
Direct and Base Excision Repair-Mediated Regulation of a GC-Rich cis-Element in Response to 5-Formylcytosine and 5-Carboxycytosine


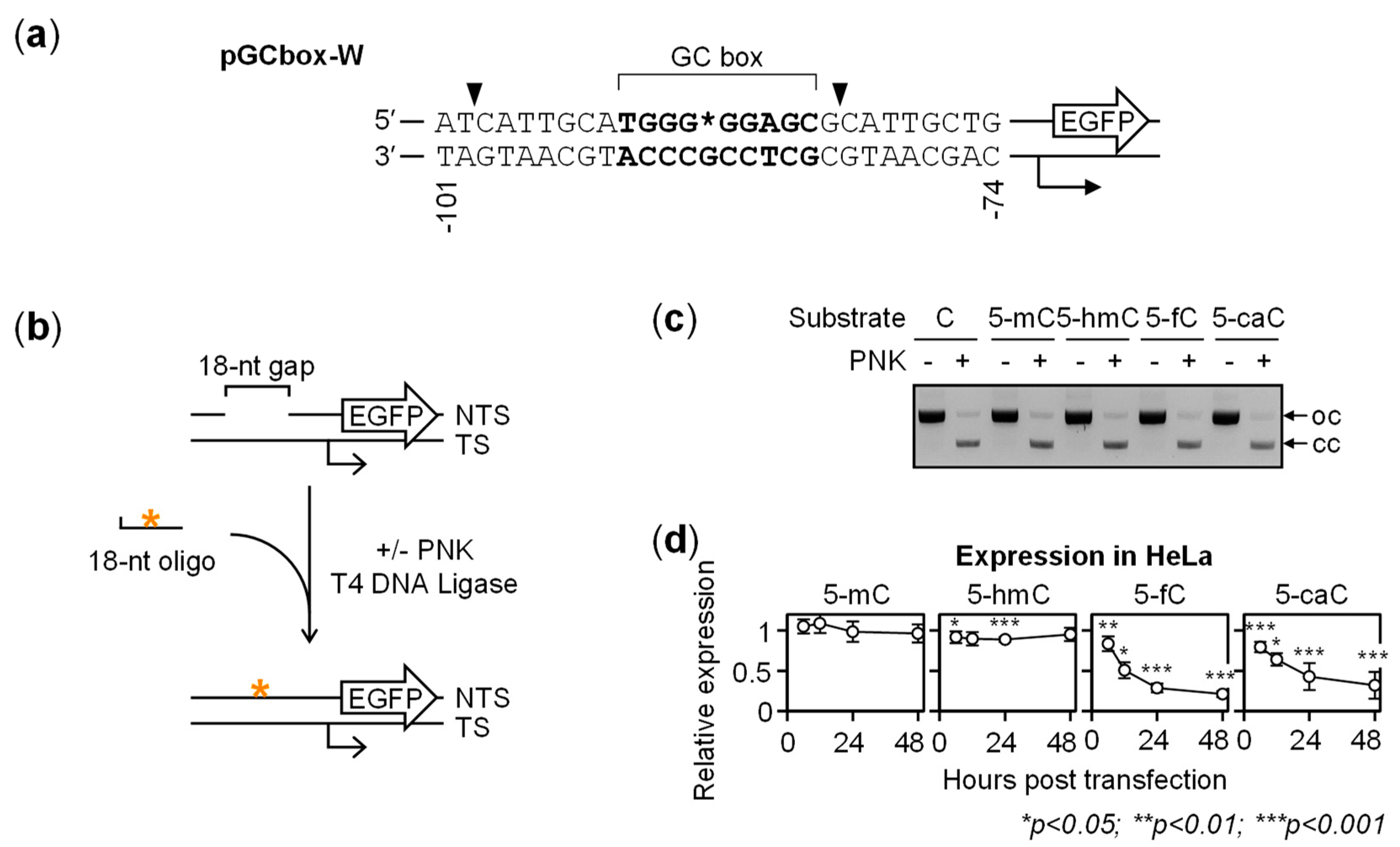{kind=link}
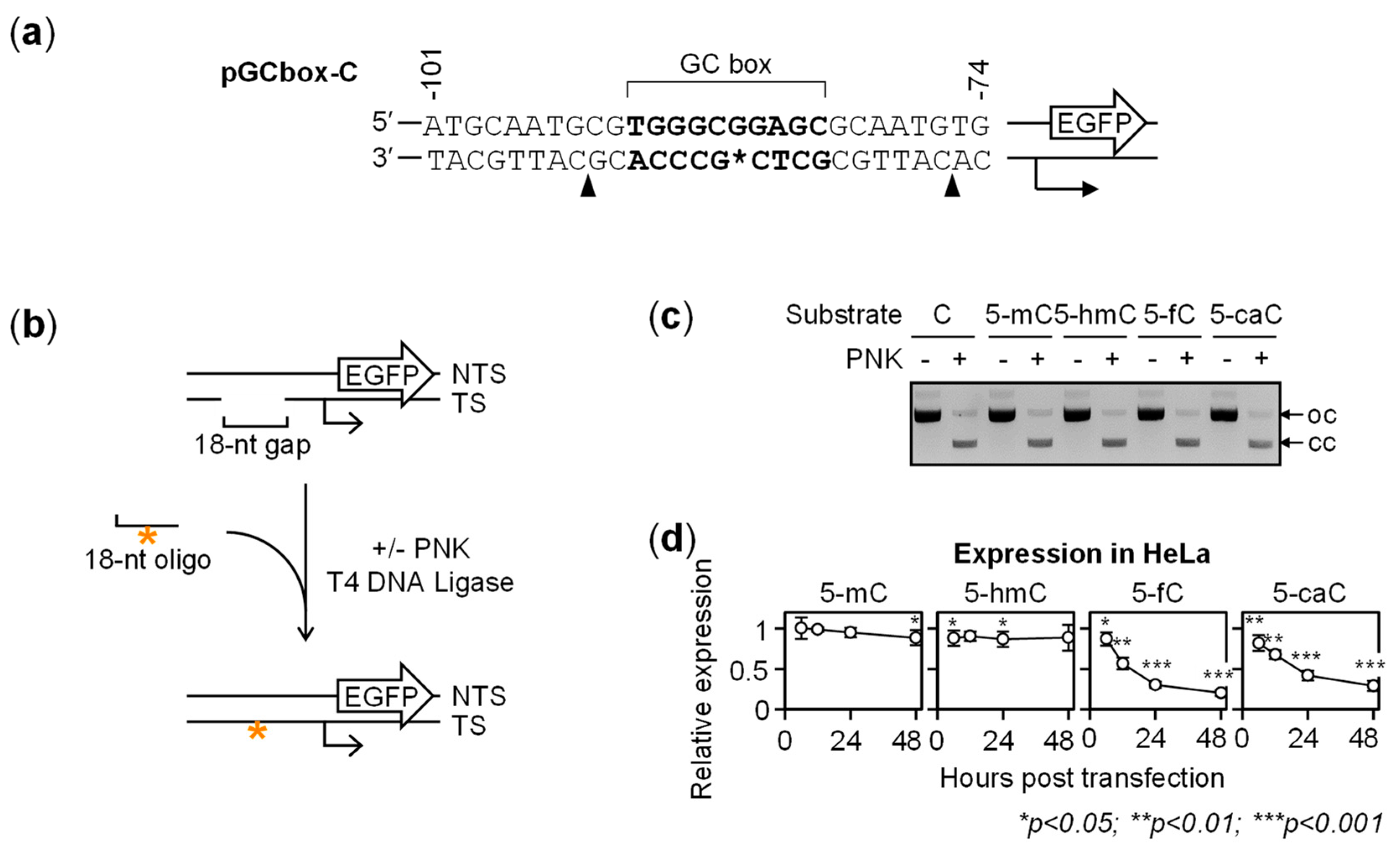{kind=link}
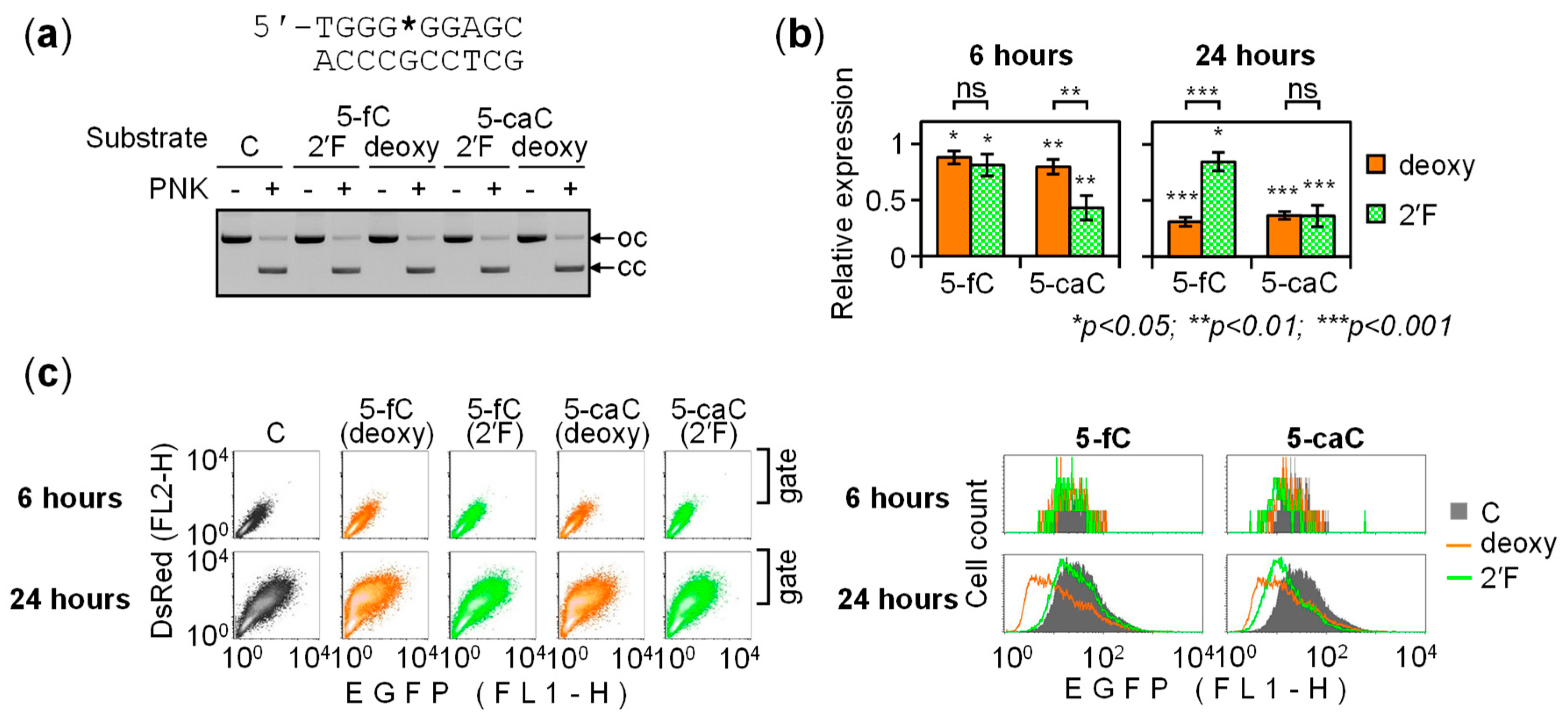{kind=link}
{kind=link}
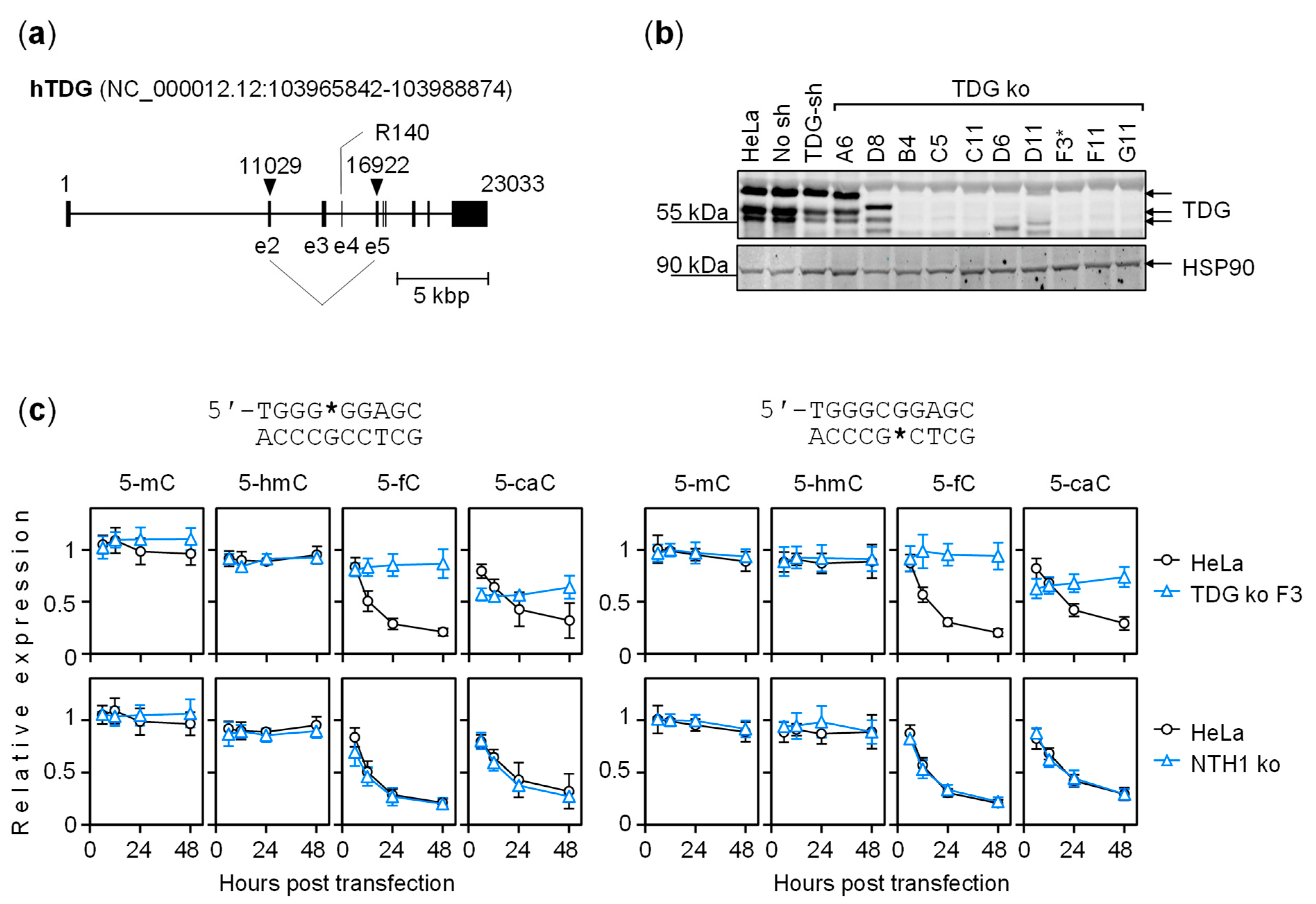{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
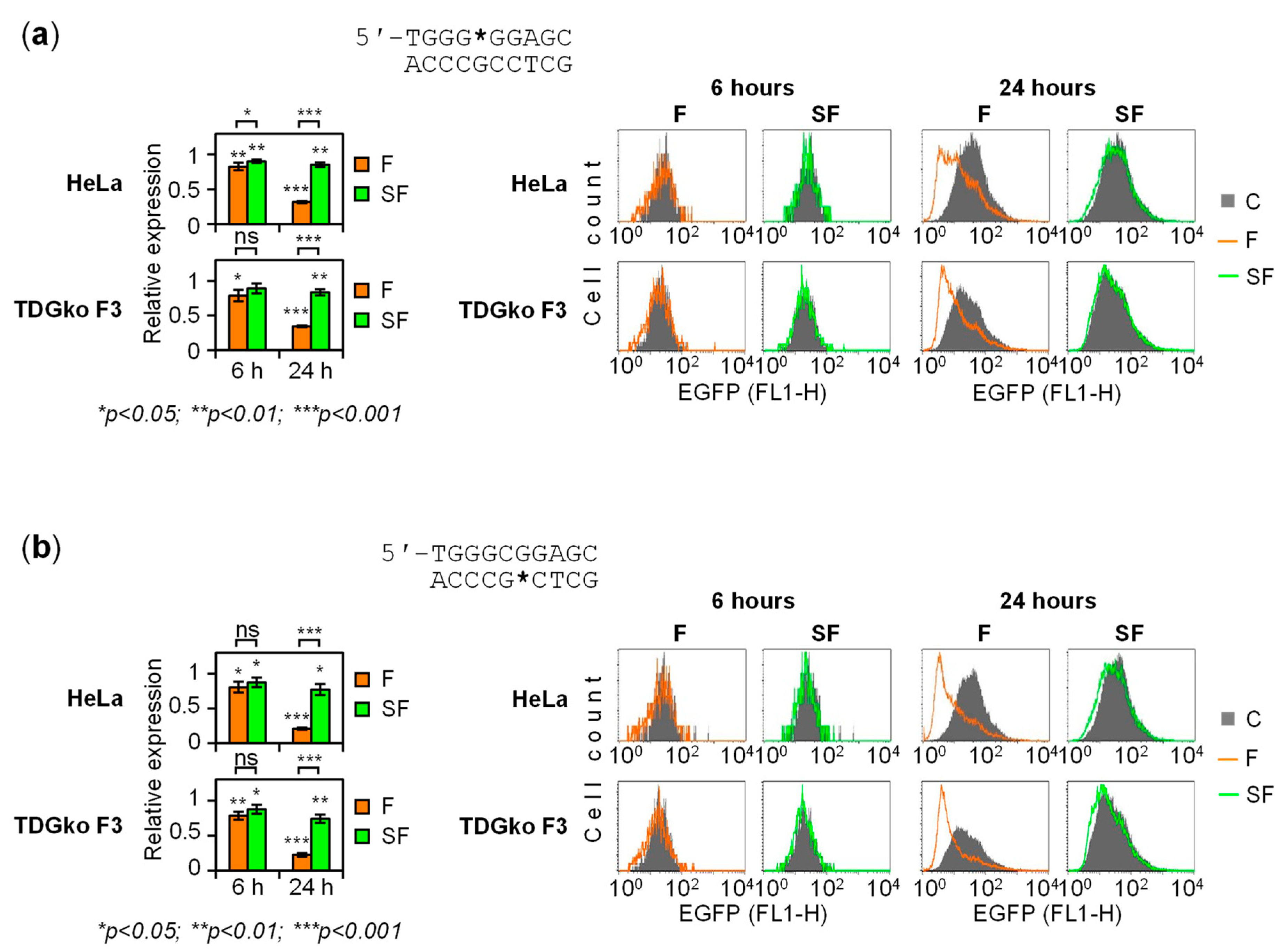
2.1. Gene Repression in HeLa Cells Induced by 5-fC and 5-caC in the GC Box CpG Dinucleotide
2.2. BER-Resistant 5-caC and, to Some Extent, 5-fC Directly Diminish the GC Box Activity
2.3. BER of 5-caC in the GC Box Induces a Transient Promoter Activation
2.4. The Dynamics of Transcriptional Regulation by 5-fC and 5-caC Is Entirely TDG-Dependent
2.5. Regulation of the GC Box Promoter by Acytosinic Sites and the Effect of Strand Cleavage
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Synthetic Oligonucleotides Carrying Cytosine Modifications
5.2. Generation of Reporter Constructs Containing Cytosine Modifications in the GC Box CpG Dinucleotide
5.3. Quantitative Analyses of EGFP Expression in Transfected Cells
5.4. TDG Gene Knockout in HeLa Cells
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bernstein, B.E.; Meissner, A.; Lander, E.S. The mammalian epigenome. Cell 2007, 128, 669–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tate, P.H.; Bird, A.P. Effects of DNA methylation on DNA-binding proteins and gene expression. Curr. Opin. Genet. Dev. 1993, 3, 226–231. [Google Scholar] [CrossRef]
