
The Interplay between Insulin Resistance, Inflammation, Oxidative Stress, Base Excision Repair and Metabolic Syndrome in Nonalcoholic Fatty Liver Disease

 , and
, and
Abstract
:
1. Introduction
2. Development of NAFLD
2.1. Epidemiology and Etiology
2.2. FFA Accumulation
2.3. Inflammation
2.4. ER Stress
2.5. Oxidative Stress
2.6. Mitochondrial Dysfunction
3. The Interplay between NAFLD, MS, IR, and DNA Repair
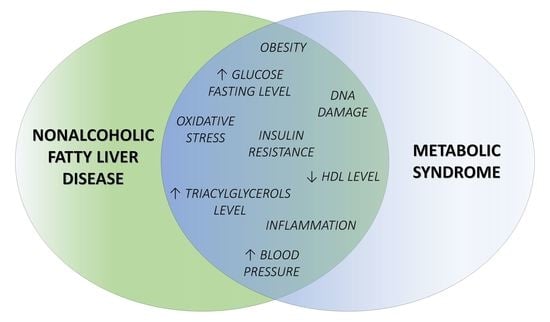
3.1. Common Features of NAFLD and MS
3.1.1. Obesity
3.1.2. T2DM
3.2. The Role of BER in NAFLD
3.2.1. BER and Adipose Tissue Metabolism
3.2.2. DNA Repair in NAFLD
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Paschos, P.; Paletas, K. Non alcoholic fatty liver disease and metabolic syndrome. Hippokratia 2009, 13, 9–19. [Google Scholar]
- Abd El-Kader, S.M.; El-Den Ashmawy, E.M.S. Non-alcoholic fatty liver disease: The diagnosis and management. World J. Hepatol. 2015, 7, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Dharmalingam, M.; Yamasandhi, P.G. Nonalcoholic Fatty Liver Disease and Type 2 Diabetes Mellitus. Indian J. Endocrinol. Metab. 2018, 22, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Yasutake, K.; Kohjima, M.; Kotoh, K.; Nakashima, M.; Nakamuta, M.; Enjoji, M. Dietary habits and behaviors associated with nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 1756–1767. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.L. A comprehensive definition for metabolic syndrome. Dis. Model. Mech. 2009, 2, 231–237. [Google Scholar] [CrossRef] [Green Version]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism. 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Hurrle, S.; Hsu, W.H. The etiology of oxidative stress in insulin resistance. Biomed. J. 2017, 40, 257–262. [Google Scholar] [CrossRef]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef]
- Czarny, P.; Merecz-Sadowska, A.; Majchrzak, K.; Jablkowski, M.; Szemraj, J.; Sliwinski, T.; Karwowski, B. The Influence of Hepatitis C Virus Therapy on the DNA Base Excision Repair System of Peripheral Blood Mononuclear Cells. DNA Cell Biol. 2017, 36, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Asrih, M.; Jornayvaz, F.R. Metabolic syndrome and nonalcoholic fatty liver disease: Is insulin resistance the link? Mol. Cell. Endocrinol. 2015, 418 Pt 1, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Hübscher, S.G. Histological assessment of non-alcoholic fatty liver disease. Histopathology 2006, 49, 450–465. [Google Scholar] [CrossRef]
- Gariani, K.; Philippe, J.; Jornayvaz, F.R. Non-alcoholic fatty liver disease and insulin resistance: From bench to bedside. Diabetes Metab. 2013, 39, 16–26. [Google Scholar] [CrossRef]
- López-Velázquez, J.A.; Silva-Vidal, K.V.; Ponciano-Rodríguez, G.; Chávez-Tapia, N.C.; Arrese, M.; Uribe, M.; Méndez-Sánchez, N. The prevalence of nonalcoholic fatty liver disease in the Americas. Ann. Hepatol. 2014, 13, 166–178. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.-C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Yang, J.; Fernández-Galilea, M.; Martínez-Fernández, L.; González-Muniesa, P.; Pérez-Chávez, A.; Martínez, J.A.; Moreno-Aliaga, M.J. Oxidative Stress and Non-Alcoholic Fatty Liver Disease: Effects of Omega-3 Fatty Acid Supplementation. Nutrients 2019, 11, 872. [Google Scholar] [CrossRef] [Green Version]
- Schwimmer, J.B. Definitive diagnosis and assessment of risk for nonalcoholic fatty liver disease in children and adolescents. Semin. Liver Dis. 2007, 27, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Zheng, L.; Wang, M.; Du, Y.; Jiang, J. Prevalence trends in non-alcoholic fatty liver disease at the global, regional and national levels, 1990–2017: A population-based observational study. BMJ Open 2020, 10, e036663. [Google Scholar] [CrossRef]
- Basaranoglu, M.; Neuschwander-Tetri, B.A. Nonalcoholic Fatty Liver Disease: Clinical Features and Pathogenesis. Gastroenterol. Hepatol. 2006, 2, 282–291. [Google Scholar]
- Neuschwander-Tetri, B.A.; Caldwell, S.H. Nonalcoholic steatohepatitis: Summary of an AASLD Single Topic Conference. Hepatology 2003, 37, 1202–1219. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, G.; Celsa, C.; Giammanco, A.; Spatola, F.; Petta, S. The Relevance of Noninvasive Tools To Assess Fibrosis in Non-Alcoholic Fatty Liver Disease. Curr. Pharm. Des. 2020, 26, 3928–3938. [Google Scholar] [CrossRef]
- Lin, S.; Huang, J.; Wang, M.; Kumar, R.; Liu, Y.; Liu, S.; Wu, Y.; Wang, X.; Zhu, Y. Comparison of MAFLD and NAFLD diagnostic criteria in real world. Liver Int. Off. J. Int. Assoc. Study Liver 2020, 40, 2082–2089. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mignery, G.A.; Pikaard, C.S.; Park, W.D. Molecular characterization of the patatin multigene family of potato. Gene 1988, 62, 27–44. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Donati, B.; Fares, R.; Lombardi, R.; Mancina, R.M.; Romeo, S.; Valenti, L. PNPLA3 I148M polymorphism and progressive liver disease. World J. Gastroenterol. 2013, 19, 6969–6978. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Patman, G.L.; Leathart, J.B.S.; Piguet, A.-C.; Burt, A.D.; Dufour, J.-F.; Day, C.P.; Daly, A.K.; Reeves, H.L.; Anstee, Q.M. Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J. Hepatol. 2014, 61, 75–81. [Google Scholar] [CrossRef]
- Pennisi, G.; Celsa, C.; Giammanco, A.; Spatola, F.; Petta, S. The Burden of Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease: Screening Issue and Future Perspectives. Int. J. Mol. Sci. 2019, 20, 5613. [Google Scholar] [CrossRef] [Green Version]
- Eslam, M.; George, J. Genetic contributions to NAFLD: Leveraging shared genetics to uncover systems biology. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 40–52. [Google Scholar] [CrossRef]
- Simoes, I.C.M.; Karkucinska-wieckowska, A.; Janikiewicz, J.; Szymanska, S.; Pronicki, M.; Dobrzyn, P.; Dabrowski, M.; Dobrzyn, A.; Oliveira, P.J.; Zischka, H.; et al. Western diet causes obesity-induced nonalcoholic fatty liver disease development by differentially compromising the autophagic response. Antioxidants 2020, 9, 995. [Google Scholar] [CrossRef]
- Wu, W.; Tsuchida, H.; Kato, T.; Niwa, H.; Horikawa, Y.; Takeda, J.; Iizuka, K. Fat and carbohydrate in western diet contribute differently to hepatic lipid accumulation. Biochem. Biophys. Res. Commun. 2015, 461, 681–686. [Google Scholar] [CrossRef]
- Feng, R.; Luo, C.; Li, C.; Du, S.; Okekunle, A.P.; Li, Y.; Chen, Y.; Zi, T.; Niu, Y. Free fatty acids profile among lean, overweight and obese non-alcoholic fatty liver disease patients: A case—Control study. Lipids Health Dis. 2017, 16, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Diraison, F.; Moulin, P.; Beylot, M. Contribution of hepatic de novo lipogenesis and reesterification of plasma non esterified fatty acids to plasma triglyceride synthesis during non-alcoholic fatty liver disease. Diabetes Metab. 2003, 29, 478–485. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos–Roman, M.A.; Browning, J.D.; Parks, E.J. Increased De novo Lipogenesis Is a Distinct Characteristic of Individuals With Nonalcoholic Fatty Liver Disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Shimano, H.; Yahagi, N.; Amemiya-Kudo, M.; Hasty, A.H.; Osuga, J.I.; Tamura, Y.; Shionoiri, F.; Iizuka, Y.; Ohashi, K.; Harada, K.; et al. Sterol regulatory element-binding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes. J. Biol. Chem. 1999, 274, 35832–35839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferré, P.; Foufelle, F. SREBP-1c transcription factor and lipid homeostasis: Clinical perspective. Horm. Res. 2007, 68, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Prieto, P.; Postic, C. Carbohydrate sensing through the transcription factor ChREBP. Front. Genet. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foretz, M.; Pacot, C.; Dugail, I.; Lemarchand, P.; Guichard, C.; le Lièpvre, X.; Berthelier-Lubrano, C.; Spiegelman, B.; Kim, J.B.; Ferré, P.; et al. ADD1/SREBP-1c Is Required in the Activation of Hepatic Lipogenic Gene Expression by Glucose. Mol. Cell. Biol. 1999, 19, 3760–3768. [Google Scholar] [CrossRef] [Green Version]
