
Extracellular Matrix in Calcific Aortic Valve Disease: Architecture, Dynamic and Perspectives

 , , , ,
, , , ,  and
and 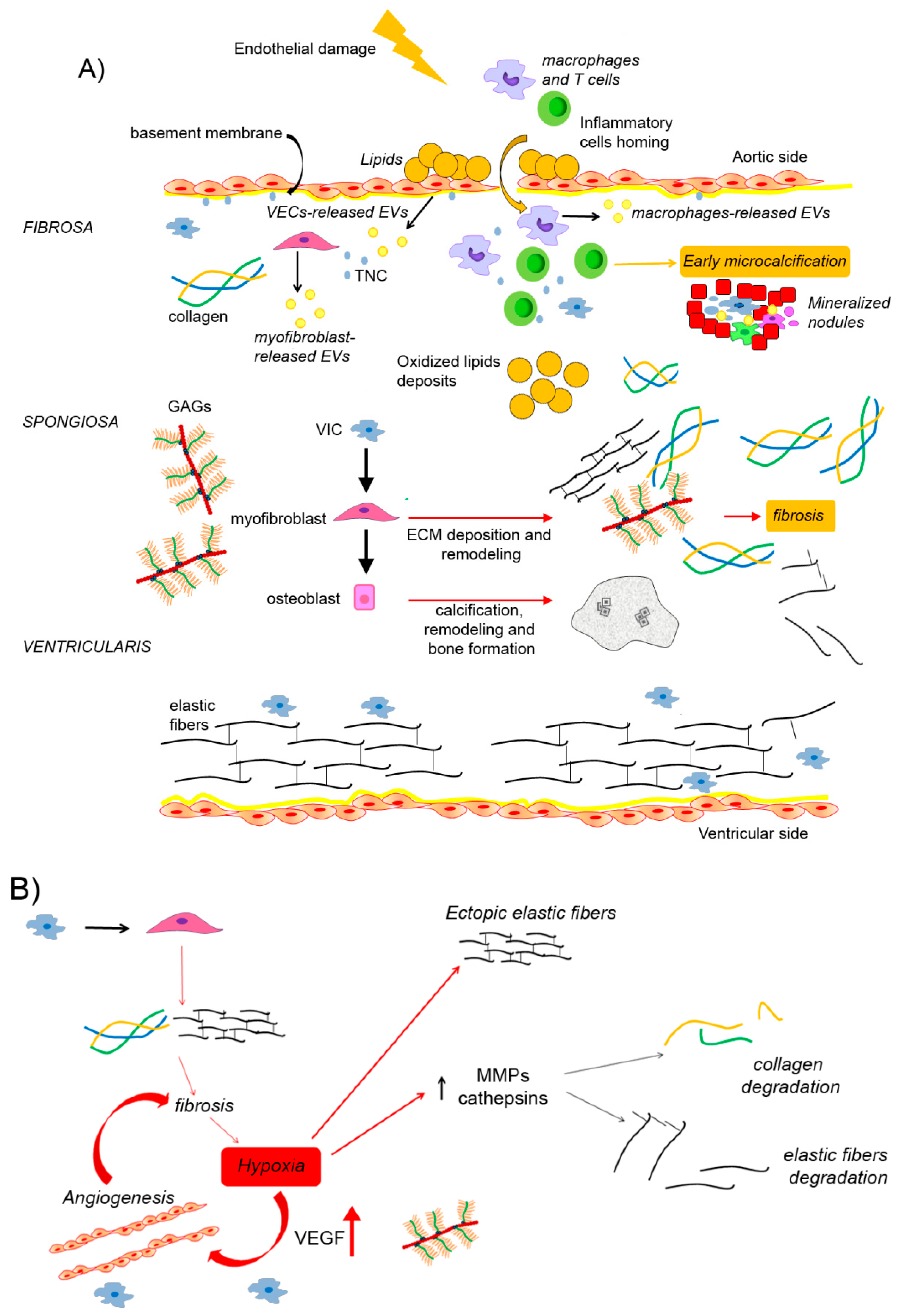{kind=link}
{kind=link}
Abstract
:1. Calcific Aortic Valve Disease
2. An Overview of the Cellular Processes Involved in Aortic Valve Calcification
3. The Role of Inflammation and Oxidative Stress in the Onset of Aortic Valve Calcification
4. Architecture of Extracellular Matrix in CAVD
4.1. Dystrophic Calcification and Biomineralization
4.2. The Extracellular Vesicles as Mediators of Aortic Valve Calcification
4.3. Production and Distribution of Collagen Fibers and Elastic Fibers in CAVD
4.3.1. The Role of Hypoxia in Collagen and Elastin Remodeling
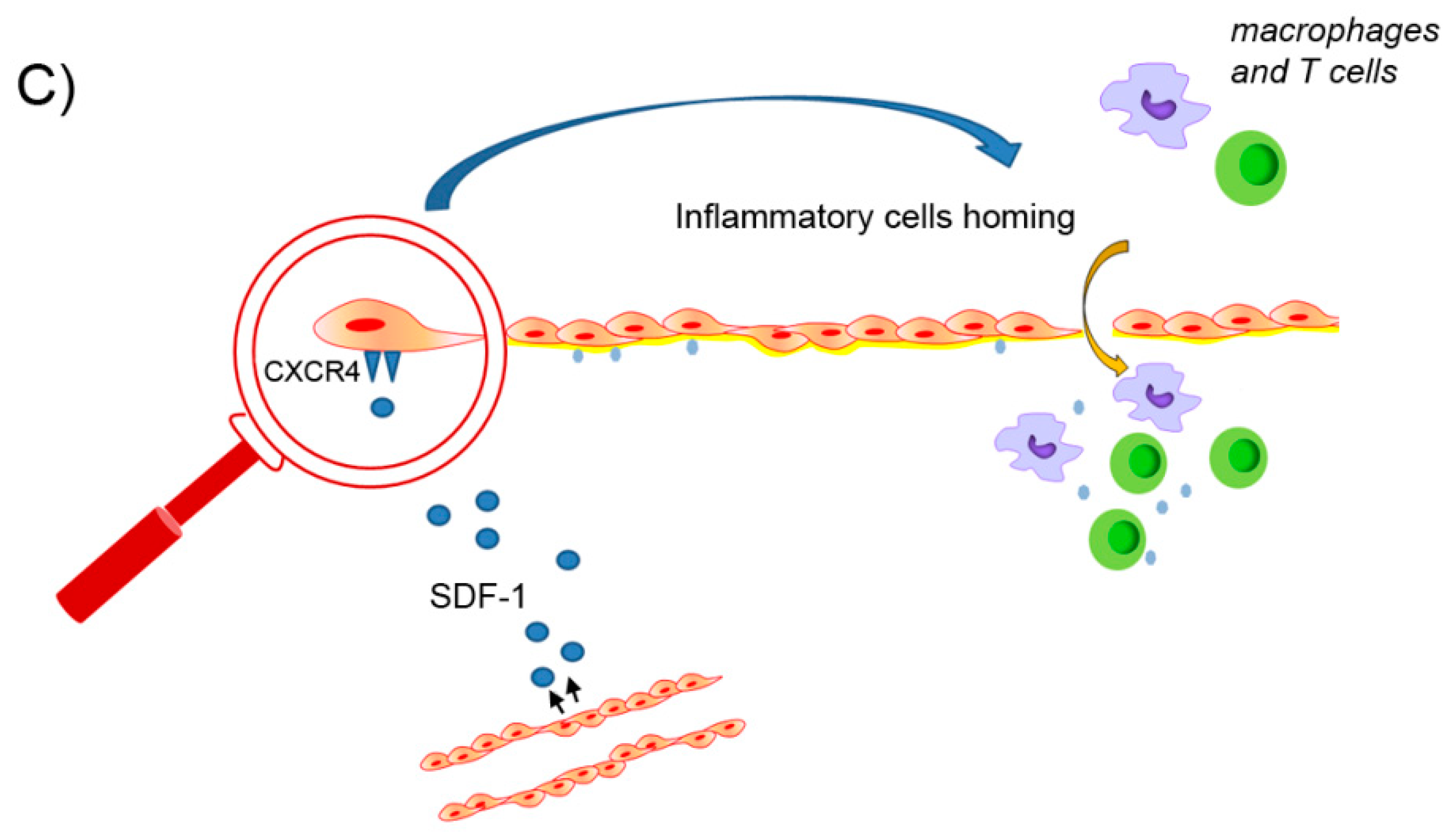
4.3.2. The Role of the SDF-1/CXCR4 Axis in Collagen and Elastin Remodeling
4.4. Production and Distribution of Glycosaminoglycans and Proteoglycans in CAVD
4.5. Production and Distribution of Tenascin-C and Periostin in CAVD
5. Discussion and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADAMTS | A disintegrin-like and metalloprotease domain with thrombospondin type 1 motifs ALPAlkaline Phosphatase |
| AS | aortic stenosis |
| BMP | bone morphogenetic protein |
| CAVD | calcific aortic valve disease |
| ECM | extracellular matrix |
| EMT | epithelial-mesenchymal transition |
| EndMT | endothelial-mesenchymal transition |
| EVs | extracellular vesicles |
| GAGs | glycosaminoglycans |
| HA | hyaluronic acid |
| HIF-1α | hypoxia inducible factor-1 α |
| ICAM-1 | intercellular adhesion molecule 1 |
| LOX | lysyl oxidase |
| LPS | lipopolysaccharides |
| MMPs | matrix metalloproteinases |
| Msx2 | MshHomeobox 2 |
| MVs | matrix vesicles |
| NGAL | neutrophil gelatinase-associated lipocalin |
| PCSK9 | proprotein convertase subtilisin/kexin type 9 |
| PGC-1α | peroxisome proliferator-activated receptor-γ coactivator 1-α |
| PGs | proteoglycans |
| PO | periostin |
| RHAMM | receptor for hyaluronate-mediated motility |
| RUNX2 | runt-related transcription factor 2 |
| SMCs | smooth muscle cells |
| TAVI | trans-catheter aortic valve implantation |
| TAVR | trans-catheter aortic valve replacement |
| TGF-β | transforming growth factor-β |
| TIMPs | tissue inhibitors of metalloproteinases |
| TNC | tenascin-C |
| TSG-6 | TNF-α stimulated gene-6 |
| VECs | vascular endothelial cells |
| VEGF | vascular endothelial growth factor |
| VICs | vascular interstitial cells |
| α-SMA | α-smooth muscle actin |
References
- Virmani, R.; Burke, A.; Farb, A.; Atkinson, J.B. Cardiovascular Pathology; Saunders: Philadelphia, PA, USA, 2001; pp. 248–249. [Google Scholar]
- Fishbein, G.A.; Fishbein, M.C. Pathology of the Aortic Valve: Aortic Valve Stenosis/Aortic Regurgitation. Curr. Cardiol. Rep. 2019, 21, 81. [Google Scholar] [CrossRef]
- Liu, T.; Xie, M.; Lv, Q.; Li, Y.; Fang, L.; Zhang, L.; Deng, W.; Wang, J. Bicuspid Aortic Valve: An Update in Morphology, Genetics, Biomarker, Complications, Imaging Diagnosis and Treatment. Front. Physiol. 2019, 9, 1921. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Wang, Y.; Xiao, F.; Butcher, J.T.; Yutzey, K.E.; Zhou, B. Developmental Mechanisms of Aortic Valve Malformation and Disease. Annu. Rev. Physiol. 2017, 79, 21–41. [Google Scholar] [CrossRef]
- Dutta, P.; Lincoln, J. Calcific Aortic Valve Disease: A Developmental Biology Perspective. Curr. Cardiol. Rep. 2018, 20, 21. [Google Scholar] [CrossRef] [Green Version]
- Rajput, F.A.; Zeltser, R. Aortic Valve Replacement; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Leopold, J.A. Cellular mechanisms of aortic valve calcification. Circ. Cardiovasc. Interv. 2012, 5, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Hadji, F.; Boulanger, M.C.; Guay, S.P.; Gaudreault, N.; Amellah, S.; Mkannez, G.; Bouchareb, R.; Marchand, J.T.; Nsaibia, M.J.; Guauque-Olarte, S.; et al. Altered DNA Methylation of Long Noncoding RNA H19 in Calcific Aortic Valve Disease Promotes Mineralization by Silencing NOTCH1. Circulation 2016, 134, 1848–1862. [Google Scholar] [CrossRef]
- García-Rodriguez, C.; Parra-Izquierdo, I.; Castaños-Mollor, I.; López, J.; San Román, J.A.; Sánchez Crespo, M. Toll-like receptors, inflammation, and calcific aortic valve disease. Front. Physiol. 2018, 9, 201. [Google Scholar] [CrossRef] [Green Version]
- Latif, N.; Sarathchandra, P.; Chester, A.H.; Yacoub, M.H. Expression of smooth muscle cell markers and co-activators in calcified aortic valves. Eur. Heart J. 2015, 36, 1335–1345. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Zhao, D.; Yuan, P.; Li, J.; Yun, Y.; Cui, Y.; Zhang, T.; Ma, J.; Sun, L.; Ma, H.; et al. Endothelial-to-Mesenchymal Transition in Calcific Aortic Valve Disease. Acta Cardiol. Sin. 2020, 36, 183–194. [Google Scholar] [CrossRef]
- Aikawa, E.; Whittaker, P.; Farber, M.; Mendelson, K.; Padera, R.F.; Aikawa, M.; Schoen, F.J. Human semilunar cardiac valve remodeling by activated cells from fetus to adult: Implications for postnatal adaptation, pathology, and tissue engineering. Circulation 2006, 113, 1344–1352. [Google Scholar] [CrossRef]
