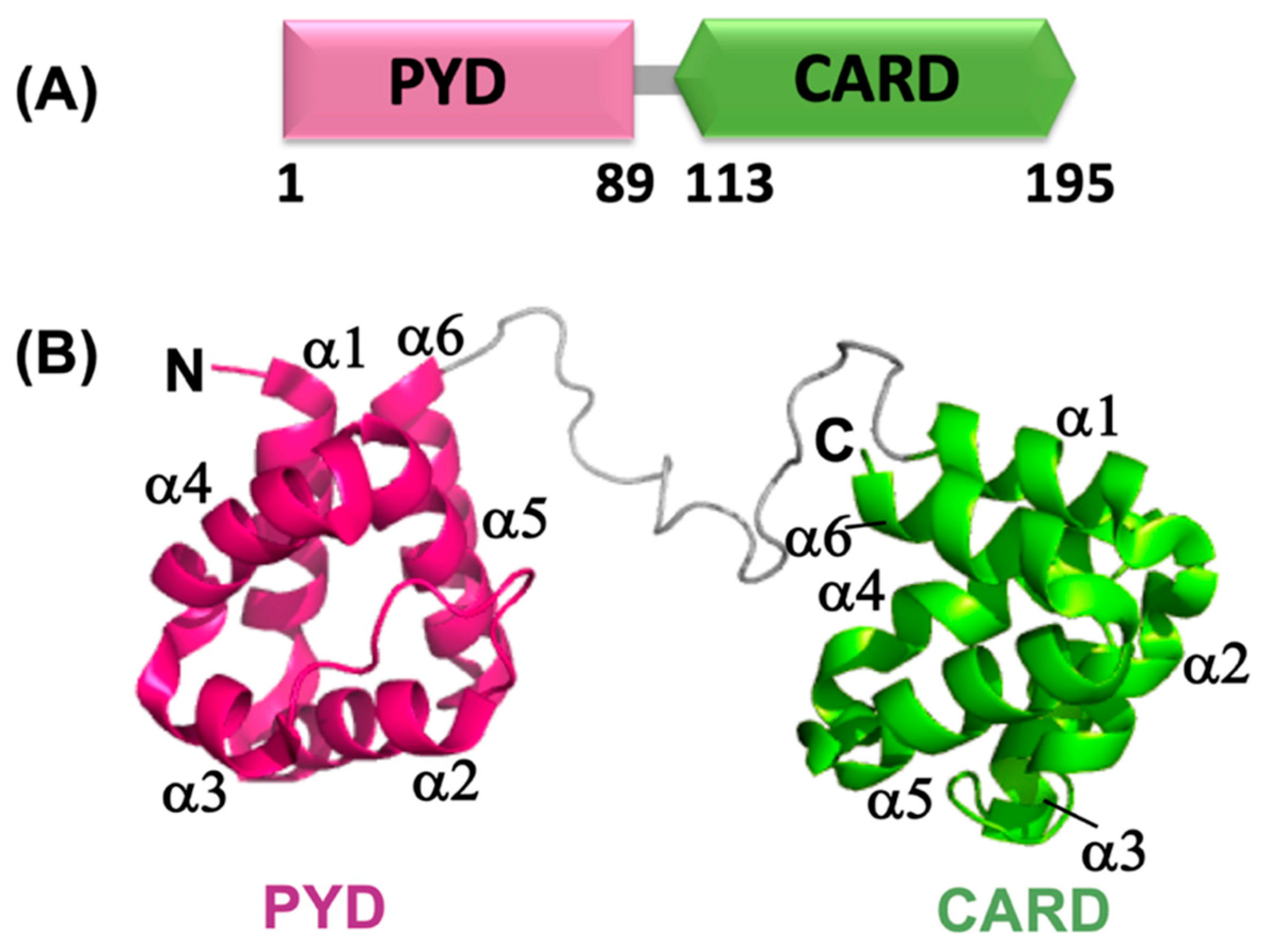
4.1. Structural Details of ASCPYD and Its Self-Association
4.2. Structural Details of ASCCARD and Its Self-Association
4.3. ASC Filament Formation
4.4. Regulation of ASC
4.4.1. Regulation of ASC Mediated by ASC2
4.4.2. Post-Translational Modifications of ASC
5. Caspase-1
5.1. Structure and Activation of Caspase-1
5.2. Negative Regulation of Caspase-1 Activation
6. Formation of Inflammasome Assembly
6.1. NLRP3-ASC Interaction
6.2. Interaction of ASC with Procaspase-1 and Formation of the NLRP3 Inflammasome
6.3. Interaction of AIM2 with ASC and Formation of the AIM2 Inflammasome
6.4. AIM2 Ternary Complex: AIM2PYD:ASCFL:GFP-Casp1CARD
7. Concluding Remarks and Perspective
Funding
Acknowledgments
Conflicts of Interest
Acronyms and Abbreviations
| ADP | Adenosine diphosphate |
| AIM2 | Absent in melanoma 2 |
| ALRs | (AIM2)-like receptors |
| Apaf-1 | Apoptotic protease activating factor |
| ASC | Apoptosis-associated speck-like protein containing a caspase-activation and recruitment domain (CARD) |
| Bcl | B-cell lymphoma |
| BIR | Baculovirus IAP (inhibitor of apoptosis protein) repeat |
| BMDCs | Bone marrow derived dendritic cells |
| BRCA1 | Breast cancer type-1 |
| BRCC3 | BRCA1/BRCA2- containing complex, subunit 3 |
| c-FLIP | FLICE-like inhibitory protein |
| CAPS | Cryopyrin-associated periodic syndrome |
| CARD | Caspase-activation and recruitment domain |
| CDL | CARD domain linker |
| CIAS1 | cold induced autoinflammatory syndrome 1 |
| CLR | C-type lectin receptor |
| COPs | CARD-only proteins |
| COS-1 | CV-1 in Origin with SV40 genes |
| COVID-19 | Coronavirus disease 2019 |
| DAMPs | Danger-associated molecular patterns |
| DD | Death domain |
| DEDs | Death-effector domain |
| DEFCAP | Death Effector Filament-forming Ced-4-likeApoptosis Protein |
| DUBs | Deubiquitinating enzyme |
| EM | Electron microscopy |
| FADD | Fas-associated protein with death domain |
| FBXL2 | F-box L |
| FBXO3 | F-box O3 |
| FLICE | Caspase-8/FADD-like IL-1β-converting enzyme |
| FP | Fluorescence polarization |
| FRET | Fluorescence resonance energy transfer |
| GFP | Green-fluorescent protein |
| GLMN | Glomulin |
| GSDMD | Gasdermin D |
| HD | helical domain |
| HDAC | Histone deacetylases |
| HIN | Hematopoietic, Interferon-inducible, Nuclear localization |
| IAPs | Inhibitor of apoptosis proteins |
| IC50 | Inhibitory concentration |
| ICE | Interleukin 1β-converting enzyme |
| ICE | Interleukin-1beta converting enzyme |
| IFI16 | Gamma-interferon-inducible protein |
| IFN | Interferon |
| IKK-γ | Inhibitor of nuclear factor kappa-B kinase subunit gamma |
| IL | Interleukin |
| INCA | Inhibitors of NFAT-calcineurin association |
| IPAF | Ice protease activating factor |
| IRF4 | Interferon regulatory factor |
| ITC | Isothermal titration calorimetry |
| JNK | Jun N-terminal kinase |
| LPS | Lipopolysaccharide |
| LRRs | Leucine- rich repeat |
| LTA | Lipoteichoic acid |
| LUBAC | linear ubiquitin chain assembly complex |
| MALS | Multiangle light scattering |
| MALT1 | Mucosa-associated lymphoid tissue lymphoma translocation protein 1 |
| MAPK | Mitogen-activated protein kinase |
| MAPL | Mitochondria-associated protein ligase |
| MARCH7 | Membrane associated ring finger (C3HC4) 7 |
| MAVS | Mitochondrial antiviral-signaling protein |
| MBP | Maltose-binding protein |
| MLKL | Mixed lineage kinase domain like pseudokinase |
| MNDA | Myeloid cell nuclear differentiation antigen |
| MSU | Monosodium urate |
| Mul1 | Mitochondrial E3 Ubiquitin Protein Ligase 1 |
| NAIP | NLR family of apoptosis inhibitory protein |
| NALP | NACHT, LRR and PYD domains-containing proteins |
| NBD | Nucleotide binding domain |
| NEK7 | NIMA-related kinase 7 |
| NEMO | NF-kappa-B essential modulator |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRC | NLR family CARD domain-containing protein |
| NLRP | Nod-like receptor protein |
| NLRP3 | NLR family pyrin domain containing 3 |
| NLRs | NOD-like receptors |
| NMR | Nuclear magnetic resonance |
| NOD | Nucleotide-binding oligomerization domain |
| OB | Oligonucleotide/oligosaccharide binding) |
| PAMPs | Pathogen-associated molecular patterns |
| PAO | Phenylarsine oxide |
| PKA | protein kinase A |
| PKD | protein kinase D |
| POPs | PYD-only proteins |
| PP2A | protein phosphatase 2 A |
| PRRs | Pathogen recognition receptors |
| PTM | Post-translation modifications |
| PTPase | Protein tyrosine phosphatase |
| PTPN22 | Protein tyrosine phosphatase nonreceptor type 22 |
| PYCARD | PYD and CARD domain-containing |
| PYD | Pyrin domain |
| PYHIN | Pyrin + HIN |
| PYPAF1 | PYRIN-containing APAF1-like protein 1 |
| RAIDD | RIP-associated ICH1/CED3-homologous protein with a death domain |
| RICK | RIP-like interacting CLARP kinase |
| RIP2 | Receptor-interacting protein 2 |
| RMSD | Root-mean-square deviation |
| RNO | Regulated by nitric oxide |
| ROS | Reactive oxygen species |
| SARS-Cov-2 | Severe acute respiratory syndrome coronavirus 2 |
| SCF | Skp-Cullin-F box |
| SEC | Size-exclusion chromatography |
| SHARPIN | Shank-associated RH domain-interacting protein |
| STAT1 | Signal transducer and activator of transcription |
| TEV | Tobacco etch virus. |
| TH2 | T-helper cell type 2 |
| TLR | Toll-like receptors |
| TMS | Target of Methylation-induced Silencing-1 |
| TNF | Tumor necrosis factor |
| TPA | 12-O-tetradecanoylphorbol-13-acetate |
| TRAF3 | TNF receptor associated factor 3 |
| TRIF | TIR-domain-containing adapter-inducing interferon-β |
| TRIM | Tripartite motif |
| WHD | winged helical domain |
| WHO | World health organization |
References
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauernfeind, F.; Hornung, V. Of inflammasomes and pathogens–sensing of microbes by the inflammasome. EMBO Mol. Med. 2013, 5, 814–826. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, I.; Tan, Y.; Di Gioia, M.; Broggi, A.; Ruan, J.; Shi, J.; Donado, C.A.; Shao, F.; Wu, H.; Springstead, J.R.; et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 2016, 352, 1232–1236. [Google Scholar] [CrossRef] [Green Version]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszyński, A. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef]
- Rathinam, V.A.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.J.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395. [Google Scholar] [CrossRef] [Green Version]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Bottero, V.; Sadagopan, S.; Otageri, P.; Chandran, B. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 2011, 9, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [Green Version]
- Harton, J.A.; Linhoff, M.W.; Zhang, J.; Ting, J.P. Cutting edge: CATERPILLER: A large family of mammalian genes containing CARD, pyrin, nucleotide-binding, and leucine-rich repeat domains. J. Immunol. 2002, 169, 4088–4093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inohara, N.; Nuñez, G. The NOD: A signaling module that regulates apoptosis and host defense against pathogens. Oncogene 2001, 20, 6473–6481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khare, S.; Dorfleutner, A.; Bryan, N.B.; Yun, C.; Radian, A.D.; de Almeida, L.; Rojanasakul, Y.; Stehlik, C. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity 2012, 36, 464–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vladimer, G.I.; Weng, D.; Paquette, S.W.M.; Vanaja, S.K.; Rathinam, V.A.; Aune, M.H.; Conlon, J.E.; Burbage, J.J.; Proulx, M.K.; Liu, Q.; et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity 2012, 37, 96–107. [Google Scholar] [CrossRef] [Green Version]
- Chavarría-Smith, J.; Vance, R.E. The NLRP 1 inflammasomes. Immunol. Rev. 2015, 265, 22–34. [Google Scholar] [CrossRef]
- Abderrazak, A.; Syrovets, T.; Couchie, D.; El Hadri, K.; Friguet, B.; Simmet, T.; Rouis, M. NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015, 4, 296–307. [Google Scholar] [CrossRef]
- Bauer, R.; Rauch, I. The NAIP/NLRC4 inflammasome in infection and pathology. Mol. Aspects Med. 2020, 76, 100863. [Google Scholar] [CrossRef]
