Congenital Diseases of DNA Replication: Clinical Phenotypes and Molecular Mechanisms



Abstract
:1. Introduction
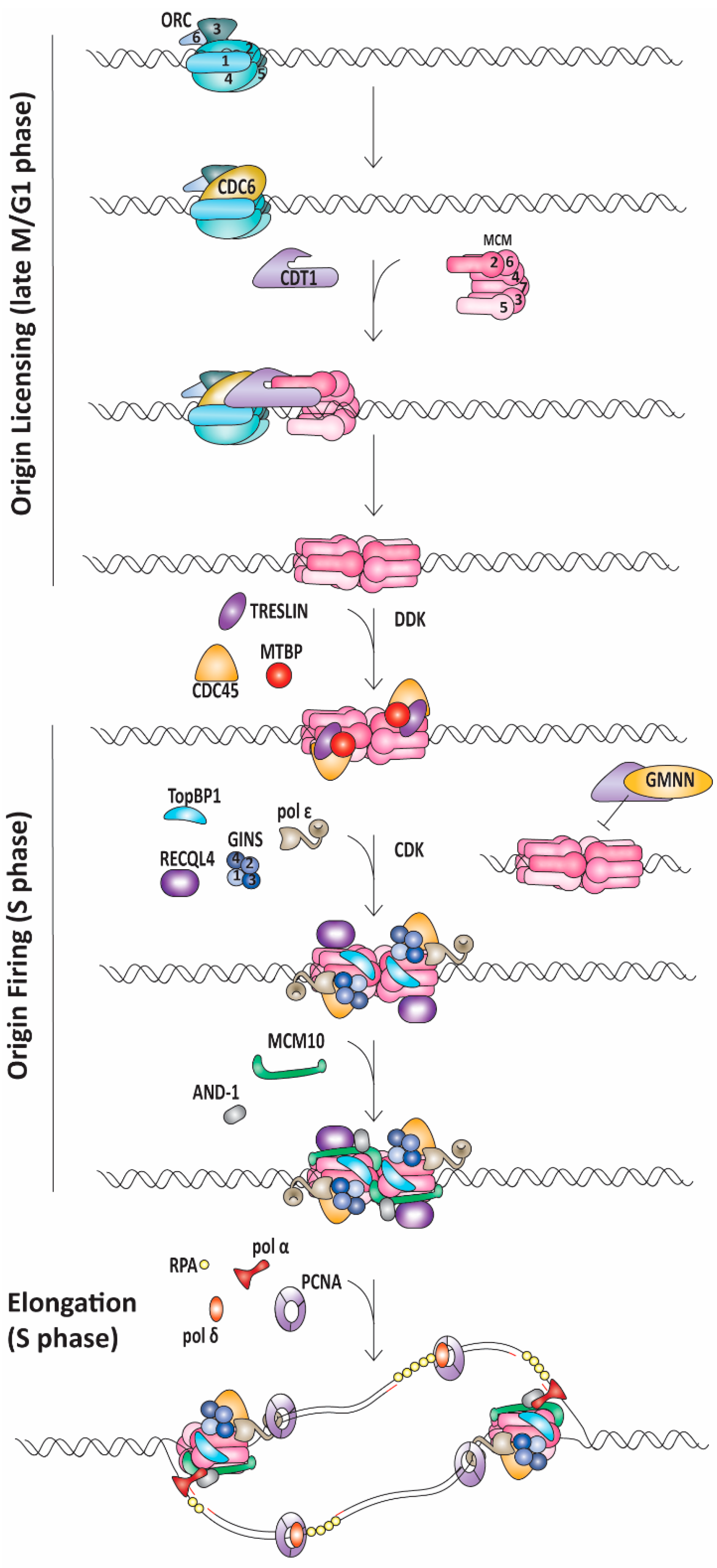
1.1. Replication Initiation
1.2. Replication Elongation
1.3. Replication Termination
2. Diseases
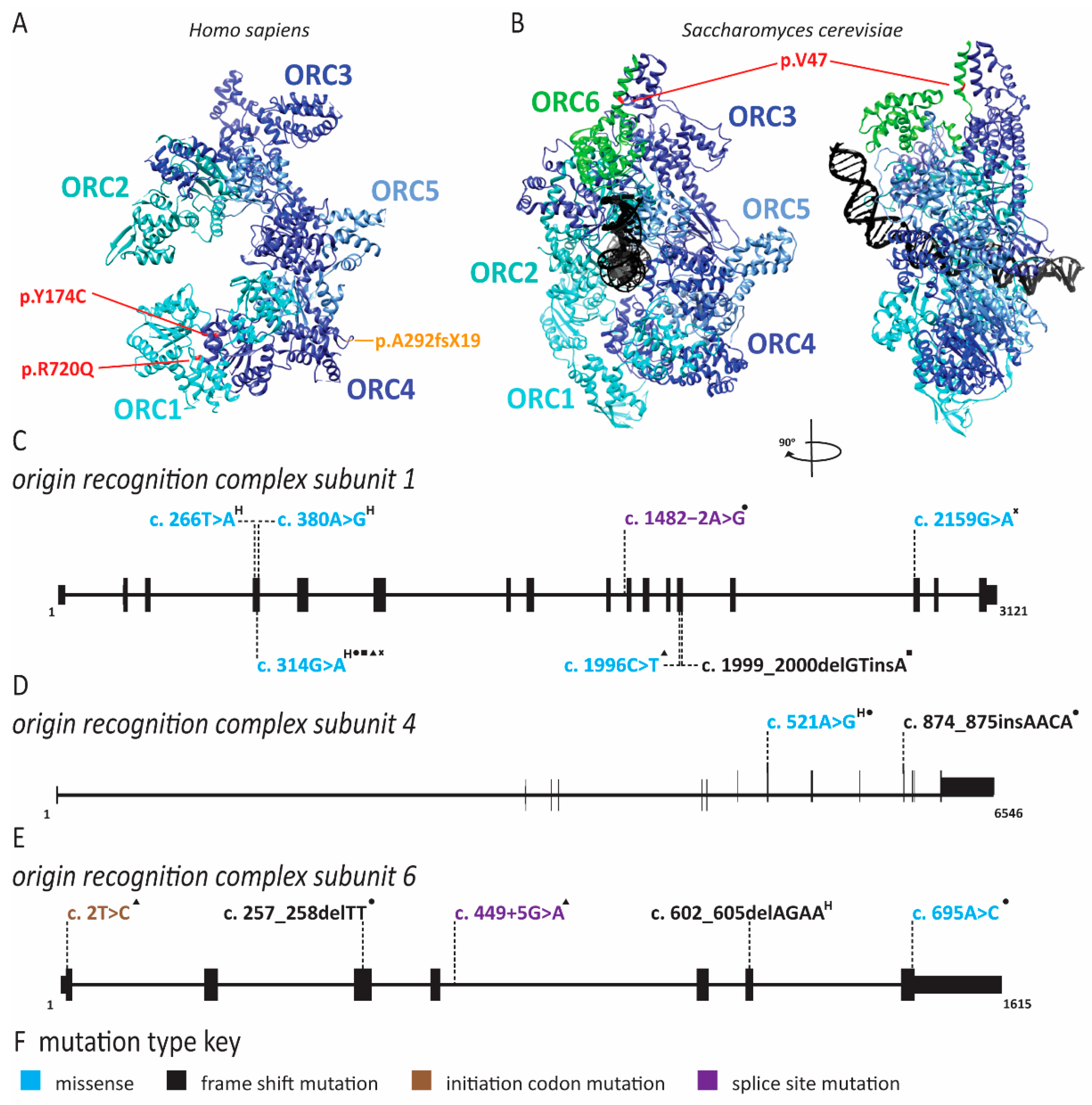
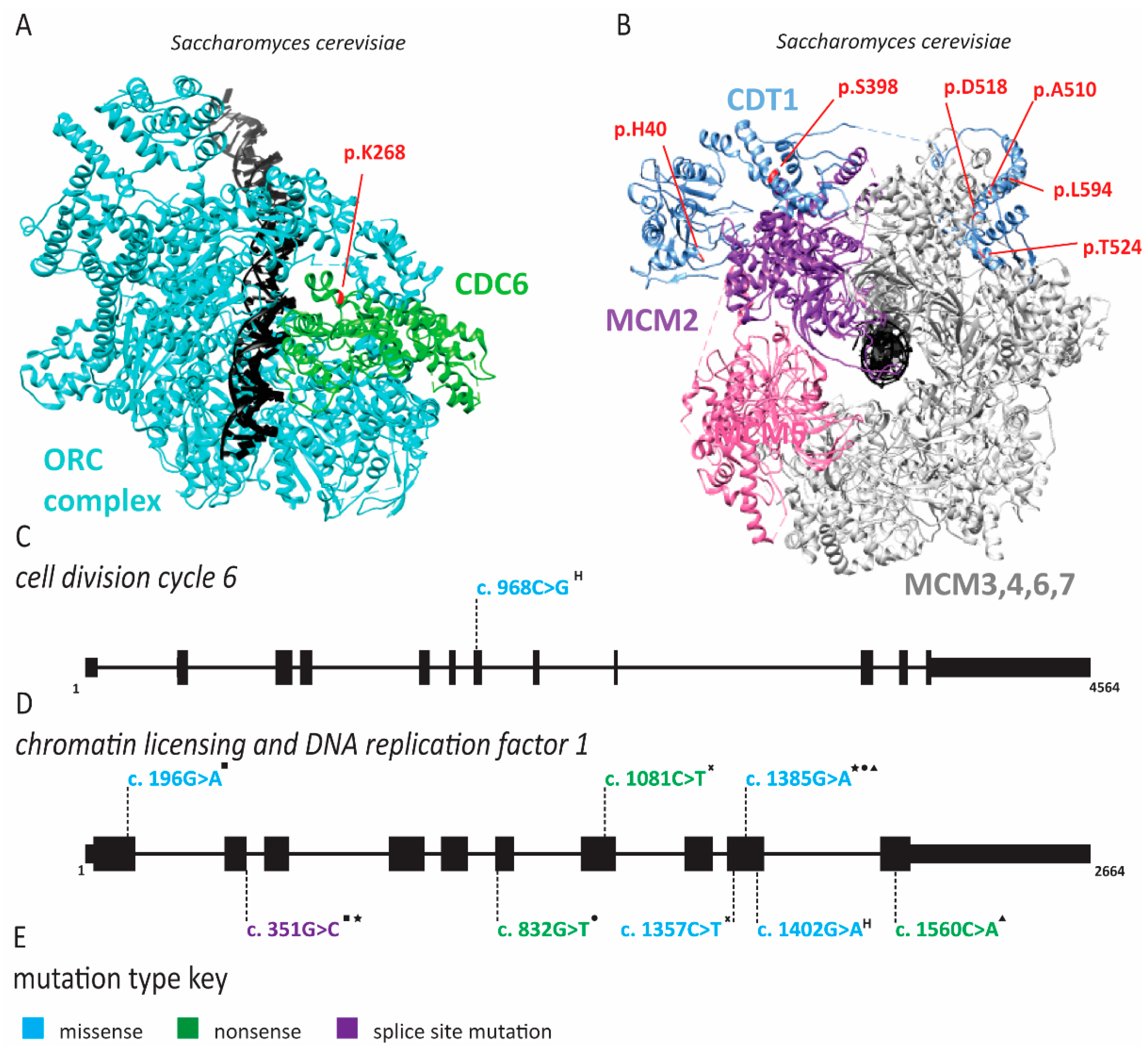
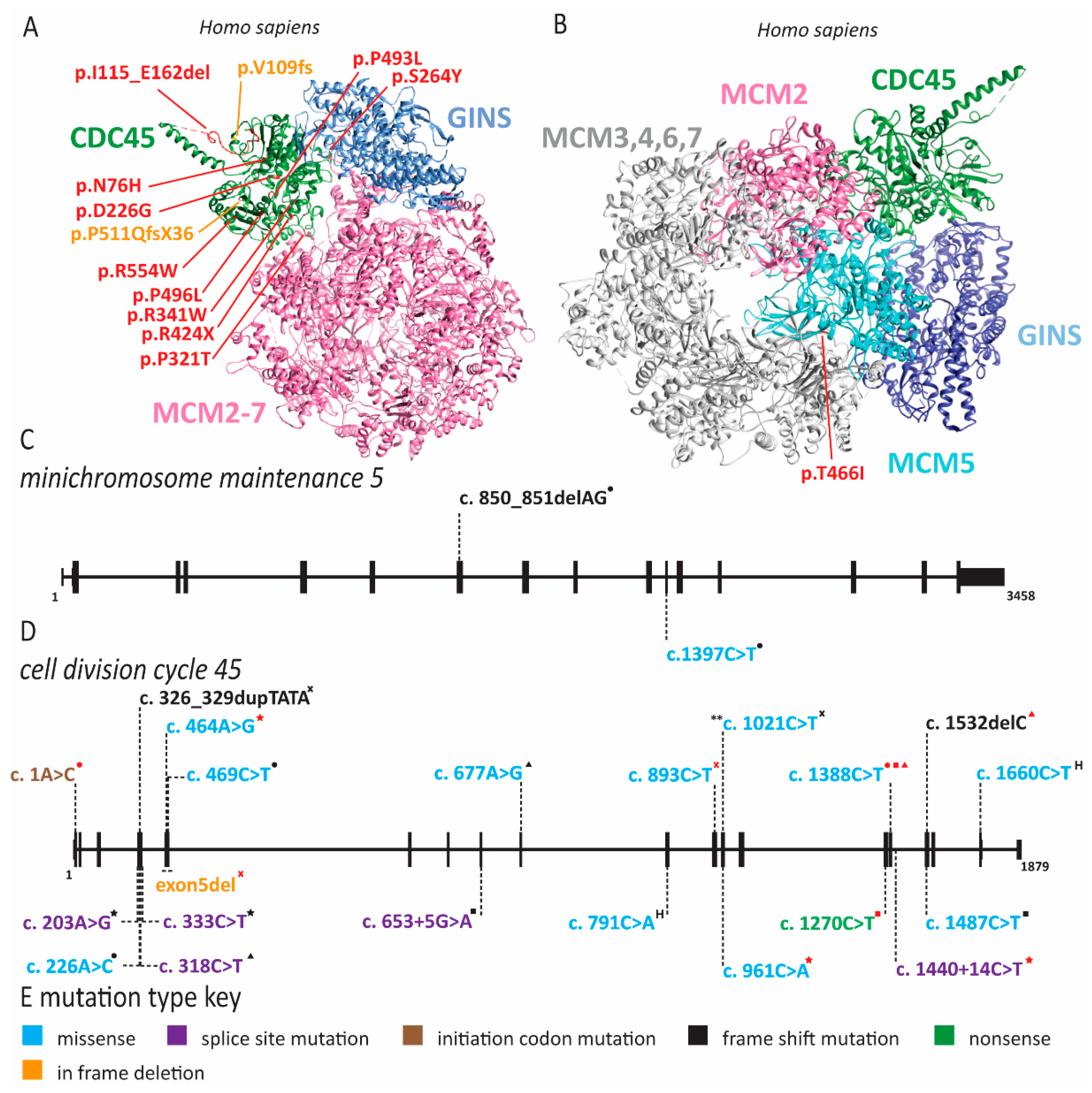
2.1. Meier-Gorlin Syndrome Is Caused by Defects in Origin Licensing
2.2. MCM2 Deficiency and Familial Deafness
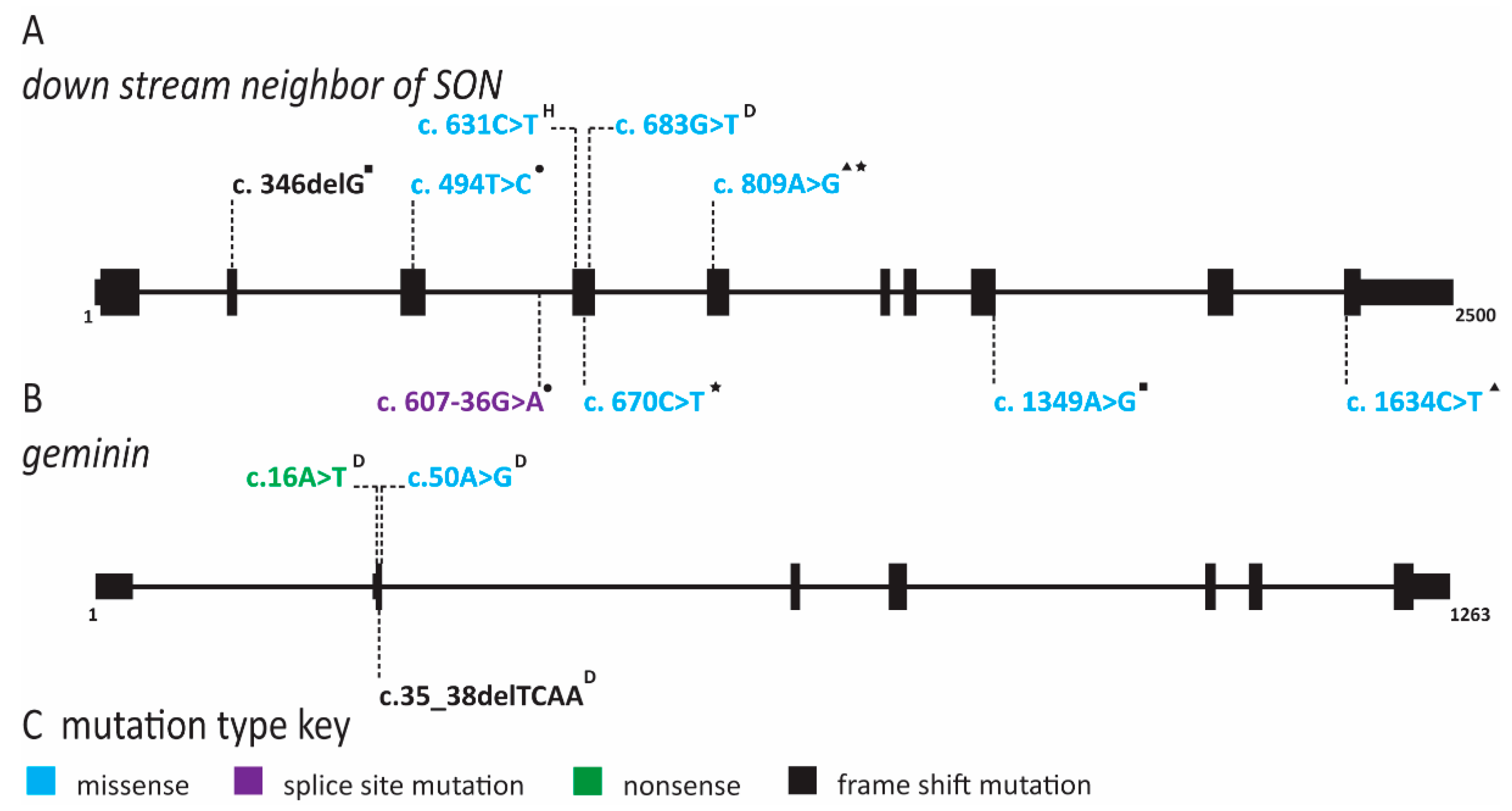
2.3. Natural Killer Cell Deficiency Is Caused by Defects in Origin Licensing or Initiation Factors and Is Associated with Genome Instability
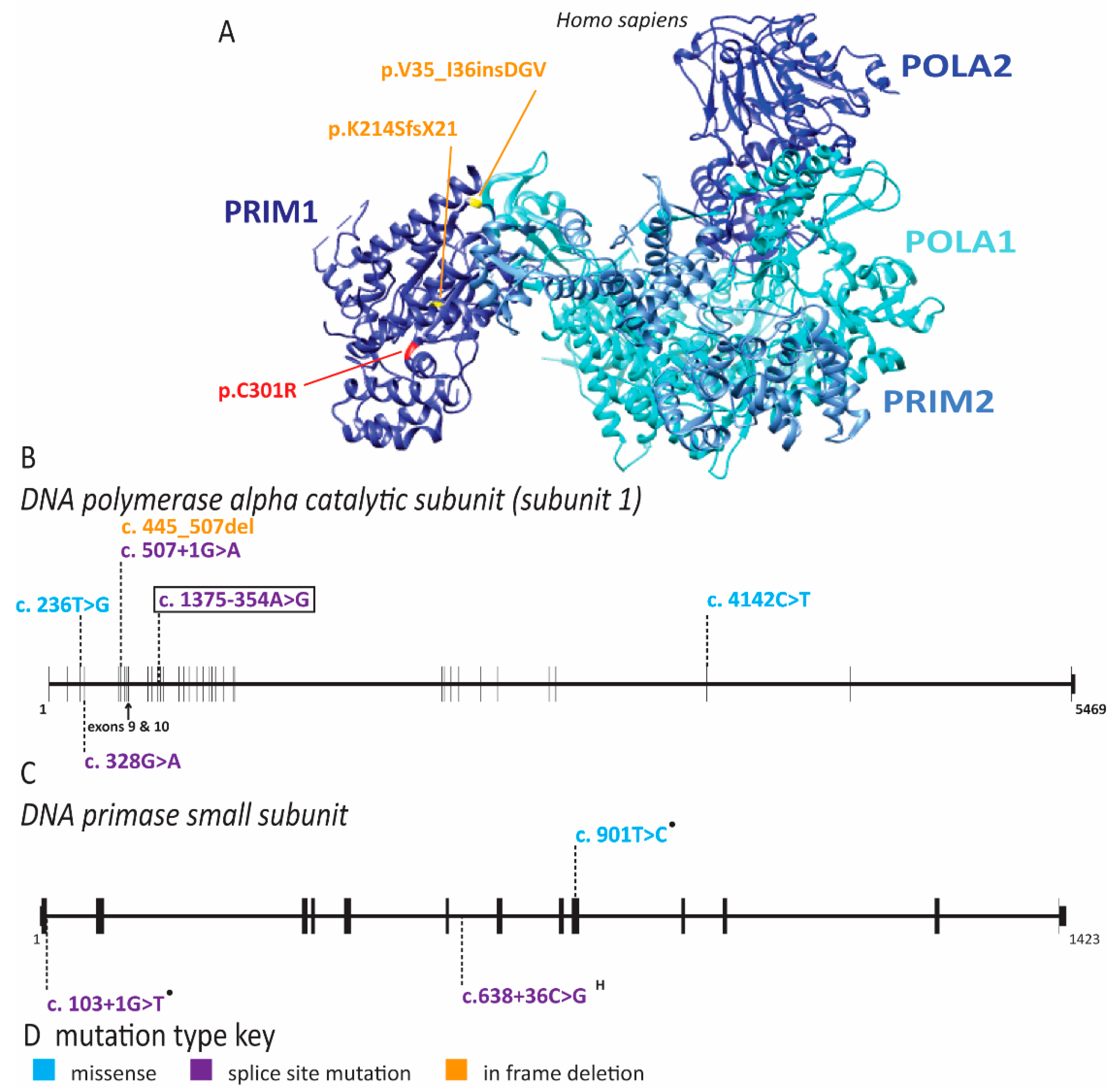
2.4. Multiple Pol α Subunits Are Linked to Human Disease
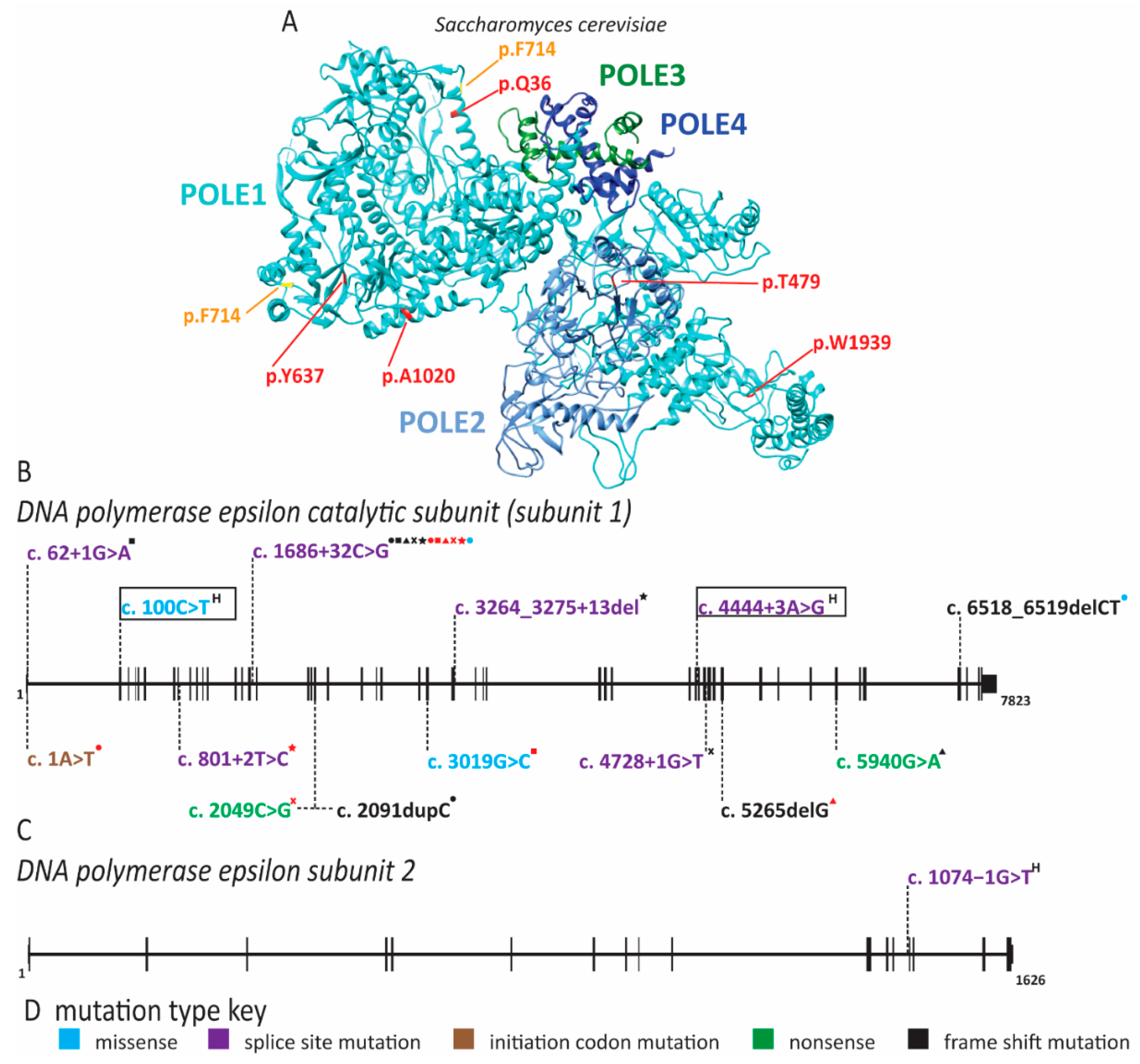
2.5. Pol ε Dysfunction Causes Immunodeficiency
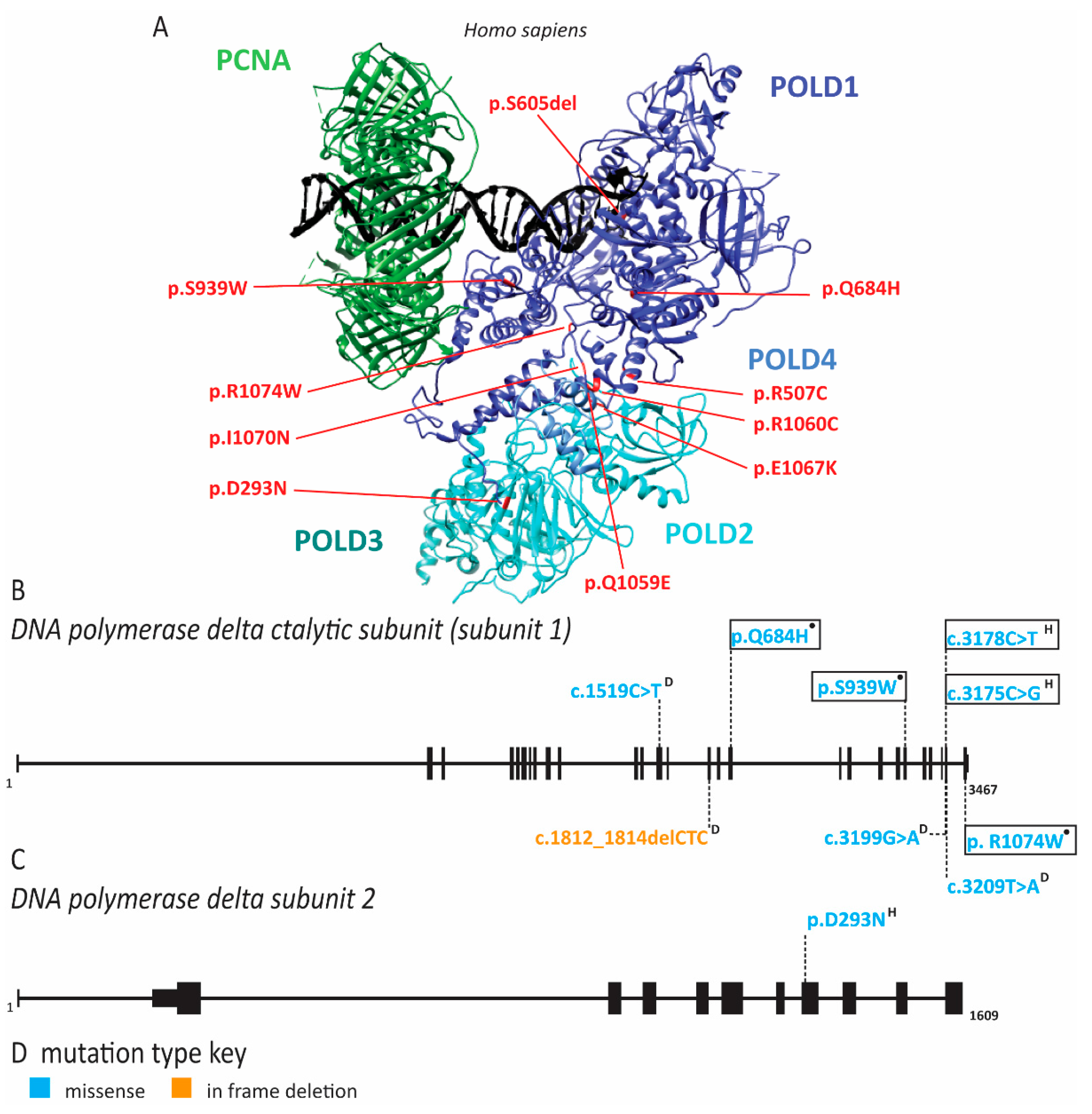
2.6. Pol δ Associated Progeroid Syndrome and Immune Deficiency
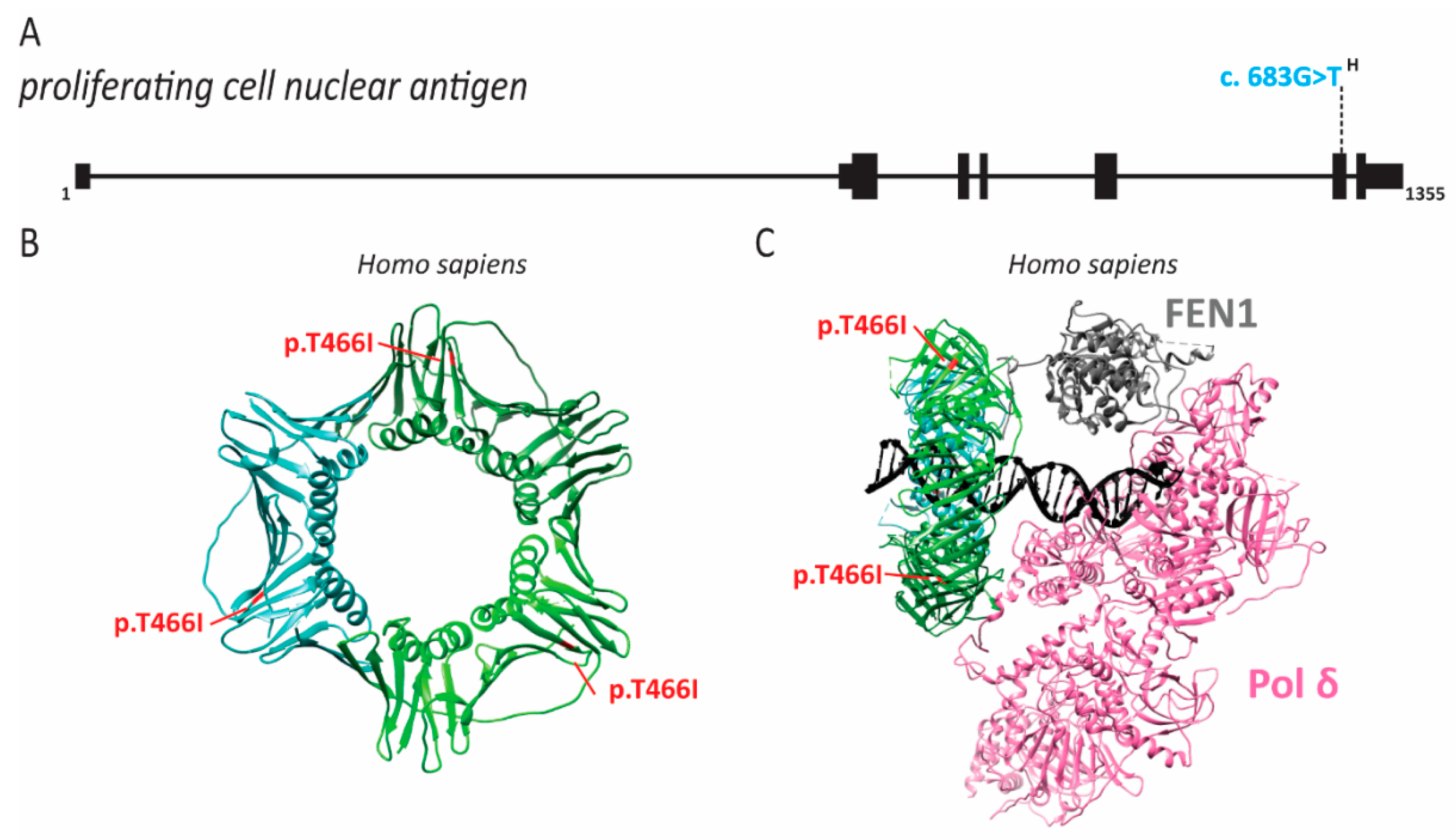
2.7. PCNA Associated Human Disease
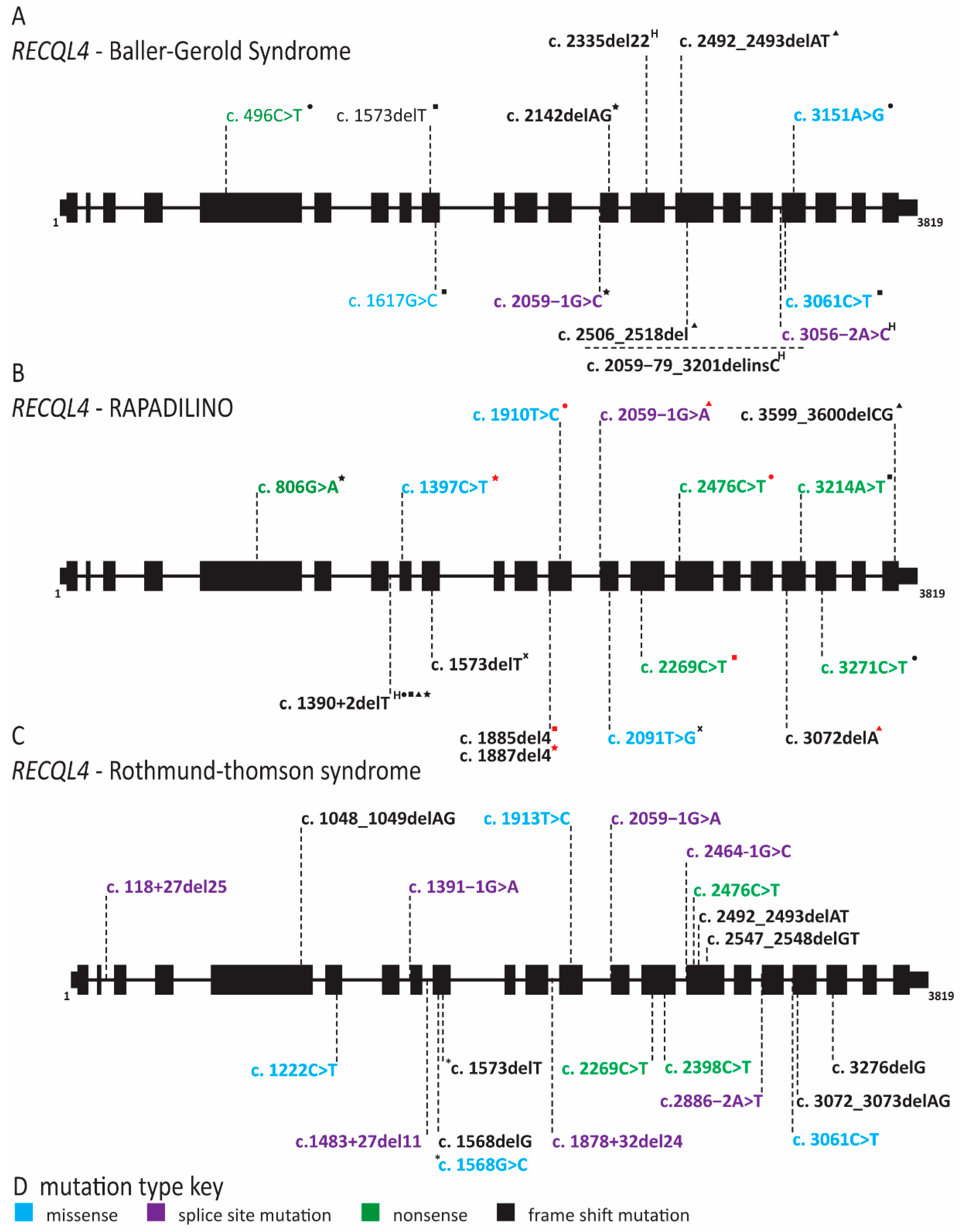
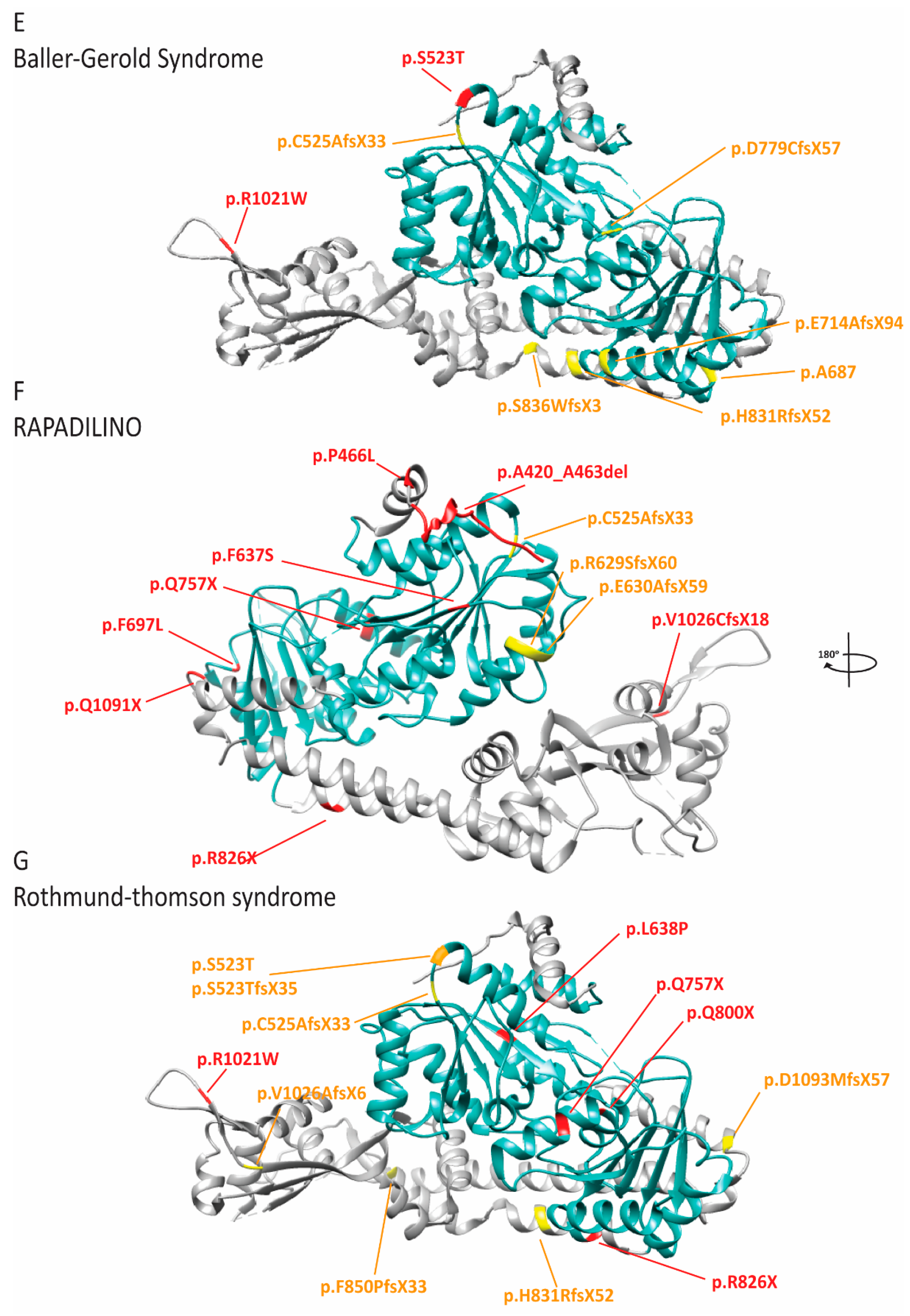
2.8. RECQL4 Dysfunction Causes Three Overlapping Syndromes
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chandrasekaran, S.; Reidy, T.K.; Gowen, J. Regulation of DNA Replication Origin Licensing. In Fundamental Aspects of DNA Replication; IntechOpen: London, UK, 2011. [Google Scholar] [CrossRef] [Green Version]
- Courtot, L.; Hoffmann, J.-S.; Bergoglio, V. The protective role of dormant origins in response to replicative stress. Int. J. Mol. Sci. 2018, 19, 3569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiratani, I.; Takahashi, S. DNA replication timing enters the single-cell era. Genes 2019, 10, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Bellani, M.A.; James, R.C.; Pokharel, D.; Zhang, Y.; Reynolds, J.J.; McNee, G.S.; Jackson, A.P.; Stewart, G.S.; Seidman, M.M. DONSON and FANCM associate with different replisomes distinguished by replication timing and chromatin domain. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Reynolds, J.J.; Bicknell, L.S.; Carroll, P.; Higgs, M.R.; Shaheen, R.; Murray, J.E.; Papadopoulos, D.K.; Leitch, A.; Murina, O.; Tarnauskaitė, Ž.; et al. Mutations in DONSON disrupt replication fork stability and cause microcephalic dwarfism. Nat. Genet. 2017, 49, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Bandura, J.L.; Beall, E.L.; Bell, M.; Silver, H.R.; Botchan, M.R.; Calvi, B.R. humpty dumpty Is Required for Developmental DNA Amplification and Cell Proliferation in Drosophila. Curr. Biol. 2005, 15, 755–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaremko, M.J.; On, K.F.; Thomas, D.R.; Stillman, B.; Joshua-Tor, L. The dynamic nature of the human Origin Recognition Complex revealed through five cryoEM structures. eLife 2020, 9, e58622. [Google Scholar] [CrossRef]
- Li, N.; Lam, W.H.; Zhai, Y.; Cheng, J.; Cheng, E.; Zhao, Y.; Gao, N.; Tye, B.K. Structure of the origin recognition complex bound to DNA replication origin. Nat. Cell Biol. 2018, 559, 217–222. [Google Scholar] [CrossRef]
- Yuan, Z.; Riera, A.; Bai, L.; Sun, J.; Nandi, S.; Spanos, C.; Chen, Z.A.; Barbon, M.; Rappsilber, J.; Stillman, B.; et al. Structural basis of Mcm2–7 replicative helicase loading by ORC–Cdc6 and Cdt1. Nat. Struct. Mol. Biol. 2017, 24, 316–324. [Google Scholar] [CrossRef] [Green Version]
- Pozo, P.N.; Matson, J.P.; Cole, Y.; Kedziora, K.M.; Grant, G.D.; Temple, B.; Cook, J.G. Cdt1 variants reveal unanticipated aspects of interactions with cyclin/CDK and MCM important for normal genome replication. Mol. Biol. Cell 2018, 29, 2989–3002. [Google Scholar] [CrossRef]
- Zhou, Y.; Pozo, P.N.; Oh, S.; Stone, H.M.; Cook, J.G. Distinct and sequential re-replication barriers ensure precise genome duplication. PLoS Genet. 2020, 16, e1008988. [Google Scholar] [CrossRef]
- Frigola, J.; He, J.; Kinkelin, K.; Pye, V.E.; Renault, L.; Douglas, M.E.; Remus, D.; Cherepanov, P.; Costa, A.; Diffley, J.F. Cdt1 stabilizes an open MCM ring for helicase loading. Nat. Commun. 2017, 8, 15720. [Google Scholar] [CrossRef] [PubMed]
- Remus, D.; Beuron, F.; Tolun, G.; Griffith, J.D.; Morris, E.P.; Diffley, J.F. Concerted Loading of Mcm2–7 Double Hexamers around DNA during DNA Replication Origin Licensing. Cell 2009, 139, 719–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evrin, C.; Clarke, P.; Zech, J.; Lurz, R.; Sun, J.; Uhle, S.; Li, H.; Stillman, B.; Speck, C. A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc. Natl. Acad. Sci. USA 2009, 106, 20240–20245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ticau, S.; Friedman, L.J.; Ivica, N.A.; Gelles, J.; Bell, S.P. Single-molecule studies of origin licensing reveal mechanisms ensuring bidirectional helicase loading. Cell 2015, 161, 513–525. [Google Scholar] [CrossRef] [Green Version]
- Georgescu, R.; Yuan, Z.; Bai, L.; Santos, R.D.L.A.; Sun, J.; Zhang, D.; Yurieva, O.; Li, H.; O’Donnell, M.E. Structure of eukaryotic CMG helicase at a replication fork and implications to replisome architecture and origin initiation. Proc. Natl. Acad. Sci. USA 2017, 114, E697–E706. [Google Scholar] [CrossRef] [Green Version]
- Wohlschlegel, J.A.; Dwyer, B.T.; Dhar, S.K.; Cvetic, C.; Walter, J.C.; Dutta, A. Inhibition of Eukaryotic DNA Replication by Geminin Binding to Cdt1. Science 2000, 290, 2309–2312. [Google Scholar] [CrossRef]
- McGarry, T.J.; Kirschner, M.W. Geminin, an Inhibitor of DNA Replication, Is Degraded during Mitosis. Cell 1998, 93, 1043–1053. [Google Scholar] [CrossRef] [Green Version]
- Yeeles, J.T.P.; Deegan, T.D.; Janska, A.; Early, A.; Diffley, J.F.X. Regulated eukaryotic DNA replication origin firing with purified proteins. Nat. Cell Biol. 2015, 519, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Mueller, A.C.; Keaton, M.A.; Dutta, A. DNA replication: Mammalian treslin–topbp1 interaction mirrors yeast Sld3–Dpb11. Curr. Biol. 2011, 21, R638–R640. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Pulido, L.; Diffley, J.F.; Ponting, C.P. Homology explains the functional similarities of Treslin/Ticrr and Sld3. Curr. Biol. 2010, 20, R509–R510. [Google Scholar] [CrossRef] [Green Version]
- Matsuno, K.; Kumano, M.; Kubota, Y.; Hashimoto, Y.; Takisawa, H. The N-Terminal Noncatalytic Region of Xenopus RecQ4 Is Required for Chromatin Binding of DNA Polymerase α in the Initiation of DNA Replication. Mol. Cell. Biol. 2006, 26, 4843–4852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köhler, K.; Sanchez-Pulido, L.; Höfer, V.; Marko, A.; Ponting, C.P.; Snijders, A.P.; Feederle, R.; Schepers, A.; Boos, D. The Cdk8/19-cyclin C transcription regulator functions in genome replication through metazoan Sld7. PLoS Biol. 2019, 17, e2006767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumagai, A.; Dunphy, W.G. MTBP, the partner of Treslin, contains a novel DNA-binding domain that is essential for proper initiation of DNA replication. Mol. Biol. Cell 2017, 28, 2998–3012. [Google Scholar] [CrossRef] [PubMed]
- Labib, K. How do Cdc7 and cyclin-dependent kinases trigger the initiation of chromosome replication in eukaryotic cells? Genes Dev. 2010, 24, 1208–1219. [Google Scholar] [CrossRef] [Green Version]
- Larasati, B.; Duncker, P. Mechanisms Governing DDK Regulation of the Initiation of DNA Replication. Genes 2016, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Dhingra, N.; Bruck, I.; Smith, S.; Ning, B.; Kaplan, D.L. Dpb11 Protein Helps Control Assembly of the Cdc45·Mcm2-7·GINS Replication Fork Helicase. J. Biol. Chem. 2015, 290, 7586–7601. [Google Scholar] [CrossRef] [Green Version]