- Boyes, J.; Bird, A. DNA methylation inhibits transcription indirectly via a methyl-CpG binding protein. Cell 1991, 64, 1123–1134. [Google Scholar] [CrossRef]
- Lewis, J.D.; Meehan, R.R.; Henzel, W.J.; Maurer-Fogy, I.; Jeppesen, P.; Klein, F.; Bird, A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 1992, 69, 905–914. [Google Scholar] [CrossRef]
- Jones, P.L.; Veenstra, G.J.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef]
- Fuks, F.; Burgers, W.A.; Brehm, A.; Hughes-Davies, L.; Kouzarides, T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat. Genet. 2000, 24, 88–91. [Google Scholar] [CrossRef]
- Iguchi-Ariga, S.M.; Schaffner, W. CpG methylation of the cAMP-responsive enhancer/promoter sequence TGACGTCA abolishes specific factor binding as well as transcriptional activation. Genes Dev. 1989, 3, 612–619. [Google Scholar] [CrossRef] [Green Version]
- Spruijt, C.G.; Gnerlich, F.; Smits, A.H.; Pfaffeneder, T.; Jansen, P.W.; Bauer, C.; Munzel, M.; Wagner, M.; Muller, M.; Khan, F.; et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 2013, 152, 1146–1159. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Morgunova, E.; Jolma, A.; Kaasinen, E.; Sahu, B.; Khund-Sayeed, S.; Das, P.K.; Kivioja, T.; Dave, K.; Zhong, F.; et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 2017, 356. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [Green Version]
- Pfaffeneder, T.; Hackner, B.; Truss, M.; Munzel, M.; Muller, M.; Deiml, C.A.; Hagemeier, C.; Carell, T. The discovery of 5-formylcytosine in embryonic stem cell DNA. Angew. Chem. Int. Ed. Engl. 2011, 50, 7008–7012. [Google Scholar] [CrossRef] [PubMed]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiti, A.; Drohat, A.C. