
Dynamics of Choline-Containing Phospholipids in Traumatic Brain Injury and Associated Comorbidities


 ,
,  , ,
, ,  ,
,  , ,
, , 
Abstract
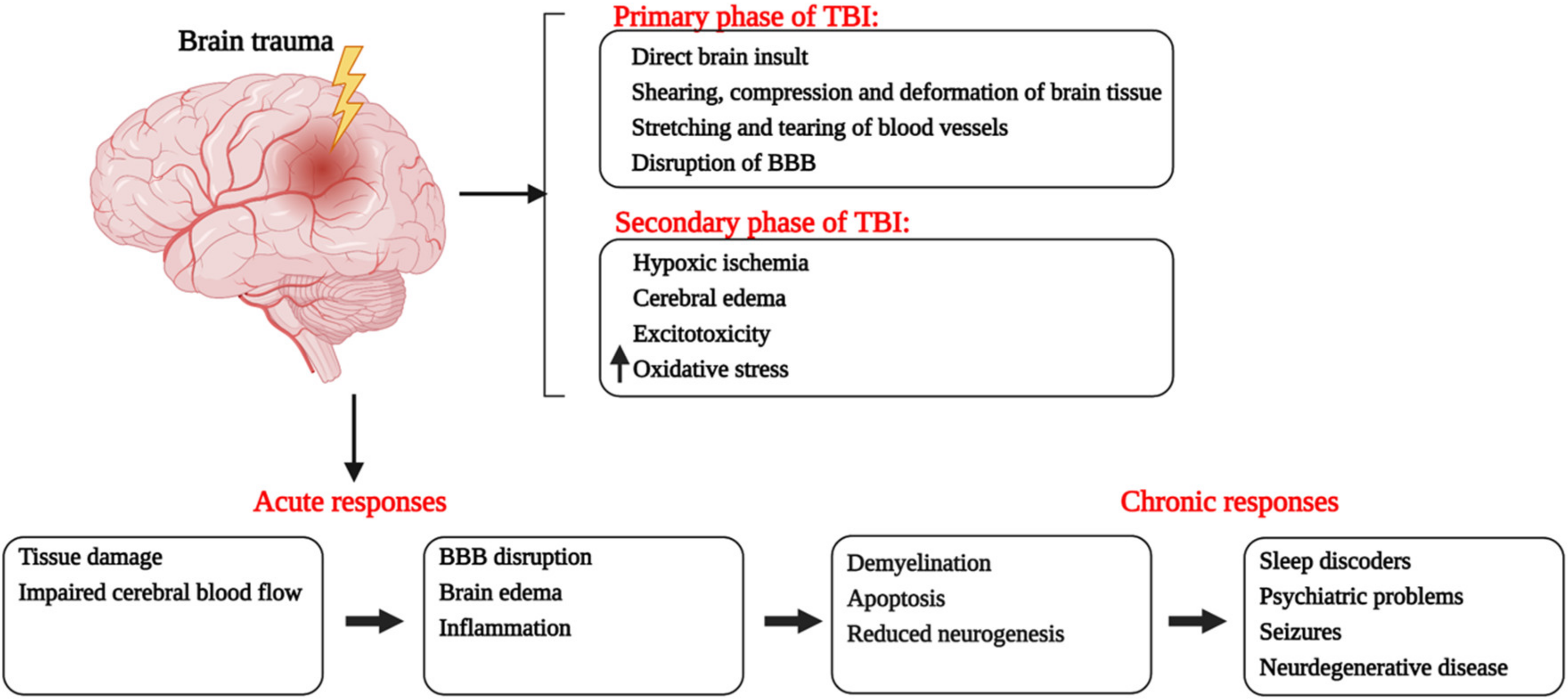
:1. Introduction
2. Neurobiological Significance of Lipids
3. TBI-Induced Pathophysiological Changes in Brain Phospholipids
4. Importance of Choline-Containing Phospholipids in Brain
5. Changes in Choline-Containing Phospholipids after TBI

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Preclinical Studies | ||||
| Animal | Brain Insult | Choline-Phospholipids Levels | Authors | Ref. |
| SD rats | Controlled cortical impact injury | ↓ PCh and GPC in the pericontusional zone at 2 and 4 h after injury | Xu et al. | [51] |
| SD rats | Controlled cortical impact injury | ↑ Free choline in surrounding of injured area after 24 h of injury | Scremin et al. | [45] |
| C57BL6 mice | Controlled cortical impact injury | ↓ cortical and cerebellar PC and SM ↑ hippocampal PC and SM after 3 months of injury | Abdullah et al. | [47] |
| C57BL6 mice | Closed head injury | ↓ plasma PC and lyso-PC after 3, 12 and 24 months of injury | Emmerich et al. | [49] |
| C57BL6 mice | Closed head injury | ↑ cortical and hippocampal PC, lyso-PC and SM after 24 h, and 3, 6, 9 and 12 months of injury | Ojo et al. | [48] |
| C57BL6 mice | Controlled cortical impact injury | ↑ SM in brains after 2 and 7 days of injury | Novgorodov et al. | [52] |
| C57BL6 mice | Controlled cortical impact injury | ↑ Lyso-PC in lysosomal membranes of injured cortices after 1 h f injury | Sarkar et al. | [53] |
| Sabra rats | Weight drop method | 75, 81, and 245% ↑ PLA2 activity after 15 min, 4 and 24 h of injury resulted in respective elevation of fatty acid release after aminocaproylphosphatidylcholine catalysis | Shohami et al. | [54] |
| Rats | Controlled cortical impact injury | ↑ PC in mid brain and thalamus after 14 days of injury | Li et al. | [55] |
| SD rats | Controlled cortical impact injury | ↑ PC and lyso-PC in white and grey matter after 1 and 3 h of injury | McDonald et al. | [56] |
| Clinical Studies | ||||
| Patients | Brain Injury | Observations | Authors | Ref. |
| 10 | Fall/vehicle crash | Highest lyso-PC on day 1 and highest PC on day 4 was detected in CSF | Pasvogel et al. | [50] |
| 40 | Vehicle accidents | ↑ regional choline/creatinine ratio estimated during 1–16 days after injury | Holshouser et al. | [57] |
| 26 | Accidental head injuries | ↑ choline/creatinine and ↓ NAA/choline ratios in white matter during 3–38 (mean 11 days) days after injury | Garnett et al. | [58] |
| 25 | Mild head injuries | ↑ NAA/choline ratio capsula interna and cerebral peduncles estimated during 1–20 days after injury | Kubas et al. | [59] |
| 45 | Fall/vehicle accidents | ↑ choline/creatinine and ↓ NAA/choline ratios during 6–12 months after injury | Holshouser et al. | [60] |
| 42 | Severe brain injuries | ↑ choline levels in occipital gray matter and parietal white matter after initial 7 days of injury | Eisele et al. | [61] |
| NA | Vehicle accidents | Highest PC within 24 h was found in CSF | Parsons et al. | [62] |
| 10 | Fall/vehicle accidents | ↑ choline ratios in central brain after 48–72 h of injury | Marino et al. | [63] |
| 8 | Severe brain injuries | ↑ choline/creatinine and ↓ NAA/choline ratios in occipital gray matter and parietal white matter after 5 months of injury | Yoon et al. | [64] |
5.1. Post-TBI Choline Changes during Subacute, Acute and Chronic Phases Evident from Neuroimaging


5.2. Post-TBI Alternation in the Central Cholinergic System
6. TBI-Associated Neurological Comorbidities
6.1. Alzheimer’s Disease (AD)
6.2. Parkinson’s Disease
6.3. Epilepsy
6.4. Depression
7. Choline-Specific Therapeutic Strategies for the Amelioration of TBI and Coexisting Neurological Diseases
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Nizamutdinov, D.; Shapiro, L.A. Overview of traumatic brain injury: An immunological context. Brain Sci. 2017, 7, 11. [Google Scholar] [CrossRef]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.-C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018, 130, 1080–1097. [Google Scholar] [CrossRef] [Green Version]
- Roozenbeek, B.; Maas, A.I.R.; Menon, D.K. Changing patterns in the epidemiology of traumatic brain injury. Nat. Rev. Neurol. 2013, 9, 231–236. [Google Scholar] [CrossRef]
- Adibhatla, R.M.; Hatcher, J.F. Role of lipids in brain injury and diseases. Future Lipidol. 2007, 2, 403–422. [Google Scholar] [CrossRef] [Green Version]
- Alqahtani, F.; Assiri, M.A.; Mohany, M.; Imran, I.; Javaid, S.; Rasool, M.F.; Shakeel, W.; Sivandzade, F.; Alanazi, A.Z.; Al-Rejaie, S.S.; et al. Coadministration of ketamine and perampanel improves behavioral function and reduces inflammation in acute traumatic brain injury mouse model. BioMed Res. Int. 2020, 2020, 3193725. [Google Scholar] [CrossRef]
- Graham, D.I. Neurpathology of Head Injury. Semin. Clin. Neuropsychiatry 1998, 3, 160–175. [Google Scholar]
- Armstead, W.M. Cerebral blood flow autoregulation and dysautoregulation. Anesthesiol. Clin. 2016, 34, 465–477. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeld, J.V.; Maas, A.I.; Bragge, P.; Morganti-Kossmann, M.C.; Manley, G.T.; Gruen, R.L. Early management of severe traumatic brain injury. Lancet 2012, 380, 1088–1098. [Google Scholar] [CrossRef]
- Thal, S.C.; Neuhaus, W. The blood–brain barrier as a target in traumatic brain injury treatment. Arch. Med. Res. 2014, 45, 698–710. [Google Scholar] [CrossRef]
- Guerriero, R.M.; Giza, C.C.; Rotenberg, A. Glutamate and GABA imbalance following traumatic brain injury. Curr. Neurol. Neurosci. Rep. 2015, 15, 27. [Google Scholar] [CrossRef]