- Simon, A.C.; Sannino, V.; Costanzo, V.; Pellegrini, L. Structure of human Cdc45 and implications for CMG helicase function. Nat. Commun. 2016, 7, 11638. [Google Scholar] [CrossRef] [Green Version]
- Bruck, I.; Kaplan, D.L. GINS and Sld3 Compete with One Another for Mcm2-7 and Cdc45 Binding. J. Biol. Chem. 2011, 286, 14157–14167. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, U.; Wollmann, Y.; Franke, C.; Grosse, F.; Saluz, H.-P.; Hänel, F. Characterization of the interaction between the human DNA topoisomerase IIβ-binding protein 1 (TopBP1) and the cell division cycle 45 (Cdc45) protein. Biochem. J. 2007, 409, 169–177. [Google Scholar] [CrossRef]
- Shin, G.; Jeong, D.; Kim, H.; Im, J.-S.; Lee, J.-K. RecQL4 tethering on the pre-replicative complex induces unscheduled origin activation and replication stress in human cells. J. Biol. Chem. 2019, 294, 16255–16265. [Google Scholar] [CrossRef]
- Sangrithi, M.N.; Bernal, J.A.; Madine, M.; Philpott, A.; Lee, J.; Dunphy, W.G.; Venkitaraman, A.R. Initiation of DNA Replication Requires the RECQL4 Protein Mutated in Rothmund-Thomson Syndrome. Cell 2005, 121, 887–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirbu, B.M.; McDonald, W.H.; Dungrawala, H.; Badu-Nkansah, A.; Kavanaugh, G.M.; Chen, Y.; Tabb, D.L.; Cortez, D. Identification of Proteins at Active, Stalled, and Collapsed Replication Forks Using Isolation of Proteins on Nascent DNA (iPOND) Coupled with Mass Spectrometry. J. Biol. Chem. 2013, 288, 31458–31467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, Y.; Yuan, Z.; Bai, L.; Schneider, S.; Zhao, G.; Stillman, B.; Speck, C.; Li, H. Cryo-EM structure of Mcm2-7 double hexamer on DNA suggests a lagging-strand DNA extrusion model. Proc. Natl. Acad. Sci. USA 2017, 114, E9529–E9538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasserman, M.R.; Schauer, G.D.; O’Donnell, M.E.; Liu, S. Replication Fork Activation Is Enabled by a Single-Stranded DNA Gate in CMG Helicase. Cell 2019, 178, 600–611.e16. [Google Scholar] [CrossRef] [PubMed]
- Petojevic, T.; Pesavento, J.J.; Costa, A.; Liang, J.; Wang, Z.; Berger, J.M.; Botchan, M.R. Cdc45 (cell division cycle protein 45) guards the gate of the Eukaryote Replisome helicase stabilizing leading strand engagement. Proc. Natl. Acad. Sci. USA 2015, 112, E249–E258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Arnaiz, P.; Bruck, I.; Colbert, M.K.; Kaplan, D.L. An intact Mcm10 coiled-coil interaction surface is important for origin melting, helicase assembly and the recruitment of Pol-α to Mcm2–7. Nucleic Acids Res. 2017, 45, 7261–7275. [Google Scholar] [CrossRef] [Green Version]
- Lõoke, M.; Maloney, M.F.; Bell, S.P. Mcm10 regulates DNA replication elongation by stimulating the CMG replicative helicase. Genes Dev. 2017, 31, 291–305. [Google Scholar] [CrossRef] [Green Version]
- Perez-Arnaiz, P.; Kaplan, D.L. An Mcm10 Mutant Defective in ssDNA Binding Shows Defects in DNA Replication Initiation. J. Mol. Biol. 2016, 428, 4608–4625. [Google Scholar] [CrossRef] [Green Version]
- Langston, L.D.; Mayle, R.; Schauer, G.D.; Yurieva, O.; Zhang, D.; Yao, N.Y.; Georgescu, R.E.; O’Donnell, M.E. Mcm10 promotes rapid isomerization of CMG-DNA for replisome bypass of lagging strand DNA blocks. eLife 2017, 6, e29118. [Google Scholar] [CrossRef]
- Baxley, R.M.; Bielinsky, A.-K. Mcm10: A Dynamic Scaffold at Eukaryotic Replication Forks. Genes 2017, 8, 73. [Google Scholar] [CrossRef]
- Lujan, S.A.; Williams, J.S.; Kunkel, T.A. DNA Polymerases Divide the Labor of Genome Replication. Trends Cell Biol. 2016, 26, 640–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgescu, R.E.; Schauer, G.D.; Yao, N.Y.; Langston, L.D.; Yurieva, O.; Zhang, D.; Finkelstein, J.; O’Donnell, M.E. Reconstitution of a eukaryotic replisome reveals suppression mechanisms that define leading/lagging strand operation. eLife 2015, 4, e04988. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Ukomadu, C.; Jha, S.; Senga, T.; Dhar, S.K.; Wohlschlegel, J.A.; Nutt, L.K.; Kornbluth, S.; Dutta, A. Mcm10 and And-1/CTF4 recruit DNA polymerase to chromatin for initiation of DNA replication. Genes Dev. 2007, 21, 2288–2299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chattopadhyay, S.; Bielinsky, A.-K. Human Mcm10 Regulates the Catalytic Subunit of DNA Polymerase-α and Prevents DNA Damage during Replication. Mol. Biol. Cell 2007, 18, 4085–4095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, G.D.; O’Donnell, M.; Kuriyan, J. Structural analysis of a eukaryotic sliding DNA clamp–clamp loader complex. Nat. Cell Biol. 2004, 429, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Tsurimoto, T.; Stillman, B. Replication factors required for SV40 DNA replication in vitro. I. DNA structure-specific recognition of a primer-template junction by eukaryotic DNA polymerases and their accessory proteins. J. Biol. Chem. 1991, 266, 1950–1960. [Google Scholar] [CrossRef]
- Liu, B.; Hu, J.; Wang, J.; Kong, D. Direct Visualization of RNA-DNA Primer Removal from Okazaki Fragments Provides Support for Flap Cleavage and Exonucleolytic Pathways in Eukaryotic Cells. J. Biol. Chem. 2017, 292, 4777–4788. [Google Scholar] [CrossRef] [Green Version]
- Stodola, J.L.; Burgers, P.M. Resolving individual steps of Okazaki-fragment maturation at a millisecond timescale. Nat. Struct. Mol. Biol. 2016, 23, 402–408. [Google Scholar] [CrossRef] [Green Version]
- Stodola, J.L.; Burgers, P.M. Mechanism of lagging-strand dna replication in eukaryotes. In Advances in Experimental Medicine and Biology; Springer: Singapore, 2017; Volume 1042, pp. 117–133. [Google Scholar]
- Rossi, M.L.; Bambara, R.A. Reconstituted Okazaki Fragment Processing Indicates Two Pathways of Primer Removal. J. Biol. Chem. 2006, 281, 26051–26061. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.A.; Campbell, J.L.; Bambara, R.A. DNA2 is a structure-specific nuclease, with affinity for 5′-flap intermediates. Nucleic Acids Res. 2009, 38, 920–930. [Google Scholar] [CrossRef]
- Howes, T.R.L.; Tomkinson, A.E. DNA Ligase I, the Replicative DNA Ligase. Macromol. Protein Complexes Struct. Funct. 2012, 62, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Moreno, S.P.; Gambus, A. Mechanisms of eukaryotic replisome disassembly. Biochem. Soc. Trans. 2020, 48, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Dewar, J.M.; Walter, J.C. Mechanisms of DNA replication termination. Nat. Rev. Mol. Cell Biol. 2017, 18, 507–516. [Google Scholar] [CrossRef]
- Dewar, J.M.; Low, E.; Mann, M.; Räschle, M.; Walter, J.C. CRL2Lrr1promotes unloading of the vertebrate replisome from chromatin during replication termination. Genes Dev. 2017, 31, 275–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, S.P.; Bailey, R.; Campion, N.; Herron, S.; Gambus, A. Polyubiquitylation drives replisome disassembly at the termination of DNA replication. Science 2014, 346, 477–481. [Google Scholar] [CrossRef]
- Sedlackova, H.; Rask, M.-B.; Gupta, R.; Choudhary, C.; Somyajit, K.; Lukas, J. Equilibrium between nascent and parental MCM proteins protects replicating genomes. Nat. Cell Biol. 2020, 587, 297–302. [Google Scholar] [CrossRef]