- Galeone, A.; Brunetti, G.; Oranger, A.; Greco, G.; Di Benedetto, A.; Mori, G.; Colucci, S.; Zallone, A.; Paparella, D.; Grano, M. Aortic valvular interstitial cells apoptosis and calcification are mediated by TNF-related apoptosis-inducing ligand. Int. J. Cardiol. 2013, 169, 296–304. [Google Scholar] [CrossRef]
- Sun, C.; Liu, H.; Si, K.; Wu, Y.; Zhao, K.; Xu, R.; Zhou, Z.; Zheng, Z. Meis2 represses the osteoblastic transdifferentiation of aortic valve interstitial cells through the Notch1/Twist1 pathway. Biochem. Biophys. Res. Commun. 2019, 509, 455–461. [Google Scholar] [CrossRef]
- Nagy, E.; Eriksson, P.; Yousry, M.; Caidahl, K.; Ingelsson, E.; Hansson, G.K.; Franco-Cereceda, A.; Bäck, M. Valvular osteoclasts in calcification and aortic valve stenosis severity. Int. J. Cardiol. 2013, 168, 2264–2271. [Google Scholar] [CrossRef] [Green Version]
- Rutkovskiy, A.; Malashicheva, A.; Sullivan, G.; Bogdanova, M.; Kostareva, A.; Stensløkken, K.O.; Fiane, A.; Vaage, J. Valve Interstitial Cells: The Key to Understanding the Pathophysiology of Heart Valve Calcification. J. Am. Heart Assoc. 2017, 6, e006339. [Google Scholar] [CrossRef]
- Xu, K.; Xie, S.; Huang, Y.; Zhou, T.; Liu, M.; Zhu, P.; Wang, C.; Shi, J.; Li, F.; Sellke, F.W.; et al. Cell-Type Transcriptome Atlas of Human Aortic Valves Reveal Cell Heterogeneity and Endothelial to Mesenchymal Transition Involved in Calcific Aortic Valve Disease. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2910–2921. [Google Scholar] [CrossRef]
- Huang, Y.; Xu, K.; Zhou, T.; Zhu, P.; Dong, N.; Shi, J. Comparison of Rapidly Proliferating, Multipotent Aortic Valve-Derived Stromal Cells and Valve Interstitial Cells in the Human Aortic Valve. Stem Cells Int. 2019, 2019, 7671638. [Google Scholar] [CrossRef]
- Lindman, B.R.; Clavel, M.A.; Mathieu, P.; Iung, B.; Lancellotti, P.; Otto, C.M.; Pibarot, P. Calcific aortic stenosis. Nat. Rev. Dis. Primers 2016, 2, 16006. [Google Scholar] [CrossRef] [Green Version]
- Pedriali, G.; Morciano, G.; Patergnani, S.; Cimaglia, P.; Morelli, C.; Mikus, E.; Ferrari, R.; Gasbarro, V.; Giorgi, C.; Wieckowski, M.R.; et al. Aortic Valve Stenosis and MitochondrialDysfunctions: Clinical and MolecularPerspectives. Int. J. Mol. Sci. 2020, 21, 4899. [Google Scholar] [CrossRef]
- O’Brien, K.D.; Reichenbach, D.D.; Marcovina, S.M.; Kuusisto, J.; Alpers, C.E.; Otto, C.M. Apolipoproteins B, (a), and E accumulate in the morphologically early lesion of ‘degenerative’ valvular aortic stenosis. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 523–532. [Google Scholar] [CrossRef]
- Boffa, M.B.; Koschinsky, M.L. Oxidized phospholipids as a unifying theory for lipoprotein(a) and cardiovascular disease. Nat. Rev. Cardiol. 2019, 16, 305–318. [Google Scholar] [CrossRef]
- Cho, K.I.; Sakuma, I.; Sohn, I.S.; Jo, S.H.; Koh, K.K. Inflammatory and metabolic mechanisms underlying the calcific aortic valve disease. Atherosclerosis 2018, 277, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Mahmut, A.; Boulanger, M.C.; Fournier, D.; Couture, C.; Trahan, S.; Pagé, S.; Arsenault, B.; Després, J.P.; Pibarot, P.; Mathieu, P. Lipoprotein lipase in aortic valve stenosis is associated with lipid retention and remodelling. Eur. J. Clin. Investig. 2013, 43, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Yao, Q.; Song, R.; Zhai, Y.; Wang, W.; Fullerton, D.A.; Meng, X. Lysophosphatidylcholine activates the Akt pathway to upregulate extracellular matrix protein production in human aortic valve cells. J. Surg. Res. 2017, 213, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.T.; Chen, Y.Y.; Mao, J.Y.; Wang, Y.P.; Chen, Y.F.; Hu, X.; Yang, K.; Liu, Y. Oxidized HDL, as a Novel Biomarker for Calcific Aortic Valve Disease, Promotes the Calcification of Aortic Valve Interstitial Cells. J. Cardiovasc. Transl. Res. 2019, 12, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Hofmanis, J.; Hofmane, D.; Svirskis, S.; Mackevics, V.; Tretjakovs, P.; Lejnieks, A.; Signorelli, S.S. HDL-C Role in Acquired Aortic Valve Stenosis Patients and Its Relationship with Oxidative Stress. Medicina 2019, 55, 416. [Google Scholar] [CrossRef] [Green Version]
- Farrar, E.J.; Huntley, G.D.; Butcher, J. Endothelial-derived oxidative stress drives myofibroblastic activation and calcification of the aortic valve. PLoS ONE 2015, 10, e0123257. [Google Scholar] [CrossRef]
- Zhong, S.; Li, L.; Shen, X.; Li, Q.; Xu, W.; Wang, X.; Tao, Y.; Yin, H. An update on lipid oxidation and inflammation in cardiovascular diseases. Free Radic. Biol. Med. 2019, 144, 266–278. [Google Scholar] [CrossRef]
- Nsaibia, M.J.; Boulanger, M.C.; Bouchareb, R.; Mkannez, G.; Le Quang, K.; Hadji, F.; Argaud, D.; Dahou, A.; Bossé, Y.; Koschinsky, M.L.; et al. OxLDL-derived lysophosphatidic acid promotes the progression of aortic valve stenosis through a LPAR1-RhoA-NF-κB pathway. Cardiovasc. Res. 2017, 113, 1351–1363. [Google Scholar] [CrossRef] [Green Version]
- Pawade, T.A.; Newby, D.E.; Dweck, M.R. Calcification in Aortic Stenosis: The Skeleton Key. J. Am. Coll. Cardiol. 2015, 66, 561–577. [Google Scholar] [CrossRef] [Green Version]