- Liang, G.; Yang, J.; Horton, J.D.; Hammer, R.E.; Goldstein, J.L.; Brown, M.S. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J. Biol. Chem. 2002, 277, 9520–9528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knebel, B.; Haas, J.; Hartwig, S.; Jacob, S.; Köllmer, C.; Nitzgen, U.; Muller-Wieland, D.; Kotzka, J. Liver-specific expression of transcriptionally active srebp-1c is associated with fatty liver and increased visceral fat mass. PLoS ONE 2012, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Kohjima, M.; Enjoji, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2007, 20, 351–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. From The Cover: Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Iizuka, K.; Takao, K.; Horikawa, Y.; Kitamura, T.; Takeda, J. ChREBP-knockout mice show sucrose intolerance and fructose malabsorption. Nutrients 2018, 10, 340. [Google Scholar] [CrossRef]
- Diraison, F.; Dusserre, E.; Vidal, H.; Sothier, M.; Beylot, M. Increased hepatic lipogenesis but decreased expression of lipogenic gene in adipose tissue in human obesity. Am. J. Physiol. Endocrinol. Metab. 2002, 282, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Forcheron, F.; Cachefo, A.; Thevenon, S.; Pinteur, C.; Beylot, M. Mechanisms of the triglyceride- and cholesterol-lowering effect of fenofibrate in hyperlipidemic type 2 diabetic patients. Diabetes 2002, 51, 3486–3491. [Google Scholar] [CrossRef] [Green Version]
- Dentin, R.; Benhamed, F.; Hainault, I.; Fauveau, V.; Foufelle, F.; Dyck, J.R.B.; Girard, J.; Postic, C. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes 2006, 55, 2159–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benhamed, F.; Denechaud, P.D.; Lemoine, M.; Robichon, C.; Moldes, M.; Bertrand-Michel, J.; Ratziu, V.; Serfaty, L.; Housset, C.; Capeau, J.; et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J. Clin. Invest. 2012, 122, 2176–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, J.; DeMayo, F.J.; Li, H.; Abu-Elheiga, L.; Gu, Z.; Shaikenov, T.E.; Kordari, P.; Chirala, S.S.; Heird, W.C.; Wakil, S.J. Liver-specific deletion of acetyl-CoA carboxylase 1 reduces hepatic triglyceride accumulation without affecting glucose homeostasis. Proc. Natl. Acad. Sci. USA 2006, 103, 8552–8557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.A. Peroxisome proliferator-activated receptors and the regulation of mammalian lipid metabolism. Biochem. Soc. Trans. 2002, 30, 1086–1090. [Google Scholar] [CrossRef] [PubMed]
- Ip, E.; Farrell, G.; Hall, P.; Robertson, G.; Leclercq, I. Administration of the Potent PPARα Agonist, Wy-14,643, Reverses Nutritional Fibrosis and Steatohepatitis in Mice. Hepatology 2004, 39, 1286–1296. [Google Scholar] [CrossRef] [PubMed]
- Ip, E.; Farrell, G.C.; Robertson, G.; Hall, P.; Kirsch, R.; Leclercq, I. Central role of PPARα-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology 2003, 38, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Larter, C.Z.; Yeh, M.M.; van Rooyen, D.M.; Brooling, J.; Ghatora, K.; Farrell, G.C. Peroxisome proliferator-activated receptor-α agonist, Wy 14643, improves metabolic indices, steatosis and ballooning in diabetic mice with non-alcoholic steatohepatitis. J. Gastroenterol. Hepatol. 2012, 27, 341–350. [Google Scholar] [CrossRef]
- Guerre-Millo, M.; Gervois, P.; Raspé, E.; Madsen, L.; Poulain, P.; Derudas, B.; Herbert, J.M.; Winegar, D.A.; Willson, T.M.; Fruchart, J.C.; et al. Peroxisome proliferator-activated receptor α activators improve insulin sensitivity and reduce adiposity. J. Biol. Chem. 2000, 275, 16638–16642. [Google Scholar] [CrossRef] [Green Version]
- Tordjman, K.; Bernal-Mizrachi, C.; Zemany, L.; Weng, S.; Feng, C.; Zhang, F.; Leone, T.C.; Coleman, T.; Kelly, D.P.; Semenkovich, C.F. PPARalpha deficiency reduces insulin resistance and atherosclerosis in apoE-null mice. J. Clin. Invest. 2001, 107, 1025–1034. [Google Scholar] [CrossRef] [Green Version]
- Vancells Lujan, P.; Viñas Esmel, E.; Sacanella Meseguer, E. Overview of non-alcoholic fatty liver disease (Nafld) and the role of sugary food consumption and other dietary components in its development. Nutrients 2021, 13, 1442. [Google Scholar] [CrossRef]
- Hydes, T.; Alam, U.; Cuthbertson, D.J. The Impact of Macronutrient Intake on Non-alcoholic Fatty Liver Disease (NAFLD): Too Much Fat, Too Much Carbohydrate, or Just Too Many Calories? Front. Nutr. 2021, 8. [Google Scholar] [CrossRef]
- Green, C.J.; Hodson, L. The influence of dietary fat on liver fat accumulation. Nutrients 2014, 6, 5018–5033. [Google Scholar] [CrossRef]
- Velázquez, K.T.; Enos, R.T.; Bader, J.E.; Sougiannis, A.T.; Carson, M.S.; Chatzistamou, I.; Carson, J.A.; Nagarkatti, P.S.; Nagarkatti, M.; Murphy, E.A. Prolonged high-fat-diet feeding promotes non-alcoholic fatty liver disease and alters gut microbiota in mice. World J. Hepatol. 2019, 11, 619–637. [Google Scholar] [CrossRef]
- Yang, P.; Wang, Y.; Tang, W.; Sun, W.; Ma, Y.; Lin, S.; Jing, J.; Jiang, L.; Shi, H.; Song, Z.; et al. Western diet induces severe nonalcoholic steatohepatitis, ductular reaction, and hepatic fibrosis in liver CGI-58 knockout mice. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Perdomo, C.M.; Frühbeck, G.; Escalada, J. Impact of nutritional changes on nonalcoholic fatty liver disease. Nutrients 2019, 11, 677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sertoglu, E.; Kayadibi, H.; Uyanik, M. A biochemical view: Increase in polyunsaturated fatty acid ω-6/ω-3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. J. Diabetes Complications 2015, 29, 157. [Google Scholar] [CrossRef] [PubMed]
- De Castro, G.S.; Calder, P.C. Non-alcoholic fatty liver disease and its treatment with n-3 polyunsaturated fatty acids. Clin. Nutr. 2018, 37, 37–55. [Google Scholar] [CrossRef] [Green Version]
- Khadge, S.; Sharp, J.G.; Thiele, G.M.; McGuire, T.R.; Klassen, L.W.; Duryee, M.J.; Britton, H.C.; Dafferner, A.J.; Beck, J.; Black, P.N.; et al. Dietary omega-3 and omega-6 polyunsaturated fatty acids modulate hepatic pathology. J. Nutr. Biochem. 2018, 52, 92–102. [Google Scholar] [CrossRef]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.-H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Noworolski, S.M.; Wen, M.J.; Dyachenko, A.; Prior, J.L.; Weinberg, M.E.; Herraiz, L.A.; Tai, V.W.; Bergeron, N.; Bersot, T.P.; et al. Effect of a high-fructose weight-maintaining diet on lipogenesis and liver fat. J. Clin. Endocrinol. Metab. 2015, 100, 2434–2442. [Google Scholar] [CrossRef]