- Weber, J.T. Altered calcium signaling following traumatic brain injury. Front. Pharmacol. 2012, 3, 60. [Google Scholar] [CrossRef] [Green Version]
- Slemmer, J.E.; Shacka, J.J.; Sweeney, M.I.; Weber, J.T. Antioxidants and free radical scavengers for the treatment of stroke, traumatic brain injury and aging. Curr. Med. Chem. 2008, 15, 404–414. [Google Scholar] [CrossRef] [Green Version]
- Gomez, D.; Byrne, J.P.; Alali, A.S.; Xiong, W.; Hoeft, C.; Neal, M.; Subacius, H.; Nathens, A.B. Inclusion of highest glasgow coma scale motor component score in mortality risk adjustment for benchmarking of trauma center performance. J. Am. Coll. Surg. 2017, 225, 755–762. [Google Scholar] [CrossRef]
- Richardson, R.N.; Sherer, M.; Seel, R.T.; Hart, T.; Hanks, R.; Arango-Lasprilla, J.C.; Yablon, S.A.; Sander, A.M.; Barnett, S.D.; Walker, W.C.; et al. Utility of post-traumatic amnesia in predicting 1-year productivity following traumatic brain injury: Comparison of the Russell and Mississippi PTA classification intervals. J. Neurol. Neurosurg. Psychiatry 2011, 82, 494–499. [Google Scholar] [CrossRef]
- Hussain, G.; Wang, J.; Rasul, A.; Anwar, H.; Imran, A.; Qasim, M.; Zafar, S.; Kamran, S.K.S.; Razzaq, A.; Aziz, N.; et al. Role of cholesterol and sphingolipids in brain development and neurological diseases. Lipids Health Dis. 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Bruce, K.D.; Zsombok, A.; Eckel, R.H. Lipid processing in the brain: A key regulator of systemic metabolism. Front. Endocrinol. 2017, 8, 60. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Liu, Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell 2015, 6, 254–264. [Google Scholar] [CrossRef] [Green Version]
- Svennerholm, L.; Boström, K.; Jungbjer, B.; Olsson, L. Membrane lipids of adult human brain: Lipid composition of frontal and temporal lobe in subjects of age 20 to 100 years. J. Neurochem. 1994, 63, 1802–1811. [Google Scholar] [CrossRef]
- Olsen, A.S.B.; Færgeman, N.J. Sphingolipids: Membrane microdomains in brain development, function and neurological diseases. Open Biol. 2017, 7, 170069. [Google Scholar] [CrossRef] [Green Version]
- Naudí, A.; Cabré, R.; Jové, M.; Ayala, V.; Gonzalo, H.; Portero-Otín, M.; Ferrer, I.; Pamplona, R. Lipidomics of human brain aging and Alzheimer’s disease pathology. Int. Rev. Neurobiol. 2015, 122, 133–189. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Horrocks, L.A.; Farooqui, T. Glycerophospholipids in brain: Their metabolism, incorporation into membranes, functions, and involvement in neurological disorders. Chem. Phys. Lipids 2000, 106, 1–29. [Google Scholar] [CrossRef]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The role of lipids in Parkinson’s disease. Cells 2019, 8, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazinet, R.P.; Layé, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.K.; Titsworth, W.; Xu, X.-M. Phospholipase A2 in CNS disorders: Implication on traumatic spinal cord and brain injuries. Handb. Neurochem. Mol. Neurobiol. 2009, 321–341. [Google Scholar] [CrossRef]
- Yui, K.; Imataka, G.; Nakamura, H.; Ohara, N.; Naito, Y. Eicosanoids derived from arachidonic acid and their family prostaglandins and cyclooxygenase in psychiatric disorders. Curr. Neuropharmacol. 2015, 13, 776–785. [Google Scholar] [CrossRef] [Green Version]
- Farooqui, A.A.; Ong, W.-Y.; Horrocks, L.A. Inhibitors of brain phospholipase A2 activity: Their neuropharmacological effects and therapeutic importance for the treatment of neurologic disorders. Pharmacol. Rev. 2006, 58, 591–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imran, I.; Hillert, M.H.; Klein, J. Early metabolic responses to lithium/pilocarpine-induced status epilepticus in rat brain. J. Neurochem. 2015, 135, 1007–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhang, W.; Hu, H.; Wang, M.; Sheng, W.; Yao, H.; Ding, W.; Chen, Z.; Wei, E. Expression patterns of 5-lipoxygenase in human brain with traumatic injury and astrocytoma. Neuropathology 2006, 26, 99–106. [Google Scholar] [CrossRef]
- Bayir, H.; Kochanek, P.M.; Kagan, V.E. Oxidative stress in immature brain after traumatic brain injury. Dev. Neurosci. 2006, 28, 420–431. [Google Scholar] [CrossRef]
- Anthonymuthu, T.S.; Kenny, E.M.; Bayir, H. Therapies targeting lipid peroxidation in traumatic brain injury. Brain Res. 2016, 1640, 57–76. [Google Scholar] [CrossRef] [Green Version]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Angeli, J.P.F.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tayebati, S.K.; Amenta, F. Choline-containing phospholipids: Relevance to brain functional pathways. Clin. Chem. Lab. Med. 2013, 51, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Jabir, N.R.; Khan, F.R.; Tabrez, S. Cholinesterase targeting by polyphenols: A therapeutic approach for the treatment of Alzheimer’s disease. CNS Neurosci. Ther. 2018, 24, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Amenta, F.; Battineni, G.; Traini, E.; Pallotta, G. Choline-containing phospholipids and treatment of adult-onset dementia disorders. Diagn. Manag. Dement. 2020, 1, 477–493. [Google Scholar] [CrossRef]
- Schneider, N.; Hauser, J.; Oliveira, M.; Cazaubon, E.; Mottaz, S.C.; O’Neill, B.V.; Steiner, P.; Deoni, S.C.L. Sphingomyelin in brain and cognitive development: Preliminary data. eNeuro 2019, 6. [Google Scholar] [CrossRef] [PubMed]
- O’Muircheartaigh, J.; Dean, D.C.; Dirks, H.; Waskiewicz, N.; Lehman, K.; Jerskey, B.A.; Deoni, S.C.L. Interactions between white matter asymmetry and language during neurodevelopment. J. Neurosci. 2013, 33, 16170–16177. [Google Scholar] [CrossRef] [Green Version]