- Bongers, E.; Van Kampen, A.; Van Bokhoven, H.; Knoers, N.V.A.M.; Van Bokhoven, H. Human syndromes with congenital patellar anomalies and the underlying gene defects. Clin. Genet. 2005, 68, 302–319. [Google Scholar] [CrossRef]
- Gorlin, R.J.; Cervenka, J.; Moller, K. A selected miscellany. Birth Defects Orig. Artic. Ser. 1975, 11, 39–50. [Google Scholar]
- Hurst, J.A.; Winter, R.M.; Baraitser, M.; Optiz, J.M.; Reynolds, J.F. Distinctive syndrome of short stature, craniosynostosis, skeletal changes, and malformed ears. Am. J. Med. Genet. 1988, 29, 107–115. [Google Scholar] [CrossRef]
- Cohen, B.; Temple, I.K.; Symons, J.C.; Hall, C.M.; Shaw, D.G.; Bhamra, M.; Jackson, A.M.; Pembrey, M.E. Microtia and short stature: A new syndrome. J. Med. Genet. 1991, 28, 786–790. [Google Scholar] [CrossRef] [Green Version]
- Gorlin, R.J. Microtia, absent patellae, short stature, micrognathia syndrome. J. Med. Genet. 1992, 29, 516–517. [Google Scholar] [PubMed]
- Boles, R.G.; Teebi, A.S.; Schwartz, D.; Harper, J.F. Further delineation of the ear, patella, short stature syndrome (Meier-Gorlin syndrome). Clin. Dysmorphol. 1994, 3, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, D.; Toutain, A.; Gorlin, R.J.; Oley, C.A.; Battin, J. Clinical identification of a human equivalent to the short ear (se) murine phenotype. Annales Génétique 1994, 37, 184–191. [Google Scholar]
- Bongers, E.M.; Opitz, J.M.; Fryer, A.; Sarda, P.; Hennekam, R.C.; Hall, B.D.; Superneau, D.W.; Harbison, M.; Poss, A.; Van Bokhoven, H.; et al. Meier-Gorlin syndrome: Report of eight additional cases and review. Am. J. Med. Genet. 2001, 102, 115–124. [Google Scholar] [CrossRef]
- Bicknell, L.S.; Bongers, E.M.; Leitch, A.; Brown, S.; Schoots, J.; Harley, M.E.; Aftimos, S.; Al-Aama, J.Y.; Bober, M.B.; Brown, P.A.J.; et al. Mutations in the pre-replication complex cause Meier-Gorlin syndrome. Nat. Genet. 2011, 43, 356–359. [Google Scholar] [CrossRef] [Green Version]
- Guernsey, D.L.; Matsuoka, M.; Jiang, H.; Evans, S.C.; MacGillivray, C.; Nightingale, M.; Perry, S.; Ferguson, M.; LeBlanc, M.A.; Paquette, J.; et al. Mutations in origin recognition complex gene ORC4 cause Meier-Gorlin syndrome. Nat. Genet. 2011, 43, 360–364. [Google Scholar] [CrossRef]
- Fenwick, A.L.; Kliszczak, M.; Cooper, F.; Murray, J.; Sanchez-Pulido, L.; Twigg, S.R.; Goriely, A.; McGowan, S.J.; Miller, K.A.; Taylor, I.B.; et al. Mutations in CDC45, Encoding an Essential Component of the Pre-initiation Complex, Cause Meier-Gorlin Syndrome and Craniosynostosis. Am. J. Hum. Genet. 2016, 99, 125–138. [Google Scholar] [CrossRef] [Green Version]
- Vetro, A.; Savasta, S.; Raucci, A.R.; Cerqua, C.; Sartori, G.; Limongelli, I.; Forlino, A.; Maruelli, S.; Perucca, P.; Vergani, D.; et al. MCM5: A new actor in the link between DNA replication and Meier-Gorlin syndrome. Eur. J. Hum. Genet. 2017, 25, 646–650. [Google Scholar] [CrossRef] [Green Version]
- Burrage, L.C.; Charng, W.-L.; Eldomery, M.K.; Willer, J.R.; Davis, E.E.; Lugtenberg, D.; Zhu, W.; LeDuc, M.S.; Akdemir, Z.C.; Azamian, M.; et al. De Novo GMNN Mutations Cause Autosomal-Dominant Primordial Dwarfism Associated with Meier-Gorlin Syndrome. Am. J. Hum. Genet. 2015, 97, 904–913. [Google Scholar] [CrossRef] [Green Version]
- A De Munnik, S.; Bicknell, L.S.; Aftimos, S.; Al-Aama, J.Y.; Van Bever, Y.; Bober, M.B.; Clayton-Smith, J.; Edrees, A.Y.; Feingold, M.; Fryer, A.; et al. Meier-Gorlin syndrome genotype–phenotype studies: 35 individuals with pre-replication complex gene mutations and 10 without molecular diagnosis. Eur. J. Hum. Genet. 2012, 20, 598–606. [Google Scholar] [CrossRef] [Green Version]
- Vojtková, J.; Čiljaková, M.; Jeseňák, M.; Bánovčin, P. Meier-Gorlin syndrome caused by ORC1 mutation associated with chromosomal breakage—Coincidental finding or new feature of known syndrome? Endokrynol. Polska 2019, 70, 457–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bicknell, L.S.; Walker, S.A.; Klingseisen, A.; Stiff, T.; Leitch, A.; Kerzendorfer, C.; Martin, C.-A.; Yeyati, P.L.; Al Sanna, N.; Bober, M.B.; et al. Mutations in ORC1, encoding the largest subunit of the origin recognition complex, cause microcephalic primordial dwarfism resembling Meier-Gorlin syndrome. Nat. Genet. 2011, 43, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Allon-Shalev, S.; Khayat, M.; Etty, D.-S.; Elpeleg, O. Further insight into the phenotype associated with a mutation in the ORC6 gene, causing Meier-Gorlin syndrome 3. Am. J. Med. Genet. 2015, 167, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Knapp, K.M.; Sullivan, R.; Murray, J.; Gimenez, G.; Arn, P.; D’Souza, P.; Gezdirici, A.; Wilson, W.G.; Jackson, A.P.; Ferreira, C.; et al. Linked-read genome sequencing identifies biallelic pathogenic variants in DONSON as a novel cause of Meier-Gorlin syndrome. J. Med. Genet. 2019, 57, 195–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ting, C.Y.; Bhatia, N.S.; Lim, J.Y.; Goh, C.-Y.J.; Vasanwala, R.F.; Ong, C.C.P.; Seow, W.T.; Yeow, V.K.-L.; Ting, T.W.; Ng, I.S.L.; et al. Further delineation of CDC45-related Meier-Gorlin syndrome with craniosynostosis and review of literature. Eur. J. Med. Genet. 2020, 63, 103652. [Google Scholar] [CrossRef]
- Karaca, E.; Posey, J.E.; Bostwick, B.; Liu, P.; Gezdirici, A.; Yesil, G.; Akdemir, Z.C.; Bayram, Y.; Harms, F.L.; Meinecke, P.; et al. Biallelic and De Novo Variants in DONSON Reveal a Clinical Spectrum of Cell Cycle-opathies with Microcephaly, Dwarfism and Skeletal Abnormalities. Am. J. Med. Genet. 2019, 179, 2056–2066. [Google Scholar] [CrossRef]
- Casey, J.P.; Nobbs, M.; Mcgettigan, P.A.; Lynch, S.; Ennis, S. Recessive mutations inMCM4/PRKDC cause a novel syndrome involving a primary immunodeficiency and a disorder of DNA repair. J. Med. Genet. 2012, 49, 242–245. [Google Scholar] [CrossRef]
- Gineau, L.; Cognet, C.; Kara, N.; Lach, F.P.; Dunne, J.; Veturi, U.; Picard, C.; Trouillet, C.; Eidenschenk, C.; Aoufouchi, S.; et al. Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J. Clin. Investig. 2012, 122, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Hughes, C.R.; Guasti, L.; Meimaridou, E.; Chuang, C.-H.; Schimenti, J.C.; King, P.J.; Costigan, C.; Clark, A.J.L.; Metherell, L.A. MCM4 mutation causes adrenal failure, short stature, and natural killer cell deficiency in humans. J. Clin. Investig. 2012, 122, 814–820. [Google Scholar] [CrossRef] [Green Version]
- Bernard, F.; Picard, C.; Cormier-Daire, V.; Eidenschenk, C.; Pinto, G.; Bustamante, J.-C.; Jouanguy, E.; Teillac-Hamel, D.; Colomb, V.; Funck-Brentano, I.; et al. A Novel Developmental and Immunodeficiency Syndrome Associated With Intrauterine Growth Retardation and a Lack of Natural Killer Cells. Pediatrics 2003, 113, 136–141. [Google Scholar] [CrossRef]
- Cottineau, J.; Kottemann, M.C.; Lach, F.P.; Kang, Y.-H.; Vély, F.; Deenick, E.K.; Lazarov, T.; Gineau, L.; Wang, Y.; Farina, A.; et al. Inherited GINS1 deficiency underlies growth retardation along with neutropenia and NK cell deficiency. J. Clin. Investig. 2017, 127, 1991–2006. [Google Scholar] [CrossRef] [PubMed]
- Mace, E.; Paust, S.; Conte, M.I.; Baxley, R.M.; Schmit, M.M.; Patil, S.L.; Guilz, N.C.; Mukherjee, M.; Pezzi, A.E.; Chmielowiec, J.; et al. Human NK cell deficiency as a result of biallelic mutations in MCM10. J. Clin. Investig. 2020, 130, 10. [Google Scholar] [CrossRef] [PubMed]