- Davis, B.K.; Wen, H.; Ting, J.P. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Heilig, R.; Broz, P. Function and mechanism of the pyrin inflammasome. Eur. J. Immunol. 2018, 48, 230–238. [Google Scholar] [CrossRef]
- Pawlowski, K.; Pio, F.; Chu, Z.L.; Reed, J.C.; Godzik, A. PAAD–a new protein domain associated with apoptosis, cancer and autoimmune diseases. Trends Biochem. Sci. 2001, 26, 85–87. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weston, C.R.; Davis, R.J. The JNK signal transduction pathway. Curr. Opin. Cell Biol. 2007, 19, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ren, W.; Jiang, Z.; Zhu, L. Regulation of the NLRP3 inflammasome and macrophage pyroptosis by the p38 MAPK signaling pathway in a mouse model of acute lung injury. Mol. Med. Rep. 2018, 18, 4399–4409. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.W.; Kayagaki, N.; Broz, P.; Henry, T.; Newton, K.; O’Rourke, K.; Chan, S.; Dong, J.; Qu, Y.; Roose-Girma, M.; et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl. Acad. Sci. USA 2010, 107, 9771–9776. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Newton, K.; Lamkanfi, M.; Mariathasan, S.; Dixit, V.M.; Monack, D.M. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against. Salmonella Exp. Med. 2010, 207, 1745–1755. [Google Scholar] [CrossRef]
- Faustin, B.; Lartigue, L.; Bruey, J.M.; Luciano, F.; Sergienko, E.; Bailly-Maitre, B.; Volkmann, N.; Hanein, D.; Rouiller, I.; Reed, J.C. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol. Cell 2007, 25, 713–724. [Google Scholar] [CrossRef]
- Poyet, J.L.; Srinivasula, S.M.; Tnani, M.; Razmara, M.; Fernandes-Alnemri, T.; Alnemri, E.S. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J. Biol. Chem. 2001, 276, 28309–28313. [Google Scholar] [CrossRef] [Green Version]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Moayeri, M.; Sastalla, I.; Leppla, S.H. infection, Anthrax and the inflammasome. Microbes Infect. 2012, 14, 392–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyden, E.D.; Dietrich, W.F. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet. 2006, 38, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Ewald, S.E.; Chavarria-Smith, J.; Boothroyd, J.C. NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect. Immun. 2014, 82, 460–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Mailloux, C.M.; Gowan, K.; Riccardi, S.L.; LaBerge, G.; Bennett, D.C.; Fain, P.R.; Spritz, R.A. NALP1 in vitiligo-associated multiple autoimmune disease. N. Engl. J. Med. 2007, 356, 1216–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandemange, S.; Sanchez, E.; Louis-Plence, P.; Mau-Them, F.T.; Bessis, D.; Coubes, C.; Frouin, E.; Seyger, M.; Girard, M.; Puechberty, J.; et al. A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis). Ann. Rheum. Dis. 2017, 76, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.L.; Mamaï, O.; Sborgi, L.; Boussofara, L.; Hopkins, R.; Robinson, K.; Szeverényi, I.; Takeichi, T.; Balaji, R.; Lau, A.; et al. Germline NLRP1 mutations cause skin inflammatory and cancer susceptibility syndromes via inflammasome activation. Cell 2016, 167, 187–202. [Google Scholar] [CrossRef] [Green Version]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef] [Green Version]
- Poeck, H.; Bscheider, M.; Gross, O.; Finger, K.; Roth, S.; Rebsamen, M.; Hannesschläger, N.; Schlee, M.; Rothenfusser, S.; Barchet, W.; et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1β production. Nat. Immunol. 2010, 11, 63–69. [Google Scholar] [CrossRef]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Duncan, J.A.; Gao, X.; Huang, M.T.H.; O’Connor, B.P.; Thomas, C.E.; Willingham, S.B.; Bergstralh, D.T.; Jarvis, G.A.; Sparling, P.F.; Ting, J.P. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J. Immunol. 2009, 182, 6460–6469. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Pétrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef]
- Keddie, S.; Parker, T.; Lachmann, H.J.; Ginsberg, L. Cryopyrin-associated periodic fever syndrome and the nervous system. Curr. Treat. Options Neurol. 2018, 20, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, P.N.; Lachmann, H.J.; Aganna, E.; McDermott, M.F. Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Off. J. Am. Coll. Rheumatol. 2004, 50, 607–612. [Google Scholar] [CrossRef]
- Aróstegui, J.I.; Lopez Saldaña, M.D.; Pascal, M.; Clemente, D.; Aymerich, M.; Balaguer, F.; Goel, A.; Fournier del Castillo, C.; Rius, J.; Plaza, S.; et al. Rheumatism, A somatic NLRP3 mutation as a cause of a sporadic case of chronic infantile neurologic, cutaneous, articular syndrome/neonatal-onset multisystem inflammatory disease: Novel evidence of the role of low-level mosaicism as the pathophysiologic mechanism underlying mendelian inherited diseases. Arthritis Rheum. 2010, 62, 1158–1166. [Google Scholar]
- Goldbach-Mansky, R.; Shroff, S.D.; Wilson, M.; Snyder, C.; Plehn, S.; Barham, B.; Pham, T.H.; Pucino, F.; Wesley, R.A.; Papadopoulos, J.H. A pilot study to evaluate the safety and efficacy of the long-acting interleukin-1 inhibitor rilonacept (interleukin-1 Trap) in patients with familial cold autoinflammatory syndrome. Off. J. Am. Coll. Rheumatol. 2008, 58, 2432–2442. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857. [Google Scholar] [CrossRef] [Green Version]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Ehses, J.A.; Donath, M.Y.; Mandrup-Poulsen, T. Sustained effects of interleukin-1 receptor antagonist treatment in type 2 diabetes. Diabetes Care 2009, 32, 1663–1668. [Google Scholar] [CrossRef] [Green Version]
- Moossavi, M.; Parsamanesh, N.; Bahrami, A.; Atkin, S.L.; Sahebkar, A. Role of the NLRP3 inflammasome in cancer. Mol. Cancer 2018, 17, 158. [Google Scholar] [CrossRef] [Green Version]
- Goldbach-Mansky, R.; Dailey, N.J.; Canna, S.W.; Gelabert, A.; Jones, J.; Rubin, B.I.; Kim, H.J.; Brewer, C.; Zalewski, C.; Wiggs, E.J.; et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1β inhibition. N. Engl. J. Med. 2006, 355, 581–592. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Franchi, L.; Toma, C.; Ashida, H.; Ogawa, M.; Yoshikawa, Y.; Mimuro, H.; Inohara, N.; Sasakawa, C.; Nuñez, G. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog 2007, 3, e111. [Google Scholar] [CrossRef] [Green Version]
- Amer, A.; Franchi, L.; Kanneganti, T.D.; Body-Malapel, M.; Özören, N.; Brady, G.; Meshinchi, S.; Jagirdar, R.; Gewirtz, A.; Akira, S.; et al. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J. Biol. Chem. 2006, 281, 35217–35223. [Google Scholar] [CrossRef] [Green Version]
- Franchi, L.; Amer, A.; Body-Malapel, M.; Kanneganti, T.D.; Özören, N.; Jagirdar, R.; Inohara, N.; Vandenabeele, P.; Bertin, J.; Coyle, A.; et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1β in salmonella-infected macrophages. Nat. Immunol. 2006, 7, 576–582. [Google Scholar] [CrossRef]
- Romberg, N.; Al Moussawi, K.; Nelson-Williams, C.; Stiegler, A.L.; Loring, E.; Choi, M.; Overton, J.; Meffre, E.; Khokha, M.K.; Huttner, A.; et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat. Genet. 2014, 46, 1135–1139. [Google Scholar] [CrossRef] [Green Version]
- Canna, S.W.; de Jesus, A.A.; Gouni, S.; Brooks, S.R.; Marrero, B.; Liu, Y.; DiMattia, M.A.; Zaal, K.J.; Sanchez, G.A.M.; Kim, H.; et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet. 2014, 46, 1140–1146. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Zhu, S.; Yang, L.; Cui, S.; Pan, W.; Jackson, R.; Zheng, Y.; Rongvaux, A.; Sun, Q.; Yang, G. Nlrp6 regulates intestinal antiviral innate immunity. 2015, 350, 826–830. Science 2015, 350, 826–830. [Google Scholar] [CrossRef] [Green Version]
- Leng, F.; Yin, H.; Qin, S.; Zhang, K.; Guan, Y.; Fang, R.; Wang, H.; Li, G.; Jiang, Z.; Sun, F.; et al. NLRP6 self-assembles into a linear molecular platform following LPS binding and ATP stimulation. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Hara, H.; Seregin, S.S.; Yang, D.; Fukase, K.; Chamaillard, M.; Alnemri, E.S.; Inohara, N.; Chen, G.Y.; Núñez, G. The NLRP6 inflammasome recognizes lipoteichoic acid and regulates Gram-positive pathogen infection. Cell 2018, 175, 1651–1664. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.Y.; Liu, M.; Wang, F.; Bertin, J.; Núñez, G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J. Immunol. 2011, 186, 7187–7194. [Google Scholar] [CrossRef]
