Antiinflammatory Actions of Klotho: Implications for Therapy of Diabetic Nephropathy


Abstract
:1. Diabetes Mellitus and Diabetic Nephropathy
2. Klotho Protein and Its Tissue Expression
3. Structure and Function of Klotho
4. Role of Klotho in Diabetes and Diabetic Nephropathy
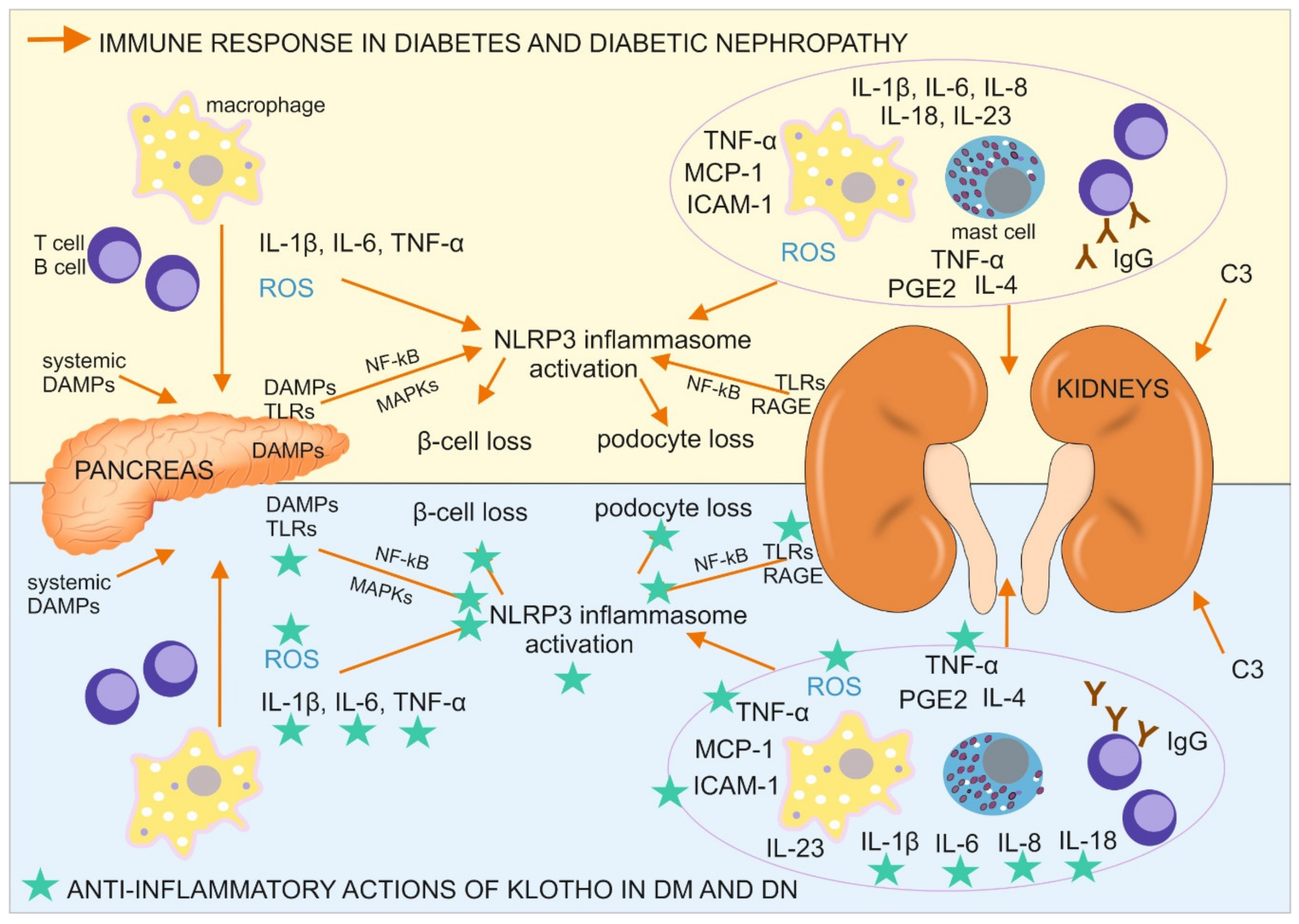
5. Immune Response in Diabetes and Diabetic Nephropathy
6. Antiinflammatory Actions of Klotho in Diabetes and Diabetic Nephropathy
6.1. Klotho Expression Is Downregulated during Inflammation
6.2. Klotho Induces Antiinflammatory Reactions
6.3. Klotho Suppresses Proinflammatory NF-κB Activation
6.4. Klotho Inhibits TLR4 Signaling and Related Oxidative Stress
6.5. Klotho Reduces Oxidative Stress by Inhibiting IGF-1 Signaling and NLRP3 Inflammasome Activation
6.6. Klotho Reduces Leukocyte Infiltration of the Kidneys, Renal Injury, and Fibrosis
7. Klotho May Serve as a Key Molecule in Immunotherapy of Diabetes and Diabetic Nephropathy
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tan, S.Y.; Wong, J.L.M.; Sim, Y.J.; Wong, S.S.; Elhassan, S.A.M.; Tan, S.H.; Lim, G.P.L.; Tay, N.W.R.; Annan, N.C.; Bhattamisra, S.K.; et al. Type 1 and 2 diabetes mellitus: A review on current treatment approach and gene therapy as potential intervention. Diabetes Metab. Syndr. 2019, 13, 364–372. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Status Report on Noncommunicable Diseases 2014; World Health Organisation: Geneva, Switzerland, 2014. [Google Scholar]
- Takiyama, Y.; Haneda, M. Hypoxia in diabetic kidneys. BioMed. Res. Int. 2014, 2014, 837421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonneijck, L.; Muskiet, M.H.; Smits, M.M.; van Bommel, E.J.; Heerspink, H.J.; van Raalte, D.H.; Joles, J.A. Glomerular Hyperfiltration in Diabetes: Mechanisms, Clinical Significance, and Treatment. J. Am. Soc. Nephrol. 2017, 28, 1023–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.H.; Susztak, K. The hyperglycemic and hyperinsulinemic combo gives you diabetic kidney disease immediately. Focus on “Combined acute hyperglycemic and hyperinsulinemic clamp induced profibrotic and proinflammatory responses in the kidney”. Am. J. Physiol. Cell Physiol. 2014, 306, C198–C199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazar, C.M. Mechanism of hypertension in diabetic nephropathy. J. Nephropharmacol. 2014, 3, 49–55. [Google Scholar]
- Guo, J.; Zheng, H.J.; Zhang, W.; Lou, W.; Xia, C.; Han, X.T.; Huang, W.J.; Zhang, F.; Wang, Y.; Liu, W.J. Accelerated Kidney Aging in Diabetes Mellitus. Oxid. Med. Cell. Longev. 2020, 2020, 1234059. [Google Scholar] [CrossRef] [PubMed]
- Tesch, G.H. Diabetic nephropathy—Is this an immune disorder? Clin. Sci. 2017, 131, 2183–2199. [Google Scholar] [CrossRef]