- Baxley, R.M.; Leung, W.; Schmit, M.M.; Matson, J.P.; Oram, M.K.; Wang, L.; Taylor, J.; Yin, L.; Hedberg, J.; Rogers, C.B.; et al. Bi-allelic MCM10 mutations cause telomere shortening with immune dysfunction and cardiomyopathy. bioRxiv 2020, 844498. [Google Scholar] [CrossRef] [Green Version]
- Starokadomskyy, P.; Gemelli, T.; Rios, J.J.; Xing, C.; Wang, R.C.; Li, H.; Pokatayev, V.; Dozmorov, I.; Khan, S.; Miyata, N.; et al. DNA polymerase-α regulates the activation of type I interferons through cytosolic RNA:DNA synthesis. Nat. Immunol. 2016, 17, 495–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, R.C.; Zinn, A.R.; Kim, J.; Carder, K.R. X-linked Reticulate Pigmentary Disorder with Systemic Manifestations: Report of a Third Family and Literature Review. Pediatr. Dermatol. 2005, 22, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Adès, L.C.; Rogers, M.; Sillence, D.O. An X-Linked Reticulate Pigmentary Disorder with Systemic Manifestations: Report of a Second Family. Pediatr. Dermatol. 1993, 10, 344–351. [Google Scholar] [CrossRef]
- Pezzani, L.; Brena, M.; Callea, M.; Colombi, M.; Tadini, G. X-linked reticulate pigmentary disorder with systemic manifestations: A new family and review of the literature. Am. J. Med. Genet. 2013, 161, 1414–1420. [Google Scholar] [CrossRef]
- Starokadomskyy, P.; Wilton, K.M.; Krzewski, K.; Lopez, A.M.; Sifuentes-Dominguez, L.; Overlee, B.; Chen, Q.; Ray, A.; Gil-Krzewska, A.; Peterson, M.; et al. NK cell defects in X-linked pigmentary reticulate disorder. JCI Insight 2019, 4, 125688. [Google Scholar] [CrossRef]
- Van Esch, H.; Colnaghi, R.; Freson, K.; Starokadomskyy, P.; Zankl, A.; Backx, L.; Abramowicz, I.; Outwin, E.; Rohena, L.; Faulkner, C.; et al. Defective DNA Polymerase α-Primase Leads to X-Linked Intellectual Disability Associated with Severe Growth Retardation, Microcephaly, and Hypogonadism. Am. J. Hum. Genet. 2019, 104, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Van Esch, H.; Zanni, G.; Holvoet, M.; Borghgraef, M.; Chelly, J.; Fryns, J.-P.; Devriendt, K. X-linked mental retardation, short stature, microcephaly and hypogonadism maps to Xp22.1-p21.3 in a Belgian family. Eur. J. Med. Genet. 2005, 48, 145–152. [Google Scholar] [CrossRef]
- Parry, D.A.; Tamayo-Orrego, L.; Carroll, P.; Marsh, J.A.; Greene, P.; Murina, O.; Uggenti, C.; Leitch, A.; Káposzta, R.; Merő, G.; et al. PRIM1 deficiency causes a distinctive primordial dwarfism syndrome. Genes Dev. 2020, 34, 1520–1533. [Google Scholar] [CrossRef] [PubMed]
- Schmid, J.P.; Lemoine, R.; Nehme, N.T.; Cormier-Daire, V.; Revy, P.; Debeurme, F.; Debré, M.; Nitschke, P.; Bole-Feysot, C.; Legeai-Mallet, L.; et al. Polymerase ε1 mutation in a human syndrome with facial dysmorphism, immunodeficiency, livedo, and short stature (“FILS syndrome”). J. Exp. Med. 2012, 209, 2323–2330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eason, C.; Aleisa, A.; Jones, J.R.; Prijoles, E.J.; Lee, L.W. Filling in the gaps on FILS syndrome: A case report and literature review. Pediatr. Dermatol. 2020, 37, 915–917. [Google Scholar] [CrossRef] [PubMed]
- Thiffault, I.; Saunders, C.; Jenkins, J.; Raje, N.; Canty, K.; Sharma, M.; Grote, L.; Welsh, H.I.; Farrow, E.; Twist, G.; et al. A patient with polymerase E1 deficiency (POLE1): Clinical features and overlap with DNA breakage/instability syndromes. BMC Med. Genet. 2015, 16, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logan, C.V.; Murray, J.E.; Parry, D.A.; Robertson, A.; Bellelli, R.; Tarnauskaitė, Ž.; Challis, R.; Cleal, L.; Borel, V.; Fluteau, A.; et al. DNA Polymerase Epsilon Deficiency Causes IMAGe Syndrome with Variable Immunodeficiency. Am. J. Hum. Genet. 2018, 103, 1038–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frugoni, F.; Dobbs, K.; Felgentreff, K.; Aldhekri, H.; Al Saud, B.K.; Arnaout, R.; Ali, A.A.; Abhyankar, A.; Alroqi, F.; Giliani, S.; et al. A novel mutation in the POLE2 gene causing combined immunodeficiency. J. Allergy Clin. Immunol. 2016, 137, 635–638.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shastry, S.; Simha, V.; Godbole, K.; Sbraccia, P.; Melancon, S.; Yajnik, C.S.; Novelli, G.; Kroiss, M.; Garg, A. A Novel Syndrome of Mandibular Hypoplasia, Deafness, and Progeroid Features Associated with Lipodystrophy, Undescended Testes, and Male Hypogonadism. J. Clin. Endocrinol. Metab. 2010, 95, E192–E197. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, H.; Yanagi, K.; Ugi, S.; Kobayashi, K.; Ohkubo, K.; Tajiri, Y.; Maegawa, H.; Kashiwagi, A.; Kaname, T. Definitive diagnosis of mandibular hypoplasia, deafness, progeroid features and lipodystrophy (MDPL) syndrome caused by a recurrent de novo mutation in the POLD1 gene. Endocr. J. 2018, 65, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Weedon, M.N.; Ellard, S.; Prindle, M.J.; Caswell, R.; Allen, H.L.; Oram, R.A.; Godbole, K.; Yajnik, C.S.; Sbraccia, P.; Novelli, G.; et al. An in-frame deletion at the polymerase active site of POLD1 causes a multisystem disorder with lipodystrophy. Nat. Genet. 2013, 45, 947–950. [Google Scholar] [CrossRef] [Green Version]
- Okada, A.; Kohmoto, T.; Naruto, T.; Yokota, I.; Kotani, Y.; Shimada, A.; Miyamoto, Y.; Takahashi, R.; Goji, A.; Masuda, K.; et al. The first Japanese patient with mandibular hypoplasia, deafness, progeroid features and lipodystrophy diagnosed via POLD1 mutation detection. Hum. Genome Var. 2017, 4, 17031. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-W.; Lu, L.-Y.; Xie, Y.; Yu, X. A Chinese girl with mandibular hypoplasia, deafness, progeroid features, and lipodystrophy (MDPL) diagnosed via POLD1 mutation detection. Chin. Med. J. 2020, 133, 2009–2011. [Google Scholar] [CrossRef] [PubMed]
- Pelosini, C.; Martinelli, S.; Ceccarini, G.; Magno, S.; Barone, I.; Basolo, A.; Fierabracci, P.; Vitti, P.; Maffei, M.; Santini, F. Identification of a novel mutation in the polymerase delta 1 (POLD1) gene in a lipodystrophic patient affected by mandibular hypoplasia, deafness, progeroid features (MDPL) syndrome. Metabolism 2014, 63, 1385–1389. [Google Scholar] [CrossRef] [PubMed]
- Lessel, D.; Hisama, F.M.; Szakszon, K.; Saha, B.; Sanjuanelo, A.B.; Salbert, B.A.; Steele, P.D.; Baldwin, J.; Brown, W.T.; Piussan, C.; et al. POLD1Germline Mutations in Patients Initially Diagnosed with Werner Syndrome. Hum. Mutat. 2015, 36, 1070–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elouej, S.; Beleza-Meireles, A.; Caswell, R.; Colclough, K.; Ellard, S.; Desvignes, J.P.; Béroud, C.; Lévy, N.; Mohammed, S.; De Sandre-Giovannoli, A. Exome sequencing reveals a de novo POLD1 mutation causing phenotypic variability in mandibular hypoplasia, deafness, progeroid features, and lipodystrophy syndrome (MDPL). Metabolism 2017, 71, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Ajluni, N.; Meral, R.; Neidert, A.H.; Brady, G.F.; Buras, E.; McKenna, B.; DiPaola, F.; Chenevert, T.L.; Horowitz, J.F.; Buggs-Saxton, C.; et al. Spectrum of disease associated with partial lipodystrophy: Lessons from a trial cohort. Clin. Endocrinol. 2017, 86, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Conde, C.D.; Petronczki, Ö.Y.; Baris, S.; Willmann, K.L.; Girardi, E.; Salzer, E.; Weitzer, S.; Ardy, R.C.; Krolo, A.; Ijspeert, H.; et al. Polymerase δ deficiency causes syndromic immunodeficiency with replicative stress. J. Clin. Investig. 2019, 129, 4194–4206. [Google Scholar] [CrossRef]
- Nichols-Vinueza, D.X.; Delmonte, O.M.; Bundy, V.; Bosticardo, M.; Zimmermann, M.T.; Dsouza, N.R.; Pala, F.; Dobbs, K.; Stoddard, J.; Niemela, J.E.; et al. POLD1 Deficiency Reveals a Role for POLD1 in DNA Repair and T and B Cell Development. J. Clin. Immunol. 2020, 1–4. [Google Scholar] [CrossRef]