- Kaden, J.J.; Dempfle, C.E.; Grobholz, R.; Fischer, C.S.; Vocke, D.C.; Kiliç, R.; Sarikoç, A.; Piñol, R.; Hagl, S.; Lang, S.; et al. Inflammatoryregulation of extracellularmatrixremodeling in calcificaortic valve stenosis. Cardiovasc. Pathol. 2005, 14, 80–87. [Google Scholar] [CrossRef]
- Donato, G.; Conforti, F.; Camastra, C.; Ammendola, M.; Donato, A.; Renzulli, A. The role of mast cell tryptases in cardiac myxoma: Histogenesis and development of a challenging tumor. Oncol. Lett. 2014, 8, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Passos, L.S.A.; Lupieri, A.; Becker-Greene, D.; Aikawa, E. Innate and adaptive immunity in cardiovascular calcification. Atherosclerosis 2020, 306, 59–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akahori, H.; Tsujino, T.; Masuyama, T.; Ishihara, M. Mechanisms of aortic stenosis. J. Cardiol. 2018, 71, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Li, F.; Huang, Y.; Zhou, T.; Chen, S.; Li, G.; Shi, J.; Dong, N.; Xu, K. Caffeic Acid Phenethyl Ester Ameliorates Calcification by Inhibiting Activation of the AKT/NF-κB/NLRP3 Inflammasome Pathway in Human Aortic Valve Interstitial Cells. Front. Pharmacol. 2020, 11, 826. [Google Scholar] [CrossRef]
- Li, X.F.; Wang, Y.; Zheng, D.D.; Xu, H.X.; Wang, T.; Pan, M.; Shi, J.H.; Zhu, J.H. M1 macrophages promote aortic valve calcification mediated by microRNA-214/TWIST1 pathway in valvular interstitial cells. Am. J. Transl. Res. 2016, 8, 5773–5783. [Google Scholar]
- Perrotta, I.; Carito, V.; Russo, E.; Tripepi, S.; Aquila, S.; Donato, G. Macrophage autophagy and oxidative stress: An ultrastructural and immunoelectron microscopical study. Oxid. Med. Cell Longev. 2011, 2011, 282739. [Google Scholar] [CrossRef]
- Schoen, F.J. Evolving concepts of cardiac valve dynamics: The continuum of development, functional structure, pathobiology, and tissue engineering. Circulation 2008, 118, 1864–1880. [Google Scholar] [CrossRef]
- Miller, J.D.; Weiss, R.M.; Heistad, D.D. Calcific aortic valve stenosis: Methods, models and mechanisms. Circ. Res. 2011, 108, 1392–1412. [Google Scholar] [CrossRef] [Green Version]
- Hutcheson, J.D.; Goettsch, C.; Rogers, M.A.; Aikawa, E. Revisiting cardiovascular calcification: A multifaceted disease requiring a multidisciplinary approach. Semin. Cell Dev. Biol. 2015, 46, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Stallons, M.V.; Tretter, J.T.; Hassel, K.; Gonzalez-Ramos, O.; Amofa, D.; Ollberding, N.J.; Mazur, W.; Choo, J.K.; Smith, J.M.; Kereiakes, D.J.; et al. Calcification and extracellularmatrixdysregulation in human postmortem and surgicalaorticvalves. Heart 2019, 105, 1616–1621. [Google Scholar] [CrossRef]
- Yip, Y.Y.C.; Chen, J.H.; Zhao, R.; Simmons, C.A. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 936–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertazzo, S.; Gentleman, E.; Cloyd, K.L.; Chester, A.H.; Yacoub, M.H.; Stevens, M.M. Nano-analytical electron microscopy reveals fundamental insights into human cardiovascular tissue calcification. Nat. Mater. 2013, 12, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Bouchareb, R.; Boulanger, M.C.; Fournier, D.; Pibarot, P.; Messaddeq, Y.; Mathieu, P. Mechanical strain induces the production of spheroid mineralized microparticles in the aortic valve through a RhoA/ROCK-dependent mechanism. J. Mol. Cell Cardiol. 2014, 67, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.T.; Johnstone, R.M. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: Selective externalization of the receptor. Cell 1983, 33, 967–978. [Google Scholar] [CrossRef]
- Gronowicz, G.; Swift, H.; Steck, T.L. Maturation of the reticulocyte in vitro. J. Cell Sci. 1984, 71, 177–197. [Google Scholar]
- McGough, I.J.; Vincent, J.P. Exosomes in developmental signaling. Development 2016, 143, 2482–2493. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Piao, H.; Wang, Y.; Zheng, D.; Wang, W. Circulating exosomes in cardiovascular disease: Novel carriers of biological information. Biomed. Pharmacother. 2021, 135, 111148. [Google Scholar] [CrossRef]
- Schubert, A.; Boutros, M. Extracellular vesicles and oncogenic signaling. Mol. Oncol. 2021, 15, 3–26. [Google Scholar] [CrossRef]
- Mancuso, T.; Barone, A.; Salatino, A.; Molinaro, C.; Marino, F.; Scalise, M.; Torella, M.; De Angelis, A.; Urbanek, K.; Torella, D.; et al. Unravelling the Biology of Adult Cardiac Stem Cell-Derived Exosomes to Foster Endogenous Cardiac Regeneration and Repair. Int. J. Mol. Sci. 2020, 21, 3725. [Google Scholar] [CrossRef]
- Cozzolino, M.; Ciceri, P.; Galassi, A.; Mangano, M.; Carugo, S.; Capelli, I.; Cianciolo, G. The Key Role of Phosphate on Vascular Calcification. Toxins 2019, 11, 213. [Google Scholar] [CrossRef] [Green Version]