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: Potential implications for active demethylation of CpG sites. J. Biol. Chem. 2011, 286, 35334–35338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Lu, X.; Lu, J.; Liang, H.; Dai, Q.; Xu, G.L.; Luo, C.; Jiang, H.; He, C. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat. Chem. Biol. 2012, 8, 328–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neri, F.; Incarnato, D.; Krepelova, A.; Rapelli, S.; Anselmi, F.; Parlato, C.; Medana, C.; Dal Bello, F.; Oliviero, S. Single-Base Resolution Analysis of 5-Formyl and 5-Carboxyl Cytosine Reveals Promoter DNA Methylation Dynamics. Cell Rep. 2015, 10, 674–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iurlaro, M.; McInroy, G.R.; Burgess, H.E.; Dean, W.; Raiber, E.A.; Bachman, M.; Beraldi, D.; Balasubramanian, S.; Reik, W. In vivo genome-wide profiling reveals a tissue-specific role for 5-formylcytosine. Genome Biol. 2016, 17, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licyte, J.; Gibas, P.; Skardziute, K.; Stankevicius, V.; Ruksenaite, A.; Kriukiene, E. A Bisulfite-free Approach for Base-Resolution Analysis of Genomic 5-Carboxylcytosine. Cell Rep. 2020, 32, 108155. [Google Scholar] [CrossRef]
- Cadet, J.; Wagner, J.R. TET enzymatic oxidation of 5-methylcytosine, 5-hydroxymethylcytosine and 5-formylcytosine. Mutat. Res. Genet. Toxicol. Environ. Mutagen 2014, 764–765, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Carell, T.; Kurz, M.Q.; Muller, M.; Rossa, M.; Spada, F. Non-canonical Bases in the Genome: The Regulatory Information Layer in DNA. Angew. Chem.-Int. Ed. 2018, 57, 4296–4312. [Google Scholar] [CrossRef] [PubMed]