- Cui, Y.; Keles, S.; Charbonnier, L.-M.; Julé, A.M.; Henderson, L.; Celik, S.C.; Reisli, I.; Shen, C.; Xie, W.J.; Schmitz-Abe, K.; et al. Combined immunodeficiency caused by a loss-of-function mutation in DNA polymerase delta 1. J. Allergy Clin. Immunol. 2020, 145, 391–401.e8. [Google Scholar] [CrossRef] [Green Version]
- Baple, E.L.; Chambers, H.; Cross, H.E.; Fawcett, H.; Nakazawa, Y.; Chioza, B.A.; Harlalka, G.V.; Mansour, S.; Sreekantan-Nair, A.; Patton, M.A.; et al. Hypomorphic PCNA mutation underlies a human DNA repair disorder. J. Clin. Investig. 2014, 124, 3137–3146. [Google Scholar] [CrossRef] [Green Version]
- Siitonen, H.A.; Sotkasiira, J.; Biervliet, M.; Benmansour, A.; Capri, Y.; Cormier-Daire, V.; Crandall, B.; Hannula-Jouppi, K.; Hennekam, R.; Herzog, D.; et al. The mutation spectrum in RECQL4 diseases. Eur. J. Hum. Genet. 2008, 17, 151–158. [Google Scholar] [CrossRef]
- Van Maldergem, L.; Siitonen, H.A.; Jalkh, N.; Chouery, E.; De Roy, M.; Delague, V.; Muenke, M.; Jabs, E.W.; Cai, J.; Wang, L.L.; et al. Revisiting the craniosynostosis-radial ray hypoplasia association: Baller-Gerold syndrome caused by mutations in the RECQL4 gene. J. Med Genet. 2005, 43, 148–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, D.-H.; Mu, K.; Liu, D.; Sun, J.; Bai, X.; Zhang, N.; Qiu, G.; Ma, X. Case Report Identification of novel compound heterozygous RECQL4 mutations and prenatal diagnosis of Baller-Gerold syndrome: A case report. Genet. Mol. Res. 2015, 14, 4757–4766. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, H.; Izumi, R.; Oda, H.; Ohara, O.; Sameshima, K.; Ohnishi, H.; Fukao, T.; Funato, M. Nationwide survey of Baller-Gerold syndrome in Japanese population. Mol. Med. Rep. 2017, 15, 3222–3224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debeljak, M.; Zver, A.; Jazbec, J. A patient with Baller-Gerold syndrome and midline NK/T lymphoma. Am. J. Med. Genet. 2009, 149, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Gannavarapu, A.; Kozinetz, C.A.; Levy, M.L.; Lewis, R.A.; Chintagumpala, M.M.; Ruiz-Maldanado, R.; Contreras-Ruiz, J.; Cunniff, C.; Erickson, R.P.; et al. Association Between Osteosarcoma and Deleterious Mutations in the RECQL4 Gene in Rothmund-Thomson Syndrome. J. Natl. Cancer Instig. 2003, 95, 669–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piard, J.; Aral, B.; Vabres, P.; Holder-Espinasse, M.; Mégarbané, A.; Gauthier, S.; Capra, V.; Pierquin, G.; Callier, P.; Baumann, C.; et al. Search forReCQL4mutations in 39 patients genotyped for suspected Rothmund-Thomson/Baller-Gerold syndromes. Clin. Genet. 2014, 87, 244–251. [Google Scholar] [CrossRef]
- Cao, F.; Lu, L.; Abrams, S.A.; Hawthorne, K.M.; Tam, A.; Jin, W.; Dawson, B.; Shypailo, R.; Liu, H.; Lee, B.; et al. Generalized metabolic bone disease and fracture risk in Rothmund-Thomson syndrome. Hum. Mol. Genet. 2017, 26, 3046–3055. [Google Scholar] [CrossRef] [Green Version]
- Kitao, S.; Shimamoto, A.; Goto, M.; Miller, R.W.; Smithson, W.A.; Lindor, N.M.; Furuichi, Y. Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat. Genet. 1999, 22, 82–84. [Google Scholar] [CrossRef]
- Cabral, R.E.C.; Queille, S.; Bodemer, C.; De Prost, Y.; Neto, J.B.C.; Sarasin, A.; Daya-Grosjean, L. Identification of new RECQL4 mutations in Caucasian Rothmund–Thomson patients and analysis of sensitivity to a wide range of genotoxic agents. Mutat. Res. Mol. Mech. Mutagen. 2008, 643, 41–47. [Google Scholar] [CrossRef]
- Kellermayer, R.; Siitonen, H.A.; Hadzsiev, K.; Kestilä, M.; Kosztolányi, G. A Patient with Rothmund-Thomson Syndrome and All Features of RAPADILINO. Arch. Dermatol. 2005, 141, 617–620. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Geng, S.; Zheng, Y. Rare presentation of Rothmund-Thomson syndrome with novel compound heterozygous mutations of the RECQL4 gene. Bras. Dermatol. 2020, 95, 538–540. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Thakur, S.; Kohlhase, J.; Bhari, N.; Kabra, M.; Gupta, N. Report of Two Novel Mutations in Indian Patients with Rothmund–Thomson Syndrome. J. Pediatr. Genet. 2019, 8, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chen, L.; She, Q.; Dong, Y.; Deng, Y. Four novel RECQL4 mutations in four Chinese patients with Rothmund-Thomson syndrome and analysis of RECQL4 mRNA expression level in one typical patient. J. Dermatol. Sci. 2018, 91, 335–337. [Google Scholar] [CrossRef] [PubMed]
- Lindor, N.M.; Furuichi, Y.; Kitao, S.; Shimamoto, A.; Arndt, C.; Jalal, S. Rothmund-Thomson syndrome due toRECQ4 helicase mutations: Report and clinical and molecular comparisons with Bloom syndrome and Werner syndrome. Am. J. Med. Genet. 2000, 90, 223–228. [Google Scholar] [CrossRef]
- Wang, L.L.; Worley, K.C.; Gannavarapu, A.; Chintagumpala, M.M.; Levy, M.L.; Plon, S.E. Intron-Size Constraint as a Mutational Mechanism in Rothmund-Thomson Syndrome. Am. J. Hum. Genet. 2002, 71, 165–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balraj, P.; Concannon, P.; Jamal, R.; Beghini, A.; Hoe, T.; Khoo, A.S.; Volpi, L. An unusual mutation in RECQ4 gene leading to Rothmund–Thomson syndrome. Mutat. Res. Mol. Mech. Mutagen. 2002, 508, 99–105. [Google Scholar] [CrossRef]
- Beghini, A.; Castorina, P.; Roversi, G.; Modiano, P.; Larizza, L. RNA processing defects of the helicase gene RECQL4 in a compound heterozygous Rothmund-Thomson patient. Am. J. Med. Genet. 2003, 120, 395–399. [Google Scholar] [CrossRef]
- Van Rij, M.C.; Grijsen, M.L.; Appelman-Dijkstra, N.M.; Hansson, K.; Al Ruivenkamp, C.; Mulder, K.; Van Doorn, R.; Oranje, A.P.; Kant, S.G. Rothmund-Thomson syndrome and osteoma cutis in a patient previously diagnosed as COPS syndrome. Eur. J. Nucl. Med. Mol. Imaging 2016, 176, 279–283. [Google Scholar] [CrossRef] [Green Version]
- Gui, B.; Song, Y.; Hu, X.; Li, H.; Qin, Z.; Su, J.; Li, C.; Fan, X.; Li, M.; Luo, J.; et al. Novel pathogenic RECQL4 variants in Chinese patients with Rothmund-Thomson syndrome. Gene 2018, 654, 110–115. [Google Scholar] [CrossRef]
- Salih, A.; Inoue, S.; Onwuzurike, N. Rothmund-Thomson syndrome (RTS) with osteosarcoma due to RECQL4 mutation. BMJ Case Rep. 2018, 2018, 2017222384. [Google Scholar] [CrossRef]
- Bhoyrul, B.; Lindsay, H.; Robinson, R.; Stahlschmidt, J.; Palmer, T.; Edward, S.; Clark, S. Pili annulati in a case of Rothmund-Thomson syndrome with a novel frameshift mutation in RECQL4. J. Eur. Acad. Dermatol. Venereol. 2017, 32, e221–e223. [Google Scholar] [CrossRef] [PubMed]
- Suter, A.; Itin, P.; Heinimann, K.; Ahmed, M.; Ashraf, T.; Fryssira, H.; Kini, U.; Lapunzina, P.; Miny, P.; Sommerlund, M.; et al. Rothmund-Thomson Syndrome: Novel pathogenic mutations and frequencies of variants in the RECQL4 and USB1 (C16orf57) gene. Mol. Genet. Genom. Med. 2016, 4, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-González, G.A.; Martínez-Cabriales, S.A.; Hernández-Juárez, A.A.; Lugo-Trampe, J.D.J.; Espinoza-González, N.A.; Gómez-Flores, M.; Ocampo-Candiani, J. Rothmund-thomson syndrome: A 13-year follow-up. Case Rep. Dermatol. 2014, 6, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.; Kohlhase, J.; Wilhelm, C.; Kochanek, M.; De Carolis, B.; Berthold, F. Multiple malignant diseases in a patient with Rothmund-Thomson syndrome with RECQL4 mutations: Case report and literature review. Am. J. Med. Genet. 2010, 152, 1575–1579. [Google Scholar] [CrossRef]