- Chen, H.; Wang, J.; Li, Z.; Kei Lam, C.W.; Xiao, Y.; Wu, Q.; Zhang, W. Consumption of sugar-sweetened beverages has a dose-dependent effect on the risk of non-alcoholic fatty liver disease: An updated systematic review and dose-response meta-analysis. Int. J. Environ. Res. Public Health 2019, 16, 2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijarnpreecha, K.; Thongprayoon, C.; Edmonds, P.J.; Cheungpasitporn, W. Associations of sugar- and artificially sweetened soda with nonalcoholic fatty liver disease: A systematic review and meta-analysis. QJM 2016, 109, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Sevastianova, K.; Santos, A.; Kotronen, A.; Hakkarainen, A.; Makkonen, J.; Silander, K.; Peltonen, M.; Romeo, S.; Lundbom, J.; Lundbom, N.; et al. Effect of short-term carbohydrate overfeeding and long-term weight loss on liver fat in overweight humans. Am. J. Clin. Nutr. 2012, 96, 727–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Than, N.N.; Newsome, P.N. A concise review of non-alcoholic fatty liver disease. Atherosclerosis 2015, 239, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Bilzer, M.; Roggel, F.; Gerbes, A.L. Role of Kupffer cells in host defense and liver disease. Liver Int. Off. J. Int. Assoc. Study Liver 2006, 26, 1175–1186. [Google Scholar] [CrossRef]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Invernizzi, P.; Mantovani, A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology 2014, 59, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Louvet, A.; Teixeira-Clerc, F.; Chobert, M.-N.; Deveaux, V.; Pavoine, C.; Zimmer, A.; Pecker, F.; Mallat, A.; Lotersztajn, S. Cannabinoid CB2 receptors protect against alcoholic liver disease by regulating Kupffer cell polarization in mice. Hepatology 2011, 54, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; Hunter, D.; Huber, R.; Lemieux, J.; Slaymaker, S.; Vaddi, K.; Charo, I.; Leibel, R.L.; Ferrante, A.W.J. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J. Clin. Invest. 2006, 116, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Wasmuth, H.E.; Lammert, F.; Zaldivar, M.M.; Weiskirchen, R.; Hellerbrand, C.; Scholten, D.; Berres, M.-L.; Zimmermann, H.; Streetz, K.L.; Tacke, F.; et al. Antifibrotic effects of CXCL9 and its receptor CXCR3 in livers of mice and humans. Gastroenterology 2009, 137, 309–319.e3. [Google Scholar] [CrossRef] [Green Version]
- Kitade, H.; Sawamoto, K.; Nagashimada, M.; Inoue, H.; Yamamoto, Y.; Sai, Y.; Takamura, T.; Yamamoto, H.; Miyamoto, K.; Ginsberg, H.N.; et al. CCR5 plays a critical role in obesity-induced adipose tissue inflammation and insulin resistance by regulating both macrophage recruitment and M1/M2 status. Diabetes 2012, 61, 1680–1690. [Google Scholar] [CrossRef] [Green Version]
- Braunersreuther, V.; Viviani, G.L.; Mach, F.; Montecucco, F. Role of cytokines and chemokines in non-alcoholic fatty liver disease. World J. Gastroenterol. 2012, 18, 727–735. [Google Scholar] [CrossRef]
- Kitade, H.; Chen, G.; Ni, Y.; Ota, T. Nonalcoholic Fatty Liver Disease and Insulin Resistance: New Insights and Potential New Treatments. Nutrients 2017, 9, 387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, P.S.; Cortez-Pinto, H.; Solá, S.; Castro, R.E.; Ramalho, R.M.; Baptista, A.; Moura, M.C.; Camilo, M.E.; Rodrigues, C.M.P. Hepatocyte apoptosis, expression of death receptors, and activation of NF-kappaB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am. J. Gastroenterol. 2004, 99, 1708–1717. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-Q.; Xu, C.-F.; Yu, C.-H.; Chen, W.-X.; Li, Y.-M. Role of endoplasmic reticulum stress in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wei, Y.; Pagliassotti, M.J. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology 2006, 147, 943–951. [Google Scholar] [CrossRef] [Green Version]
- Lenna, S.; Trojanowska, M. The role of endoplasmic reticulum stress and the unfolded protein response in fibrosis. Curr. Opin. Rheumatol. 2012, 24, 663–668. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Wagner, M.; Moore, D.D. Endoplasmic reticulum stress and glucose homeostasis. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 367–373. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [Green Version]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006, 38, 317–332. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef] [Green Version]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef] [Green Version]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.-H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [Green Version]
- Du, K.; Herzig, S.; Kulkarni, R.N.; Montminy, M. TRB3: A tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 2003, 300, 1574–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S.M.; Schroeder-Gloeckler, J.M.; Janssen, R.C.; Jiang, H.; Qadri, I.; Maclean, K.N.; Friedman, J.E. CCAAT/enhancing binding protein beta deletion in mice attenuates inflammation, endoplasmic reticulum stress, and lipid accumulation in diet-induced nonalcoholic steatohepatitis. Hepatology 2007, 45, 1108–1117. [Google Scholar] [CrossRef] [PubMed]
- Sreejayan, N.; Dong, F.; Kandadi, M.R.; Yang, X.; Ren, J. Chromium alleviates glucose intolerance, insulin resistance, and hepatic ER stress in obese mice. Obesity 2008, 16, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Mirshahi, F.; Cheung, O.; Natarajan, R.; Maher, J.W.; Kellum, J.M.; Sanyal, A.J. Activation and Dysregulation of the Unfolded Protein Response in Nonalcoholic Fatty Liver Disease. Gastroenterology 2008, 134, 568–576. [Google Scholar] [CrossRef]
- Simopoulos, A.P. Human requirement for N-3 polyunsaturated fatty acids. Poult. Sci. 2000, 79, 961–970. [Google Scholar] [CrossRef]
- Colagar, A.H.; Marzony, E.T. Ascorbic Acid in Human Seminal Plasma: Determination and Its Relationship to Sperm Quality. J. Clin. Biochem. Nutr. 2009, 45, 144–149. [Google Scholar] [CrossRef] [Green Version]
- Farzanegi, P.; Dana, A.; Ebrahimpoor, Z.; Asadi, M.; Azarbayjani, M.A. Mechanisms of beneficial effects of exercise training on non-alcoholic fatty liver disease (NAFLD): Roles of oxidative stress and inflammation. Eur. J. Sport Sci. 2019, 19, 994–1003. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Invest. 2004, 114, 1752–1761. [Google Scholar] [CrossRef]
- Polimeni, L.; Del Ben, M.; Baratta, F.; Perri, L.; Albanese, F.; Pastori, D.; Violi, F.; Angelico, F. Oxidative stress: New insights on the association of non-alcoholic fatty liver disease and atherosclerosis. World J. Hepatol. 2015, 7, 1325–1336. [Google Scholar] [CrossRef]
- Hançer, N.J.; Qiu, W.; Cherella, C.; Li, Y.; Copps, K.D.; White, M.F. Insulin and metabolic stress stimulate multisite serine/threonine phosphorylation of insulin receptor substrate 1 and inhibit tyrosine phosphorylation. J. Biol. Chem. 2014, 289, 12467–12484. [Google Scholar] [CrossRef] [Green Version]
- Blanco, C.L.; McGill-Vargas, L.L.; Gastaldelli, A.; Seidner, S.R.; McCurnin, D.C.; Leland, M.M.; Anzueto, D.G.; Johnson, M.C.; Liang, H.; DeFronzo, R.A.; et al. Peripheral insulin resistance and impaired insulin signaling contribute to abnormal glucose metabolism in preterm baboons. Endocrinology 2015, 156, 813–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Nakagawa, Y.; Kojima, I.; Shibata, H. Prolonged insulin stimulation down-regulates GLUT4 through oxidative stress-mediated retromer inhibition by a protein kinase CK2-dependent mechanism in 3T3-L1 adipocytes. J. Biol. Chem. 2014, 289, 133–142. [Google Scholar] [CrossRef] [Green Version]