- Zheng, Z.; Zheng, F. Immune Cells and Inflammation in Diabetic Nephropathy. J. Diabetes Res. 2016, 2016, 1841690. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Zhou, L. The Signaling of Cellular Senescence in Diabetic Nephropathy. Oxid. Med. Cell. Longev. 2019, 2019, 7495629. [Google Scholar] [CrossRef]
- Hu, M.C.; Kuro-o, M.; Moe, O.W. Secreted klotho and chronic kidney disease. Adv. Exp. Med. Biol. 2012, 728, 126–157. [Google Scholar] [CrossRef] [Green Version]
- Buchanan, S.; Combet, E.; Stenvinkel, P.; Shiels, P.G. Klotho, Aging, and the Failing Kidney. Front. Endocrinol. 2020, 11, 560. [Google Scholar] [CrossRef] [PubMed]
- Buendía, P.; Ramírez, R.; Aljama, P.; Carracedo, J. Klotho Prevents Translocation of NFκB. Vitam. Horm. 2016, 101, 119–150. [Google Scholar] [CrossRef] [PubMed]
- Mencke, R.; Harms, G.; Moser, J.; van Meurs, M.; Diepstra, A.; Leuvenink, H.G.; Hillebrands, J.L. Human alternative Klotho mRNA is a nonsense-mediated mRNA decay target inefficiently spliced in renal disease. JCI Insight 2017, 2, 10117294375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Xie, J.; Hwang, K.H.; Wu, Y.L.; Oliver, N.; Eom, M.; Park, K.S.; Barrezueta, N.; Kong, I.D.; Fracasso, R.P.; et al. Klotho May Ameliorate Proteinuria by Targeting TRPC6 Channels in Podocytes. J. Am. Soc. Nephrol. 2017, 28, 140–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.C.; Shi, M.; Zhang, J.; Pastor, J.; Nakatani, T.; Lanske, B.; Razzaque, M.S.; Rosenblatt, K.P.; Baum, M.G.; Kuro-o, M.; et al. Klotho: A novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 2010, 24, 3438–3450. [Google Scholar] [CrossRef] [Green Version]
- Richter, B.; Faul, C. FGF23 Actions on Target Tissues—With and Without Klotho. Front. Endocrinol. 2018, 9, 189. [Google Scholar] [CrossRef] [PubMed]
- Bian, A.; Neyra, J.A.; Zhan, M.; Hu, M.C. Klotho, stem cells, and aging. Clin. Interv. Aging 2015, 10, 1233–1243. [Google Scholar] [CrossRef] [Green Version]
- Quarles, L.D. Fibroblast growth factor 23 and α-Klotho co-dependent and independent functions. Curr. Opin. Nephrol. Hypertens. 2019, 28, 16–25. [Google Scholar] [CrossRef]
- Lim, S.W.; Jin, L.; Luo, K.; Jin, J.; Shin, Y.J.; Hong, S.Y.; Yang, C.W. Klotho enhances FoxO3-mediated manganese superoxide dismutase expression by negatively regulating PI3K/AKT pathway during tacrolimus-induced oxidative stress. Cell Death Dis. 2017, 8, e2972. [Google Scholar] [CrossRef]
- Bob, F.; Schiller, A.; Timar, R.; Lighezan, D.; Schiller, O.; Timar, B.; Bujor, C.G.; Munteanu, M.; Gadalean, F.; Mihaescu, A.; et al. Rapid decline of kidney function in diabetic kidney disease is associated with high soluble klotho levels. Nefrologia 2019, 39, 250–257. [Google Scholar] [CrossRef]