- Zaki, M.H.; Man, S.M.; Vogel, P.; Lamkanfi, M.; Kanneganti, T.D. Salmonella exploits NLRP12-dependent innate immune signaling to suppress host defenses during infection. Proc. Natl. Acad. Sci. USA 2014, 111, 385–390. [Google Scholar] [CrossRef] [Green Version]
- Vanaja, S.K.; Rathinam, V.A.; Fitzgerald, K.A. Mechanisms of inflammasome activation: Recent advances and novel insights. Trends Cell Biol. 2015, 25, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Jeru, I.; Duquesnoy, P.; Fernandes-Alnemri, T.; Cochet, E.; Yu, J.; Lackmy-Port-Lis, M.; Grimprel, E.; Landman-Parker, J.; Hentgen, V.; Marlin, S.; et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc. Natl. Acad. Sci. USA 2008, 105, 1614–1619. [Google Scholar] [CrossRef] [Green Version]
- Bürckstümmer, T.; Baumann, C.; Blüml, S.; Dixit, E.; Dürnberger, G.; Jahn, H.; Planyavsky, M.; Bilban, M.; Colinge, J.; Bennett, K.L.; et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 2009, 10, 266. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Yu, J.W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [Green Version]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [Green Version]
- Roberts, T.L.; Idris, A.; Dunn, J.A.; Kelly, G.M.; Burnton, C.M.; Hodgson, S.; Hardy, L.L.; Garceau, V.; Sweet, M.J.; Ross, I.L. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 2009, 323, 1057–1060. [Google Scholar] [CrossRef] [Green Version]
- de Koning, H.D.; Bergboer, J.G.; van den Bogaard, E.H.; van Vlijmen-Willems, I.M.; Rodijk-Olthuis, D.; Simon, A.; Zeeuwen, P.L.; Schalkwijk, J. Strong induction of AIM 2 expression in human epidermis in acute and chronic inflammatory skin conditions. Exp. Dermatol. 2012, 21, 961–964. [Google Scholar] [CrossRef]
- Dihlmann, S.; Erhart, P.; Mehrabi, A.; Nickkholgh, A.; Lasitschka, F.; Böckler, D.; Hakimi, M. Increased expression and activation of absent in melanoma 2 inflammasome components in lymphocytic infiltrates of abdominal aortic aneurysms. Mol. Med. 2014, 20, 230–237. [Google Scholar] [CrossRef]
- Choubey, D.; Panchanathan, R. Absent in Melanoma 2 proteins in SLE. Clin. Immunol. 2017, 176, 42–48. [Google Scholar] [CrossRef] [Green Version]
- Man, S.M.; Karki, R.; Kanneganti, T.D. AIM2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur. J. Immunol. 2016, 46, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Diner, B.A.; Lum, K.K.; Toettcher, J.E.; Cristea, I.M. Viral DNA sensors IFI16 and cyclic GMP-AMP synthase possess distinct functions in regulating viral gene expression, immune defenses, and apoptotic responses during herpesvirus infection. MBio 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.E.; Chikoti, L.; Chandran, B. Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J. Virol. 2013, 87, 5005–5018. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.V.; Kerur, N.; Bottero, V.; Dutta, S.; Chakraborty, S.; Ansari, M.A.; Paudel, N.; Chikoti, L.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus latency in endothelial and B cells activates gamma interferon-inducible protein 16-mediated inflammasomes. J. Virol. 2013, 87, 4417–4431. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Dutta, D.; Iqbal, J.; Pisano, G.; Gjyshi, O.; Ansari, M.A.; Kumar, B.; Chandran, B. Nuclear innate immune DNA sensor IFI16 is degraded during lytic reactivation of Kaposi’s sarcoma-associated herpesvirus (KSHV): Role of IFI16 in maintenance of KSHV latency. J. Virol. 2016, 90, 8822–8841. [Google Scholar] [CrossRef] [Green Version]
- Jakobsen, M.R.; Bak, R.O.; Andersen, A.; Berg, R.K.; Jensen, S.B.; Jin, T.; Laustsen, A.; Hansen, K.; Østergaard, L.; Fitzgerald, K.A.; et al. IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc. Natl. Acad. Sci. USA 2013, 110, E4571–E4580. [Google Scholar] [CrossRef] [Green Version]
- Monroe, K.M.; Yang, Z.; Johnson, J.R.; Geng, X.; Doitsh, G.; Krogan, N.J.; Greene, W.C. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 2014, 343, 428–432. [Google Scholar] [CrossRef] [Green Version]
- Caneparo, V.; Landolfo, S.; Gariglio, M.; De Andrea, M. The absent in melanoma 2-like receptor IFN-inducible protein 16 as an inflammasome regulator in systemic lupus erythematosus: The dark side of sensing microbes. Front. Immunol. 2018, 9, 1180. [Google Scholar] [CrossRef]
- Loeven, N.A.; Medici, N.P.; Bliska, J.B. The pyrin inflammasome in host–microbe interactions. Curr. Opin. Microbiol. 2020, 54, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Wood, G.; Kastner, D.L.; Chae, J.J. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat. Immunol. 2016, 17, 914. [Google Scholar] [CrossRef] [PubMed]
- Jamilloux, Y.; Magnotti, F.; Belot, A.; Henry, T. The pyrin inflammasome: From sensing RhoA GTPases-inhibiting toxins to triggering autoinflammatory syndromes. Pathog. Dis. 2018, 76, fty020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belkhir, R.; Moulonguet-Doleris, L.; Hachulla, E.; Prinseau, J.; Baglin, A.; Hanslik, T. Treatment of familial Mediterranean fever with anakinra. Ann. Intern. Med. 2007, 146, 825–826. [Google Scholar] [CrossRef] [PubMed]
- Moghaddas, F.; Llamas, R.; De Nardo, D.; Martinez-Banaclocha, H.; Martinez-Garcia, J.J.; Mesa-del-Castillo, P.; Baker, P.J.; Gargallo, V.; Mensa-Vilaro, A.; Canna, S.; et al. A novel Pyrin-Associated Autoinflammation with Neutrophilic Dermatosis mutation further defines 14–3-3 binding of pyrin and distinction to Familial Mediterranean Fever. Ann. Rheum. Dis. 2017, 76, 2085–2094. [Google Scholar] [CrossRef]
- Lacey, C.A.; Mitchell, W.J.; Dadelahi, A.S.; Skyberg, J.A. Caspase-1 and caspase-11 mediate pyroptosis, inflammation, and control of Brucella joint infection. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef] [Green Version]
- Flood, B.; Oficjalska, K.; Laukens, D.; Fay, J.; O’Grady, A.; Caiazza, F.; Heetun, Z.; Mills, K.; Sheahan, K.; Ryan, E.J.; et al. Altered expression of caspases-4 and-5 during inflammatory bowel disease and colorectal cancer: Diagnostic and therapeutic potential. Clin. Exp. Immunol. 2015, 181, 39–50. [Google Scholar] [CrossRef] [Green Version]
- de Carvalho, R.V.; Andrade, W.A.; Lima-Junior, D.S.; Dilucca, M.; de Oliveira, C.V.; Wang, K.; Nogueira, P.M.; Rugani, J.N.; Soares, R.P.; Beverley, S.M.; et al. Leishmania lipophosphoglycan triggers caspase-11 and the non-canonical activation of the NLRP3 inflammasome. Cell Rep. 2019, 26, 429–437. [Google Scholar] [CrossRef] [Green Version]
- Yi, Y.S.; Son, Y.J.; Ryou, C.; Sung, G.H.; Kim, J.H.; Cho, J.Y. Functional roles of Syk in macrophage-mediated inflammatory responses. Mediat. Inflamm. 2014, 270302. [Google Scholar] [CrossRef] [Green Version]
- Gabrielli, E.; Pericolini, E.; Luciano, E.; Sabbatini, S.; Roselletti, E.; Perito, S.; Kasper, L.; Hube, B.; Vecchiarelli, A. Induction of caspase-11 by aspartyl proteinases of Candida albicans and implication in promoting inflammatory response. Infect. Immun. 2015, 83, 1940–1948. [Google Scholar] [CrossRef] [Green Version]
- Hisahara, S.; Yuan, J.; Momoi, T.; Okano, H.; Miura, M. Caspase-11 mediates oligodendrocyte cell death and pathogenesis of autoimmune-mediated demyelination. J. Exp. Med. 2001, 193, 111–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.J.; Sanchez, I.; Jing, N.; Yuan, J. Dissociation between neurodegeneration and caspase-11-mediated activation of caspase-1 and caspase-3 in a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 2003, 23, 5455–5460. [Google Scholar] [CrossRef] [PubMed]
- Furuya, T.; Hayakawa, H.; Yamada, M.; Yoshimi, K.; Hisahara, S.; Miura, M.; Mizuno, Y.; Mochizuki, H. Caspase-11 mediates inflammatory dopaminergic cell death in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. J. Neurosci. 2004, 24, 1865–1872. [Google Scholar] [CrossRef] [PubMed]