- Branco, M.R.; Ficz, G.; Reik, W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat. Rev. Genet. 2012, 13, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Kitsera, N.; Allgayer, J.; Parsa, E.; Geier, N.; Rossa, M.; Carell, T.; Khobta, A. Functional impacts of 5-hydroxymethylcytosine, 5-formylcytosine, and 5-carboxycytosine at a single hemi-modified CpG dinucleotide in a gene promoter. Nucleic Acids Res. 2017, 45, 11033–11042. [Google Scholar] [CrossRef] [Green Version]
- Kitsera, N.; Rodriguez-Alvarez, M.; Emmert, S.; Carell, T.; Khobta, A. Nucleotide excision repair of abasic DNA lesions. Nucleic Acids Res. 2019, 47, 8537–8547. [Google Scholar] [CrossRef]
- Luhnsdorf, B.; Epe, B.; Khobta, A. Excision of uracil from transcribed DNA negatively affects gene expression. J. Biol. Chem. 2014, 289, 22008–22018. [Google Scholar] [CrossRef] [Green Version]
- Allgayer, J.; Kitsera, N.; Bartelt, S.; Epe, B.; Khobta, A. Widespread transcriptional gene inactivation initiated by a repair intermediate of 8-oxoguanine. Nucleic Acids Res. 2016, 44, 7267–7280. [Google Scholar] [CrossRef] [Green Version]
- Kribelbauer, J.F.; Lu, X.J.; Rohs, R.; Mann, R.S.; Bussemaker, H.J. Toward a Mechanistic Understanding of DNA Methylation Readout by Transcription Factors. J. Mol. Biol. 2020, 432, 1801–1815. [Google Scholar] [CrossRef]
- Muller, N.; Khobta, A. Regulation of GC box activity by 8-oxoguanine. Redox Biol. 2021, 43, 101997. [Google Scholar] [CrossRef]