- Kim, S.S.; Song, S.H.; Kim, I.J.; Lee, E.Y.; Lee, S.M.; Chung, C.H.; Kwak, I.S.; Lee, E.K.; Kim, Y.K. Decreased plasma α-Klotho predict progression of nephropathy with type 2 diabetic patients. J. Diabetes Complicat. 2016, 30, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Xiao, T.; Han, W.; Xiong, J.; He, T.; Liu, Y.; Huang, Y.; Yang, K.; Bi, X.; Xu, X.; et al. Klotho inhibits PKCα/p66SHC-mediated podocyte injury in diabetic nephropathy. Mol. Cell. Endocrinol. 2019, 494, 110490. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.J.; Ro, H.; Kim, H.; Chang, J.H.; Lee, H.H.; Chung, W.; Jung, J.Y. Klotho and S100A8/A9 as Discriminative Markers between Pre-Renal and Intrinsic Acute Kidney Injury. PLoS ONE 2016, 11, e0147255. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Lei, C.-T.; Zhang, C. Interleukin-6 Signaling Pathway and Its Role in Kidney Disease: An Update. Front. Immunol. 2017, 8, 405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, S.; Xu, S.; Ma, Y.; Liu, G.; Jiang, H.; Fang, J. Modulatory mechanisms of the nlrp3 inflammasomes in diabetes. Biomolecules 2019, 9, 850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrass, C.K. Fc-receptor-mediated phagocytosis: Abnormalities associated with diabetes mellitus. Clin. Immunol. Immunopathol. 1991, 58, 1–17. [Google Scholar] [CrossRef]
- Xia, H.; Bao, W.; Shi, S. Innate Immune Activity in Glomerular Podocytes. Front. Immunol. 2017, 8, 122. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, R.; Tsokos, G.C. The immune podocyte. Curr. Opin. Rheumatol. 2019, 31, 167–174. [Google Scholar] [CrossRef]
- Wang, L.; Wang, J.; Fang, J.; Zhou, H.; Liu, X.; Su, S.B. High glucose induces and activates Toll-like receptor 4 in endothelial cells of diabetic retinopathy. Diabetol. Metab. Syndr. 2015, 7, 89. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xia, W.; Zhao, F.; Wen, Z.; Zhang, A.; Huang, S.; Jia, Z.; Zhang, Y. Prostaglandins in the pathogenesis of kidney diseases. Oncotarget 2018, 9, 26586–26602. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Banerjee, S.; Dey, N.; LeJeune, W.S.; Sarkar, P.S.; Brobey, R.; Rosenblatt, K.P.; Tilton, R.G.; Choudhary, S. Klotho depletion contributes to increased inflammation in kidney of the db/db mouse model of diabetes via RelA (serine)536 phosphorylation. Diabetes 2011, 60, 1907–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 1997, 389, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Balkwill, F.; Chonchol, M.; Cominelli, F.; Donath, M.Y.; Giamarellos-Bourboulis, E.J.; Golenbock, D.; Gresnigt, M.S.; Heneka, M.T.; Hoffman, H.M.; et al. A guiding map for inflammation. Nat. Immunol. 