- Demon, D.; Kuchmiy, A.; Fossoul, A.; Zhu, Q.; Kanneganti, T.D.; Lamkanfi, M. Caspase-11 is expressed in the colonic mucosa and protects against dextran sodium sulfate-induced colitis. Mucosal Immunol. 2014, 7, 1480–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, T.M.; Leeth, R.A.; Rothschild, D.E.; McDaniel, D.K.; Coutermarsh-Ott, S.L.; Simmons, A.E.; Kable, K.H.; Heid, B.; Allen, I.C. Caspase-11 attenuates gastrointestinal inflammation and experimental colitis pathogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G139–G150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, K.T.; Xiong, S.; Ye, Z.; Hong, Z.; Di, A.; Tsang, K.M.; Gao, X.; An, S.; Mittal, M.; Vogel, S.M.; et al. Caspase-11–mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J. Clin. Investig. 2017, 127, 4124–4135. [Google Scholar] [CrossRef]
- Eltom, S.; Belvisi, M.G.; Stevenson, C.S.; Maher, S.A.; Dubuis, E.; Fitzgerald, K.A.; Birrell, M.A. Role of the inflammasome-caspase1/11-IL-1/18 axis in cigarette smoke driven airway inflammation: An insight into the pathogenesis of COPD. PLoS ONE 2014, 9, e112829. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, W.; Harton, J.A.; Zhu, X.; Linhoff, M.W.; Ting, J.P. Cutting edge: CIAS1/cryopyrin/PYPAF1/NALP3/CATERPILLER 1.1 is an inducible inflammatory mediator with NF-κB suppressive properties. J. Immunol. 2003, 171, 6329–6333. [Google Scholar] [CrossRef] [Green Version]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A. Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- He, Q.; You, H.; Li, X.M.; Liu, T.H.; Wang, P.; Wang, B.E. HMGB1 promotes the synthesis of pro-IL-1β and pro-IL-18 by activation of p38 MAPK and NF-κB through receptors for advanced glycation end-products in macrophages. Asian Pac. J. Cancer Prev. 2012, 13, 1365–1370. [Google Scholar] [CrossRef] [Green Version]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghonime, M.G.; Shamaa, O.R.; Das, S.; Eldomany, R.A.; Fernandes-Alnemri, T.; Alnemri, E.S.; Gavrilin, M.A.; Wewers, M.D. Inflammasome priming by lipopolysaccharide is dependent upon ERK signaling and proteasome function. J. Immunol. 2014, 192, 3881–3888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Py, B.F.; Kim, M.-S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 2013, 49, 331–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization, Weekly Operational Update-14 December 2020. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update (accessed on 14 December 2020).
- Wu, Z.; McGoogan, J.M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: Summary of a report of 72 314 cases from the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef]
- Chi, Y.; Ge, Y.; Wu, B.; Zhang, W.; Wu, T.; Wen, T.; Liu, J.; Guo, X.; Huang, C.; Jiao, Y.; et al. Serum cytokine and chemokine profile in relation to the severity of coronavirus disease 2019 in China. J. Infect. Dis. 2020, 222, 746–754. [Google Scholar] [CrossRef]
- Han, Y.; Zhang, H.; Mu, S.; Wei, W.; Jin, C.; Tong, C.; Song, Z.; Zha, Y.; Xue, Y.; Gu, G. Lactate dehydrogenase, an independent risk factor of severe COVID-19 patients: A retrospective and observational study. Aging 2020, 12, 11245. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, T.S.; de Sá, K.S.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Gonçalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2020, 218, e20201707. [Google Scholar] [CrossRef]
- Bae, J.Y.; Park, H.H. Crystal structure of NALP3 protein pyrin domain (PYD) and its implications in inflammasome assembly. J. Biol. Chem. 2011, 286, 39528–39536. [Google Scholar] [CrossRef] [Green Version]
- Oroz, J.; Barrera-Vilarmau, S.; Alfonso, C.; Rivas, G.; de Alba, E. ASC pyrin domain self-associates and binds NLRP3 protein using equivalent binding interfaces. J. Biol. Chem. 2016, 291, 19487–19501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinheiro, A.S.; Eibl, C.; Ekman-Vural, Z.; Schwarzenbacher, R.; Peti, W. The NLRP12 pyrin domain: Structure, dynamics, and functional insights. J. Mol. Biol. 2011, 413, 790–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiller, S.; Kohl, A.; Fiorito, F.; Herrmann, T.; Wider, G.; Tschopp, J.; Grütter, M.G.; Wüthrich, K. NMR structure of the apoptosis-and inflammation-related NALP1 pyrin domain. Structure 2003, 11, 1199–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinheiro, A.S.; Proell, M.; Eibl, C.; Page, R.; Schwarzenbacher, R.; Peti, W. Three-dimensional Structure of the NLRP7 pyrin domain insight into pyrin-pyrin-mediated effector domain signaling in innate immunity. J. Biol. Chem. 2010, 285, 27402–27410. [Google Scholar] [CrossRef] [Green Version]
- Eibl, C.; Grigoriu, S.; Hessenberger, M.; Wenger, J.; Puehringer, S.; Pinheiro, A.S.; Wagner, R.N.; Proell, M.; Reed, J.C.; Page, R. Structural and functional analysis of the NLRP4 pyrin domain. Biochemistry 2012, 51, 7330–7341. [Google Scholar] [CrossRef]
- Su, M.Y.; Kuo, C.I.; Chang, C.F.; Chang, C.I. Three-dimensional structure of human NLRP10/PYNOD pyrin domain reveals a homotypic interaction site distinct from its mouse homologue. PLoS ONE 2013, 8, e67843. [Google Scholar] [CrossRef]
- Sharif, H.; Wang, L.; Wang, W.L.; Magupalli, V.G.; Andreeva, L.; Qiao, Q.; Hauenstein, A.V.; Wu, Z.; Nunez, G.; Mao, Y.; et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 2019, 570, 338–343. [Google Scholar] [CrossRef]
- Sušjan, P.; Roškar, S.; Hafner-Bratkovič, I. The mechanism of NLRP3 inflammasome initiation: Trimerization but not dimerization of the NLRP3 pyrin domain induces robust activation of IL-1β. Biochem. Biophys. Res. Commun. 2017, 483, 823–828. [Google Scholar] [CrossRef]
- Jin, T.; Huang, M.; Jiang, J.; Smith, P.; Xiao, T.S. Crystal structure of human NLRP12 PYD domain and implication in homotypic interaction. PLoS ONE 2018, 13, e0190547. [Google Scholar] [CrossRef] [Green Version]
- Hafner-Bratkovič, I.; Sušjan, P.; Lainšček, D.; Tapia-Abellán, A.; Cerović, K.; Kadunc, L.; Angosto-Bazarra, D.; Pelegrin, P.; Jerala, R. NLRP3 lacking the leucine-rich repeat domain can be fully activated via the canonical inflammasome pathway. Nat. Commun. 2018, 9, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Stutz, A.; Kolbe, C.C.; Stahl, R.; Horvath, G.L.; Franklin, B.S.; van Ray, O.; Brinkschulte, R.; Geyer, M.; Meissner, F.; Latz, E. NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J. Exp. Med. 2017, 214, 1725–1736. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Lear, T.B.; Jerome, J.A.; Rajbhandari, S.; Snavely, C.A.; Gulick, D.L.; Gibson, K.F.; Zou, C.; Chen, B.B.; Mallampalli, R.K. Lipopolysaccharide primes the NALP3 inflammasome by inhibiting its ubiquitination and degradation mediated by the SCFFBXL2 E3 ligase. J. Biol. Chem. 2015, 290, 18124–18133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spalinger, M.R.; Kasper, S.; Gottier, C.; Lang, S.; Atrott, K.; Vavricka, S.R.; Scharl, S.; Gutte, P.M.; Grütter, M.G.; Beer, H.D.; et al. NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J. Clin. Investig. 2016, 126, 1783–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruchard, M.; Rebé, C.; Derangère, V.; Togbé, D.; Ryffel, B.; Boidot, R.; Humblin, E.; Hamman, A.; Chalmin, F.; Berger, H.; et al. The receptor NLRP3 is a transcriptional regulator of TH 2 differentiation. Nat. Immunol. 2015, 16, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Liu, Z.S.; Xue, W.; Bai, Z.F.; Wang, Q.Y.; Dai, J.; Liu, X.; Huang, Y.J.; Cai, H.; Zhan, X.Y.; et al. NLRP3 phosphorylation is an essential priming event for inflammasome activation. Mol. Cell 2017, 68, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Meszaros, G.; He, W.; Xu, Y.; de Fatima Magliarelli, H.; Mailly, L.; Mihlan, M.; Liu, Y.; Puig Gámez, M.; Goginashvili, A.; et al. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J. Exp. Med. 2017, 214, 2671–2693. [Google Scholar] [CrossRef]
- Guo, C.; Xie, S.; Chi, Z.; Zhang, J.; Liu, Y.; Zhang, L.; Zheng, M.; Zhang, X.; Xia, D.; Ke, Y.; et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity 2016, 45, 802–816. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Jiang, W.; Liu, L.; Wang, X.; Ding, C.; Tian, Z.; Zhou, R. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 2015, 160, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Humphries, F.; Bergin, R.; Jackson, R.; Delagic, N.; Wang, B.; Yang, S.; Dubois, A.V.; Ingram, R.J.; Moynagh, P.N. The E3 ubiquitin ligase Pellino2 mediates priming of the NLRP3 inflammasome. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Yen, H.; Sugimoto, N.; Tobe, T. Enteropathogenic Escherichia coli uses NleA to inhibit NLRP3 inflammasome activation. PLoS Pathog 2015, 11, e1005121. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Shi, X.; Qian, T.; Li, J.; Tian, Z.; Ni, B.; Hao, F. A20 overexpression alleviates pristine-induced lupus nephritis by inhibiting the NF-κB and NLRP3 inflammasome activation in macrophages of mice. Int. J. Clin. Exp. Med. 2015, 8, 17430. [Google Scholar] [PubMed]