- Campa, C.C.; Ciraolo, E.; Ghigo, A.; Germena, G.; Hirsch, E. Crossroads of PI3K and Rac pathways. Small GTPases 2015, 6, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Henriksen, E.J.; Diamond-Stanic, M.K.; Marchionne, E.M. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic. Biol. Med. 2011, 51, 993–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, K.-H.; Wang, W.-J.; Lin, C.-W.; Pai, P.; Lai, T.-Y.; Tsai, C.-Y.; Kuo, W.-W. NADPH oxidase-derived superoxide anion-induced apoptosis is mediated via the JNK-dependent activation of NF-κB in cardiomyocytes exposed to high glucose. J. Cell. Physiol. 2012, 227, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Grattagliano, I.; de Bari, O.; Bernardo, T.C.; Oliveira, P.J.; Wang, D.Q.-H.; Portincasa, P. Role of mitochondria in nonalcoholic fatty liver disease--from origin to propagation. Clin. Biochem. 2012, 45, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.H.; Swerdlow, R.H.; Khan, E.M.; Iezzoni, J.C.; Hespenheide, E.E.; Parks, J.K.; Parker, W.D.J. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J. Hepatol. 1999, 31, 430–434. [Google Scholar] [CrossRef]
- Eaton, S.; Zaitoun, A.M.; Record, C.O.; Bartlett, K. beta-Oxidation in human alcoholic and non-alcoholic hepatic steatosis. Clin. Sci. 1996, 90, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Massart, J.; Robin, M.-A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef]
- Cusi, K. Role of Insulin Resistance and Lipotoxicity in Non-Alcoholic Steatohepatitis. Clin. Liver Dis. 2009, 13, 545–563. [Google Scholar] [CrossRef]
- Silva, M.F.B.; Aires, C.C.P.; Luis, P.B.M.; Ruiter, J.P.N.; IJlst, L.; Duran, M.; Wanders, R.J.A.; Tavares de Almeida, I. Valproic acid metabolism and its effects on mitochondrial fatty acid oxidation: A review. J. Inherit. Metab. Dis. 2008, 31, 205–216. [Google Scholar] [CrossRef]
- Ding, R.-B.; Bao, J.; Deng, C.-X. Emerging roles of SIRT1 in fatty liver diseases. Int. J. Biol. Sci. 2017, 13, 852–867. [Google Scholar] [CrossRef]
- Li, M.; Guo, K.; Vanella, L.; Taketani, S.; Adachi, Y.; Ikehara, S. Stem cell transplantation upregulates Sirt1 and antioxidant expression, ameliorating fatty liver in type 2 diabetic mice. Int. J. Biol. Sci. 2015, 11, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, A.A.; Choudhury, M.; Rahman, S.M.; McCurdy, C.E.; Friederich, M.; Van Hove, J.L.K.; Watson, P.A.; Birdsey, N.; Bao, J.; Gius, D.; et al. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem. J. 2011, 433, 505–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gariani, K.; Menzies, K.J.; Ryu, D.; Wegner, C.J.; Wang, X.; Ropelle, E.R.; Moullan, N.; Zhang, H.; Perino, A.; Lemos, V.; et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 2016, 63, 1190–1204. [Google Scholar] [CrossRef]
- Tan, M.; Mosaoa, R.; Graham, G.T.; Kasprzyk-Pawelec, A.; Gadre, S.; Parasido, E.; Catalina-Rodriguez, O.; Foley, P.; Giaccone, G.; Cheema, A.; et al. Inhibition of the mitochondrial citrate carrier, Slc25a1, reverts steatosis, glucose intolerance, and inflammation in preclinical models of NAFLD/NASH. Cell Death Differ. 2020, 27, 2143–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Zhao, H. Role of Carnitine in Non-alcoholic Fatty Liver Disease and Other Related Diseases: An Update. Front. Med. 2021, 8, 689042. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.-X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Patterson, R.E.; Kalavalapalli, S.; Williams, C.M.; Nautiyal, M.; Mathew, J.T.; Martinez, J.; Reinhard, M.K.; McDougall, D.J.; Rocca, J.R.; Yost, R.A.; et al. Lipotoxicity in steatohepatitis occurs despite an increase in tricarboxylic acid cycle activity. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E484–E494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 14205–14218. [Google Scholar] [CrossRef]
- Cheng, Z.; Tseng, Y.; White, M.F. Insulin signaling meets mitochondria in metabolism. Trends Endocrinol. Metab. 2010, 21, 589–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maassen, J.A.; ’T Hart, L.M.; Van Essen, E.; Heine, R.J.; Nijpels, G.; Jahangir Tafrechi, R.S.; Raap, A.K.; Janssen, G.M.C.; Lemkes, H.H.P.J. Mitochondrial diabetes: Molecular mechanisms and clinical presentation. Diabetes 2004, 53 (Suppl. S1), S103–S109. [Google Scholar] [CrossRef] [Green Version]
- Chalasani, N.; Gorski, J.C.; Asghar, M.S.; Asghar, A.; Foresman, B.; Hall, S.D.; Crabb, D.W. Hepatic cytochrome P450 2E1 activity in nondiabetic patients with nonalcoholic steatohepatitis. Hepatology 2003, 37, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Weltman, M.D.; Farrell, G.C.; Liddle, C. Increased hepatocyte CYP2E1 expression in a rat nutritional model of hepatic steatosis with inflammation. Gastroenterology 1996, 111, 1645–1653. [Google Scholar] [CrossRef]
- Varela, N.M.; Quiñones, L.A.; Orellana, M.; Poniachik, J.; Csendes, A.; Smok, G.; Rodrigo, R.; Cáceres, D.D.; Videla, L.A. Study of cytochrome P450 2E1 and its allele variants in liver injury of nondiabetic, nonalcoholic steatohepatitis obese women. Biol. Res. 2008, 41, 81–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caballero, F.; Fernández, A.; Matías, N.; Martínez, L.; Fucho, R.; Elena, M.; Caballeria, J.; Morales, A.; Fernández-Checa, J.C.; García-Ruiz, C. Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: Impact on mitochondrial S-adenosyl-L-methionine and glutathione. J. Biol. Chem. 2010, 285, 18528–18536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobili, V.; Donati, B.; Panera, N.; Vongsakulyanon, A.; Alisi, A.; Dallapiccola, B.; Valenti, L. A 4-polymorphism risk score predicts steatohepatitis in children with nonalcoholic fatty liver disease. J. Pediatr. Gastroenterol. Nutr. 2014, 58, 632–636. [Google Scholar] [CrossRef]
- Reynolds, K.; He, J. Epidemiology of the metabolic syndrome. Am. J. Med. Sci. 2005, 330, 273–279. [Google Scholar] [CrossRef]
- Grundy, S.M.; Cleeman, J.I.; Daniels, S.R.; Donato, K.A.; Eckel, R.H.; Franklin, B.A.; Gordon, D.J.; Krauss, R.M.; Savage, P.J.; Smith, S.C.J.; et al. Diagnosis and management of the metabolic syndrome: An American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 2005, 112, 2735–2752. [Google Scholar] [CrossRef] [Green Version]
- Reaven, G. Why a cluster is truly a cluster: Insulin resistance and cardiovascular disease. Clin. Chem. 2008, 54, 785–787. [Google Scholar] [CrossRef]
- Kashyap, S.R.; Defronzo, R.A. The insulin resistance syndrome: Physiological considerations. Diabetes Vasc. Dis. Res. 2007, 4, 13–19. [Google Scholar] [CrossRef]
- Kahn, B.B.; Flier, J.S. Obesity and insulin resistance. J. Clin. Invest. 2000, 106, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Katagiri, H.; Yamada, T.; Oka, Y. Adiposity and cardiovascular disorders: Disturbance of the regulatory system consisting of humoral and neuronal signals. Circ. Res. 2007, 101, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Sarafidis, P.A.; Bakris, G.L. Review: Insulin and endothelin: An interplay contributing to hypertension development? J. Clin. Endocrinol. Metab. 2007, 92, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Soresi, M.; Giannitrapani, L.; Noto, D.; Terranova, A.; Campagna, M.E.; Cefalù, A.B.; Giammanco, A.; Montalto, G. Effects of steatosis on hepatic hemodynamics in patients with metabolic syndrome. Ultrasound Med. Biol. 2015, 41, 1545–1552. [Google Scholar] [CrossRef]
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Bugianesi, E.; Lenzi, M.; McCullough, A.J.; Natale, S.; Forlani, G.; Melchionda, N. Nonalcoholic fatty liver disease: A feature of the metabolic syndrome. Diabetes 2001, 50, 1844–1850. [Google Scholar] [CrossRef] [Green Version]
- Kotronen, A.; Westerbacka, J.; Bergholm, R.; Pietiläinen, K.H.; Yki-Järvinen, H. Liver fat in the metabolic syndrome. J. Clin. Endocrinol. Metab. 2007, 92, 3490–3497. [Google Scholar] [CrossRef]