- Schroder, A.S.; Kotljarova, O.; Parsa, E.; Iwan, K.; Raddaoui, N.; Carell, T. Synthesis of (R)-Configured 2’-Fluorinated mC, hmC, fC, and caC Phosphoramidites and Oligonucleotides. Org. Lett. 2016, 18, 4368–4371. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Ding, Y.; Burrows, C.J. Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc. Natl. Acad. Sci. USA 2017, 114, 2604–2609. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.M., 3rd; Takeshita, M.; Grollman, A.P.; Demple, B. Incision activity of human apurinic endonuclease (Ape) at abasic site analogs in DNA. J. Biol. Chem. 1995, 270, 16002–16007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valinluck, V.; Tsai, H.H.; Rogstad, D.K.; Burdzy, A.; Bird, A.; Sowers, L.C. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res. 2004, 32, 4100–4108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valinluck, V.; Sowers, L.C. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007, 67, 946–950. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, H.; Liu, Y.; Upadhyay, A.K.; Chang, Y.; Howerton, S.B.; Vertino, P.M.; Zhang, X.; Cheng, X. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res. 2012, 40, 4841–4849. [Google Scholar] [CrossRef]
- Giehr, P.; Kyriakopoulos, C.; Ficz, G.; Wolf, V.; Walter, J. The Influence of Hydroxylation on Maintaining CpG Methylation Patterns: A Hidden Markov Model Approach. PLoS Comput. Biol. 2016, 12, e1004905. [Google Scholar] [CrossRef]
- Ji, D.; Lin, K.; Song, J.; Wang, Y. Effects of Tet-induced oxidation products of 5-methylcytosine on Dnmt1- and DNMT3a-mediated cytosine methylation. Mol. Biosyst. 2014, 10, 1749–1752. [Google Scholar] [CrossRef] [Green Version]