- Fournier, D.E.; Norley, C.J.D.; Pollmann, S.I.; Bailey, C.S.; Al Helal, F.; Willmore, K.E.; Holdsworth, D.W.; Dixon, S.J.; Séguin, C.A. Ectopic spinal calcification associated with diffuse idiopathic skeletal hyperostosis (DISH): A quantitative micro-ct analysis. J. Orthop. Res. 2019, 37, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Priante, G.; Mezzabotta, F.; Cristofaro, R.; Quaggio, F.; Ceol, M.; Gianesello, L.; Del Prete, D.; Anglani, F. Cell death in ectopiccalcification of the kidney. Cell Death Dis. 2019, 10, 466. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, I.M.; Landis, W.J.; Risbud, M.V. Matrix vesicles: Are they anchored exosomes? Bone 2015, 79, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuthier, R.E.; Lipscomb, G.F. Matrix vesicles: Structure, composition, formation and function in calcification. Front. Biosci. 2011, 16, 2812–2902. [Google Scholar] [CrossRef] [PubMed]
- Blaser, M.C.; Aikawa, E. Roles and Regulation of Extracellular Vesicles in Cardiovascular Mineral Metabolism. Front. Cardiovasc. Med. 2018, 5, 187. [Google Scholar] [CrossRef] [Green Version]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Bottini, M.; Mebarek, S.; Anderson, K.L.; Strzelecka-Kiliszek, A.; Bozycki, L.; Simão, A.M.S.; Bolean, M.; Ciancaglini, P.; Pikula, J.B.; Pikula, S.; et al. Matrix vesicles from chondrocytes and osteoblasts: Their biogenesis, properties, functions and biomimetic models. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 532–546. [Google Scholar] [CrossRef]
- Schmidt, J.R.; Kliemt, S.; Preissler, C.; Moeller, S.; von Bergen, M.; Hempel, U.; Kalkhof, S. Osteoblast-released matrix vesicles, regulation of activity and composition by sulfated and non-sulfated glycosaminoglycans. Mol. Cell Proteom. 2016, 15, 558–572. [Google Scholar] [CrossRef] [Green Version]
- Mahmut, A.; Boulanger, M.C.; Bouchareb, R.; Hadji, F.; Mathieu, P. Adenosine derived from ecto-nucleotidases in calcific aortic valve disease promotes mineralization through A2a adenosine receptor. Cardiovasc. Res. 2015, 106, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, L.; Weinbaum, S. Microcalcifications, Their Genesis, Growth, and Biomechanical Stability in Fibrous Cap Rupture. Adv. Exp. Med. Biol. 2018, 1097, 129–155. [Google Scholar] [CrossRef]
- Zazzeroni, L.; Faggioli, G.; Pasquinelli, G. Mechanisms of Arterial Calcification: The Role of Matrix Vesicles. Eur. J. Vasc. Endovasc. Surg. 2018, 55, 425–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- New, S.E.; Goettsch, C.; Aikawa, M.; Marchini, J.F.; Shibasaki, M.; Yabusaki, K.; Libby, P.; Shanahan, C.M.; Croce, K.; Aikawa, E. Macrophage-derived matrix vesicles: An alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ. Res. 2013, 113, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Kapustin, A.N.; Chatrou, M.L.; Drozdov, I.; Zheng, Y.; Davidson, S.M.; Soong, D.; Furmanik, M.; Sanchis, P.; De Rosales, R.T.; Alvarez-Hernandez, D.; et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ. Res. 2015, 116, 1312–1323. [Google Scholar] [CrossRef] [Green Version]
- Kirsch, T.; Ishikawa, Y.; Mwale, F.; Wuthier, R.E. Roles of the nucleational core complex and collagens (types II and X) in calcification of growth plate cartilage matrix vesicles. J. Biol. Chem. 1994, 269, 20103–20109. [Google Scholar] [CrossRef]
- Wu, L.N.; Genge, B.R.; Sauer, G.R.; Wuthier, R.E. Characterization and reconstitution of the nucleational complex responsible for mineral formation by growth plate cartilage matrix vesicles. Connect. Tissue Res. 1996, 35, 309–315. [Google Scholar] [CrossRef]
- Boonrungsiman, S.; Gentleman, E.; Carzaniga, R.; Evans, N.D.; McComb, D.W.; Porter, A.E.; Stevens, M.M. The role of intracellular calcium phosphate in osteoblast-mediated bone apatite formation. Proc. Natl. Acad. Sci. USA 2012, 109, 14170–14175. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, T. Ultrastructure and biological function of matrix vesicles in bone mineralization. Histochem. Cell Biol. 2018, 149, 289–304. [Google Scholar] [CrossRef]
- Can Demirdöğen, B. Potential role of calcifying nanoparticles in the etiology of multiple sclerosis. Med. Hypotheses 2019, 128, 25–27. [Google Scholar] [CrossRef]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef] [Green Version]
- Ricard-Blum, S.; Ruggiero, F. The collagen superfamily: From the extracellular matrix to the cell membrane. Pathol. Biol. 2005, 53, 430–442. [Google Scholar] [CrossRef]
- Schoen, F.J. Aortic valve structure-function correlations: Role of elastic fibers no longer a stretch of the imagination. J. Heart Valve Dis. 1997, 6, 1–6. [Google Scholar] [PubMed]
- Eriksen, H.A.; Satta, J.; Risteli, J.; Veijola, M.; Väre, P.; Soini, Y. Type I and type III collagen synthesis and composition in the valve matrix in aortic valve stenosis. Atherosclerosis 2006, 189, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Latif, N.; Sarathchandra, P.; Taylor, P.M.; Antoniw, J.; Yacoub, M.H. Localization and pattern of expression of extracellular matrix components in human heart valves. J. Heart Valve Dis. 2005, 14, 218–227. [Google Scholar] [PubMed]