2017, 18, 826–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlsson, A.C.; Östgren, C.J.; Nystrom, F.H.; Länne, T.; Jennersjö, P.; Larsson, A.; Ärnlöv, J. Association of soluble tumor necrosis factor receptors 1 and 2 with nephropathy, cardiovascular events, and total mortality in type 2 diabetes. Cardiovasc. Diabetol. 2016, 15, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, E.A.; Han, X.; Xiao, Z.; Quarles, L.D. Role of Fibroblast Growth Factor-23 in Innate Immune Responses. Front. Endocrinol. 2018, 9, 320. [Google Scholar] [CrossRef]
- Zhou, X.; Lei, H.; Sun, Z. Participation of Immune Cells in Klotho Deficiency-induced Salt-sensitive Hypertension. FASEB J. 2015, 29, 667.3. [Google Scholar] [CrossRef]
- Witkowski, J.M.; Soroczyńska-Cybula, M.; Bryl, E.; Smoleńska, Z.; Jóźwik, A. Klotho—A common link in physiological and rheumatoid arthritis-related aging of human CD4+ lymphocytes. J. Immunol. 2007, 178, 771–777. [Google Scholar] [CrossRef] [Green Version]
- Hui, H.; Zhai, Y.; Ao, L.; Cleveland, J.C., Jr.; Liu, H.; Fullerton, D.A.; Meng, X. Klotho suppresses the inflammatory responses and ameliorates cardiac dysfunction in aging endotoxemic mice. Oncotarget 2017, 8, 15663–15676. [Google Scholar] [CrossRef]
- Mytych, J.; Romerowicz-Misielak, M.; Koziorowski, M. Klotho protects human monocytes from LPS-induced immune impairment associated with immunosenescent-like phenotype. Mol. Cell. Endocrinol. 2018, 470, 1–13. [Google Scholar] [CrossRef]
- Oh, H.J.; Nam, B.Y.; Wu, M.; Kim, S.; Park, J.; Kang, S.; Park, J.T.; Yoo, T.H.; Kang, S.W.; Han, S.H. Klotho plays a protective role against glomerular hypertrophy in a cell cycle-dependent manner in diabetic nephropathy. Am. J. Physiol. Renal. Physiol. 2018, 315, F791–F805. [Google Scholar] [CrossRef]
- Moreno, J.A.; Izquierdo, M.C.; Sanchez-Niño, M.D.; Suárez-Alvarez, B.; Lopez-Larrea, C.; Jakubowski, A.; Blanco, J.; Ramirez, R.; Selgas, R.; Ruiz-Ortega, M.; et al. The inflammatory cytokines TWEAK and TNFα reduce renal klotho expression through NFκB. J. Am. Soc. Nephrol. 2011, 22, 1315–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Wang, Y.; Gao, W.; Yuan, C.; Zhang, S.; Zhou, H.; Huang, M.; Yao, X. Klotho Reduction in Alveolar Macrophages Contributes to Cigarette Smoke Extract-Induced Inflammation in Chronic Obstructive Pulmonary Disease. J. Biol. Chem. 2015, 290, 27890–27900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Ma, J.; Ren, Y.; Xiang, S.; Jia, R. Secreted klotho from exosomes alleviates inflammation and apoptosis in acute pancreatitis. Am. J. Transl. Res. 2019, 11, 3375–3383. [Google Scholar] [PubMed]
- Bi, F.; Chen, F.; Li, Y.; Wei, A.; Cao, W. Klotho preservation by Rhein promotes toll-like receptor 4 proteolysis and attenuates lipopolysaccharide-induced acute kidney injury. J. Mol. Med. 2018, 96, 915–927. [Google Scholar] [CrossRef]