- Fradin, M.; Merklen-Djafri, C.; Perrigouard, C.; Aral, B.; Muller, J.; Stoetzel, C.; Frouin, E.; Flori, E.; Doray, B.; Dollfus, H.; et al. Long-Term Follow-Up and Molecular Characterization of a Patient with a RECQL4 Mutation Spectrum Disorder. Dermatolog 2013, 226, 353–357. [Google Scholar] [CrossRef]
- Colombo, E.A.; Fontana, L.; Roversi, G.; Negri, G.; Castiglia, D.; Paradisi, M.; Zambruno, G.; Larizza, L. Novel physiological RECQL4 alternative transcript disclosed by molecular characterisation of Rothmund–Thomson Syndrome sibs with mild phenotype. Eur. J. Hum. Genet. 2014, 22, 1298–1304. [Google Scholar] [CrossRef] [Green Version]
- Colombo, E.A.; Locatelli, A.; Sánchez, L.C.; Romeo, S.; Elcioglu, N.H.; Maystadt, I.; Esteve-Martínez, A.; Sironi, A.; Fontana, L.; Finelli, P.; et al. Rothmund-Thomson Syndrome: Insights from New Patients on the Genetic Variability Underpinning Clinical Presentation and Cancer Outcome. Int. J. Mol. Sci. 2018, 19, 1103. [Google Scholar] [CrossRef] [Green Version]
- Sznajer, Y.; Siitonen, H.A.; Roversi, G.; Dangoisse, C.; Scaillon, M.; Ziereisen, F.; Tenoutasse, S.; Kestilä, M.; Larizza, L. Atypical Rothmund-Thomson syndrome in a patient with compound Heterozygous Mutations in RECQL4 Gene and phenotypic features in RECQL4 syndromes. Eur. J. Nucl. Med. Mol. Imaging 2007, 167, 175–181. [Google Scholar] [CrossRef]
- Broom, M.A.; Wang, L.; Otta, S.; Knutsen, A.; Siegfried, E.; Batanian, J.; Kelly, M.; Shah, M. Successful umbilical cord blood stem cell transplantation in a patient with Rothmund-Thomson syndrome and combined immunodeficiency. Clin. Genet. 2006, 69, 337–343. [Google Scholar] [CrossRef]
- Gao, J.; Wang, Q.; Dong, C.; Chen, S.; Qi, Y.; Liu, Y. Whole Exome Sequencing Identified MCM2 as a Novel Causative Gene for Autosomal Dominant Nonsyndromic Deafness in a Chinese Family. PLoS ONE 2015, 10, e0133522. [Google Scholar] [CrossRef]
- De Munnik, S.A.; Hoefsloot, L.H.; Roukema, J.; Schoots, J.; Knoers, N.V.A.M.; Brunner, H.G.; Jackson, A.P.; Bongers, E.M.H.F. Meier-Gorlin syndrome. Orphanet J. Rare Dis. 2015, 10, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Munnik, S.A.; Otten, B.J.; Schoots, J.; Bicknell, L.S.; Aftimos, S.; Al-Aama, J.Y.; Van Bever, Y.; Bober, M.B.; Borm, G.F.; Clayton-Smith, J.; et al. Meier-Gorlin syndrome: Growth and secondary sexual development of a microcephalic primordial dwarfism disorder. Am. J. Med. Genet. 2012, 158, 2733–2742. [Google Scholar] [CrossRef] [PubMed]
- Stiff, T.; Alagoz, M.; Alcantara, D.; Outwin, E.; Brunner, H.G.; Bongers, E.M.H.F.; O’Driscoll, M.; Jeggo, P.A. Deficiency in Origin Licensing Proteins Impairs Cilia Formation: Implications for the Aetiology of Meier-Gorlin Syndrome. PLoS Genet. 2013, 9, e1003360. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Chen, J.; Wu, X.; Jia, S.; Meng, A. Zebrafish cdc6 hypomorphic mutation causes Meier-Gorlin syndrome-like phenotype. Hum. Mol. Genet. 2017, 26, 4168–4180. [Google Scholar] [CrossRef] [Green Version]
- Bleichert, F.; Balasov, M.; Chesnokov, I.; Nogales, E.; Botchan, M.R.; Berger, J.M. A Meier-Gorlin syndrome mutation in a conserved C-terminal helix of Orc6 impedes origin recognition complex formation. eLife 2013, 2, e00882. [Google Scholar] [CrossRef]
- Kuo, A.J.; Song, J.; Cheung, P.; Ishibe-Murakami, S.; Yamazoe, S.; Chen, J.K.; Patel, D.J.; Gozani, O. The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier-Gorlin syndrome. Nat. Cell Biol. 2012, 484, 115–119. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Sankaran, S.; Gozani, O.; Song, J. A Meier-Gorlin Syndrome Mutation Impairs the ORC1-Nucleosome Association. ACS Chem. Biol. 2015, 10, 1176–1180. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.A.; Collis, S.J. Ciliogenesis and the DNA damage response: A stressful relationship. Cilia 2016, 5, 19. [Google Scholar] [CrossRef] [Green Version]
- O’Driscoll, M.; Ruiz-Perez, V.L.; Woods, C.G.; Jeggo, P.A.; Goodship, J.A. Goodship, A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat. Genet. 2003, 33, 497–501. [Google Scholar] [CrossRef] [Green Version]
- Guemez-Gamboa, A.; Coufal, N.G.; Gleeson, J.G. Primary Cilia in the Developing and Mature Brain. Neuron 2014, 82, 511–521. [Google Scholar] [CrossRef] [Green Version]
- Waters, A.M.; Beales, P.L. Ciliopathies: An expanding disease spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, K.; Oyaizu, N.; Dutta, A.; Inoue, I. The destruction box of human Geminin is critical for proliferation and tumor growth in human colon cancer cells. Oncogene 2004, 23, 58–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrill, S.; He, D.Z. Apoptosis in inner ear sensory hair cells. J. Otol. 2017, 12, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shilling, H.G.; McQueen, K.L.; Cheng, N.W.; Shizuru, J.A.; Negrin, R.S.; Parham, P. Reconstitution of NK cell receptor repertoire following HLA-matched hematopoietic cell transplantation. Blood 2003, 101, 3730–3740. [Google Scholar] [CrossRef] [Green Version]
- Chan, A.; Hong, D.-L.; Atzberger, A.; Kollnberger, S.; Filer, A.D.; Buckley, C.D.; McMichael, A.; Enver, T.; Bowness, P. CD56bright Human NK Cells Differentiate into CD56dim Cells: Role of Contact with Peripheral Fibroblasts. J. Immunol. 2007, 179, 89–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romagnani, C.; Juelke, K.; Falco, M.; Morandi, B.; D’Agostino, A.; Costa, R.; Ratto, G.; Forte, G.; Carrega, P.; Lui, G.; et al. CD56brightCD16− Killer Ig-Like Receptor− NK Cells Display Longer Telomeres and Acquire Features of CD56dim NK Cells upon Activation. J. Immunol. 2007, 178, 4947–4955. [Google Scholar] [CrossRef] [Green Version]
- Orange, J.S. Natural Killer Cell Deficiency. In Stiehm’s Immune Deficiencies; Elsevier: London, UK, 2014; pp. 765–774. [Google Scholar]
- Valiathan, R.; Deeb, K.; Diamante, M.; Ashman, M.; Sachdeva, N.; Asthana, D. Reference ranges of lymphocyte subsets in healthy adults and adolescents with special mention of T cell maturation subsets in adults of South Florida. Immunobiology 2014, 219, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469. [Google Scholar] [CrossRef]
- Orange, J.S. Natural killer cell deficiency. J. Allergy Clin. Immunol. 2013, 132, 515–525. [Google Scholar] [CrossRef] [Green Version]
- Eidenschenk, C.; Dunne, J.; Jouanguy, E.; Fourlinnie, C.; Gineau, L.; Bacq, D.; McMahon, C.; Smith, O.; Casanova, J.-L.; Abel, L.; et al. A Novel Primary Immunodeficiency with Specific Natural-Killer Cell Deficiency Maps to the Centromeric Region of Chromosome 8. Am. J. Hum. Genet. 2006, 78, 721–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shima, N.; Alcaraz, A.; Liachko, I.; Buske, T.R.; Andrews, C.A.; Munroe, R.J.; Hartford, S.A.; Tye, B.K.; Schimenti, J.C. A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat. Genet. 2006, 39, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Glover, T.W.; Berger, C.; Coyle, J.; Echo, B. DNA polymerase α inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum. Genet. 1984, 67, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Moon, W.Y.; Powis, S.J. Does Natural Killer Cell Deficiency (NKD) Increase the Risk of Cancer? NKD May Increase the Risk of Some Virus Induced Cancer. Front. Immunol. 2019, 10, 1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franchitto, A.; Pichierri, P. Replication fork recovery and regulation of common fragile sites stability. Cell. Mol. Life Sci. 2014, 71, 4507–4517. [Google Scholar] [CrossRef]