- Holthius, J.C.M.; Menon, A.K. Lipid landscape and pipelines in membrane homeostasis. Nature 2014, 510, 48–57. [Google Scholar] [CrossRef]
- Klein, J. Membrane breakdown in acute and chronic neurodegeneration: Focus on choline-containing phospholipids. J. Neural Transm. 2000, 107, 1027–1063. [Google Scholar] [CrossRef]
- Travers, J.B.; Rohan, J.G.; Sahu, R.P. New insights into the pathologic roles of the Platelet-Activating Factor system. Front. Endocrinol. 2021, 12, 624132. [Google Scholar] [CrossRef]
- Anthonymuthu, T.S.; Kenny, E.M.; Lamade, A.M.; Kagan, V.E.; Bayır, H. Oxidized phospholipid signaling in traumatic brain injury. Free Radic. Biol. Med. 2018, 124, 493–503. [Google Scholar] [CrossRef]
- Jenkins, G.M.; Frohman, M.A. Phospholipase D: A lipid centric review. Cell. Mol. Life Sci. 2005, 62, 2305–2316. [Google Scholar] [CrossRef]
- Young, M.M.; Kester, M.; Wang, H.-G. Sphingolipids: Regulators of crosstalk between apoptosis and autophagy. J. Lipid Res. 2013, 54, 5–19. [Google Scholar] [CrossRef] [Green Version]
- Scremin, O.U.; Jenden, D.J. Time-dependent changes in cerebral choline and acetylcholine induced by transient global ischemia in rats. Stroke 1991, 22, 643–647. [Google Scholar] [CrossRef] [Green Version]
- Scremin, O.U.; Li, M.G.; Roch, M.; Booth, R.; Jenden, D.J. Acetylcholine and choline dynamics provide early and late markers of traumatic brain injury. Brain Res. 2006, 1124, 155–166. [Google Scholar] [CrossRef]
- Homayoun, P.; Parkins, N.E.; Soblosky, J.; Carey, M.E.; De Turco, E.B.R.; Bazan, N.G. Cortical impact injury in rats promotes a rapid and sustained increase in polyunsaturated free fatty acids and diacylglycerols. Neurochem. Res. 2000, 25, 269–276. [Google Scholar] [CrossRef]
- Abdullah, L.; Evans, J.E.; Ferguson, S.; Mouzon, B.; Montague, H.; Reed, J.; Crynen, G.; Emmerich, T.; Crocker, M.; Pelot, R.; et al. Lipidomic analyses identify injury-specific phospholipid changes 3 mo after traumatic brain injury. FASEB J. 2014, 28, 5311–5321. [Google Scholar] [CrossRef]
- Ojo, J.O.; Algamal, M.; Leary, P.; Abdullah, L.; Mouzon, B.; Evans, J.E.; Mullan, M.; Crawford, F. Converging and differential brain phospholipid dysregulation in the pathogenesis of repetitive mild traumatic brain injury and Alzheimer’s Disease. Front. Neurosci. 2019, 13, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emmerich, T.; Abdullah, L.; Ojo, J.; Mouzon, B.; Nguyen, T.; Crynen, G.; Evans, J.E.; Reed, J.; Mullan, M.; Crawford, F. Mild TBI results in a long-term decrease in circulating phospholipids in a mouse model of injury. Neuromol. Med. 2017, 19, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Pasvogel, A.E.; Miketova, P.; Moore, I.M. Differences in CSF phospholipid concentration by traumatic brain injury outcome. Biol. Res. Nurs. 2010, 11, 325–331. [Google Scholar] [CrossRef]
- Xu, S.; Zhuo, J.; Racz, J.; Shi, D.; Roys, S.; Fiskum, G.; Gullapalli, R. Early microstructural and metabolic changes following controlled cortical impact injury in rat: A magnetic resonance imaging and spectroscopy study. J. Neurotrauma 2011, 28, 2091–2102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novgorodov, S.A.; Riley, C.L.; Yu, J.; Borg, K.T.; Hannun, Y.A.; Proia, R.L.; Kindy, M.S.; Gudz, T.I. Essential roles of neutral ceramidase and sphingosine in mitochondrial dysfunction due to traumatic brain injury. J. Biol. Chem. 2014, 289, 13142–13154. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, C.; Jones, J.W.; Hegdekar, N.; Thayer, J.A.; Kumar, A.; Faden, A.I.; Kane, M.A.; Lipinski, M.M. PLA2G4A/cPLA2-mediated lysosomal membrane damage leads to inhibition of autophagy and neurodegeneration after brain trauma. Autophagy 2019, 16, 466–485. [Google Scholar] [CrossRef] [Green Version]
- Shohami, E.; Shapira, Y.; Yadid, G.; Reisfeld, N.; Yedgar, S. Brain phospholipase A2 is activated after experimental closed head injury in the rat. J. Neurochem. 1989, 53, 1541–1546. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, Q.; Hu, E.; Wang, Y.; Lu, H. Quantitative mass spectrometry imaging of metabolomes and lipidomes for tracking changes and therapeutic response in traumatic brain injury surrounding injured area at chronic phase. ACS Chem. Neurosci. 2021, 12, 1363–1375. [Google Scholar] [CrossRef] [PubMed]
- McDonald, W.S.; Jones, E.E.; Wojciak, J.M.; Drake, R.R.; Sabbadini, R.A.; Harris, N.G. Matrix-assisted laser desorption ionization mapping of lysophosphatidic acid changes after traumatic brain injury and the relationship to cellular pathology. Am. J. Pathol. 2018, 188, 1779–1793. [Google Scholar] [CrossRef] [Green Version]
- Holshouser, B.A.; Tong, K.A.; Ashwal, S. Proton MR spectroscopic imaging depicts diffuse axonal injury in children with traumatic brain injury. Am. J. Neuroradiol. 2005, 26, 1276–1285. [Google Scholar]
- Garnett, M.R.; Blamire, A.M.; Rajagopalan, B.; Styles, P.; Cadoux-Hudson, T.A. Evidence for cellular damage in normal-appearing white matter correlates with injury severity in patients following traumatic brain injury: A magnetic resonance spectroscopy study. Brain 2000, 123, 1403–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubas, B.; Łebkowski, W.; Łebkowska, U.; Kułak, W.; Tarasow, E.; Walecki, J. Proton MR spectroscopy in mild traumatic brain injury. Polish J. Radiol. 2010, 75, 7–10. [Google Scholar]
- Holshouser, B.A.; Tong, K.A.; Ashwal, S.; Oyoyo, U.; Ghamsary, M.; Saunders, D.; Shutter, L. Prospective longitudinal proton magnetic resonance spectroscopic imaging in adult traumatic brain injury. J. Magn. Reson. Imaging 2006, 24, 33–40. [Google Scholar] [CrossRef]