- Harrington, M.A.; Jones, P.A.; Imagawa, M.; Karin, M. Cytosine methylation does not affect binding of transcription factor Sp1. Proc. Natl. Acad. Sci. USA 1988, 85, 2066–2070. [Google Scholar] [CrossRef] [Green Version]
- Holler, M.; Westin, G.; Jiricny, J.; Schaffner, W. Sp1 transcription factor binds DNA and activates transcription even when the binding site is CpG methylated. Genes Dev. 1988, 2, 1127–1135. [Google Scholar] [CrossRef] [Green Version]
- Khobta, A.; Anderhub, S.; Kitsera, N.; Epe, B. Gene silencing induced by oxidative DNA base damage: Association with local decrease of histone H4 acetylation in the promoter region. Nucleic Acids Res. 2010, 38, 4285–4295. [Google Scholar] [CrossRef]
- Broxson, C.; Hayner, J.N.; Beckett, J.; Bloom, L.B.; Tornaletti, S. Human AP endonuclease inefficiently removes abasic sites within G4 structures compared to duplex DNA. Nucleic Acids Res. 2014, 42, 7708–7719. [Google Scholar] [CrossRef] [Green Version]
- Burra, S.; Marasco, D.; Malfatti, M.C.; Antoniali, G.; Virgilio, A.; Esposito, V.; Demple, B.; Galeone, A.; Tell, G. Human AP-endonuclease (Ape1) activity on telomeric G4 structures is modulated by acetylatable lysine residues in the N-terminal sequence. DNA Repair 2019, 73, 129–143. [Google Scholar] [CrossRef]
- Fleming, A.M.; Zhu, J.; Howpay Manage, S.A.; Burrows, C.J. Human NEIL3 Gene Expression Regulated by Epigenetic-Like Oxidative DNA Modification. J. Am. Chem. Soc. 2019, 141, 11036–11049. [Google Scholar] [CrossRef] [Green Version]