- Hamaguchi, M.; Kojima, T.; Takeda, N.; Nakagawa, T.; Taniguchi, H.; Fujii, K.; Omatsu, T.; Nakajima, T.; Sarui, H.; Shimazaki, M.; et al. The metabolic syndrome as a predictor of nonalcoholic fatty liver disease. Ann. Intern. Med. 2005, 143, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Sanyal, A.J.; George, J. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef] [PubMed]
- EASL; EASD; EASO. EASL-EASD-EASO Clinical Practice Guidelines for the Management of Non-Alcoholic Fatty Liver Disease. Obes. Facts 2016, 9, 65–90. [Google Scholar] [CrossRef] [Green Version]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Sinn, D.H.; Gwak, G.-Y.; Park, H.N.; Kim, J.E.; Min, Y.W.; Kim, K.M.; Kim, Y.J.; Choi, M.S.; Lee, J.H.; Koh, K.C.; et al. Ultrasonographically detected non-alcoholic fatty liver disease is an independent predictor for identifying patients with insulin resistance in non-obese, non-diabetic middle-aged Asian adults. Am. J. Gastroenterol. 2012, 107, 561–567. [Google Scholar] [CrossRef]
- Jeong, S.-K.; Kim, Y.-K.; Park, J.-W.; Shin, Y.-J.; Kim, D.-S. Impact of visceral fat on the metabolic syndrome and nonalcoholic fatty liver disease. J. Korean Med. Sci. 2008, 23, 789–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eguchi, Y.; Eguchi, T.; Mizuta, T.; Ide, Y.; Yasutake, T.; Iwakiri, R.; Hisatomi, A.; Ozaki, I.; Yamamoto, K.; Kitajima, Y.; et al. Visceral fat accumulation and insulin resistance are important factors in nonalcoholic fatty liver disease. J. Gastroenterol. 2006, 41, 462–469. [Google Scholar] [CrossRef]
- Petersen, K.F.; Dufour, S.; Savage, D.B.; Bilz, S.; Solomon, G.; Yonemitsu, S.; Cline, G.W.; Befroy, D.; Zemany, L.; Kahn, B.B.; et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 12587–12594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef]
- Mittendorfer, B.; Magkos, F.; Fabbrini, E.; Mohammed, B.S.; Klein, S. Relationship between body fat mass and free fatty acid kinetics in men and women. Obesity 2009, 17, 1872–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, K.; Wildman, R.P. Update on the metabolic syndrome: Hypertension. Curr. Hypertens. Rep. 2009, 11, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Diehl, A.M. Hepatic triglyceride synthesis and nonalcoholic fatty liver disease. Curr. Opin. Lipidol. 2008, 19, 295–300. [Google Scholar] [CrossRef]
- Parker, R. The role of adipose tissue in fatty liver diseases. Liver Res. 2018, 2, 35–42. [Google Scholar] [CrossRef]
- Sharma, M.; Mitnala, S.; Vishnubhotla, R.K.; Mukherjee, R.; Reddy, D.N.; Rao, P.N. The Riddle of Nonalcoholic Fatty Liver Disease: Progression From Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis. J. Clin. Exp. Hepatol. 2015, 5, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Calder, P.C. Polyunsaturated fatty acids, inflammatory processes and inflammatory bowel diseases. Mol. Nutr. Food Res. 2008, 52, 885–897. [Google Scholar] [CrossRef]
- Haffner, S.M. Insulin resistance, inflammation, and the prediabetic state. Am. J. Cardiol. 2003, 92, 18J–26J. [Google Scholar] [CrossRef]
- Rogero, M.M.; Calder, P.C. Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids. Nutrients 2018, 10, 432. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.K.; O’Brien, M.; Mau, T.; Yung, R. Toll-like receptor 4 (TLR4) deficient mice are protected from adipose tissue inflammation in aging. Aging 2017, 9, 1971–1982. [Google Scholar] [CrossRef] [Green Version]
- Reyna, S.M.; Ghosh, S.; Tantiwong, P.; Meka, C.S.R.; Eagan, P.; Jenkinson, C.P.; Cersosimo, E.; Defronzo, R.A.; Coletta, D.K.; Sriwijitkamol, A.; et al. Elevated toll-like receptor 4 expression and signaling in muscle from insulin-resistant subjects. Diabetes 2008, 57, 2595–2602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Könner, A.C.; Brüning, J.C. Toll-like receptors: Linking inflammation to metabolism. Trends Endocrinol. Metab. 2011, 22, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Fernández Rodríguez, C.M.; Aller, R.; Gutiérrez García, M.L.; Ampuero, J.; Gómez-Camarero, J.; Martín-Mateos, R.M.; Burgos-Santamaría, D.; Rosales, J.M.; Aspichueta, P.; Buque, X.; et al. Higher levels of serum uric acid influences hepatic damage in patients with non-alcoholic fatty liver disease (NAFLD). Rev. Esp. Enfermedades Dig. Organo Of. la Soc. Esp. Patol. Dig. 2019, 111, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Castaño, G.O.; Scian, R.; Fernández Gianotti, T.; Dopazo, H.; Rohr, C.; Gaj, G.; San Martino, J.; Sevic, I.; Flichman, D.; et al. Serum aminotransferases in nonalcoholic fatty liver disease are a signature of liver metabolic perturbations at the amino acid and Krebs cycle level. Am. J. Clin. Nutr. 2016, 103, 422–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacon, B.R.; Farahvash, M.J.; Janney, C.G.; Neuschwander-Tetri, B.A. Nonalcoholic steatohepatitis: An expanded clinical entity. Gastroenterology 1994, 107, 1103–1109. [Google Scholar] [CrossRef]
- Hagström, H.; Nasr, P.; Bottai, M.; Ekstedt, M.; Kechagias, S.; Hultcrantz, R.; Stål, P. Elevated serum ferritin is associated with increased mortality in non-alcoholic fatty liver disease after 16 years of follow-up. Liver Int. Off. J. Int. Assoc. Study Liver 2016, 36, 1688–1695. [Google Scholar] [CrossRef]
- Yokota, S.; Oda, T.; Fahimi, H.D. The role of 15-lipoxygenase in disruption of the peroxisomal membrane and in programmed degradation of peroxisomes in normal rat liver. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2001, 49, 613–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dongiovanni, P.; Fracanzani, A.L.; Fargion, S.; Valenti, L. Iron in fatty liver and in the metabolic syndrome: A promising therapeutic target. J. Hepatol. 2011, 55, 920–932. [Google Scholar] [CrossRef]
- Cornejo, P.; Varela, P.; Videla, L.A.; Fernández, V. Chronic iron overload enhances inducible nitric oxide synthase expression in rat liver. Nitric Oxide Biol. Chem. 2005, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Minamiyama, Y.; Takemura, S.; Kodai, S.; Shinkawa, H.; Tsukioka, T.; Ichikawa, H.; Naito, Y.; Yoshikawa, T.; Okada, S. Iron restriction improves type 2 diabetes mellitus in Otsuka Long-Evans Tokushima fatty rats. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1140–E1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palermo, A.; Maggi, D.; Maurizi, A.R.; Pozzilli, P.; Buzzetti, R. Prevention of type 2 diabetes mellitus: Is it feasible? Diabetes. Metab. Res. Rev. 2014, 30 (Suppl. S1), 4–12. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [Green Version]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.W.M.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916. [Google Scholar] [CrossRef]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Mattace Raso, G.; Simeoli, R.; Russo, R.; Iacono, A.; Santoro, A.; Paciello, O.; Ferrante, M.C.; Canani, R.B.; Calignano, A.; Meli, R. Effects of sodium butyrate and its synthetic amide derivative on liver inflammation and glucose tolerance in an animal model of steatosis induced by high fat diet. PLoS ONE 2013, 8, e68626. [Google Scholar] [CrossRef] [Green Version]
- Mitra, S.; De, A.; Chowdhury, A. Epidemiology of non-alcoholic and alcoholic fatty liver diseases. Transl. Gastroenterol. Hepatol. 2020, 5, 16. [Google Scholar] [CrossRef]
- Subichin, M.; Clanton, J.; Makuszewski, M.; Bohon, A.; Zografakis, J.G.; Dan, A. Liver disease in the morbidly obese: A review of 1000 consecutive patients undergoing weight loss surgery. Surg. Obes. Relat. Dis. Off. J. Am. Soc. Bariatr. Surg. 2015, 11, 137–141. [Google Scholar] [CrossRef]