- Eisele, A.; Hill-Strathy, M.; Michels, L.; Rauen, K. Magnetic resonance spectroscopy following mild traumatic brain injury: A systematic review and meta-analysis on the potential to detect posttraumatic neurodegeneration. Neurodegener. Dis. 2020, 20, 2–11. [Google Scholar] [CrossRef]
- Parsons, L.; Dupakova, M.; Miketova, P.; Hamilton, A. High performance liquid chromatography analysis of cerebrospinal fluid for selected phospholipids following severe traumatic brain injury. J. Neurotrauma 1997, 14, 784. [Google Scholar]
- Marino, S.; Zei, E.; Battaglini, M.; Vittori, C.; Buscalferri, A.; Bramanti, P.; Federico, A. Acute metabolic brain changes following traumatic brain injury and their relevance to clinical severity and outcome. J. Neurol. Neurosurg. Psychiatry 2007, 78, 501–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.J.; Lee, J.H.; Kim, S.T.; Chun, M.H. Evaluation of traumatic brain injured patients in correlation with functional status by localized 1H-MR spectroscopy. Clin. Rehabil. 2005, 19, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Haseler, L.J.; Arcinue, E.; Danielsen, E.R.; Bluml, S.; Ross, B.D. Evidence from proton magnetic resonance spectroscopy for a metabolic cascade of neuronal damage in shaken baby syndrome. Pediatrics 1997, 99, 4–14. [Google Scholar] [CrossRef]
- Stovell, M.G.; Yan, J.-L.; Sleigh, A.; Mada, M.O.; Carpenter, T.A.; Hutchinson, P.J.A.; Carpenter, K.L.H. Assessing metabolism and injury in acute human traumatic brain Injury with magnetic resonance spectroscopy: Current and future applications. Front. Neurol. 2017, 8, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podo, F. Tumour phospholipid metabolism. NMR Biomed. Int. J. Devoted Dev. Appl. Magn. Reson. In Vivo 1999, 12, 413–439. [Google Scholar] [CrossRef]
- Lin, A.P.; Liao, H.J.; Merugumala, S.K.; Prabhu, S.P.; Meehan, W.P.; Ross, B.D. Metabolic imaging of mild traumatic brain injury. Brain Imaging Behav. 2012, 6, 208–223. [Google Scholar] [CrossRef]
- Ashwal, S.; Holshouser, B.A.; Shu, S.K.; Simmons, P.L.; Perkin, R.M.; Tomasi, L.G.; Knierim, D.S.; Sheridan, C.; Craig, K.; Andrews, G.H.; et al. Predictive value of proton magnetic resonance spectroscopy in pediatric closed head injury. Pediatr. Neurol. 2000, 23, 114–125. [Google Scholar] [CrossRef]
- Shutter, L.; Tong, K.A.; Holshouser, B.A. Proton MRS in acute traumatic brain injury: Role for glutamate/glutamine and choline for outcome prediction. J. Neurotrauma 2004, 21, 1693–1705. [Google Scholar] [CrossRef]
- Friedman, S.D.; Brooks, W.M.; Jung, R.E.; Hart, B.L.; Yeo, R.A. Proton MR spectroscopic findings correspond to neuropsychological function in traumatic brain injury. Am. J. Neuroradiol. 1998, 19, 1979–1985. [Google Scholar]
- Ashwal, S.; Holshouser, B.; Tong, K.; Serna, T.; Osterdock, R.; Gross, M.; Kido, D. Proton spectroscopy detected myoinositol in children with traumatic brain injury. Pediatr. Res. 2004, 56, 630–638. [Google Scholar] [CrossRef] [Green Version]
- Ross, B.D.; Ernst, T.; Kreis, R.; Haseler, L.J.; Bayer, S.; Danielsen, E.; Blüml, S.; Shonk, T.; Mandigo, J.C.; Caton, W.; et al. 1H MRS in acute traumatic brain injury. J. Magn. Reson. Imaging 1998, 8, 829–840. [Google Scholar] [CrossRef]
- Mallah, K.; Quanico, J.; Trede, D.; Kobeissy, F.; Zibara, K.; Salzet, M.; Fournier, I. Lipid changes associated with traumatic brain injury revealed by 3D MALDI-MSI. Anal. Chem. 2018, 90, 10568–10576. [Google Scholar] [CrossRef]
- Mallah, K.; Quanico, J.; Raffo-Romero, A.; Cardon, T.; Aboulouard, S.; Kobeissy, F.; Zibara, K.; Salzet, M.; Fournier, I. Matrix-Assisted Laser Desorption/Ionization-Mass Spectrometry Imaging of lipids in experimental model of traumatic brain injury detecting acylcarnitines as injury related markers. Anal. Chem. 2019, 91, 11879–11887. [Google Scholar] [CrossRef]
- Guo, S.; Zhou, D.; Zhang, M.; Li, T.; Liu, Y.; Xu, Y.; Chen, T.; Li, Z. Monitoring changes of docosahexaenoic acid-containing lipids during the recovery process of traumatic brain injury in rat using mass spectrometry imaging. Sci. Rep. 2017, 7, 5054. [Google Scholar] [CrossRef]
- Lin, A.; Tran, T.; Bluml, S.; Merugumala, S.; Liao, H.J.; Ross, B.D. Guidelines for acquiring and reporting clinical neurospectroscopy. Semin. Neurol. 2012, 32, 432–453. [Google Scholar] [PubMed]
- Shin, S.S.; Dixon, C.E. Alterations in Cholinergic Pathways and Therapeutic Strategies Targeting Cholinergic System after Traumatic Brain Injury. J. Neurotrauma 2015, 32, 1429–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuire, J.L.; Ngwenya, L.B.; McCullumsmith, R.E. Neurotransmitter changes after traumatic brain injury: An update for new treatment strategies. Mol. Psychiatry 2019, 24, 995–1012. [Google Scholar] [CrossRef]
- Verbois, S.L.; Scheff, S.W.; Pauly, J.R. Time-dependent changes in rat brain cholinergic receptor expression after experimental brain injury. J. Neurotrauma 2002, 19, 1569–1585. [Google Scholar] [CrossRef] [PubMed]
- Dewar, D.; Graham, D.I. Depletion of choline acetyltransferase activity but preservation of M1 and M2 muscarinic receptor binding sites in temporal cortex following head injury: A preliminary human postmortem study. J. Neurotrauma 1996, 13, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.R.; Maris, D.O.; Grady, M.S. Fluid percussion injury causes loss of forebrain choline acetyltransferase and nerve growth factor receptor immunoreactive cells in the rat. J. Neurotrauma 1994, 11, 379–392. [Google Scholar] [CrossRef]