- Hutson, H.N.; Marohl, T.; Anderson, M.; Eliceiri, K.; Campagnola, P.; Masters, K.S. Calcific Aortic Valve Disease Is Associated with Layer-Specific Alterations in Collagen Architecture. PLoS ONE 2016, 11, e0163858. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, K.J.; Masters, K.S. Regulation of valvular interstitial cell calcification by components of the extracellular matrix. J. Biomed. Mater. Res. A 2009, 90, 1043–1053. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, K.J.; Piechura, L.M.; Porras, A.M.; Masters, K.S. Manipulation of valve composition to elucidate the role of collagen in aortic valve calcification. BMC Cardiovasc. Disord. 2014, 14, 29. [Google Scholar] [CrossRef] [Green Version]
- Kierszenbaum, A.L.; Tres, L.L. Histology and Cell Biology: An Introduction to Pathology, 5th ed.; Elsevier: Philadelphia, PA, USA, 2020; pp. 140–142, 148. [Google Scholar]
- Tseng, H.; Grande-Allen, K.J. Elastic fibers in the aortic valve spongiosa: A fresh perspective on its structure and role in overall tissue function. Acta Biomater. 2011, 7, 2101–2108. [Google Scholar] [CrossRef] [Green Version]
- Hinton, R.B., Jr.; Lincoln, J.; Deutsch, G.H.; Osinska, H.; Manning, P.B.; Benson, D.W.; Yutzey, K.E. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ. Res. 2006, 98, 1431–1438. [Google Scholar] [CrossRef] [Green Version]
- Bunton, T.E.; Biery, N.J.; Myers, L.; Gayraud, B.; Ramirez, F.; Dietz, H.C. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ. Res. 2001, 88, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Perrotta, I.; Russo, E.; Camastra, C.; Filice, G.; Di Mizio, G.; Colosimo, F.; Ricci, P.; Tripepi, S.; Amorosi, A.; Triumbari, F.; et al. New evidence for a critical role of elastin in calcification of native heart valves: Immunohistochemical and ultrastructural study with literature review. Histopathology 2011, 59, 504–513. [Google Scholar] [CrossRef]
- Helske, S.; Syväranta, S.; Lindstedt, K.A.; Lappalainen, J.; Oörni, K.; Mäyränpää, M.I.; Lommi, J.; Turto, H.; Werkkala, K.; Kupari, M.; et al. Increased expression of elastolyticcathepsins S, K, and V and their inhibitor cystatin C in stenotic aortic valves. Arterioscler.Thromb. Vasc. Biol. 2006, 26, 1791–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bossé, Y.; Miqdad, A.; Fournier, D.; Pépin, A.; Pibarot, P.; Mathieu, P. Refining molecular pathways leading to calcific aortic valve stenosis by studying gene expression profile of normal and calcified stenotic human aortic valves. Circ. Cardiovasc. Genet. 2009, 2, 489–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salhiyyah, K.; Sarathchandra, P.; Latif, N.; Yacoub, M.H.; Chester, A.H. Hypoxia-mediated regulation of the secretory properties of mitral valve interstitial cells. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H14–H23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Ignazio, L.; Bandarra, D.; Rocha, S. NF-κB and HIF crosstalk in immune responses. FEBS J. 2016, 283, 413–424. [Google Scholar] [CrossRef] [Green Version]
- Malara, N.M.; Leotta, A.; Sidoti, A.; Lio, S.; D’Angelo, R.; Caparello, B.; Munao, F.; Pino, F.; Amato, A. Ageing, hormonal behaviour and cyclin D1 in ductal breast carcinomas. Breast 2006, 15, 81–89. [Google Scholar] [CrossRef]
- Swaminathan, G.; Krishnamurthy, V.K.; Sridhar, S.; Robson, D.C.; Ning, Y.; Grande-Allen, K.J. Hypoxia Stimulates Synthesis of Neutrophil Gelatinase-Associated Lipocalin in Aortic Valve Disease. Front. Cardiovasc. Med. 2019, 6, 156. [Google Scholar] [CrossRef]
- Gassmann, P.; Haier, J.; Schlüter, K.; Domikowsky, B.; Wendel, C.; Wiesner, U.; Kubitza, R.; Engers, R.; Schneider, S.W.; Homey, B.; et al. CXCR4 regulates the early extravasation of metastatic tumor cells in vivo. Neoplasia 2009, 11, 651–661. [Google Scholar] [CrossRef] [Green Version]
- Malara, N.; Trunzo, V.; Foresta, U.; Amodio, N.; De Vitis, S.; Roveda, L.; Fava, M.; Coluccio, M.; Macrì, R.; Di Vito, A.; et al. Ex-vivo characterization of circulating colon cancer cells distinguished in stem and differentiated subset provides useful biomarker for personalized metastatic risk assessment. J. Transl. Med. 2016, 14, 133. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.H.; Kim, A.R.; Nam, J.K.; Kim, J.M.; Kim, J.Y.; Seo, H.R.; Lee, H.J.; Cho, J.; Lee, Y.J. Tumour-vasculature development via endothelial-to-mesenchymal transition after radiotherapy controls CD44v6 + cancer cell and macrophage polarization. Nat. Commun. 2018, 9, 5108. [Google Scholar] [CrossRef] [Green Version]
- Chirillo, R.; Aversa, I.; Di Vito, A.; Salatino, A.; Battaglia, A.M.; Sacco, A.; Di Sanzo, M.A.; Faniello, M.C.; Quaresima, B.; Palmieri, C.; et al. FtH-Mediated ROS DysregulationPromotes CXCL12/CXCR4 AxisActivation and EMT-Like Trans-Differentiation in Erythroleukemia K562. Cells Front. Oncol. 2020, 10, 698. [Google Scholar] [CrossRef]
- Herrmann, M.; Verrier, S.; Alini, M. Strategies to Stimulate Mobilization and Homing of Endogenous Stem and Progenitor Cells for Bone Tissue Repair. Front. Bioeng. Biotechnol. 2015, 3, 79. [Google Scholar] [CrossRef] [PubMed]
- Dorfmüller, P.; Bazin, D.; Aubert, S.; Weil, R.; Brisset, F.; Daudon, M.; Capron, F.; Brochériou, I. Crystalline ultrastructures, inflammatory elements, and neoangiogenesis are present in inconspicuous aortic valve tissue. Cardiol. Res. Pract. 2010, 2010, 685926. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef]