- Van Vliet-Ostaptchouk, J.V.; Nuotio, M.-L.; Slagter, S.N.; Doiron, D.; Fischer, K.; Foco, L.; Gaye, A.; Gögele, M.; Heier, M.; Hiekkalinna, T.; et al. The prevalence of metabolic syndrome and metabolically healthy obesity in Europe: A collaborative analysis of ten large cohort studies. BMC Endocr. Disord. 2014, 14, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angulo, P.; Keach, J.C.; Batts, K.P.; Lindor, K.D. Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology 1999, 30, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- Moretto, M.; Kupski, C.; Mottin, C.C.; Repetto, G.; Garcia Toneto, M.; Rizzolli, J.; Berleze, D.; de Souza Brito, C.L.; Casagrande, D.; Colossi, F. Hepatic steatosis in patients undergoing bariatric surgery and its relationship to body mass index and co-morbidities. Obes. Surg. 2003, 13, 622–624. [Google Scholar] [CrossRef]
- Andersen, T.; Christoffersen, P.; Gluud, C. The liver in consecutive patients with morbid obesity: A clinical, morphological, and biochemical study. Int. J. Obes. 1984, 8, 107–115. [Google Scholar] [PubMed]
- Lotta, L.A.; Gulati, P.; Day, F.R.; Payne, F.; Ongen, H.; van de Bunt, M.; Gaulton, K.J.; Eicher, J.D.; Sharp, S.J.; Luan, J.; et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat. Genet. 2017, 49, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Das, K.; Mukherjee, P.S.; Ghosh, A.; Ghosh, S.; Mridha, A.R.; Dhibar, T.; Bhattacharya, B.; Bhattacharya, D.; Manna, B.; et al. Nonobese population in a developing country has a high prevalence of nonalcoholic fatty liver and significant liver disease. Hepatology 2010, 51, 1593–1602. [Google Scholar] [CrossRef]
- Godoy-Matos, A.F.; Silva Júnior, W.S.; Valerio, C.M. NAFLD as a continuum: From obesity to metabolic syndrome and diabetes. Diabetol. Metab. Syndr. 2020, 12, 60. [Google Scholar] [CrossRef]
- Pellegrinelli, V.; Carobbio, S.; Vidal-Puig, A. Adipose tissue plasticity: How fat depots respond differently to pathophysiological cues. Diabetologia 2016, 59, 1075–1088. [Google Scholar] [CrossRef] [Green Version]
- Chiyanika, C.; Wong, V.W.-S.; Wong, G.L.-H.; Chan, H.L.-Y.; Hui, S.C.N.; Yeung, D.K.W.; Chu, W.C.W. Implications of Abdominal Adipose Tissue Distribution on Nonalcoholic Fatty Liver Disease and Metabolic Syndrome: A Chinese General Population Study. Clin. Transl. Gastroenterol. 2021, 12, e00300. [Google Scholar] [CrossRef]
- Asrih, M.; Jornayvaz, F.R. Inflammation as a potential link between nonalcoholic fatty liver disease and insulin resistance. J. Endocrinol. 2013, 218, R25–R36. [Google Scholar] [CrossRef]
- Holland, W.L.; Miller, R.A.; Wang, Z.V.; Sun, K.; Barth, B.M.; Bui, H.H.; Davis, K.E.; Bikman, B.T.; Halberg, N.; Rutkowski, J.M.; et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat. Med. 2011, 17, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Lang, C.H.; Dobrescu, C.; Bagby, G.J. Tumor necrosis factor impairs insulin action on peripheral glucose disposal and hepatic glucose output. Endocrinology 1992, 130, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Uysal, K.T.; Becherer, J.D.; Arner, P.; Hotamisligil, G.S. Altered Tumor Necrosis Factor-α (TNF-α) Processing in Adipocytes and Increased Expression of Transmembrane TNF-α in Obesity. Diabetes 2002, 51, 1876–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arner, P. The adipocyte in insulin resistance: Key molecules and the impact of the thiazolidinediones. Trends Endocrinol. Metab. 2003, 14, 137–145. [Google Scholar] [CrossRef]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-α function. Nature 1997, 389, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Wieckowska, A.; Papouchado, B.G.; Li, Z.; Lopez, R.; Zein, N.N.; Feldstein, A.E. Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2008, 103, 1372–1379. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Itoh, Y.; Yokomizo, C.; Nishimura, T.; Niimi, T.; Fujii, H.; Okanoue, T.; Yoshikawa, T. Blockade of interleukin-6 signaling enhances hepatic steatosis but improves liver injury in methionine choline-deficient diet-fed mice. Lab. Investig. 2010, 90, 1169–1178. [Google Scholar] [CrossRef]
- Honda, H.; Ikejima, K.; Hirose, M.; Yoshikawa, M.; Lang, T.; Enomoto, N.; Kitamura, T.; Takei, Y.; Sato, N. Leptin is required for fibrogenic responses induced by thioacetamide in the murine liver. Hepatology 2002, 36, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Margetic, S.; Gazzola, C.; Pegg, G.G.; Hill, R.A. Leptin: A review of its peripheral actions and interactions. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 2002, 26, 1407–1433. [Google Scholar] [CrossRef] [Green Version]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism 2019, 92, 82–97. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin resistance: Review of the underlying molecular mechanisms. J. Cell. Physiol. 2019, 234, 8152–8161. [Google Scholar] [CrossRef]
- Mikolasevic, I.; Milic, S.; Turk Wensveen, T.; Grgic, I.; Jakopcic, I.; Stimac, D.; Wensveen, F.; Orlic, L. Nonalcoholic fatty liver disease—A multisystem disease? World J. Gastroenterol. 2016, 22, 9488–9505. [Google Scholar] [CrossRef]
- Dewidar, B.; Kahl, S.; Pafili, K.; Roden, M. Metabolic liver disease in diabetes—From mechanisms to clinical trials. Metabolism. 2020, 111S, 154299. [Google Scholar] [CrossRef]
- Bazick, J.; Donithan, M.; Neuschwander-Tetri, B.A.; Kleiner, D.; Brunt, E.M.; Wilson, L.; Doo, E.; Lavine, J.; Tonascia, J.; Loomba, R. Clinical Model for NASH and Advanced Fibrosis in Adult Patients With Diabetes and NAFLD: Guidelines for Referral in NAFLD. Diabetes Care 2015, 38, 1347–1355. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef]
- Lonardo, A.; Ballestri, S.; Marchesini, G.; Angulo, P.; Loria, P. Nonalcoholic fatty liver disease: A precursor of the metabolic syndrome. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2015, 47, 181–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonardo, A.; Bellentani, S.; Argo, C.K.; Ballestri, S.; Byrne, C.D.; Caldwell, S.H.; Cortez-Pinto, H.; Grieco, A.; Machado, M.V.; Miele, L.; et al. Epidemiological modifiers of non-alcoholic fatty liver disease: Focus on high-risk groups. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2015, 47, 997–1006. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Ye, L.; Liu, A.; Wen, S.W.; Deng, J.; Wu, X.; Lai, Z. Prevalence of nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus: A meta-analysis. Medicine 2017, 96, e8179. [Google Scholar] [CrossRef] [PubMed]
- Doycheva, I.; Cui, J.; Nguyen, P.; Costa, E.A.; Hooker, J.; Hofflich, H.; Bettencourt, R.; Brouha, S.; Sirlin, C.B.; Loomba, R. Non-invasive screening of diabetics in primary care for NAFLD and advanced fibrosis by MRI and MRE. Aliment. Pharmacol. Ther. 2016, 43, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Kwok, R.; Choi, K.C.; Wong, G.L.-H.; Zhang, Y.; Chan, H.L.-Y.; Luk, A.O.-Y.; Shu, S.S.-T.; Chan, A.W.-H.; Yeung, M.-W.; Chan, J.C.-N.; et al. Screening diabetic patients for non-alcoholic fatty liver disease with controlled attenuation parameter and liver stiffness measurements: A prospective cohort study. Gut 2016, 65, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Castera, L.; Vilgrain, V.; Angulo, P. Noninvasive evaluation of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D.; Szczepaniak, L.S.; Dobbins, R.; Nuremberg, P.; Horton, J.D.; Cohen, J.C.; Grundy, S.M.; Hobbs, H.H. Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology 2004, 40, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, M.; Franzén, L.E.; Mathiesen, U.L.; Thorelius, L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006, 44, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Rhee, E.J. Nonalcoholic Fatty Liver Disease and Diabetes: An Epidemiological Perspective. Endocrinol. Metab. 2019, 34, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Hazlehurst, J.M.; Woods, C.; Marjot, T.; Cobbold, J.F.; Tomlinson, J.W. Non-alcoholic fatty liver disease and diabetes. Metabolism. 2016, 65, 1096–1108. [Google Scholar] [CrossRef] [Green Version]