- Schmidt, R.H.; Grady, M.S. Loss of forebrain cholinergic neurons following fluid-percussion injury: Implications for cognitive impairment in closed head injury. J. Neurosurg. 1995, 83, 496–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donat, C.K.; Schuhmann, M.U.; Voigt, C.; Neiber, K.; Deuther-Conrad, W.; Brust, P. Time-dependent alterations of cholinergic markers after experimental traumatic brain injury. Brain Res. 2008, 1246, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Sihver, S.; Marklund, N.; Hillered, L.; Långström, B.; Watanabe, Y.; Bergström, M. Changes in mACh, NMDA and GABA(A) receptor binding after lateral fluid-percussion injury: In vitro autoradiography of rat brain frozen sections. J. Neurochem. 2001, 78, 417–423. [Google Scholar] [CrossRef]
- Dixon, C.E.; Kochanek, P.M.; Yan, H.Q.; Schiding, J.K.; Griffith, R.G.; Baum, E.; Marion, D.W.; DeKosky, S.T. One-year study of spatial memory performance, brain morphology, and cholinergic markers after moderate controlled cortical impact in rats. J. Neurotrauma 1999, 16, 109–122. [Google Scholar] [CrossRef]
- Kaufer, D.; Friedman, A.; Seidman, S.; Soreq, H. Acute stress facilitates long-lasting changes in cholinergic gene expression. Nature 1998, 393, 373–377. [Google Scholar] [CrossRef]
- Bramlett, H.M.; Dietrich, W.D. Long-Term Consequences of Traumatic Brain Injury: Current Status of Potential Mechanisms of Injury and Neurological Outcomes. J. Neurotrauma 2015, 32, 1834. [Google Scholar] [CrossRef]
- McAllistar, T.W. Neurobiological consequences of traumatic brain injury. Dialogues Clin. Neurosci. 2011, 13, 287–300. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive review on Alzheimer’s disease: Causes and treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Fleminger, S.; Oliver, D.; Lovestone, S.; Rabe-Hesketh, S.; Giora, A. Head injury as a risk factor for Alzheimer’s disease: The evidence 10 years on; a partial replication. J. Neurol. Neurosurg. Psychiatry 2003, 74, 857. [Google Scholar] [CrossRef]
- Shetty, A.K. Hippocampal injury-induced cognitive and mood dysfunction, altered neurogenesis, and epilepsy: Can early neural stem cell grafting intervention provide protection? Epilepsy Behav. 2014, 38, 117–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulder, C.; Wahlund, L.O.; Teerlink, T.; Blomberg, M.; Veerhuis, R.; Van Kamp, G.J.; Scheltens, P.; Scheffer, P.G. Decreased lysophosphatidylcholine/phosphatidylcholine ratio in cerebrospinal fluid in Alzheimer’s disease. J. Neural Transm. 2003, 110, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Mapstone, M.; Cheema, A.K.; Fiandaca, M.S.; Zhong, X.; Mhyre, T.R.; MacArthur, L.H.; Hall, W.J.; Fisher, S.G.; Peterson, D.R.; Haley, J.M.; et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 2014, 20, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Whiley, L.; Sen, A.; Heaton, J.; Proitsi, P.; García-Gómez, D.; Leung, R.; Smith, N.; Thambisetty, M.; Kloszewska, I.; Mecocci, P.; et al. Evidence of altered phosphatidylcholine metabolism in Alzheimer’s disease. Neurobiol. Aging 2014, 35, 271. [Google Scholar] [CrossRef]
- Griffin, S.L.; Van Reekum, R.; Masanic, C. A review of cholinergic agents in the treatment of neurobehavioral deficits following traumatic brain injury. J. Neuropsychiatry Clin. Neurosci. 2003, 15, 17–26. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Traumatic brain injury and amyloid-β pathology: A link to Alzheimer’s disease? Nat. Rev. Neurosci. 2010, 11, 361. [Google Scholar] [CrossRef] [Green Version]
- Ponsford, J.; McLaren, A.; Schönberger, M.; Burke, R.; Rudzki, D.; Olver, J.; Ponsford, M. The association between apolipoprotein E and traumatic brain injury severity and functional outcome in a rehabilitation sample. J. Neurotrauma 2011, 28, 1683–1692. [Google Scholar] [CrossRef]
- Gaudin, M.; Panchal, M.; Auzeil, N.; Duyckaerts, C.; Brunelle, A.; Laprévote, O.; Touboul, D. Choline-containing phospholipids in microdissected human Alzheimer’s disease brain senile plaque versus neuropil. Bioanalysis 2012, 4, 2153–2159. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Hylin, M.J.; Kobori, N.; Hood, K.N.; Moore, A.N.; Dash, P.K. Post-injury administration of galantamine reduces traumatic brain injury pathology and improves outcome. J. Neurotrauma 2018, 35, 362. [Google Scholar] [CrossRef]
- Gardner, R.C.; Byers, A.L.; Barnes, D.E.; Li, Y.; Boscardin, J.; Yaffe, K. Mild TBI and risk of Parkinson disease: A chronic effects of neurotrauma consortium study. Neurology 2018, 90, e1771. [Google Scholar] [CrossRef] [PubMed]
- Delic, V.; Beck, K.D.; Pang, K.C.H.; Citron, B.A. Biological links between traumatic brain injury and Parkinson’s disease. Acta Neuropathol. Commun. 2020, 8, 45. [Google Scholar] [CrossRef]
- Jiang, H.; He, P.; Adler, C.H.; Shill, H.; Beach, T.G.; Li, R.; Shen, Y. Bid signal pathway components are identified in the temporal cortex with Parkinson disease. Neurology 2012, 79, 1767. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Guo, Y.; An, S.; Kuang, Y.; He, X.; Ma, H.; Li, J.; Lv, J.; Zhang, N.; Jiang, C. Targeting caspase-3 as dual therapeutic benefits by rnai facilitating brain-targeted nanoparticles in a rat model of Parkinson’s disease. PLoS ONE 2013, 8, e62905. [Google Scholar] [CrossRef]
- Hartmann, A. Postmortem studies in Parkinson’s disease. Dialogues Clin. Neurosci. 2004, 6, 281. [Google Scholar] [CrossRef] [PubMed]
- Farmer, K.; Smith, C.A.; Hayley, S.; Smith, J. Major alterations of phosphatidylcholine and lysophosphotidylcholine lipids in the substantia nigra using an early stage model of Parkinson’s disease. Int. J. Mol. Sci. 2015, 16, 18865–18877. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, A.; Timothy, J.; Pandit, L.; Manju, M. Post-traumatic epilepsy: An overview. Clin. Neurol. Neurosurg. 2006, 108, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Payan, H.; Toga, M.; Bérard-Badier, M. The pathology of post-traumatic epilepsies. Epilepsia 1970, 11, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, F.; Imran, I.; Pervaiz, H.; Ashraf, W.; Parveen, N.; Rasool, M.F.; Alasmari, A.F.; Alharbi, M.; Samad, N.; Alqarni, S.A.; et al. Non-pharmacological interventions for intractable Epilepsy. Saudi Pharm. J. SPJ Off. Publ. Saudi Pharm. Soc. 2020, 28, 951–962. [Google Scholar] [CrossRef]