- Walle, L.V.; Van Opdenbosch, N.; Jacques, P.; Fossoul, A.; Verheugen, E.; Vogel, P.; Beyaert, R.; Elewaut, D.; Kanneganti, T.D.; van Loo, G.; et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 2014, 512, 69–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Liu, B.; Huai, W.; Yu, Z.; Wang, W.; Zhao, J.; Han, L.; Jiang, G.; Zhang, L.; Gao, C.; et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Mimuro, H.; Kim, M.; Ogawa, M.; Ashida, H.; Toyotome, T.; Franchi, L.; Suzuki, M.; Sanada, T.; Suzuki, T.; et al. Shigella IpaH7. 8 E3 ubiquitin ligase targets glomulin and activates inflammasomes to demolish macrophages. Proc. Natl. Acad. Sci. USA 2014, 111, E4254–E4263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Wang, Y.; Li, X.; Zhan, X.; Tang, M.; Fina, M.; Su, L.; Pratt, D.; Bu, C.H.; Hildebrand, S.; et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 2016, 17, 250–258. [Google Scholar] [CrossRef]
- Hu, Z.; Zhou, Q.; Zhang, C.; Fan, S.; Cheng, W.; Zhao, Y.; Shao, F.; Wang, H.W.; Sui, S.F.; Chai, J. Structural and biochemical basis for induced self-propagation of NLRC4. Science 2015, 350, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, S.; Ruan, J.; Wu, J.; Tong, A.B.; Yin, Q.; Li, Y.; David, L.; Lu, A.; Wang, W.L.; et al. Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science 2015, 350, 404–409. [Google Scholar] [CrossRef] [Green Version]
- Feltham, R.; Vince, J.E.; Lawlor, K.E. Caspase-8: Not so silently deadly. Clin. Transl. Immunol. 2017, 6, e124. [Google Scholar] [CrossRef]
- Lee, K.H.; Kang, T.B. The molecular links between cell death and inflammasome. Cells 2019, 8, 1057. [Google Scholar] [CrossRef] [Green Version]
- Maelfait, J.; Vercammen, E.; Janssens, S.; Schotte, P.; Haegman, M.; Magez, S.; Beyaert, R. Stimulation of Toll-like receptor 3 and 4 induces interleukin-1β maturation by caspase-8. J. Exp. Med. 2008, 205, 1967–1973. [Google Scholar] [CrossRef] [Green Version]
- Moriwaki, K.; Bertin, J.; Gough, P.J.; Chan, F.K. A RIPK3–caspase 8 complex mediates atypical pro–IL-1β processing. J. Immunol. 2015, 194, 1938–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stammler, D.; Eigenbrod, T.; Menz, S.; Frick, J.S.; Sweet, M.J.; Shakespear, M.R.; Jantsch, J.; Siegert, I.; Wölfle, S.; Langer, J.D.; et al. Inhibition of Histone Deacetylases Permits Lipopolysaccharide-Mediated Secretion of Bioactive IL-1β via a Caspase-1–Independent Mechanism. J. Immunol. 2015, 195, 5421–5431. [Google Scholar] [CrossRef] [PubMed]
- Bossaller, L.; Chiang, P.I.; Schmidt-Lauber, C.; Ganesan, S.; Kaiser, W.J.; Rathinam, V.A.; Mocarski, E.S.; Subramanian, D.; Green, D.R.; Silverman, N.; et al. Cutting edge: FAS (CD95) mediates noncanonical IL-1β and IL-18 maturation via caspase-8 in an RIP3-independent manner. J. Immunol. 2012, 189, 5508–5512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gringhuis, S.I.; Kaptein, T.M.; Wevers, B.A.; Theelen, B.; Van Der Vlist, M.; Boekhout, T.; Geijtenbeek, T.B. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat. Immunol. 2012, 13, 246. [Google Scholar] [CrossRef] [PubMed]
- Shenderov, K.; Riteau, N.; Yip, R.; Mayer-Barber, K.D.; Oland, S.; Hieny, S.; Fitzgerald, P.; Oberst, A.; Dillon, C.P.; Green, D.R.; et al. ER stress licenses macrophages to produce mature IL-1β in response to TLR4 stimulation through a caspase-8-and TRIF-dependent pathway. J. Immunol. 2014, 192, 2029. [Google Scholar] [CrossRef]
- Antonopoulos, C.; El Sanadi, C.; Kaiser, W.J.; Mocarski, E.S.; Dubyak, G.R. Proapoptotic chemotherapeutic drugs induce noncanonical processing and release of IL-1β via caspase-8 in dendritic cells. J. Immunol. 2013, 191, 4789–4803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.H.; Kuo, W.C.; Wu, Y.; Yang, K.; Chen, S.T.; Jiang, S.T.; Gordy, C.; He, Y.W.; Lai, M.Z. Differentiation, Participation of c-FLIP in NLRP3 and AIM2 inflammasome activation. Cell Death Differ. 2014, 21, 451–461. [Google Scholar] [CrossRef] [Green Version]
- Gurung, P.; Kanneganti, T.D. Novel roles for caspase-8 in IL-1β and inflammasome regulation. Am. J. Pathol. 2015, 185, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Gurung, P.; Anand, P.K.; Malireddi, R.S.; Walle, L.V.; Van Opdenbosch, N.; Dillon, C.P.; Weinlich, R.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.D. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 2014, 192, 1835–1846. [Google Scholar] [CrossRef] [Green Version]
- Vince, J.E.; Wong, W.W.L.; Gentle, I.; Lawlor, K.E.; Allam, R.; O’Reilly, L.; Mason, K.; Gross, O.; Ma, S.; Guarda, G.; et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 2012, 36, 215–227. [Google Scholar] [CrossRef] [Green Version]
- Lawlor, K.E.; Khan, N.; Mildenhall, A.; Gerlic, M.; Croker, B.A.; D’Cruz, A.A.; Hall, C.; Spall, S.K.; Anderton, H.; Masters, S.L.; et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 2015, 6, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.; Fernandes-Alnemri, T.; Rogers, C.; Mayes, L.; Wang, Y.; Dillon, C.; Roback, L.; Kaiser, W.; Oberst, A.; Sagara, J.; et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat. Commun. 2015, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.B.; Yang, S.H.; Toth, B.; Kovalenko, A.; Wallach, D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 2013, 38, 27–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Schmidt, T.; Schmid-Burgk, J.L.; Rapino, F.; Robertson, A.A.; Cooper, M.A.; Graf, T.; Hornung, V. Human monocytes engage an alternative inflammasome pathway. Immunity 2016, 44, 833–846. [Google Scholar] [CrossRef] [Green Version]
- Conos, S.A.; Chen, K.W.; De Nardo, D.; Hara, H.; Whitehead, L.; Núñez, G.; Masters, S.L.; Murphy, J.M.; Schroder, K.; Vaux, D.L.; et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. USA 2017, 114, E961–E969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A.; et al. Pathogen blockade of TAK1 triggers caspase-8–dependent cleavage of gasdermin D and cell death. Science 2018, 362, 1064–1069. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.W.; Demarco, B.; Heilig, R.; Shkarina, K.; Boettcher, A.; Farady, C.J.; Pelczar, P.; Broz, P. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP 3 inflammasome assembly. EMBO J. 2019, 38, e101638. [Google Scholar] [CrossRef]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef] [Green Version]
- Vajjhala, P.R.; Lu, A.; Brown, D.L.; Pang, S.W.; Sagulenko, V.; Sester, D.P.; Cridland, S.O.; Hill, J.M.; Schroder, K.; Stow, J.L.; et al. The inflammasome adaptor ASC induces procaspase-8 death effector domain filaments. J. Biol. Chem. 2015, 290, 29217–29230. [Google Scholar] [CrossRef] [Green Version]
- Jin, T.; Perry, A.; Smith, P.; Jiang, J.; Xiao, T.S. Structure of the absent in melanoma 2 (AIM2) pyrin domain provides insights into the mechanisms of AIM2 autoinhibition and inflammasome assembly. J. Biol. Chem. 2013, 288, 13225–13235. [Google Scholar] [CrossRef] [Green Version]
- Jin, T.; Perry, A.; Jiang, J.; Smith, P.; Curry, J.A.; Unterholzner, L.; Jiang, Z.; Horvath, G.; Rathinam, V.A.; Johnstone, R.W.; et al. Structures of the HIN domain: DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity 2012, 36, 561–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, N.; Liu, Z.J. Role of the HIN domain in regulation of innate immune responses. Cell. Biol. 2014, 34, 2–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veeranki, S.; Duan, X.; Panchanathan, R.; Liu, H.; Choubey, D. IFI16 protein mediates the anti-inflammatory actions of the type-I interferons through suppression of activation of caspase-1 by inflammasomes. PLoS ONE 2011, 6, e27040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cresswell, K.S.; Clarke, C.J.; Jackson, J.T.; Darcy, P.K.; Trapani, J.A.; Johnstone, R.W. Biochemical and growth regulatory activities of the HIN-200 family member and putative tumor suppressor protein, AIM2. Biochem. Biophys. Res. Commun. 2005, 326, 417–424. [Google Scholar] [CrossRef]