- Wild, S.H.; Morling, J.R.; McAllister, D.A.; Kerssens, J.; Fischbacher, C.; Parkes, J.; Roderick, P.J.; Sattar, N.; Byrne, C.D. Type 2 diabetes and risk of hospital admission or death for chronic liver diseases. J. Hepatol. 2016, 64, 1358–1364. [Google Scholar] [CrossRef] [Green Version]
- Lonardo, A.; Nascimbeni, F.; Mantovani, A.; Targher, G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J. Hepatol. 2018, 68, 335–352. [Google Scholar] [CrossRef] [PubMed]
- McPherson, S.; Hardy, T.; Henderson, E.; Burt, A.D.; Day, C.P.; Anstee, Q.M. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J. Hepatol. 2015, 62, 1148–1155. [Google Scholar] [CrossRef]
- Adams, L.A.; Waters, O.R.; Knuiman, M.W.; Elliott, R.R.; Olynyk, J.K. NAFLD as a risk factor for the development of diabetes and the metabolic syndrome: An eleven-year follow-up study. Am. J. Gastroenterol. 2009, 104, 861–867. [Google Scholar] [CrossRef]
- Xia, M.-F.; Bian, H.; Gao, X. NAFLD and Diabetes: Two Sides of the Same Coin? Rationale for Gene-Based Personalized NAFLD Treatment. Front. Pharmacol. 2019, 10, 877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fracanzani, A.L.; Valenti, L.; Bugianesi, E.; Andreoletti, M.; Colli, A.; Vanni, E.; Bertelli, C.; Fatta, E.; Bignamini, D.; Marchesini, G.; et al. Risk of severe liver disease in nonalcoholic fatty liver disease with normal aminotransferase levels: A role for insulin resistance and diabetes. Hepatology 2008, 48, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.-F.; Bian, H.; Yan, H.-M.; Lin, H.-D.; Chang, X.-X.; Li, X.-M.; Ma, H.; He, W.-Y.; Zhao, N.-Q.; Xia, P.; et al. Assessment of liver fat content using quantitative ultrasonography to evaluate risks for metabolic diseases. Obesity 2015, 23, 1929–1937. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.N.A.; Maglio, C.; Pirazzi, C.; Burza, M.A.; Adiels, M.; Burch, L.; Donnelly, L.A.; Colhoun, H.; Doney, A.S.; Dillon, J.F.; et al. Paradoxical lower serum triglyceride levels and higher type 2 diabetes mellitus susceptibility in obese individuals with the PNPLA3 148M variant. PLoS ONE 2012, 7, e39362. [Google Scholar] [CrossRef] [PubMed]
- Dubuquoy, C.; Burnol, A.-F.; Moldes, M. PNPLA3, a genetic marker of progressive liver disease, still hiding its metabolic function? Clin. Res. Hepatol. Gastroenterol. 2013, 37, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.M. Subcutaneous and visceral adipose tissue: Structural and functional differences. Obes. Rev. an Off. J. Int. Assoc. Study Obes. 2010, 11, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Wróblewski, A.; Strycharz, J.; Świderska, E.; Balcerczyk, A.; Szemraj, J.; Drzewoski, J.; Śliwińska, A. Chronic and Transient Hyperglycemia Induces Changes in the Expression Patterns of IL6 and ADIPOQ Genes and Their Associated Epigenetic Modifications in Differentiating Human Visceral Adipocytes. Int. J. Mol. Sci. 2021, 22, 6964. [Google Scholar] [CrossRef]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbæk, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, Y.; An, W.; Song, J.; Zhang, Y.; Zhao, X. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J. Clin. Invest. 2019, 129, 546–555. [Google Scholar] [CrossRef]
- Lomonaco, R.; Bril, F.; Portillo-Sanchez, P.; Ortiz-Lopez, C.; Orsak, B.; Biernacki, D.; Lo, M.; Suman, A.; Weber, M.H.; Cusi, K. Metabolic Impact of Nonalcoholic Steatohepatitis in Obese Patients With Type 2 Diabetes. Diabetes Care 2016, 39, 632–638. [Google Scholar] [CrossRef] [Green Version]
- Erridge, C. Diet, commensals and the intestine as sources of pathogen-associated molecular patterns in atherosclerosis, type 2 diabetes and non-alcoholic fatty liver disease. Atherosclerosis 2011, 216, 1–6. [Google Scholar] [CrossRef]
- Firneisz, G. Non-alcoholic fatty liver disease and type 2 diabetes mellitus: The liver disease of our age? World J. Gastroenterol. 2014, 20, 9072–9089. [Google Scholar] [CrossRef] [Green Version]
- Sergi, D.; Naumovski, N.N.; Heilbronn, L.H.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe-Marsh, N.L.-M. Mitochondrial (Dys)function and Insulin Resistance: From Pathophysiological Molecular Mechanisms to the Impact of Diet. Front. Physiol. 2019, 10, 532. [Google Scholar] [CrossRef]
- Kim, D.; Touros, A.; Kim, W.R. Nonalcoholic Fatty Liver Disease and Metabolic Syndrome. Clin. Liver Dis. 2018, 22, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Mabalirajan, U.; Ghosh, B. Mitochondrial dysfunction in metabolic syndrome and asthma. J. Allergy 2013, 2013, 340476. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.-T.; Price, J.W., 3rd; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Invest. 2009, 119, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity--critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-J.; Wilso, D.M., 3rd. Overview of base excision repair biochemistry. Curr. Mol. Pharmacol. 2012, 5, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czarny, P.; Wigner, P.; Galecki, P.; Sliwinski, T. The interplay between inflammation, oxidative stress, DNA damage, DNA repair and mitochondrial dysfunction in depression. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2018, 80, 309–321. [Google Scholar] [CrossRef]
- Sampath, H.; Vartanian, V.; Rollins, M.R.; Sakumi, K.; Nakabeppu, Y.; Lloyd, R.S. 8-Oxoguanine DNA glycosylase (OGG1) deficiency increases susceptibility to obesity and metabolic dysfunction. PLoS ONE 2012, 7, e51697. [Google Scholar] [CrossRef]
- Vartanian, V.; Tumova, J.; Dobrzyn, P.; Dobrzyn, A.; Nakabeppu, Y.; Lloyd, R.S.; Sampath, H. 8-oxoguanine DNA glycosylase (OGG1) deficiency elicits coordinated changes in lipid and mitochondrial metabolism in muscle. PLoS ONE 2017, 12, e0181687. [Google Scholar] [CrossRef] [Green Version]
- Scheffler, K.; Rachek, L.; You, P.; Rowe, A.D.; Wang, W.; Kuśnierczyk, A.; Kittelsen, L.; Bjørås, M.; Eide, L. 8-oxoguanine DNA glycosylase (Ogg1) controls hepatic gluconeogenesis. DNA Repair 2018, 61, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Komakula, S.S.B.; Tumova, J.; Kumaraswamy, D.; Burchat, N.; Vartanian, V.; Ye, H.; Dobrzyn, A.; Lloyd, R.S.; Sampath, H. The DNA Repair Protein OGG1 Protects Against Obesity by Altering Mitochondrial Energetics in White Adipose Tissue. Sci. Rep. 2018, 8, 14886. [Google Scholar] [CrossRef]
- Komakula, S.S.B.; Blaze, B.; Ye, H.; Dobrzyn, A.; Sampath, H. A Novel Role for the DNA Repair Enzyme 8-Oxoguanine DNA Glycosylase in Adipogenesis. Int. J. Mol. Sci. 2021, 22, 1152. [Google Scholar] [CrossRef]
- Sampath, H.; Lloyd, R.S. Roles of OGG1 in transcriptional regulation and maintenance of metabolic homeostasis. DNA Repair 2019, 81, 102667. [Google Scholar] [CrossRef] [PubMed]