- Artiach, G.; Carracedo, M.; Seime, T.; Plunde, O.; Laguna-Fernandez, A.; Matic, L.; Franco-Cereceda, A.; Bäck, M. Proteoglycan 4 is Increased in Human Calcified Aortic Valves and Enhances Valvular Interstitial Cell Calcification. Cells 2020, 9, 684. [Google Scholar] [CrossRef] [Green Version]
- Koch, C.D.; Lee, C.M.; Apte, S.S. Aggrecan in Cardiovascular Development and Disease. J. Histochem. Cytochem. 2020, 68, 777–795. [Google Scholar] [CrossRef]
- Polley, A.; Khanam, R.; Sengupta, A.; Chakraborty, S. Asporin Reduces Adult Aortic Valve Interstitial Cell Mineralization Induced by Osteogenic Media and Wnt Signaling Manipulation In Vitro. Int. J. Cell Biol. 2020, 2020, 2045969. [Google Scholar] [CrossRef] [Green Version]
- Deckx, S.; Heggermont, W.; Carai, P.; Rienks, M.; Dresselaers, T.; Himmelreich, U.; van Leeuwen, R.; Lommen, W.; van der Velden, J.; Gonzalez, A.; et al. Osteoglycin prevents the development of age-related diastolic dysfunction during pressure overload by reducing cardiac fibrosis and inflammation. Matrix Biol. 2018, 66, 110–124. [Google Scholar] [CrossRef]
- Barth, M.; Selig, J.I.; Klose, S.; Schomakers, A.; Kiene, L.S.; Raschke, S.; Boeken, U.; Akhyari, P.; Fischer, J.W.; Lichtenberg, A. Degenerative aortic valve disease and diabetes: Implications for a link between proteoglycans and diabetic disorders in the aortic valve. Diabetes Vasc. Dis. Res. 2019, 16, 254–269. [Google Scholar] [CrossRef]
- Suzuki, H.; Chikada, M.; Yokoyama, M.K.; Kurokawa, M.S.; Ando, T.; Furukawa, H.; Arito, M.; Miyairi, T.; Kato, T. AberrantGlycosylation of Lumican in Aortic Valve StenosisRevealed by Proteomic Analysis. Int. Heart J. 2016, 57, 104–111. [Google Scholar] [CrossRef] [Green Version]
- Grande-Allen, K.J.; Osman, N.; Ballinger, M.L.; Dadlani, H.; Marasco, S.; Little, P.J. Glycosaminoglycan synthesis and structure as targets for the prevention of calcific aortic valve disease. Cardiovasc. Res. 2007, 76, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Stephens, E.H.; Saltarrelli, J.G.; Baggett, L.S.; Nandi, I.; Kuo, J.J.; Davis, A.R.; Olmsted-Davis, E.A.; Reardon, M.J.; Morrisett, J.D.; Grande-Allen, K.J. Differential proteoglycan and hyaluronan distribution in calcified aortic valves. Cardiovasc. Pathol. 2011, 20, 334–342. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.E.; Ishihara, S.; Tran, P.B.; Golub, S.B.; Last, K.; Miller, R.J.; Fosang, A.J.; Malfait, A.M. An aggrecan fragment drives osteoarthritis pain through Toll-like receptor 2. JCI Insight 2018, 3, e95704. [Google Scholar] [CrossRef]
- Li, F.; Song, R.; Ao, L.; Reece, T.B.; Cleveland, J.C., Jr.; Dong, N.; Fullerton, D.A.; Meng, X. ADAMTS5 Deficiency in Calcified Aortic Valves Is Associated With Elevated Pro-Osteogenic Activity in Valvular Interstitial Cells. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1339–1351. [Google Scholar] [CrossRef] [Green Version]
- Stephens, E.H.; Chu, C.K.; Grande-Allen, K.J. Valve proteoglycan content and glycosaminoglycan fine structure are unique to microstructure, mechanical load and age: Relevance to an age-specific tissue-engineered heart valve. Acta Biomater. 2008, 4, 1148–1160. [Google Scholar] [CrossRef]
- Derbali, H.; Bossé, Y.; Côté, N.; Pibarot, P.; Audet, A.; Pépin, A.; Arsenault, B.; Couture, C.; Després, J.P.; Mathieu, P. Increased biglycan in aortic valve stenosis leads to the overexpression of phospholipid transfer protein via Toll-like receptor 2. Am. J. Pathol. 2010, 176, 2638–2645. [Google Scholar] [CrossRef]
- Tomoeda, M.; Yamada, S.; Shirai, H.; Ozawa, Y.; Yanagita, M.; Murakami, S. PLAP-1/asporin inhibits activation of BMP receptor via its leucine-rich repeat motif. Biochem. Biophys. Res. Commun. 2008, 371, 191–196. [Google Scholar] [CrossRef]
- Donato, G.; Conforti, F.; Zuccalà, V.; Russo, E.; Maltese, L.; Perrotta, I.; Amorosi, A. Expression of tenascin-c and CD44 receptors in cardiac myxomas. Cardiovasc. Pathol. 2009, 18, 173–177. [Google Scholar] [CrossRef]
- Poggio, P.; Branchetti, E.; Grau, J.B.; Lai, E.K.; Gorman, R.C.; Gorman, J.H., 3rd; Sacks, M.S.; Bavaria, J.E.; Ferrari, G. Osteopontin-CD44v6 interaction mediates calcium deposition via phospho-Akt in valve interstitial cells from patients with noncalcified aortic valve sclerosis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2086–2094. [Google Scholar] [CrossRef] [Green Version]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells. Front. Cell Dev. Biol. 2017, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Baranova, N.S.; Nilebäck, E.; Haller, F.M.; Briggs, D.C.; Svedhem, S.; Day, A.J.; Richter, R.P. The inflammation-associated protein TSG-6 cross-links hyaluronan via hyaluronan-induced TSG-6 oligomers. J. Biol. Chem. 2011, 286, 25675–25686. [Google Scholar] [CrossRef] [Green Version]
- Stephens, E.H.; Saltarrelli, J.G., Jr.; Balaoing, L.R.; Baggett, L.S.; Nandi, I.; Anderson, K.M.; Morrisett, J.D.; Reardon, M.J.; Simpson, M.A.; Weigel, P.H.; et al. Hyaluronan turnover and hypoxic brown adipocytic differentiation are co-localized with ossification in calcified human aortic valves. Pathol. Res. Pract. 2012, 208, 642–650. [Google Scholar] [CrossRef] [Green Version]