- Yin, Q.; Sester, D.P.; Tian, Y.; Hsiao, Y.S.; Lu, A.; Cridland, J.A.; Sagulenko, V.; Thygesen, S.J.; Choubey, D.; Hornung, V.; et al. Molecular mechanism for p202-mediated specific inhibition of AIM2 inflammasome activation. Cell Rep. 2013, 4, 327–339. [Google Scholar] [CrossRef] [Green Version]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schröder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef] [Green Version]
- Sagulenko, V.; Thygesen, S.J.; Sester, D.P.; Idris, A.; Cridland, J.A.; Vajjhala, P.R.; Roberts, T.L.; Schroder, K.; Vince, J.E.; Hill, J.M.; et al. Differentiation, AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013, 20, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Lu, A.; Kabaleeswaran, V.; Fu, T.; Magupalli, V.G.; Wu, H. Crystal structure of the F27G AIM2 PYD mutant and similarities of its self-association to DED/DED interactions. J. Mol. Biol. 2014, 426, 1420–1427. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Niu, X. The NMR solution structure of AIM2 PYD domain from Mus musculus reveals a distinct α2–α3 helix conformation from its human homologues. Biochem. Biophys. Res. Commun. 2015, 461, 396–400. [Google Scholar] [CrossRef]
- Morrone, S.R.; Matyszewski, M.; Yu, X.; Delannoy, M.; Egelman, E.H.; Sohn, J. Assembly-driven activation of the AIM2 foreign-dsDNA sensor provides a polymerization template for downstream ASC. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef]
- Lu, A.; Li, Y.; Yin, Q.; Ruan, J.; Yu, X.; Egelman, E.; Wu, H. Plasticity in PYD assembly revealed by cryo-EM structure of the PYD filament of AIM2. Cell Discov. 2015, 1, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ru, H.; Ni, X.; Zhao, L.; Crowley, C.; Ding, W.; Hung, L.W.; Shaw, N.; Cheng, G.; Liu, Z.J. Structural basis for termination of AIM2-mediated signaling by p202. Cell Res. 2013, 23, 855–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matyszewski, M.; Morrone, S.R.; Sohn, J. Digital signaling network drives the assembly of the AIM2-ASC inflammasome. Proc. Natl. Acad. Sci. USA 2018, 115, E1963–E1972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choubey, D.; Walter, S.; Geng, Y.; Xin, H. Cytoplasmic localization of the interferon-inducible protein that is encoded by the AIM2 (absent in melanoma) gene from the 200-gene family. FEBS Lett. 2000, 474, 38–42. [Google Scholar] [CrossRef] [Green Version]
- Koul, D.; Obeyesekere, N.U.; Gutterman, J.U.; Mills, G.B.; Choubey, D. p202 self-associates through a sequence conserved among the members of the 200-family proteins. FEBS Lett. 1998, 438, 21–24. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.H.; Ye, Z.W.; Deng, J.J.; Siu, K.L.; Gao, W.W.; Chaudhary, V.; Cheng, Y.; Fung, S.Y.; Yuen, K.S.; Ho, T.H.; et al. Inhibition of AIM 2 inflammasome activation by a novel transcript isoform of IFI 16. EMBO Rep. 2018, 19, e45737. [Google Scholar] [CrossRef]
- Liu, T.; Tang, Q.; Liu, K.; Xie, W.; Liu, X.; Wang, H.; Wang, R.F.; Cui, J. TRIM11 suppresses AIM2 inflammasome by degrading AIM2 via p62-dependent selective autophagy. Cell Rep. 2016, 16, 1988–2002. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Liu, Z.; Xiao, T.S. Post-translational regulation of inflammasomes. Cel. Mol. Immunol. 2017, 14, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.J.; Zhao, Q.C.; Xia, M.X.; Chen, J.; Chen, Y.T.; Cao, X.; Liu, Y.; Yuan, Z.Q.; Wang, X.Y.; Xu, Y. The HDAC3 inhibitor RGFP966 ameliorated ischemic brain damage by downregulating the AIM2 inflammasome. FASEB J. 2020, 34, 648–662. [Google Scholar] [CrossRef] [Green Version]
- Masumoto, J.; Taniguchi, S.I.; Ayukawa, K.; Sarvotham, H.; Kishino, T.; Niikawa, N.; Hidaka, E.; Katsuyama, T.; Higuchi, T.; Sagara, J. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J. Biol. Chem. 1999, 274, 33835–33838. [Google Scholar] [CrossRef] [Green Version]
- Sahillioglu, A.C.; Sumbul, F.; Ozoren, N.; Haliloglu, T. Structural and dynamics aspects of ASC speck assembly. Structure 2014, 22, 1722–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Alba, E. Structure and interdomain dynamics of apoptosis-associated speck-like protein containing a CARD (ASC). J. Biol. Chem. 2009, 284, 32932–32941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConnell, B.B.; Vertino, P.M. Activation of a caspase-9-mediated apoptotic pathway by subcellular redistribution of the novel caspase recruitment domain protein TMS1. Cancer Res. 2000, 60, 6243–6247. [Google Scholar] [PubMed]
- Fernandes-Alnemri, T.; Wu, J.; Yu, J.; Datta, P.; Miller, B.; Jankowski, W.; Rosenberg, S.; Zhang, J.; Alnemri, E.S. Differentiation, The pyroptosome: A supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007, 14, 1590–1604. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Salvesen, G. Regulated cell death: Signaling and mechanisms. Cell Dev. Biol. 2014, 30, 337–356. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef]
- Bryan, N.B.; Dorfleutner, A.; Kramer, S.J.; Yun, C.; Rojanasakul, Y.; Stehlik, C. Differential splicing of the apoptosis-associated speck like protein containing a caspase recruitment domain (ASC) regulates inflammasomes. J. Inflamm. 2010, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Liepinsh, E.; Barbals, R.; Dahl, E.; Sharipo, A.; Staub, E.; Otting, G. The death-domain fold of the ASC PYRIN domain, presenting a basis for PYRIN/PYRIN recognition. J. Mol. Biol. 2003, 332, 1155–1163. [Google Scholar] [CrossRef] [Green Version]
- de Alba, E. Structure, interactions and self-assembly of ASC-dependent inflammasomes. Arch. Biochem. Biophys. 2019, 670, 15–31. [Google Scholar] [CrossRef]
- Moriya, M.; Taniguchi, S.; Wu, P.; Liepinsh, E.; Otting, G.; Sagara, J. Role of charged and hydrophobic residues in the oligomerization of the PYRIN domain of ASC. Biochemistry 2005, 44, 575–583. [Google Scholar] [CrossRef]
- Dick, M.S.; Sborgi, L.; Rühl, S.; Hiller, S.; Broz, P. ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, A.; Wu, H. Structural mechanisms of inflammasome assembly. FEBS J. 2015, 282, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Sborgi, L.; Ravotti, F.; Dandey, V.P.; Dick, M.S.; Mazur, A.; Reckel, S.; Chami, M.; Scherer, S.; Huber, M.; Böckmann, A.; et al. Structure and assembly of the mouse ASC inflammasome by combined NMR spectroscopy and cryo-electron microscopy. Proc. Natl. Acad. Sci. USA 2015, 112, 13237–13242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Chou, J.; Olea, R.S.; Yuan, J.; Wagner, G. Solution structure of Apaf-1 CARD and its interaction with caspase-9 CARD: A structural basis for specific adaptor/caspase interaction. Proc. Natl. Acad. Sci. USA 1999, 96, 11265–11270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, H.; Srinivasula, S.M.; Wu, G.; Fernandes-Alnemri, T.; Alnemri, E.S.; Shi, Y. Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature 1999, 399, 549–557. [Google Scholar] [CrossRef]
- Park, H.H.; Lo, Y.C.; Lin, S.C.; Wang, L.; Yang, J.K.; Wu, H. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu. Rev. Immunol. 2007, 25, 561–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nambayan, R.J.T.; Sandin, S.I.; Quint, D.A.; Satyadi, D.M.; de Alba, E. The inflammasome adapter ASC assembles into filaments with integral participation of its two Death Domains, PYD and CARD. J. Biol. Chem. 2019, 294, 439–452. [Google Scholar] [CrossRef] [Green Version]
- Humke, E.W.; Shriver, S.K.; Starovasnik, M.A.; Fairbrother, W.J.; Dixit, V.M. ICEBERG: A novel inhibitor of interleukin-1β generation. Cell 2000, 103, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Chou, J.J.; Matsuo, H.; Duan, H.; Wagner, G. Solution structure of the RAIDD CARD and model for CARD/CARD interaction in caspase-2 and caspase-9 recruitment. Cell 1998, 94, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Manon, F.; Favier, A.; Núñez, G.; Simorre, J.P.; Cusack, S. Solution structure of NOD1 CARD and mutational analysis of its interaction with the CARD of downstream kinase RICK. J. Mol. Biol. 2007, 365, 160–174. [Google Scholar] [CrossRef]