- Davletgildeeva, A.T.; Kuznetsova, A.A.; Fedorova, O.S.; Kuznetsov, N.A. Activity of Human Apurinic/Apyrimidinic Endonuclease APE1 toward Damaged DNA and Native RNA with Non-canonical Structures. Front. Cell Dev. Biol. 2020, 8, 590848. [Google Scholar] [CrossRef]
- Mohni, K.N.; Wessel, S.R.; Zhao, R.; Wojciechowski, A.C.; Luzwick, J.W.; Layden, H.; Eichman, B.F.; Thompson, P.S.; Mehta, K.P.M.; Cortez, D. HMCES Maintains Genome Integrity by Shielding Abasic Sites in Single-Strand DNA. Cell 2019, 176, 144–153.e13. [Google Scholar] [CrossRef] [Green Version]
- Thompson, P.S.; Amidon, K.M.; Mohni, K.N.; Cortez, D.; Eichman, B.F. Protection of abasic sites during DNA replication by a stable thiazolidine protein-DNA cross-link. Nat. Struct. Mol. Biol. 2019, 26, 613–618. [Google Scholar] [CrossRef]
- Schroder, A.S.; Steinbacher, J.; Steigenberger, B.; Gnerlich, F.A.; Schiesser, S.; Pfaffeneder, T.; Carell, T. Synthesis of a DNA promoter segment containing all four epigenetic nucleosides: 5-methyl-, 5-hydroxymethyl-, 5-formyl-, and 5-carboxy-2′-deoxycytidine. Angew. Chem. Int. Ed. Engl. 2014, 53, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Luhnsdorf, B.; Kitsera, N.; Warken, D.; Lingg, T.; Epe, B.; Khobta, A. Generation of reporter plasmids containing defined base modifications in the DNA strand of choice. Anal. Biochem. 2012, 425, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Bauer, D.E.; Canver, M.C.; Orkin, S.H. Generation of genomic deletions in mammalian cell lines via CRISPR/Cas9. J. Vis. Exp. 2015, 95, e52118. [Google Scholar] [CrossRef] [PubMed]