- Taha, I.M.; Allah, A.M.A.; El Gayed, E.M.A. Expression of toll-like receptor 4 and its connection with type 2 diabetes mellitus. Cell. Mol. Biol. 2018, 64, 15–20. [Google Scholar] [CrossRef]
- Kim, J.J.; Sears, D.D. TLR4 and Insulin Resistance. Gastroenterol. Res. Pract. 2010, 2010, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sopjani, M.; Rinnerthaler, M.; Kruja, J.; Dermaku-Sopjani, M. Intracellular signaling of the aging suppressor protein Klotho. Curr. Mol. Med. 2015, 15, 27–37. [Google Scholar] [CrossRef]
- Zhu, L.; Stein, L.R.; Kim, D.; Ho, K.; Yu, G.Q.; Zhan, L.; Larsson, T.E.; Mucke, L. Klotho controls the brain-immune system interface in the choroid plexus. Proc. Natl. Acad. Sci. USA 2018, 115, E11388–E11396. [Google Scholar] [CrossRef] [Green Version]
- Jou-Valencia, D.; Molema, G.; Popa, E.; Aslan, A.; van Dijk, F.; Mencke, R.; Hillebrands, J.L.; Heeringa, P.; Hoenderop, J.G.; Zijlstra, J.G.; et al. Renal Klotho is Reduced in Septic Patients and Pretreatment with Recombinant Klotho Attenuates Organ Injury in Lipopolysaccharide-Challenged Mice. Crit. Care Med. 2018, 46, e1196–e1203. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Wu, S.; Ren, H.; Gu, J. Klotho suppresses RIG-I-mediated senescence-associated inflammation. Nat. Cell Biol. 2011, 13, 254–262. [Google Scholar] [CrossRef]
- Lonnemann, G.; Engler-Blum, G.; Müller, G.A.; Koch, K.M.; Dinarello, C.A. Cytokines in human renal interstitial fibrosis. II. Intrinsic interleukin (IL)-1 synthesis and IL-1-dependent production of IL-6 and IL-8 by cultured kidney fibroblasts. Kidney Int. 1995, 47, 845–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellano, G.; Intini, A.; Stasi, A.; Divella, C.; Gigante, M.; Pontrelli, P.; Franzin, R.; Accetturo, M.; Zito, A.; Fiorentino, M.; et al. Complement Modulation of Anti-Aging Factor Klotho in Ischemia/Reperfusion Injury and Delayed Graft Function. Am. J. Transplant. 2016, 16, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Sun, Z. Molecular basis of Klotho: From gene to function in aging. Endocr. Rev. 2015, 36, 174–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prié, D.; Friedlander, G. Reciprocal control of 1,25-dihydroxyvitamin d and fgf23 formation involving the fgf23/klotho system. Clin. J. Am. Soc. Nephrol. 2010, 5, 1717–1722. [Google Scholar] [CrossRef]
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-β Mediated SMAD Signaling for the Prevention of Fibrosis. Front. Pharmacol. 2017, 8, 461. [Google Scholar] [CrossRef] [Green Version]
- Gu, L.Y.; Yun, S.; Tang, H.T.; Xu, Z.X. Huangkui capsule in combination with metformin ameliorates diabetic nephropathy via the Klotho/TGF-β1/p38MAPK signaling pathway. J. Ethnopharmacol. 2020, 113548, in press. [Google Scholar] [CrossRef]