- Flynn, C.J.; Wecker, L. Concomitant increases in the levels of choline and free fatty acids in rat brain: Evidence supporting the seizure-induced hydrolysis of phosphatidylcholine. J. Neurochem. 1987, 48, 1178–1184. [Google Scholar] [CrossRef]
- Hillert, M.H.; Imran, I.; Zimmermann, M.; Lau, H.; Weinfurter, S.; Klein, J. Dynamics of hippocampal acetylcholine release during lithium-pilocarpine-induced status epilepticus in rats. J. Neurochem. 2014, 131, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Jope, R.S.; Jenden, D.J. Choline and phospholipid metabolism and the synthesis of acetylcholine in rat brain. J. Neurosci. Res. 1979, 4, 69–82. [Google Scholar] [CrossRef]
- Jorge, R.E.; Robinson, R.G.; Moser, D.; Tateno, A.; Crespo-Facorro, B.; Arndt, S. Major depression following traumatic brain injury. Arch. Gen. Psychiatry 2004, 61, 42–50. [Google Scholar] [CrossRef] [Green Version]
- Dikmen, S.S.; Bombardier, C.H.; Machamer, J.E.; Fann, J.R.; Temkin, N.R. Natural history of depression in traumatic brain injury. Arch. Phys. Med. Rehabil. 2004, 85, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Rapoport, S.I.; Rao, J.S. Altered arachidonic acid cascade enzymes in postmortem brain from bipolar disorder patients. Mol. Psychiatry 2009, 16, 419–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demirkan, A.; Isaacs, A.; Ugocsai, P.; Liebisch, G.; Struchalin, M.; Rudan, I.; Wilson, J.F.; Pramstaller, P.P.; Gyllensten, U.; Campbell, H.; et al. Plasma phosphatidylcholine and sphingomyelin concentrations are associated with depression and anxiety symptoms in a Dutch family-based lipidomics study. J. Psychiatr. Res. 2013, 47, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.C.; Jones, C.R. Chronic lithium treatment decreases brain phospholipase A2 activity. Neurochem. Res. 1998, 23, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Kölzer, M.; Werth, N.; Sandhoff, K. Interactions of acid sphingomyelinase and lipid bilayers in the presence of the tricyclic antidepressant desipramine. FEBS Lett. 2004, 559, 96–98. [Google Scholar] [CrossRef] [Green Version]
- Derbyshire, E.; Obeid, R. Choline, Neurological development and brain function: A systematic review focusing on the first 1000 days. Nutrients 2020, 12, 1731. [Google Scholar] [CrossRef]
- Kapalka, G.M. Mania and Agitation. In Nutritional and Herbal Therapies for Children and Adolescents; Academic Press: Cambridge, MA, USA, 2010; pp. 189–218. ISBN 978-0-12-374927-7. [Google Scholar]
- Zeisel, S.H.; Growdon, J.H.; Wurtman, R.J.; Magil, S.G.; Logue, M. Normal plasma choline responses to ingested lecithin. Neurology 1980, 30, 1226–1229. [Google Scholar] [CrossRef]
- Magil, S.G.; Zeisel, S.H.; Wurtman, R.J. Effects of ingesting soy or egg lecithins on serum choline, brain choline and brain acetylcholine. J. Nutr. 1981, 111, 166–170. [Google Scholar] [CrossRef]
- Secades, J.J. Role of Citicoline in the management of traumatic brain injury. Pharmaceuticals 2021, 14, 410. [Google Scholar] [CrossRef] [PubMed]
- Bazan, N.G.; de Turco, E.B.R.; Allan, G. Mediators of injury in neurotrauma: Intracellular signal transduction and gene expression. J. Neurotrauma 1995, 12, 791–814. [Google Scholar] [CrossRef]
- Jacotte-Simancas, A.; Costa-Miserachs, D.; Coll-Andreu, M.; Torras-Garcia, M.; Borlongan, C.V.; Portell-Cortés, I. Effects of voluntary physical exercise, citicoline, and combined treatment on object recognition memory, neurogenesis, and neuroprotection after traumatic brain injury in rats. J. Neurotrauma 2015, 32, 739–751. [Google Scholar] [CrossRef] [Green Version]
- Dempsey, R.J.; Rao, V.L.R. Cytidinediphosphocholine treatment to decrease traumatic brain injury-induced hippocampal neuronal death, cortical contusion volume, and neurological dysfunction in rats. J. Neurosurg. 2003, 98, 867–873. [Google Scholar] [CrossRef] [Green Version]
- Adibhatla, R.M.; Hatcher, J.F. Cytidine 5′-diphosphocholine (CDP-choline) in stroke and other CNS disorders. Neurochem. Res. 2005, 30, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Dixon, C.E.; Ma, X.; Marion, D.W. Effects of CDP-choline treatment on neurobehavioral deficits after TBI and on hippocampal and neocortical acetylcholine release. J. Neurotrauma 1997, 14, 161–169. [Google Scholar] [CrossRef]
- Misbach, J.; Andradi, S.; Harahap, T.P.; Soemargo, S.; Markam, S. Double-blind trial of nicholin (CDP-choline) on the patients with severe head injury. In Proceedings of the Biannual Meeting of Neurology, Psychiatry and Neurosurgery, Surabaya, Indonesia, 6–8 November 1978. [Google Scholar]
- Cohadon, F.; Richer, E.; Poletto, B. A precursor of phospholipids in the treatment of severe traumatic comas. Neurochirurgie 1982, 28, 287–290. [Google Scholar] [PubMed]
- Lozano, R. CDP-choline in the treatment of cranio-encephalic traumata. J. Neurol. Sci. 1991, 103, 43–47. [Google Scholar] [CrossRef]
- Shokouhi, G.; Haghjoo, A.G.; Sattarnezhad, N.; Asghari, M.; Sattarnezhad, A.; Asghari, A.; Pezeshki, A. Effects of citicoline on level of consciousness, serum level of fetuin-A and matrix Gla-protein (MGP) in trauma patients with diffuse axonal injury (DAI) and GCS≤8. Turk. J. Trauma Emerg. Surg. 2014, 20, 410–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFina, P.A.; Moser, R.S.; Glenn, M.; Lichtenstein, J.D.; Fellus, J. Alzheimer’s disease clinical and research update for health care practitioners. J. Aging Res. 2013, 2013. [Google Scholar] [CrossRef]