- Labun, K.; Montague, T.G.; Gagnon, J.A.; Thyme, S.B.; Valen, E. CHOPCHOP v2: A web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 2016, 44, W272–W276. [Google Scholar] [CrossRef] [PubMed]
- Allgayer, J.; Kitsera, N.; von der Lippen, C.; Epe, B.; Khobta, A. Modulation of base excision repair of 8-oxoguanine by the nucleotide sequence. Nucleic Acids Res. 2013, 41, 8559–8571. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, N.; Ponkkonen, E.; Carell, T.; Khobta, A. Direct and Base Excision Repair-Mediated Regulation of a GC-Rich cis-Element in Response to 5-Formylcytosine and 5-Carboxycytosine. Int. J. Mol. Sci. 2021, 22, 11025. https://doi.org/10.3390/ijms222011025
Müller N, Ponkkonen E, Carell T, Khobta A. Direct and Base Excision Repair-Mediated Regulation of a GC-Rich cis-Element in Response to 5-Formylcytosine and 5-Carboxycytosine. International Journal of Molecular Sciences. 2021; 22(20):11025. https://doi.org/10.3390/ijms222011025
Chicago/Turabian StyleMüller, Nadine, Eveliina Ponkkonen, Thomas Carell, and Andriy Khobta. 2021. "Direct and Base Excision Repair-Mediated Regulation of a GC-Rich cis-Element in Response to 5-Formylcytosine and 5-Carboxycytosine" International Journal of Molecular Sciences 22, no. 20: 11025. https://doi.org/10.3390/ijms222011025
APA StyleMüller, N., Ponkkonen, E., Carell, T., & Khobta, A. (2021). Direct and Base Excision Repair-Mediated Regulation of a GC-Rich cis-Element in Response to 5-Formylcytosine and 5-Carboxycytosine. International Journal of Molecular Sciences, 22(20), 11025. https://doi.org/10.3390/ijms222011025