- Sampath, H.; Batra, A.K.; Vartanian, V.; Carmical, J.R.; Prusak, D.; King, I.B.; Lowell, B.; Earley, L.F.; Wood, T.G.; Marks, D.L.; et al. Variable penetrance of metabolic phenotypes and development of high-fat diet-induced adiposity in NEIL1-deficient mice. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E724–E734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vartanian, V.; Lowell, B.; Minko, I.G.; Wood, T.G.; Ceci, J.D.; George, S.; Ballinger, S.W.; Corless, C.L.; McCullough, A.K.; Lloyd, R.S. The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. Proc. Natl. Acad. Sci. USA 2006, 103, 1864–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, D.; Wei, C.; Chen, L.; Huang, J.; Yang, S.; Diehl, A.M. Oxidative DNA damage and DNA repair enzyme expression are inversely related in murine models of fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1070–G1077. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, P.-J.; Hsieh, T.-J.; Kuo, K.-K.; Hung, W.-W.; Tsai, K.-B.; Yang, C.-H.; Yu, M.-L.; Shin, S.-J. Pioglitazone retrieves hepatic antioxidant DNA repair in a mice model of high fat diet. BMC Mol. Biol. 2008, 9, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schults, M.A.; Nagle, P.W.; Rensen, S.S.; Godschalk, R.W.; Munnia, A.; Peluso, M.; Claessen, S.M.; Greve, J.W.; Driessen, A.; Verdam, F.J.; et al. Decreased nucleotide excision repair in steatotic livers associates with myeloperoxidase-immunoreactivity. Mutat. Res. 2012, 736, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Takumi, S.; Okamura, K.; Yanagisawa, H.; Sano, T.; Kobayashi, Y.; Nohara, K. The effect of a methyl-deficient diet on the global DNA methylation and the DNA methylation regulatory pathways. J. Appl. Toxicol. 2015, 35, 1550–1556. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, F.; Senese, R.; Lasala, P.; Ziello, A.; Mazzoli, A.; Crescenzo, R.; Liverini, G.; Lanni, A.; Goglia, F.; Iossa, S. Fructose-Rich Diet Affects Mitochondrial DNA Damage and Repair in Rats. Nutrients 2017, 9, 323. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, N.; Takahashi, S.; Hu, X.; Lu, Y.; Fujimori, N.; Golla, S.; Fang, Z.-Z.; Aoyama, T.; Krausz, K.W.; Gonzalez, F.J. Growth arrest and DNA damage-inducible 45α protects against nonalcoholic steatohepatitis induced by methionine- and choline-deficient diet. Biochim. Biophys. Acta. Mol. Basis Dis. 2017, 1863, 3170–3182. [Google Scholar] [CrossRef]
- Yang, X.; Chen, X.; Xia, C.; Li, S.; Zhu, L.; Xu, C. Comparative analysis of the expression profiles of genes related to the Gadd45α signaling pathway in four kinds of liver diseases. Histol. Histopathol. 2020, 35, 949–960. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Research Characteristics (Studied Groups, Diet) | Main Outcomes | Paper |
|---|---|---|
| 12-week old male ob/ob and WT (C57BL/6) mice. WT animals were fed: HFD with ethanol, HFD with dextrin maltose, MCD diet, or control liquid diet. ob/ob mice were fed: HFD (n = 6 in each cohort) | Mice fed MCD diet had: decreased MUTYH expression | Gao et al. 2004 [249] |
| Male and female Neil1−/−, Neil1+/− and WT (C57BL/6) mice | Neil1−/− and Neil1+/− mice had: severe obesity, dyslipidemia, hyperinsulinemia, and fatty liver | Vartanian et al. 2006 [236] |
| 8-weeks old male C57BL/6 mice fed chow, HFD, or HFD with pioglitazone 100 mg/kg/day for 8 weeks (n = 5 in each cohort) | Mice fed HFD had: hepatic steatosis, improved by pioglitazone; increased malondialdehyde concentration and 8-oxo-dG, attenuated by pioglitazone; decreased gene expression of OGG1 and MUTYH, reversed by pioglitazone | Hsiao et al. 2008 [250] |
| Male and female Neil1+/+ and Neil1−/− mice fed chow and HFD (n = 6–10 in each cohort) | Neil1−/− mice fed HFD had: increased body weight, higher fat accumulation, moderate to severe hepatic steatosis, upregulation of hepatic expression of inflammatory genes, glucose intolerance | Sampath et al. 2011 [235] |
| Livers of 35 severely obese male and female patients with steatosis without inflammation or with NASH (each group divided into low or high MPO activity) (n = 8–9 in each cohort) | Patients with high expression of MPO had: reduced damage recognition capacity, decreased NER capacity | Schults et al. 2012 [251] |
| 12-week old male Ogg1−/− (n = 6), Ogg1+/− (n = 6) and WT (n-= 6) mice (C57BL/6J), fed 10 weeks chow or HFD (n = 6 in each cohort) | Ogg1−/− mice fed HFD had: increased adiposity and hepatic steatosis, higher plasma insulin level, impaired glucose tolerance, higher respiratory exchange ratio, dysregulation of fatty acid oxidation and TCA metabolism genes expression, reduced hepatic glycogen stores, reduced fasting plasma ketones | Sampath et al. 2012 [241] |
| 6-week old male C57BL/6J mice fed chow or MCD diet for 1 week (n = 6 in each cohort) | MCD diet-fed mice had: hepatic steatosis; higher gene expression level of thymine-DNA glycosylase and APEX1 | Takumi et al. 2015 [252] |
| About 100-day old male Sprague-Dawley rats fed chow or a fructose-rich diet (n = 4–9 in each cohort) | Mice fed fructose-rich diet had: decreased gene expression of gamma DNA polymerase, reduced mtDNA copy number | Cioffi et al. 2017 [253] |
| 12-week old male Ogg1−/− (n = 6) mice and WT (n = 6) (C57BL/6J), fed 10 weeks chow diet (n = 6 in each cohort) | Ogg1−/− mice had: dysregulation of expression of genes involved in mitochondrial fission, fatty acids oxidation, fatty acid uptake, TCA and pyruvate metabolism; increased muscle lipid content; decreased grip strength and treadmill endurance | Vartanian et al. 2017 [242] |
| Male and female Ogg1−/− and WT mice (C57BL/6N) fed or subjected to fasting for 24 h (n = 6–8 in each cohort) | Ogg1−/− mice in the fed state had: hyperglycemia, elevated insulin levels, higher liver glycogen content, increased accumulation of 8oxoG in mtDNA, reduced ETC capacity, decreased activity of PDH | Scheffler et al. 2018 [243] |
| Age-matched male Ogg1Tg and WT mice (C57BL/6 J) fed ad libitum chow or HFD for 12 weeks (n = 6 in each cohort) | Ogg1Tg mice fed HFD had: protection from diet-induced obesity, IR, and inflammation of adipose tissue | Komakula et al. 2018 [244] |
| Preadipocytes from 4–12 weeks old male WT (C57BL/6J), Ogg1Tg and Ogg1−/− mice fed chow diet (n = 5–6 in each cohort) | Preadipocytes from Ogg1−/− mice were more differentiated and accumulated more lipids than WT mice; from Ogg1Tg had reduced differentiation and lipid content | Komakula et al. 2021 [245] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ziolkowska, S.; Binienda, A.; Jabłkowski, M.; Szemraj, J.; Czarny, P. The Interplay between Insulin Resistance, Inflammation, Oxidative Stress, Base Excision Repair and Metabolic Syndrome in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2021, 22, 11128. https://doi.org/10.3390/ijms222011128
Ziolkowska S, Binienda A, Jabłkowski M, Szemraj J, Czarny P. The Interplay between Insulin Resistance, Inflammation, Oxidative Stress, Base Excision Repair and Metabolic Syndrome in Nonalcoholic Fatty Liver Disease. International Journal of Molecular Sciences. 2021; 22(20):11128. https://doi.org/10.3390/ijms222011128
Chicago/Turabian StyleZiolkowska, Sylwia, Agata Binienda, Maciej Jabłkowski, Janusz Szemraj, and Piotr Czarny. 2021. "The Interplay between Insulin Resistance, Inflammation, Oxidative Stress, Base Excision Repair and Metabolic Syndrome in Nonalcoholic Fatty Liver Disease" International Journal of Molecular Sciences 22, no. 20: 11128. https://doi.org/10.3390/ijms222011128
APA StyleZiolkowska, S., Binienda, A., Jabłkowski, M., Szemraj, J., & Czarny, P. (2021). The Interplay between Insulin Resistance, Inflammation, Oxidative Stress, Base Excision Repair and Metabolic Syndrome in Nonalcoholic Fatty Liver Disease. International Journal of Molecular Sciences, 22(20), 11128. https://doi.org/10.3390/ijms222011128