- Hanna, S.; Béziat, V.; Jouanguy, E.; Casanova, J.L.; Etzioni, A. A homozygous mutation of RTEL1 in a child presenting with an apparently isolated natural killer cell deficiency. J. Allergy Clin. Immunol. 2015, 136, 1113–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speckmann, C.; Sahoo, S.S.; Rizzi, M.; Hirabayashi, S.; Karow, A.; Serwas, N.K.; Hoemberg, M.; Damatova, N.; Schindler, D.; Vannier, J.-B.; et al. Clinical and Molecular Heterogeneity of RTEL1 Deficiency. Front. Immunol. 2017, 8, 449. [Google Scholar] [CrossRef] [Green Version]
- Babushok, D.V.; Hsu, A.P.; Dokal, I. Bone marrow failure syndromes. In Stiehm’s Immune Deficiencies; Elsevier: London, UK, 2020; pp. 411–441. [Google Scholar]
- Fasth, A.; Forestier, E.; Holmberg, E.; Holmgren, G.; Nordenson, I.; Soderstrom, T.; Wahlström, J. Fragility of the Centromeric Region of Chromosome 1 Associated with Combined Immunodeficiency in Siblings A Recessively Inherited Entity? Acta Paediatr. 1990, 79, 605–612. [Google Scholar] [CrossRef]
- Armando, R.G.; Gómez, D.L.M.; Maggio, J.; Sanmartin, M.C.; Gomez, D.E. Telomeropathies: Etiology, diagnosis, treatment and follow-up. Ethical and legal considerations. Clin. Genet. 2019, 96, 3–16. [Google Scholar] [CrossRef]
- Ouyang, Q.; Baerlocher, G.; Vulto, I.; Lansdorp, P.M. Telomere Length in Human Natural Killer Cell Subsets. Ann. N. Y. Acad. Sci. 2007, 1106, 240–252. [Google Scholar] [CrossRef]
- Fali, T.; Papagno, L.; Bayard, C.; Mouloud, Y.; Boddaert, J.; Sauce, D.; Appay, V. New Insights into Lymphocyte Differentiation and Aging from Telomere Length and Telomerase Activity Measurements. J. Immunol. 2019, 202, 1962–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, A.C.Y.; Blau, H.M. Short telomeres—A hallmark of heritable cardiomyopathies. Differentiation 2018, 100, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.C.Y.; Chang, A.C.H.; Kirillova, A.; Sasagawa, K.; Su, W.; Weber, G.; Lin, J.; Termglinchan, V.; Karakikes, I.; Seeger, T.; et al. Telomere shortening is a hallmark of genetic cardiomyopathies. Proc. Natl. Acad. Sci. USA 2018, 115, 9276–9281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedreira, C.; Savarirayan, R.; Zacharin, M.R. IMAGe syndrome: A complex disorder affecting growth, adrenal and gonadal function, and skeletal development. J. Pediatr. 2004, 144, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Stampone, E.; Caldarelli, I.; Zullo, A.; Bencivenga, D.; Mancini, F.P.; Ragione, F.D.; Borriello, A. Genetic and Epigenetic Control of CDKN1C Expression: Importance in Cell Commitment and Differentiation, Tissue Homeostasis and Human Diseases. Int. J. Mol. Sci. 2018, 19, 1055. [Google Scholar] [CrossRef] [Green Version]
- Reinier, F.; Zoledziewska, M.; Hanna, D.; Smith, J.D.; Valentíni, M.; Zara, I.; Berutti, R.; Sanna, S.; Oppo, M.; Cusano, R.; et al. Mandibular hypoplasia, deafness, progeroid features and lipodystrophy (MDPL) syndrome in the context of inherited lipodystrophies. Metab. Clin. Exp. 2015, 64, 1530–1540. [Google Scholar] [CrossRef]
- Shah, S.N.; Opresko, P.L.; Meng, X.; Lee, M.Y.W.T.; Eckert, K.A. DNA structure and the Werner protein modulate human DNA polymerase delta-dependent replication dynamics within the common fragile site FRA16D. Nucleic Acids Res. 2009, 38, 1149–1162. [Google Scholar] [CrossRef] [Green Version]
- Tsurimoto, T.; Shinozaki, A.; Yano, M.; Seki, M.; Enomoto, T. Human Werner helicase interacting protein 1 (WRNIP1) functions as a novel modulator for DNA polymerase δ. Genes Cells 2004, 10, 13–22. [Google Scholar] [CrossRef]
- Li, B.; Reddy, S.; Comai, L. The Werner Syndrome Helicase Coordinates Sequential Strand Displacement and FEN1-Mediated Flap Cleavage during Polymerase δ Elongation. Mol. Cell. Biol. 2016, 37, e00560-16. [Google Scholar] [CrossRef] [Green Version]
- Feltes, B.C.; Poloni, J.D.F.; Miyamoto, K.; Bonatto, D. Human Diseases Associated With Genome Instability. In Genome Stability; Elsevier: Amsterdam, The Netherlands, 2016; pp. 447–462. [Google Scholar]
- Duffy, C.M.; Hilbert, B.J.; Kelch, B.A. A Disease-Causing Variant in PCNA Disrupts a Promiscuous Protein Binding Site. J. Mol. Biol. 2016, 428, 1023–1040. [Google Scholar] [CrossRef]
- Siitonen, H.A.; Haravuori, H. Molecular defect of RAPADILINO syndrome expands the phenotype spectrum of RECQL diseases. Hum. Mol. Genet. 2003, 12, 2837–2844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larizza, L.; Magnani, I.; Roversi, G. Rothmund–Thomson syndrome and RECQL4 defect: Splitting and lumping. Cancer Lett. 2006, 232, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Dietschy, T.; Shevelev, I.; Stagljar, I. The molecular role of the Rothmund-Thomson-, RAPADILINO- and Baller-Gerold-gene product, RECQL4: Recent progress. Cell. Mol. Life Sci. 2007, 64, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Kaariainen, H.; Ryöppy, S.; Norio, R. RAPADILINO syndrome with radial and patellar aplasia/hypoplasia as main manifestations. Am. J. Med. Genet. 1989, 33, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Lindor, N.M.; Devries, E.M.G.; Michels, V.V.; Schad, C.R.; Jalal, S.M.; Donovan, K.M.; Smithson, W.A.; Kvols, L.K.; Thibodeau, S.N.; Dewald, G.W. Rothmund-Thomson syndrome in siblings: Evidence for acquired in vivo mosaicism. Clin. Genet. 1996, 49, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Larizza, L.; Roversi, G.; Volpi, L. Rothmund-Thomson syndrome. Orphanet J. Rare Dis. 2010, 5, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Pujol, L.A.; Erickson, R.P.; Heidenreich, R.A.; Cunniff, C. Variable presentation of Rothmund-Thomson syndrome. Am. J. Med. Genet. 2000, 95, 204–207. [Google Scholar] [CrossRef]
- Tong, M. Rothmund-Thomson Syndrome in Fraternal Twins. Pediatr. Dermatol. 1995, 12, 134–137. [Google Scholar] [CrossRef]
- Kim, J.S. Baller-Gerold Syndrome in a Premature Infant with a Mutation in the RECQL4 Gene. Neonatal Med. 2019, 26, 240–245. [Google Scholar] [CrossRef]
- Ajeawung, N.F.; Nguyen, T.T.M.; Lu, L.; Kucharski, T.J.; Rousseau, J.; Molidperee, S.; Atienza, J.; Gamache, I.; Jin, W.; Plon, S.E.; et al. Mutations in ANAPC1, Encoding a Scaffold Subunit of the Anaphase-Promoting Complex, Cause Rothmund-Thomson Syndrome Type 1. Am. J. Hum. Genet. 2019, 105, 625–630. [Google Scholar] [CrossRef] [Green Version]
- Adam, M.P.; Ardinger, H.H.; Pagon, R.A.; Wallace, S.E.; Bean, L.J.; Stephens, K.; Amemiya, A. (Eds.) GeneReviews [Internet]; University of Washington: Seattle, DC, USA, 1993–2021. [Google Scholar]
- Burks, L.M.; Yin, J.; Plon, S.E. Nuclear import and retention domains in the amino terminus of RECQL4. Gene 2007, 391, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.K.; Karmakar, P.; Aamann, M.; Schurman, S.H.; May, A.; Croteau, D.L.; Burks, L.; Plon, S.E.; Bohr, V.A. The involvement of human RECQL4 in DNA double-strand break repair. Aging Cell 2010, 9, 358–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croteau, D.L.; Rossi, M.L.; Ross, J.; Dawut, L.; Dunn, C.; Kulikowicz, T.; Bohr, V.A. RAPADILINO RECQL4 Mutant Protein Lacks Helicase and ATPase Activity. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 1727–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vollebregt, M.M.G.; Malfroot, A.; De Raedemaecker, M.; Van Der Burg, M.; Bosch, J.V.D.W.T. Immunodeficiency in a Child with Rapadilino Syndrome: A Case Report and Review of the Literature. Case Rep. Immunol. 2015, 2015, 1–4. [Google Scholar] [CrossRef] [Green Version]