- Li, Y.; Fu, T.M.; Lu, A.; Witt, K.; Ruan, J.; Shen, C.; Wu, H. Cryo-EM structures of ASC and NLRC4 CARD filaments reveal a unified mechanism of nucleation and activation of caspase-1. Proc. Natl. Acad. Sci. USA 2018, 115, 10845–10852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matyszewski, M.; Zheng, W.; Lueck, J.; Antiochos, B.; Egelman, E.H.; Sohn, J. Cryo-EM structure of the NLRC4CARD filament provides insights into how symmetric and asymmetric supramolecular structures drive inflammasome assembly. J. Biol. Chem. 2018, 293, 20240–20248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambin, Y.; Giles, N.; O’Carroll, A.; Polinkovsky, M.; Hunter, D.; Sierecki, E. Single-molecule fluorescence reveals the oligomerization and folding steps driving the prion-like behavior of ASC. J. Mol. Biol. 2018, 430, 491–508. [Google Scholar] [CrossRef] [Green Version]
- Vajjhala, P.R.; Mirams, R.E.; Hill, J.M. Multiple binding sites on the pyrin domain of ASC protein allow self-association and interaction with NLRP3 protein. J. Biol. Chem. 2012, 287, 41732–41743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proell, M.; Gerlic, M.; Mace, P.D.; Reed, J.C.; Riedl, S.J. The CARD plays a critical role in ASC foci formation and inflammasome signalling. Biochem. J. 2013, 449, 613–621. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.K.; Wang, L.; Zheng, L.; Wan, F.; Ahmed, M.; Lenardo, M.J.; Wu, H. Crystal structure of MC159 reveals molecular mechanism of DISC assembly and FLIP inhibition. Mol. Cell 2005, 20, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Stehlik, C.; Krajewska, M.; Welsh, K.; Krajewski, S.; Godzik, A.; Reed, J.C. The PAAD/PYRIN-only protein POP1/ASC2 is a modulator of ASC-mediated nuclear-factor-kappaB and pro-caspase-1 regulation. Biochem. J. 2003, 373, 101–113. [Google Scholar] [CrossRef]
- Srimathi, T.; Robbins, S.L.; Dubas, R.L.; Chang, H.; Cheng, H.; Roder, H.; Park, Y.C. Mapping of POP1-binding site on pyrin domain of ASC. J. Biol. Chem. 2008, 283, 15390–15398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mambwe, B.; Neo, K.; Javanmard Khameneh, H.; Leong, K.W.K.; Vacca, M.; Muimo, R.; Mortellaro, A. Tyrosine Dephosphorylation of ASC Modulates the Activation of the NLRP3 and AIM2 Inflammasomes. Front. Immunol. 2019, 10, 1556. [Google Scholar] [CrossRef]
- Tokunaga, F.; Iwai, K. Involvement of LUBAC-mediated linear polyubiquitination of NEMO in NF-kappaB activation. Tanpakushitsu Kakusan Koso Protein Nucleic Acid Enzym. 2009, 54, 635–642. [Google Scholar]
- Rodgers, M.A.; Bowman, J.W.; Fujita, H.; Orazio, N.; Shi, M.; Liang, Q.; Amatya, R.; Kelly, T.J.; Iwai, K.; Ting, J.; et al. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J. Exp. Med. 2014, 211, 1333–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, K.; Wei, C.; Zheng, Z.; Song, T.; Wu, F.; Zhang, Y.; Cao, Y.; Ma, S.; Chen, W.; Xu, Q.; et al. MAVS promotes inflammasome activation by targeting ASC for K63-linked ubiquitination via the E3 ligase TRAF3. J. Immunol. 2015, 194, 4880–4890. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, Y.; Suh, G.Y.; Lee, Y.S. Mul1 suppresses Nlrp3 inflammasome activation through ubiquitination and degradation of ASC. BioRxiv 2019, 830380. [Google Scholar] [CrossRef] [Green Version]
- Nuñez, G.; Benedict, M.A.; Hu, Y.; Inohara, N. Caspases: The proteases of the apoptotic pathway. Oncogene 1998, 17, 3237–3245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, R.A.; Kronheim, S.R.; Merriam, J.E.; March, C.J.; Hopp, T.P. A pre-aspartate-specific protease from human leukocytes that cleaves pro-interleukin-1 beta. J. Biol. Chem. 1989, 264, 5323–5326. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Bull, H.G.; Calaycay, J.R.; Chapman, K.T.; Howard, A.D.; Kostura, M.J.; Miller, D.K.; Molineaux, S.M.; Weidner, J.R.; Aunins, J.; et al. A novel heterodimeric cysteine protease is required for interleukin-1βprocessing in monocytes. Nature 1992, 356, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.M.; Rouge, L.; Wiesmann, C.; Scheer, J.M. Crystal structure of procaspase-1 zymogen domain reveals insight into inflammatory caspase autoactivation. J. Biol. Chem. 2009, 284, 6546–6553. [Google Scholar] [CrossRef] [Green Version]
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.A.; Holley, C.L.; Bierschenk, D.; Stacey, K.J.; et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med. 2018, 215, 827–840. [Google Scholar] [CrossRef]
- Romanowski, M.J.; Scheer, J.M.; O’Brien, T.; McDowell, R.S. Crystal structures of a ligand-free and malonate-bound human caspase-1: Implications for the mechanism of substrate binding. Structure 2004, 12, 1361–1371. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Chang, H.Y.; Baltimore, D. Autoproteolytic activation of pro-caspases by oligomerization. Mol. Cell 1998, 1, 319–325. [Google Scholar] [CrossRef]
- Walker, N.; Talanian, R.; Brady, K.; Dang, L.; Bump, N.; Ferenza, C.; Franklin, S.; Ghayur, T.; Hackett, M.; Hammill, L.; et al. Crystal structure of the cysteine protease interleukin-1β-converting enzyme: A (p20/p10) 2 homodimer. Cell 1994, 78, 343–352. [Google Scholar] [CrossRef]
- Wilson, K.P.; Black, J.A.F.; Thomson, J.A.; Kim, E.E.; Griffith, J.P.; Navia, M.A.; Murcko, M.A.; Chambers, S.P.; Aldape, R.A.; Raybuck, S.A.; et al. Structure and mechanism of interleukin-lβ converting enzyme. Nature 1994, 370, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yuan, J. Caspases in apoptosis and beyond. Oncogene 2008, 27, 6194–6206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentes-Prior, P.; Salvesen, G.S. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem. J. 2004, 384, 201–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Wu, J.; Faucheu, C.; Lalanne, J.; Diu, A.; Livingston, D.; Su, M.S. Interleukin-1 beta converting enzyme requires oligomerization for activity of processed forms in vivo. EMBO J. 1995, 14, 1923–1931. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Li, P.; Allen, H.; Banerjee, S.; Franklin, S.; Herzog, L.; Johnston, C.; McDowell, J.; Paskind, M.; Rodman, L.; Salfeld, J.; et al. Mice deficient in IL-1β-converting enzyme are defective in production of mature IL-1β and resistant to endotoxic shock. Cell 1995, 80, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [Green Version]
- Lu, A.; Li, Y.; Schmidt, F.I.; Yin, Q.; Chen, S.; Fu, T.M.; Tong, A.B.; Ploegh, H.L.; Mao, Y.; Wu, H. Molecular basis of caspase-1 polymerization and its inhibition by a new capping mechanism. Nat. Struct. Mol. Biol. 2016, 23, 416–425. [Google Scholar] [CrossRef] [Green Version]
- Ghayur, T.; Banerjee, S.; Hugunin, M.; Butler, D.; Herzog, L.; Carter, A.; Quintal, L.; Sekut, L.; Talanian, R.; Paskind, M. Caspase-1 processes IFN-γ-inducing factor and regulates LPS-induced IFN-γ production. Nature 1997, 386, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Xia, S.; Liu, X.; Lieberman, J.; Wu, H. Cryo-EM structure of the gasdermin A3 membrane pore. Nature 2018, 557, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Cookson, B.T. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell. Microbiol. 2006, 8, 1812–1825. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Wang, B.; Yin, Q. AIM2 inflammasome activation and regulation: A structural perspective. J. Struct. Biol. 2017, 200, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Lo, Y.C.; Wu, H. Helical assembly in the MyD88–IRAK4–IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Peisley, A.; Tetrault, D.; Li, Z.; Egelman, E.H.; Magor, K.E.; Walz, T.; Penczek, P.A.; Hur, S. Molecular imprinting as a signal-activation mechanism of the viral RNA sensor RIG-I. Mol. Cell 2014, 55, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Yin, Q.; Fu, T.M.; Li, J.; Wu, H. Structural biology of innate immunity. Annu. Rev. Immunol. 2015, 33, 393–416. [Google Scholar] [CrossRef] [Green Version]