- Ramos, A.M.; Fernández-Fernández, B.; Pérez-Gómez, M.V.; Carriazo Julio, S.M.; Sanchez-Niño, M.D.; Sanz, A.; Ruiz-Ortega, M.; Ortiz, A. Design and optimization strategies for the development of new drugs that treat chronic kidney disease. Expert Opin. Drug Discov. 2020, 15, 101–115. [Google Scholar] [CrossRef]
- Eltablawy, N.; Ashour, H.; Rashed, L.A.; Hamza, W.M. Vitamin d protection from rat diabetic nephropathy is partly mediated through klotho expression and renin-angiotensin inhibition. Arch. Physiol. Biochem. 2018, 124, 461–467. [Google Scholar] [CrossRef]
- Zou, D.; Wu, W.; He, Y.; Ma, S.; Gao, J. The role of klotho in chronic kidney disease. BMC Nephrol. 2018, 19, 285. [Google Scholar] [CrossRef] [Green Version]
- Grange, C.; Papadimitriou, E.; Dimuccio, V.; Pastorino, C.; Molina, J.; O’Kelly, R.; Niedernhofer, L.J.; Robbins, P.D.; Camussi, G.; Bussolati, B. Urinary Extracellular Vesicles Carrying Klotho Improve the Recovery of Renal Function in an Acute Tubular Injury Model. Mol. Ther. 2020, 28, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Neyra, J.A.; Hu, M.C.; Moe, O.W. Klotho in Clinical Nephrology: Diagnostic and Therapeutic Implications. Clin. J. Am. Soc. Nephrol. 2020, 16, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Kale, A.; Sankrityayan, H.; Anders, H.J.; Gaikwad, A.B. Epigenetic and non-epigenetic regulation of Klotho in kidney disease. Life Sci. 2021, 264, 118644. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Cell/Pathway/Protein | Action of Klotho on the Cell/Pathway/Protein | Action of the Cell/Pathway/Protein on Klotho | Connections with Other Cells/Pathways/Proteins | Reference |
|---|---|---|---|---|
| T cells | Klotho downregulation causes T-cell infiltration of kidneys | [37] | ||
| T-helper cells (CD4+ lymphocytes, CD28 molecule) | Klotho downregulation in CD4+ lymphocytes is associated with lower levels of CD28 molecule through an increase in TNF-α | TNF-α | [38] | |
| Macrophages | Klotho downregulation causes macrophage infiltration of kidneys | [37] | ||
| MCP-1 (CCL2)/CCR2, ICAM-1 | Klotho downregulation increases the expression of MCP-1 and ICAM-1 in the kidneys Klotho inhibits TXNIP-mediated expression of ICAM-1 | PKC, TXNIP, IL-1β, TNF-α, RIG-I | [8,37,49,50] | |
| Complement system | Proteins of the complement system (C1, C5a), released during inflammation, inhibit Klotho expression | [53] | ||
| PI3K/Akt | Klotho blocks TNF-α-induced PI3K/Akt pathway to restrain NF-κB activation | TNF-α, NF-κB | [13] | |
| TNF-α, TWEAK | Klotho inhibits TNF- α actions on the intensification of inflammatory processes | TNF-α and TWEAK inhibit Klotho through NF-κB activation | PGC-1α, NF-κB | [39,42] |