- Baugh, L.; Watso, M.C.; Kemmerling, E.C.; Hinds, P.W.; Huggins, G.S.; Black, L.D., 3rd. Knockdown of CD44 expression decreases valve interstitial cell calcification in vitro. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H26–H36. [Google Scholar] [CrossRef]
- Adams, J.C. Matricellular Proteins: Functional Insights from Non-mammalian Animal Models. Curr. Top. Dev. Biol. 2018, 130, 39–105. [Google Scholar] [CrossRef]
- Murphy-Ullrich, J.E.; Sage, E.H. Revisiting the matricellular concept. Matrix Biol. 2014, 37, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Cardwell, R.D.; Kluge, J.A.; Thayer, P.S.; Guelcher, S.A.; Dahlgren, L.A.; Kaplan, D.L.; Goldstein, A.S. Static and cyclic mechanical loading of mesenchymal stem cells on elastomeric, electrospun polyurethane meshes. J. Biomech. Eng. 2015, 137, 071010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendaoui, I.; Tucker, R.P.; Zingg, D.; Bichet, S.; Schittny, J.; Chiquet-Ehrismann, R. Tenascin-C is required for normal Wnt/β-catenin signaling in the whisker follicle stem cell niche. Matrix Biol. 2014, 40, 46–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinhard, J.; Brösicke, N.; Theocharidis, U.; Faissner, A. The extracellular matrix niche microenvironment of neural and cancer stem cells in the brain. Int. J. Biochem. Cell Biol. 2016, 81, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Satta, J.; Melkko, J.; Pöllänen, R.; Tuukkanen, J.; Pääkkö, P.; Ohtonen, P.; Mennander, A.; Soini, Y. Progression of human aortic valve stenosis is associated with tenascin-C expression. J. Am. Coll. Cardiol. 2002, 39, 96–101. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, T.; Yamamoto, M.; Suzuki, K.; Suzuki, Y.; Kamishima, M.; Sakata, M.; Kurachi, K.; Setoh, M.; Konno, H.; Takeuchi, H. Tenascin-C Produced by Intestinal Myofibroblasts Promotes Colitis-associated Cancer Development Through Angiogenesis. Inflamm. Bowel. Dis. 2019, 25, 732–741. [Google Scholar] [CrossRef]
- Di Vito, A.; Scali, E.; Ferraro, G.; Mignogna, C.; Presta, I.; Camastra, C.; Palmieri, C.; Donato, G.; Barni, T. Elastofibroma dorsi: A histochemical and immunohistochemical study. Eur. J. Histochem. 2015, 59, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Jian, B.; Jones, P.L.; Quanyi, L.; Mohler, E.R., 3rd; Schoen, F.J.; Levy, R.J. Matrix metalloproteinase-2 is associated with tenascin-C in calcific aortic stenosis. Am. J. Pathol. 2001, 159, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, K.; Satoh, K.; Maniwa, T.; Araki, A.; Maruyama, R.; Oda, T. Noticeable decreased expression of tenascin-X in calcific aortic valves. Connect. Tissue Res. 2012, 53, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K. Vascular Calcification-Pathological Mechanism and Clinical Application-Extracellular matrix tenascin-X in calcific aortic valves. Clin. Calcium 2015, 25, 701–710. [Google Scholar]
- Takeshita, S.; Kikuno, R.; Tezuka, K.; Amann, E. Osteoblast-specific factor 2: Cloning of a putative bone adhesion protein with homology with the insect protein fasciclin I. Biochem. J. 1993, 294, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Kudo, A. Periostin in fibrillogenesis for tissue regeneration: Periostin actions inside and outside the cell. Cell Mol. Life Sci. 2011, 68, 3201–3207. [Google Scholar] [CrossRef] [Green Version]
- González-González, L.; Alonso, J. Periostin: A Matricellular Protein with Multiple Functions in Cancer Development and Progression. Front. Oncol. 2018, 8, 225. [Google Scholar] [CrossRef]
- Maruhashi, T.; Kii, I.; Saito, M.; Kudo, A. Interaction between periostin and BMP-1 promotes proteolytic activation of lysyl oxidase. J. Biol. Chem. 2010, 285, 13294–13303. [Google Scholar] [CrossRef] [Green Version]
- Norris, R.A.; Moreno-Rodriguez, R.A.; Sugi, Y.; Hoffman, S.; Amos, J.; Hart, M.M.; Potts, J.D.; Goodwin, R.L.; Markwald, R.R. Periostin regulates atrioventricular valve maturation. Dev. Biol. 2008, 316, 200–213. [Google Scholar] [CrossRef] [Green Version]
- Markwald, R.R.; Moreno-Rodriguez, R.A.; Ghatak, S.; Misra, S.; Norris, R.A.; Sugi, Y. Role of Periostin in Cardiac Valve Development. Adv. Exp. Med. Biol. 2019, 1132, 177–191. [Google Scholar] [CrossRef]
- Tkatchenko, T.V.; Moreno-Rodriguez, R.A.; Conway, S.J.; Molkentin, J.D.; Markwald, R.R.; Tkatchenko, A.V. Lack of periostin leads to suppression of Notch1 signaling and calcific aortic valve disease. Physiol. Genom. 2009, 39, 160–168. [Google Scholar] [CrossRef] [Green Version]
- Bogdanova, M.; Kostina, A.; ZihlavnikovaEnayati, K.; Zabirnyk, A.; Malashicheva, A.; Stensløkken, K.O.; Sullivan, G.J.; Kaljusto, M.L.; Kvitting, J.P.; Kostareva, A.; et al. Inflammation and Mechanical Stress Stimulate Osteogenic Differentiation of Human Aortic Valve Interstitial Cells. Front. Physiol. 2018, 9, 1635. [Google Scholar] [CrossRef] [PubMed]