- Lee, S.H.; Choi, B.Y.; Kim, J.H.; Kho, A.R.; Sohn, M.; Song, H.K.; Choi, H.C.; Suh, S.W. Late treatment with choline alfoscerate (l-alpha glycerylphosphorylcholine, α-GPC) increases hippocampal neurogenesis and provides protection against seizure-induced neuronal death and cognitive impairment. Brain Res. 2017, 1654, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Thau-Zuchman, O.; Gomes, R.N.; Dyall, S.C.; Davies, M.; Priestley, J.V.; Groenendijk, M.; De Wilde, M.C.; Tremoleda, J.L.; Michael-Titus, A.T. Brain phospholipid precursors administered post-injury reduce tissue damage and improve neurological outcome in experimental traumatic brain injury. J. Neurotrauma 2019, 36, 25. [Google Scholar] [CrossRef] [PubMed]
- Soininen, H.; Solomon, A.; Visser, P.J.; Hendrix, S.B.; Blennow, K.; Kivipelto, M.; Hartmann, T. 36-month LipiDiDiet multinutrient clinical trial in prodromal Alzheimer’s disease. Alzheimer’s Dement. 2021, 17, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Pallier, P.N.; Poddighe, L.; Zbarsky, V.; Kostusiak, M.; Choudhury, R.; Hart, T.; Burguillos, M.A.; Musbahi, O.; Groenendijk, M.; Sijben, J.W.; et al. A nutrient combination designed to enhance synapse formation and function improves outcome in experimental spinal cord injury. Neurobiol. Dis. 2015, 82, 504–515. [Google Scholar] [CrossRef]
- Cardenas, D.D.; Jr, M.A.; Roberts, F.L.; Baker, L.; Brooke, M.; Haselkorn, J. Oral physostigmine and impaired memory in adults with brain injury. Brain Inj. 1994, 8, 579–587. [Google Scholar] [CrossRef]
- Weinberg, R.M.; Auerbach, S.H.; Moore, S. Pharmacologic treatment of cognitive deficits: A case study. Brain Inj. 2009, 1, 57–59. [Google Scholar] [CrossRef]
- Taverni, J.P.; Seliger, G.; Lichtman, S.W. Donepezil medicated memory improvement in traumatic brain injury during post acute rehabilitation. Brain Inj. 1998, 12, 77–80. [Google Scholar] [CrossRef]
- Whelan, F.J.; Walker, M.S.; Schultz, S.K. Donepezil in the treatment of cognitive dysfunction associated with traumatic brain injury. Ann. Clin. Psychiatry 2000, 12, 131–135. [Google Scholar] [CrossRef]
- Guseva, M.V.; Hopkins, D.M.; Scheff, S.W.; Pauly, J.R. Dietary choline supplementation improves behavioral, histological, and neurochemical outcomes in a rat model of traumatic brain injury. J. Neurotrauma 2008, 25, 975–983. [Google Scholar] [CrossRef] [Green Version]
- Başkaya, M.K.; Doğan, A.; Rao, A.M.; Dempsey, R.J. Neuroprotective effects of citicoline on brain edema and blood-brain barrier breakdown after traumatic brain injury. J. Neurosurg. 2000, 92, 448–452. [Google Scholar] [CrossRef]
- Menku, A.; Ogden, M.; Saraymen, R. The protective effects of propofol and citicoline combination in experimental head injury in rats. Turk. Neurosurg. 2010, 20, 57–62. [Google Scholar]
- Qian, K.; Gu, Y.; Zhao, Y.; Li, Z.; Sun, M. Citicoline protects brain against closed head injury in rats through suppressing oxidative stress and calpain over-activation. Neurochem. Res. 2014, 39, 1206–1218. [Google Scholar] [CrossRef]
- Lee, H.J.; Kang, J.S.; Kim, Y.I. Citicoline protects against cognitive impairment in a rat model of chronic cerebral hypoperfusion. J. Clin. Neurol. 2009, 5, 33–38. [Google Scholar] [CrossRef]
- Maldonado, V.C.; Pérez, J.C.; Escario, J.A. Effects of CDP-choline on the recovery of patients with head injury. J. Neurol. Sci. 1991, 103, 15–18. [Google Scholar] [CrossRef]
- Tazaki, Y.; Sakai, F.; Otomo, E.; Kutsuzawa, T.; Kameyama, M.; Omae, T.; Fujishima, M.; Sakuma, A. Treatment of acute cerebral infarction with a choline precursor in a multicenter double-blind placebo-controlled study. Stroke 1988, 19, 211–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- León-Carrión, J.; Dominguez-Roldán, J.M.; Murillo-Cabezas, F.; Dominguez-Morales, M.D.R.; Muñoz-Sanchez, M.A. The role of citicholine in neuropsychological training after traumatic brain injury. NeuroRehabilitation 2000, 14, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Levin, H.S. Treatment of postconcussional symptoms with CDP-choline. J. Neurol. Sci. 1991, 103, 39–42. [Google Scholar] [CrossRef]
- Lazowski, T.; Kierul, K.; Bartnicki, M.; Mayzner-Zawadzka, E.; Toczylowska, B.; Ryba, M.; Lewandowski, Z. Effects of citicoline treatment in patients with isolated head trauma: A randomized trial. Crit. Care 2003, 7, P078. [Google Scholar] [CrossRef]
- Secades, J.J. Citicoline for the Treatment of Head Injury: A Systematic Review and Meta-analysis of Controlled Clinical Trials. J. Trauma Treat. 2014, 4, 227. [Google Scholar] [CrossRef]
- Trimmel, H.; Majdan, M.; Wodak, A.; Herzer, G.; Csomor, D.; Brazinova, A. Citicoline in severe traumatic brain injury: Indications for improved outcome A retrospective matched pair analysis from 14 Austrian trauma centers. Wien. Klin. Wochenschr. 2018, 130, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Salehpou, F.; Shokouhi, G.; Shakeri, M.; Shimia, M.; Mahdkhah, A.; Baradaran, A.; Imani, M.T.; Mirzaee, A.; Azar, A.K.; Bazzazi, A.M.; et al. Neuroprotective Effects of Citicoline in Diffuse Axonal Injuries. Adv. Biosci. Clin. Med. 2013, 1, 16–19. [Google Scholar]
- Levin, H.S.; Peters, B.H.; Kalisky, Z.; High, W.M.; Von Laufen, A.; Eisenberg, H.M.; Morrison, D.P.; Gary, H.E. Effects of Oral Physostigmine and Lecithin on Memory and Attention in Closed Head-Injured Patients. Cent. Nerv. Syst. Trauma 1986, 3, 333–342. [Google Scholar] [CrossRef] [PubMed]





| Glasgow Coma Scale Score Calculation | |||||
|---|---|---|---|---|---|
| Eye Opening Response | Score | Verbal Response | Score | Motor Response | Score |