- Le, H.T.; Harton, J.A. Pyrin-and CARD-only proteins as regulators of NLR functions. Front. Immunol. 2013, 4, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kersse, K.; Berghe, T.V.; Lamkanfi, M.; Vandenabeele, P. A phylogenetic and functional overview of inflammatory caspases and caspase-1-related CARD-only proteins. Biochem. Soc. Trans. 2007, 35, 1508–1511. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Denecker, G.; Kalai, M.; D’hondt, K.; Meeus, A.; Declercq, W.; Saelens, X.; Vandenabeele, P. INCA, a novel human caspase recruitment domain protein that inhibits interleukin-1β generation. J. Biol. Chem. 2004, 279, 51729–51738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kersse, K.; Lamkanfi, M.; Bertrand, M.J.; Berghe, T.V.; Vandenabeele, P. Interaction patches of procaspase-1 caspase recruitment domains (CARDs) are differently involved in procaspase-1 activation and receptor-interacting protein 2 (RIP2)-dependent nuclear factor κB signaling. J. Biol. Chem. 2011, 286, 35874–35882. [Google Scholar] [CrossRef] [Green Version]
- Druilhe, A.; Srinivasula, S.; Razmara, M.; Ahmad, M.; Alnemri, E. Differentiation, Regulation of IL-1β generation by Pseudo-ICE and ICEBERG, two dominant negative caspase recruitment domain proteins. Cell Death Differ. 2001, 8, 649–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufner, A.; Pownall, S.; Mak, T.W. Caspase recruitment domain protein 6 is a microtubule-interacting protein that positively modulates NF-κB activation. Proc. Natl. Acad. Sci. USA 2006, 103, 988–993. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Inohara, N.; Hernandez, L.D.; Galán, J.E.; Núñez, G.; Janeway, C.A.; Medzhitov, R.; Flavell, R.A. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature 2002, 416, 194–199. [Google Scholar] [CrossRef] [Green Version]
- Chin, A.I.; Dempsey, P.W.; Bruhn, K.; Miller, J.F.; Xu, Y.; Cheng, G. Involvement of receptor-interacting protein 2 in innate and adaptive immune responses. Nature 2002, 416, 190–194. [Google Scholar] [CrossRef]
- Yuan, S.; Akey, C.W. Apoptosome structure, assembly, and procaspase activation. Structure 2013, 21, 501–515. [Google Scholar] [CrossRef] [Green Version]
- Halff, E.F.; Diebolder, C.A.; Versteeg, M.; Schouten, A.; Brondijk, T.H.C.; Huizinga, E.G. Formation and structure of a NAIP5-NLRC4 inflammasome induced by direct interactions with conserved N-and C-terminal regions of flagellin. J. Biol. Chem. 2012, 287, 38460–38472. [Google Scholar] [CrossRef] [Green Version]
- Santiveri, C.M.; Oroz, J.; de Alba, E. A ring-like model for ASC self-association via the CARD domain. Inflammasome 2014, 1, 44–54. [Google Scholar]
- Wu, J.; Fernandes-Alnemri, T.; Alnemri, E.S. Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J. Clin. Immunol. 2010, 30, 693–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, D.; Chen, S.; Sun, R.; Zhang, X.; Wang, D. The NLRP3 inflammasome: Role in metabolic disorders and regulation by metabolic pathways. Cancer Lett. 2018, 419, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, A.; Frera, G.; Lugrin, J.; Jamilloux, Y.; Hsu, E.T.; Tardivel, A.; De Gassart, A.; Zaffalon, L.; Bujisic, B.; Siegert, S.; et al. AIM2 inflammasome is activated by pharmacological disruption of nuclear envelope integrity. Proc. Natl. Acad. Sci. USA 2016, 113, E4671–E4680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroder, K.; Muruve, D.A.; Tschopp, J. Innate immunity: Cytoplasmic DNA sensing by the AIM2 inflammasome. Curr. Biol. 2009, 19, R262–R265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Yissachar, N.; Salem, H.; Tennenbaum, T.; Motro, B. Nek7 kinase is enriched at the centrosome, and is required for proper spindle assembly and mitotic progression. FEBS Lett. 2006, 580, 6489–6495. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.J.; Liu, H.Y.; Huang, T.N.; Hsueh, Y.P. AIM 2 inflammasomes regulate neuronal morphology and influence anxiety and memory in mice. Scientific Report 2016, 6, 32405. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inflamma-some | Expression Site | Activation Signal | Diseases | Structure |
|---|---|---|---|---|
| Canonical Inflammasomes | ||||
| NLRP1 (NALP1, CARD7, CLR17.1, DEFCAP, VAMASI) | Adaptive immune cells and tissues, non- hematopoietic tissues | A/B toxin of Bacillus anthracis [31,32], Toxoplasma gondii infection [33], Muramyl dipeptide [28] | Vitiligo-associated multiple autoimmune disease [34], NLRP-1 associated autoinflammation with arthritis and dyskeratosis (NAIAD) [35], palmoplantar carcinoma and familial keratosis lichenoides chronica (FKLC) [36] |  |
| NLRP3 (Cryopyrin, NALP3, CIAS1, CLR1.1, PYPAF1) | Monocytes, neutrophils, dendritic cells, lymphocytes, osteoblasts, and epithelial cells | Pathogen-associated molecular patterns (PAMPs) (bacteria and virus) [37,38,39,40], danger-associated molecular patterns (DAMPs) such as Monosodium urate [41], alum [42], silica [43], asbestos [43], calcium/potassium efflux [44], extracellular ATP [39], reactive oxygen species (ROS) [45] | Cryopyrin-associated periodic fever syndrome (CAPS) [46], Muckle-Wells syndrome (MWS) [47], neonatal-onset multisystem inflammatory disease [48], Familial cold auto- inflammatory syndrome (FCAS) [49], Alzheimer’s disease [50] type II diabetes [51], cancer [52], chronic infantile neurological cutaneous and articular syndrome (CINCA, NOMID) [53] |  |
| NLRC4 (IPAF, CARD12, CLR2.1) | Macrophage and intestinal epithelial cells | Bacteria [54,55], Cytosolic flagellin [56] | Syndrome of enterocolitis and autoinflammation associated with mutation NLRC4 (SCAN4) [57], Macrophage activation syndrome (MAS) [58] |  |
| NLRP6 | Cells of intestine and liver | Viral RNA [59], LTA of Gram-positive bacteria [60,61] | Colitis and colitis-induced tumorigenesis [62] |  |
| NLRP12 (RNO, PYPAF7, Monarch-1) | Neutrophils, eosinophils, monocytes, macrophages, and dendritic cells | Bacterial components [15,63,64] | Familial cold auto- inflammatory syndrome 2 (FCAS2) [65] |  |
| AIM2 (PYHIN4) | Cytosol of hematopoietic cells | Bacterial and viral dsDNA [66,67,68,69] | Psoriasis [70], abdominal aortic aneurysm [71], systemic lupus erythematosus [72], prostate and colonic cancer [73] |  |
| IFI16 | Lymphocytes, monocytes, and epithelial cells | Viral and bacterial infections [74,75], Kaposi’s sarcoma-associated virus (KHSV) [76,77], HIV infections [78,79] | Systemic lupus erythematosus (SLE) [80] |  |
| Pyrin (Marenostrin, TRIM20) | Neutrophils, eosinophils, monocytes, dendritic cells and synovial fibroblast | Bacterial infection [81], RhoA-GTPase inactivation [82,83] | Familial Mediterranean fever (FMF) [84], pyrin-associated autoinflammation with neutrophilic dermatosis (PAAND) [85] |  |
| Non-Canonical Inflammasomes | ||||
| Human Caspase-4/5 | Macrophages, epithelial cells, and monocytes | LPS of Gram-negative bacteria [4], Oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (oxPAPC) [86] | Inflammatory bowel disease and colorectal cancer [87] |  |
| Mouse Caspase-11 | Macrophages, epithelial cells, endothelial cells and neutrophils | LPS of Gram-negative bacteria [4], oxPAPC [5], Lipophosphoglycan (LPG) of Leishmania parasite [88], TIR-domain-containing adapter-inducing interferon β (TRIF) [89], secreted aspartyl proteinases [90] | Multiple sclerosis [91], Amyotrophic lateral sclerosis [92], Parkinson’s disease [93]. Inflammatory bowel diseases (IBDs) [94,95], Rheumatoid arthritis [86], Inflammatory respiratory diseases [96], Chronic obstructive pulmonary disease (COPD) [97] |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, M.; de Alba, E. Structure, Activation and Regulation of NLRP3 and AIM2 Inflammasomes. Int. J. Mol. Sci. 2021, 22, 872. https://doi.org/10.3390/ijms22020872
Sharma M, de Alba E. Structure, Activation and Regulation of NLRP3 and AIM2 Inflammasomes. International Journal of Molecular Sciences. 2021; 22(2):872. https://doi.org/10.3390/ijms22020872
Chicago/Turabian StyleSharma, Meenakshi, and Eva de Alba. 2021. "Structure, Activation and Regulation of NLRP3 and AIM2 Inflammasomes" International Journal of Molecular Sciences 22, no. 2: 872. https://doi.org/10.3390/ijms22020872
APA StyleSharma, M., & de Alba, E. (2021). Structure, Activation and Regulation of NLRP3 and AIM2 Inflammasomes. International Journal of Molecular Sciences, 22(2), 872. https://doi.org/10.3390/ijms22020872