- Sathyamurthy, I.; Alex, S. Calcific aortic valve disease: Is it another face of atherosclerosis? Indian Heart J. 2015, 67, 503–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, D.H. Aortic Valve Calcification: Moving Toward the Root of the Problem. J. Am. Coll. Cardiol. 2019, 73, 315–316. [Google Scholar] [CrossRef]
- Kostyunin, A.E.; Yuzhalin, A.E.; Ovcharenko, E.A.; Kutikhin, A.G. Development of calcific aortic valve disease: Do we know enough for new clinical trials? J. Mol. Cell Cardiol. 2019, 132, 189–209. [Google Scholar] [CrossRef]
- De Pieri, A.; Rana, S.; Korntner, S.; Zeugolis, D.I. Seaweed polysaccharides as macromolecular crowding agents. Int. J. Biol. Macromol. 2020, 164, 434–446. [Google Scholar] [CrossRef]
- Musiani, F.; Giorgetti, A. Protein Aggregation and Molecular Crowding: Perspectives from Multiscale Simulations. Int. Rev. Cell Mol. Biol. 2017, 329, 49–77. [Google Scholar] [CrossRef]
- Graceffa, V.; Zeugolis, D.I. Carrageenan enhances chondrogenesis and osteogenesis in human bone marrow stem cell culture. Eur. Cell Mater. 2019, 37, 310–332. [Google Scholar] [CrossRef]
- VeDepo, M.C.; Buse, E.E.; Paul, A.; Converse, G.L.; Hopkins, R.A. Non-physiologic Bioreactor Processing Conditions for Heart Valve Tissue Engineering. Cardiovasc. Eng. Technol. 2019, 10, 628–637. [Google Scholar] [CrossRef]
- Parvin-Nejad, S.; Blaser, M.C.; Santerre, J.P.; Caldarone, C.A.; Simmons, C.A. Biomechanical conditioning of tissue engineered heart valves: Too much of a good thing? Adv. Drug Deliv. Rev. 2016, 96, 161–175. [Google Scholar] [CrossRef]
- Noble, C.; Maxson, E.L.; Lerman, A.; Young, M.D. Mechanical and finite element evaluation of a bioprinted scaffold following recellularization in a rat subcutaneous model. J. Mech. Behav. Biomed. Mater. 2020, 102, 103519. [Google Scholar] [CrossRef]
- Chen, Q.; Bruyneel, A.; Carr, C.; Czernuszka, J. Bio-mechanical properties of novel bi-layer collagen-elastin scaffolds for heart valve tissue engineering. Procedia Eng. 2013, 59, 247–254. [Google Scholar] [CrossRef] [Green Version]
- Monzack, E.L.; Masters, K.S. A time course investigation of the statin paradox among valvular interstitial cell phenotypes. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H903–H909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, M.; Ferri, N.; Lupo, M.G.; Faggin, E.; Rattazzi, M. Current Evidence and Future Perspectives on Pharmacological Treatment of Calcific Aortic Valve Stenosis. Int. J. Mol. Sci. 2020, 21, 8263. [Google Scholar] [CrossRef] [PubMed]
- Alushi, B.; Curini, L.; Christopher, M.R.; Grubitzch, H.; Landmesser, U.; Amedei, A.; Lauten, A. Calcific Aortic Valve Disease-Natural History and Future Therapeutic Strategies. Front. Pharmacol. 2020, 11, 685. [Google Scholar] [CrossRef]
- Rodriguez, K.J.; Piechura, L.M.; Masters, K.S. Regulation of valvular interstitial cell phenotype and function by hyaluronic acid in 2-D and 3-D culture environments. Matrix Biol. 2011, 30, 70–82. [Google Scholar] [CrossRef] [Green Version]
- Porras, A.M.; Westlund, J.A.; Evans, A.D.; Masters, K.S. Creation of disease-inspired biomaterial environments to mimic pathological events in early calcific aortic valve disease. Proc. Natl. Acad. Sci. USA 2018, 115, E363–E371. [Google Scholar] [CrossRef] [Green Version]
- Pereira, H.; Sousa, D.A.; Cunha, A.; Andrade, R.; Espregueira-Mendes, J.; Oliveira, J.M.; Reis, R.L. Hyaluronic Acid. In Osteochondral Tissue Engineering. Advances in Experimental Medicine and Biology; Oliveira, J., Pina, S., Reis, R., San Roman, J., Eds.; Springer: Cham, Switzerland, 2018; Volume 1059. [Google Scholar] [CrossRef]
- Shendi, D.; Marzi, J.; Linthicum, W.; Rickards, A.J.; Dolivo, D.M.; Keller, S.; Kauss, M.A.; Wen, Q.; McDevitt, T.C.; Dominko, T.; et al. Hyaluronic acid as a macromolecular crowding agent for production of cell-derived matrices. Acta Biomater. 2019, 100, 292–305. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Vito, A.; Donato, A.; Presta, I.; Mancuso, T.; Brunetti, F.S.; Mastroroberto, P.; Amorosi, A.; Malara, N.; Donato, G. Extracellular Matrix in Calcific Aortic Valve Disease: Architecture, Dynamic and Perspectives. Int. J. Mol. Sci. 2021, 22, 913. https://doi.org/10.3390/ijms22020913
Di Vito A, Donato A, Presta I, Mancuso T, Brunetti FS, Mastroroberto P, Amorosi A, Malara N, Donato G. Extracellular Matrix in Calcific Aortic Valve Disease: Architecture, Dynamic and Perspectives. International Journal of Molecular Sciences. 2021; 22(2):913. https://doi.org/10.3390/ijms22020913
Chicago/Turabian StyleDi Vito, Anna, Annalidia Donato, Ivan Presta, Teresa Mancuso, Francesco Saverio Brunetti, Pasquale Mastroroberto, Andrea Amorosi, Natalia Malara, and Giuseppe Donato. 2021. "Extracellular Matrix in Calcific Aortic Valve Disease: Architecture, Dynamic and Perspectives" International Journal of Molecular Sciences 22, no. 2: 913. https://doi.org/10.3390/ijms22020913
APA StyleDi Vito, A., Donato, A., Presta, I., Mancuso, T., Brunetti, F. S., Mastroroberto, P., Amorosi, A., Malara, N., & Donato, G. (2021). Extracellular Matrix in Calcific Aortic Valve Disease: Architecture, Dynamic and Perspectives. International Journal of Molecular Sciences, 22(2), 913. https://doi.org/10.3390/ijms22020913