| Spontaneous | 4 | Oriented | 5 | Obeys commands | 6 |
| Response to verbal command | 3 | Confused | 4 | Localizing response to pain | 5 |
| Response to pain | 2 | Inappropriate words | 3 | Withdrawal response to pain | 4 |
| No eye-opening | 1 | Incomprehensible speech | 2 | Flexion to pain | 3 |
| No verbal response | 1 | Extension to pain | 2 | ||
| No motor response | 1 | ||||
| Severity of Traumatic Brain Injury | |||
|---|---|---|---|
| Classification System | Mild | Moderate | Severe |
| GCS scale | 13–15 | 9–12 | 3–8 |
| PTA scale | Less than 1 day | From 2 to 7 days | More than 7 days |
| Pre-Clinical Studies | |||||
| Animal | TBI Model | Treatment and Schedule | Observation | Author | Ref. |
| SD rats | Controlled cortical impact | Dietary choline supplementation for 2 weeks | Improved memory and reduced neuroinflammation | Guseva et al. | [142] |
| SD rats | Controlled cortical impact | CDP-Choline 100 mg/kg i.p. for 18 days | Increase Ach release and decreased spatial memory deficit. | Dixon et al. | [128] |
| SD rats | Controlled cortical impact injury | CDP-Choline 100, 200 and 400 mg/kg given i.p. immediately and 6 h after TBI | Decrease neuronal loss and contusion volume with improved neurologic recovery | Dempsey et al. | [126] |
| SD rats | Controlled cortical impact | CDP-Choline 100 and 400 mg/kg i.p. given twice after TBI | Reduced edema in injury area with decreased BBB breakdown | Baskaya et al. | [143] |
| Wistar rats | Blunt Trauma | Citicoline 250 mg/kg i.p. | Reduced oxidative stress | Menku et al. | [144] |
| SD rats | Closed head injury | Citicoline 250 mg/kg injected i.v. 30 min and 4 h after injury | Decreased brain edema, BBB permeability, axonal and myelin sheath damage and reduced oxidative stress. | Qian et al. | [145] |
| SD rats | Controlled cortical impact injury | Citicoline 200 mg/kg i.p. Started 4 h after surgery and continued until five injections. | Reduced post-TBI cognitive impairment | Jacotte–Simancas et al. | [125] |
| Wistar rats | Chronic hypoperfusion | Citicoline 500 mg/kg i.p. for 21 days | Prevented white matter damage and enhanced cognition | Lee et al. | [146] |
| C57BL/6 mice | Controlled cortical impact injury | Fortasyn added to diet for 70 days | Improved cognition and neurogenesis with less oligodendrocyte loss | Thau–Zuchman et al. | [135] |
| Clinical Studies | |||||
| Patients | Study Design | Treatment | Treatment Schedule | Author | Ref. |
| 216 | Single-blinded randomized study | CDP-choline 4 g/day divided in 4 doses give i.v. on day 1–2 followed by 3 g/day divided in three doses for days 3–4 and continued as 200 mg orally every 8 h after discharge from ICU | Overall improvement in patient’s status, reduced physical dependency and better social reinsertion | Maldonado et al. | [147] |
| 272 | Double-blinded placebo-controlled study | CDP-choline 1000 mg CDP-choline i.v. daily for 14 days | Improved consciousness of patients as compared to placebo | Tazaki et al. | [148] |
| 10 | Placebo-controlled study design | CDP-choline 1 g/d p.o. for 3 months | Normalization of cerebral blood flow and enhanced memory | Carri ’on et al. | [149] |
| 14 | Double-blinded placebo-controlled study | CDP-choline 1 g p.o. for 1 month | Improved cognition as compared to placebo | Levin et al. | [150] |
| 28 | Placebo-controlled randomized trial | Citicoline 1 g i.v, for 14 days | Improved neuroprotection yielded in patients | Lazowsk et al. | [151] |
| 2706 | Systematic review and meta-analysis | Citicoline 250 mg to 6 g per day, administered orally or parenterally for 7–90 days | Beneficial health outcomes and with no safety concerns | Secades et al. | [152] |
| 134 | Retrospective matched pair analysis | Citicoline 3 g/day by i.v. for 21 days | The early administration of citicoline resulted in better outcomes | Trimmel et al. | [153] |
| 40 | Double-blinded randomized clinical trial | Citicoline 500 mg/6 h or 2 g/day i.v. for 15 days | Treatment of patients resulted in reduced MDA levels | Salehpour et al. | [154] |
| 16 | Double-blinded placebo-controlled study | Lecithin 16 g/day divided in two doses was given for 30 days | Improved cognition was observed | Levin et al. | [155] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Javaid, S.; Farooq, T.; Rehman, Z.; Afzal, A.; Ashraf, W.; Rasool, M.F.; Alqahtani, F.; Alsanea, S.; Alasmari, F.; Alanazi, M.M.; et al. Dynamics of Choline-Containing Phospholipids in Traumatic Brain Injury and Associated Comorbidities. Int. J. Mol. Sci. 2021, 22, 11313. https://doi.org/10.3390/ijms222111313
Javaid S, Farooq T, Rehman Z, Afzal A, Ashraf W, Rasool MF, Alqahtani F, Alsanea S, Alasmari F, Alanazi MM, et al. Dynamics of Choline-Containing Phospholipids in Traumatic Brain Injury and Associated Comorbidities. International Journal of Molecular Sciences. 2021; 22(21):11313. https://doi.org/10.3390/ijms222111313
Chicago/Turabian StyleJavaid, Sana, Talha Farooq, Zohabia Rehman, Ammara Afzal, Waseem Ashraf, Muhammad Fawad Rasool, Faleh Alqahtani, Sary Alsanea, Fawaz Alasmari, Mohammed Mufadhe Alanazi, and et al. 2021. "Dynamics of Choline-Containing Phospholipids in Traumatic Brain Injury and Associated Comorbidities" International Journal of Molecular Sciences 22, no. 21: 11313. https://doi.org/10.3390/ijms222111313
APA StyleJavaid, S., Farooq, T., Rehman, Z., Afzal, A., Ashraf, W., Rasool, M. F., Alqahtani, F., Alsanea, S., Alasmari, F., Alanazi, M. M., Alharbi, M., & Imran, I. (2021). Dynamics of Choline-Containing Phospholipids in Traumatic Brain Injury and Associated Comorbidities. International Journal of Molecular Sciences, 22(21), 11313. https://doi.org/10.3390/ijms222111313