- De Somer, L.; Wouters, C.; Morren, M.-A.; De Vos, R.; Oord, J.V.D.; Devriendt, K.; Meyts, I. Granulomatous skin lesions complicating Varicella infection in a patient with Rothmund-Thomson syndrome and immune deficiency: Case report. Orphanet J. Rare Dis. 2010, 5, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Tokura, Y.; Moriwaki, S.; Yasuda, K.; Ohnishi, A.; Furukawa, F.; Yamaizumi, K.; Takigawa, M. Rothmund-Thomson syndrome with herpes encephalitis. Eur. J. Dermatol. EJD 1999, 9, 354–356. [Google Scholar]
- Kubota, M.; Yasunaga, M.; Hashimoto, H.; Kimata, H.; Mikawa, H.; Shinya, A.; Nishigori, C. IgG4 deficiency with Rothmund-Thomson syndrome: A case report. Eur. J. Nucl. Med. Mol. Imaging 1993, 152, 406–408. [Google Scholar] [CrossRef]
- Abe, T.; Yoshimura, A.; Hosono, Y.; Tada, S.; Seki, M.; Enomoto, T. The N-terminal region of RECQL4 lacking the helicase domain is both essential and sufficient for the viability of vertebrate cells. Biochim. Biophys. Acta Bioenergy 2011, 1813, 473–479. [Google Scholar] [CrossRef] [Green Version]
- Castillo-Tandazo, W.; Smeets, M.F.; Murphy, V.; Liu, R.; Hodson, C.; Heierhorst, J.; Deans, A.J.; Walkley, C.R. ATP-dependent helicase activity is dispensable for the physiological functions of Recql4. PLoS Genet. 2019, 15, e1008266. [Google Scholar] [CrossRef]
- Matson, J.P.; Dumitru, R.; Coryell, P.; Baxley, R.M.; Chen, W.; Twaroski, K.; Webber, B.R.; Tolar, J.; Bielinsky, A.-K.; Purvis, J.E.; et al. Rapid DNA replication origin licensing protects stem cell pluripotency. eLife 2017, 6, e30473. [Google Scholar] [CrossRef]
- Rivera-Mulia, J.C.; Buckley, Q.; Sasaki, T.; Zimmerman, J.; Didier, R.A.; Nazor, K.; Loring, J.F.; Lian, Z.; Weissman, S.; Robins, A.J.; et al. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res. 2015, 25, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Besnard, E.; Babled, A.; Lapasset, L.; Milhavet, O.; Parrinello, H.; Dantec, C.; Marin, J.-M.; Lemaitre, J.-M. Unraveling cell type–specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat. Struct. Mol. Biol. 2012, 19, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Maccaroni, K.; Balzano, E.; Mirimao, F.; Giunta, S.; Pelliccia, F. Impaired Replication Timing Promotes Tissue-Specific Expression of Common Fragile Sites. Genes 2020, 11, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, S.; Díaz, M.; Flach, J.; Rodriguez-Acebes, S.; López-Contreras, A.J.; Martínez, D.; Cañamero, M.; Fernández-Capetillo, O.; Isern, J.; Passegué, E.; et al. Replication stress caused by low MCM expression limits fetal erythropoiesis and hematopoietic stem cell functionality. Nat. Commun. 2015, 6, 8548. [Google Scholar] [CrossRef] [PubMed]
- Stevens, H.; Williams, A.B.; Michael, W.M. Cell-Type Specific Responses to DNA Replication Stress in Early C. elegans Embryos. PLoS ONE 2016, 11, e0164601. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene(s) Affected | MIM ID | Inheritance | IUGR | Short Stature | Microcephaly | Craniosynostosis | Facial Features | Skin Changes | Radial Ray Defects | A/Hypoplastic Patella | Adrenal Insufficiency | GU Abnormalities (male) | ID | DD | Hearing Loss | Immune Defects | Anorectal Malformation | Feeding Difficulties | Diarrhea | Premature Aging | Lipodystrophy | Cancer Predisposition | Other symptoms | References | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MGS | ORC1 | 224690 | AR | x | x | x | microtia microstomia full lipsret rognathia micrognathia | x | x | x | x | [67,68,72,73,74] | |||||||||||||
| ORC4 | 613800 | AR | x | x | x | x | x | [67,68,72] | |||||||||||||||||
| ORC6 | 613803 | AR | x | x | x | x | x | x | [67,72,75] | ||||||||||||||||
| CDT1 | 613804 | AR | x | x | x | x | x | x | [67,68,72,76] | ||||||||||||||||
| CDC6 | 613805 | AR | x | x | x | x | x | [72,76] | |||||||||||||||||
| MCM5 | 617564 | AR | x | x | x | x | [70] | ||||||||||||||||||
| CDC45 | 617063 | AR | x | x | x | x | x | x | x | x | [69,77] | ||||||||||||||
| GMNN | 616835 | AD | x | x | x | x | x | x | [71] | ||||||||||||||||
| DONSON | AR | x | x | x | x | x | x | [76,78] | |||||||||||||||||
| NKD | MCM4 | 609981 | AR | x | x | x | large forehead, thin upper lip | x | x | x | x | +/− | [79,80,81,82] | ||||||||||||
| GINS1 | 617827 | AR | x | x | flat nasal bridge, round nose tip, blepharophimosis, posteriorly rotated ears, thin upper lip | E | x | +/− | [83] | ||||||||||||||||
| MCM10 | AR | UR | UR | none noted | x | [84,85] | |||||||||||||||||||
| XLPRD | POLA1 | 301220 | XLR | Blepharophimosis, flared eyebrows, upswept hair | x | x | sterile multiorgan inflammation | [86,87,88,89,90] | |||||||||||||||||
| VEODS | POLA1 | 301030 | XLR | x | x | inconsistent | x | x | +/− | [91,92] | |||||||||||||||
| PRIM1 | AR | x | x | prominent forehead, triangular face, blepharophimosis, micrognathia, small-low set ears | x | x | x | x | x | [93] | |||||||||||||||
| FILS | POLE1 | 615139 | AR | x | malar hypoplasia high forehead | L | x | [94,95,96] | |||||||||||||||||
| IMAGe | POLE1 | 618336 | AR | x | x | x | Micrognathia, long thin nose, short wide neck, small low-set, posteriorly rotated ears | other | x | x | x | x | +/− | [97] | |||||||||||
| POLE2 | AR | x | low anterior hairline, flat supraorbital ridges, downturned corners of mouth, short philtrum | x | [98] | ||||||||||||||||||||
| MDP | POLD1 | 615381 | AD | x | mandibular hypoplasia, pinched nose, microstomia, bird like facies, prominent eyes | T | x | x | x | x | [99,100,101,102,103,104,105,106,107] | ||||||||||||||
| POLD1 | AR | x | none noted | x | x | x | [108,109,110] | ||||||||||||||||||
| POLD2 | AR | x | x | none noted | x | x | x | [108] | |||||||||||||||||
| PCNA | 615919 | AR | x | none noted | T | x | x | x | x | neurodegeneration, ataxia | [111] | ||||||||||||||
| BGS | RECQL4 | 218600 | AR | x | x | x | present but variable | P | x | x | x | x | x | x | [112,113,114,115,116] | ||||||||||
| RAPA | RECQL4 | 266280 | AR | x | x | long face, long slender nose, narrow palpebral fissures, arched palate | x | x | +/− | x | x | x | [112,117,118,119] | ||||||||||||
| RTS | RECQL4 | 268400 | AR | x | x | x | present but variable | T, P | x | x | x | +/− | x | x | x | x | alopecia | [112,114,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141] | |||||||
| MCM2 | 616968 | AD | none noted | x | [142] | ||||||||||||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmit, M.; Bielinsky, A.-K. Congenital Diseases of DNA Replication: Clinical Phenotypes and Molecular Mechanisms. Int. J. Mol. Sci. 2021, 22, 911. https://doi.org/10.3390/ijms22020911
Schmit M, Bielinsky A-K. Congenital Diseases of DNA Replication: Clinical Phenotypes and Molecular Mechanisms. International Journal of Molecular Sciences. 2021; 22(2):911. https://doi.org/10.3390/ijms22020911
Chicago/Turabian StyleSchmit, Megan, and Anja-Katrin Bielinsky. 2021. "Congenital Diseases of DNA Replication: Clinical Phenotypes and Molecular Mechanisms" International Journal of Molecular Sciences 22, no. 2: 911. https://doi.org/10.3390/ijms22020911
APA StyleSchmit, M., & Bielinsky, A. -K. (2021). Congenital Diseases of DNA Replication: Clinical Phenotypes and Molecular Mechanisms. International Journal of Molecular Sciences, 22(2), 911. https://doi.org/10.3390/ijms22020911