| NF-κB and its p65 subunit (RelA) | Klotho inhibits NF-κB activity | NF-κB inhibits Klotho expression | p38 MAPK, JNK, PKC, TNF-α, IL-6, IL-8, IL-10, MCP-1, RANTES (CCL5) | [8,32,39,42,54] |
| IκBα | Klotho modulates IκBα function to inhibit RelA activation | NF-κB, RelA | [43] | |
| HSP70 | Klotho increases HSP70 levels, which inhibit NF-κB activation | NF-κB | [39] | |
| TLR4 | Klotho induces proteolytic degradation of TLR4 | TLR4 induces downregulation of Klotho expression | RIG-I, IL-1 β, MyD88, NF-κB, JNK, IKK, p38 MAPK, IRS | [8,45] |
| RIG-I | Klotho inhibits RIG-I | NF-κB, IL-6, IL-8, TLR4 | [50,51] | |
| IL-6, IL-8 | Klotho inhibits IL-6 and IL-8 through RIG-I and NF-κB suppression | RIG-I, NF-κB | [50,51] | |
| IL-1β, NLRP3 | Klotho inhibits vitamin D3 and TXNIP effect on IL-1β production and NLRP3-inflammasome activation | calcitriol, TXNIP | [49] | |
| IL-10 | Klotho increases IL-10 secretion | JAK2/STAT3, p38 MAPK, HuR, TNF-α, NF-κB | [40] | |
| Vitamin D3 in its bioactive form (calcitriol) | FGF23/Klotho pathway activation suppresses calcitriol production and promotes its degradation | Calcitriol stimulates expression of Klotho and FGF23 | FGF23 | [40,49,55] |
| TGF-β1 | Klotho binds to type II TGFβ receptors, thus inhibiting TGF-β1 signaling | Wnt, PGC-1α, MAPK, NF-κB, Smads | [54,56] | |
| Wnt/β-catenin | Klotho binds several Wnt protein family members, thus inhibiting canonical Wnt signaling | TGF-β1, PGE2 | [48] | |
| ROS | Klotho inhibits IGF-1 signaling cascade, thus causing the activation of FoxO transcription factors, induction of the expression of MnSOD, and removal of ROS Klotho suppresses TXNIP-dependent activation of the NLRP3 inflammasome in macrophages through the enhancement of FGF23 signaling | TLR4, PGC-1α, IGF-1, FoxO, MnSOD, TXNIP, NLRP3, FGF23 | [48] | |
| NO | Klotho modulates NO metabolism and prevents its impairment | [48] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Typiak, M.; Piwkowska, A. Antiinflammatory Actions of Klotho: Implications for Therapy of Diabetic Nephropathy. Int. J. Mol. Sci. 2021, 22, 956. https://doi.org/10.3390/ijms22020956
Typiak M, Piwkowska A. Antiinflammatory Actions of Klotho: Implications for Therapy of Diabetic Nephropathy. International Journal of Molecular Sciences. 2021; 22(2):956. https://doi.org/10.3390/ijms22020956
Chicago/Turabian StyleTypiak, Marlena, and Agnieszka Piwkowska. 2021. "Antiinflammatory Actions of Klotho: Implications for Therapy of Diabetic Nephropathy" International Journal of Molecular Sciences 22, no. 2: 956. https://doi.org/10.3390/ijms22020956
APA StyleTypiak, M., & Piwkowska, A. (2021). Antiinflammatory Actions of Klotho: Implications for Therapy of Diabetic Nephropathy. International Journal of Molecular Sciences, 22(2), 956. https://doi.org/10.3390/ijms22020956