
The Pathogenesis of End-Stage Renal Disease from the Standpoint of the Theory of General Pathological Processes of Inflammation



Abstract
:1. Introduction
2. General Characteristics of the Causes and Consequences of ESRD
- poisoning of the body with nephrotoxins (various middle molecules, derivatives of phenol and indole, homocysteine, and other molecules);
- excess in the blood of potentially toxic water-soluble drugs;
- hypoproteinemia, hyperlipidemia, hyperphosphatemia, hyperkalemia, hyponatremia, hyperuricemia, and metabolic acidosis;
- hypertension, accelerated development of atherosclerosis and heart failure, and rapid progression of diabetes mellitus (with diabetic kidney disease—DKD);
- thrombophilia and thrombocytopathy;
- systemic proinflammatory processes;
- dysfunction of the renin–angiotensin–aldosterone system, as well as other neuroendocrine dysfunctions.
3. Cellular Stress as a Common Pathogenetic Basis for General Pathological Processes Associated with Inflammation
- oxidative stress;
- cell response to DNA damage;
- mitochondrial stress, including mitochondrial unfolded protein response (UPRmt);
- the stress of the endoplasmic reticulum (ER), including calcium-dependent mechanisms and UPRER;
- response of inducible heat-shock proteins (HSPs), including their participation in the UPR;
- inhibition (during cell growth) or intensification of autophagy processes (utilization of altered organelles and macromolecules) and other manifestations of lysosomal stress;
- inflammasome formation;
- formation of stress noncoding microRNAs (miRNAs);
- formation of an intracellular network of signaling pathways of cellular stress;
- formation of proinflammatory receptor and secretory cell phenotype.
- the transcription factor HIF-1 (hypoxia-inducible factor-1) during hypoxia;
- the HSF1 factor for HSP production;
- the Nrf2 factor (to trigger the production of antioxidants through a negative feedback mechanism) in case of oxidative stress;
- ATF4 plays, along with HSF, an essential role in the development of UPRmt and UPRER.
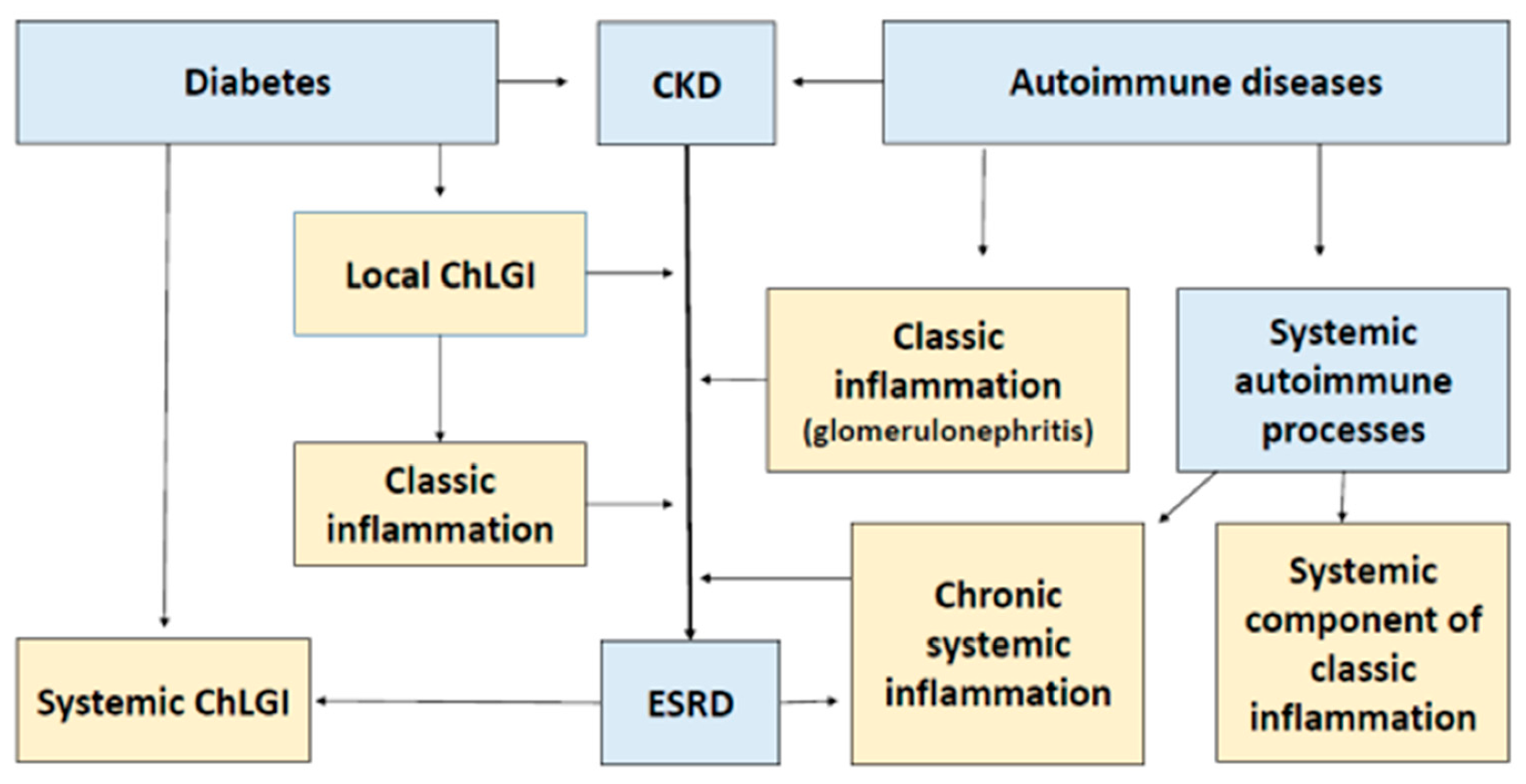
4. Typical Low-Intensity Inflammatory Processes in CKD and ESRD
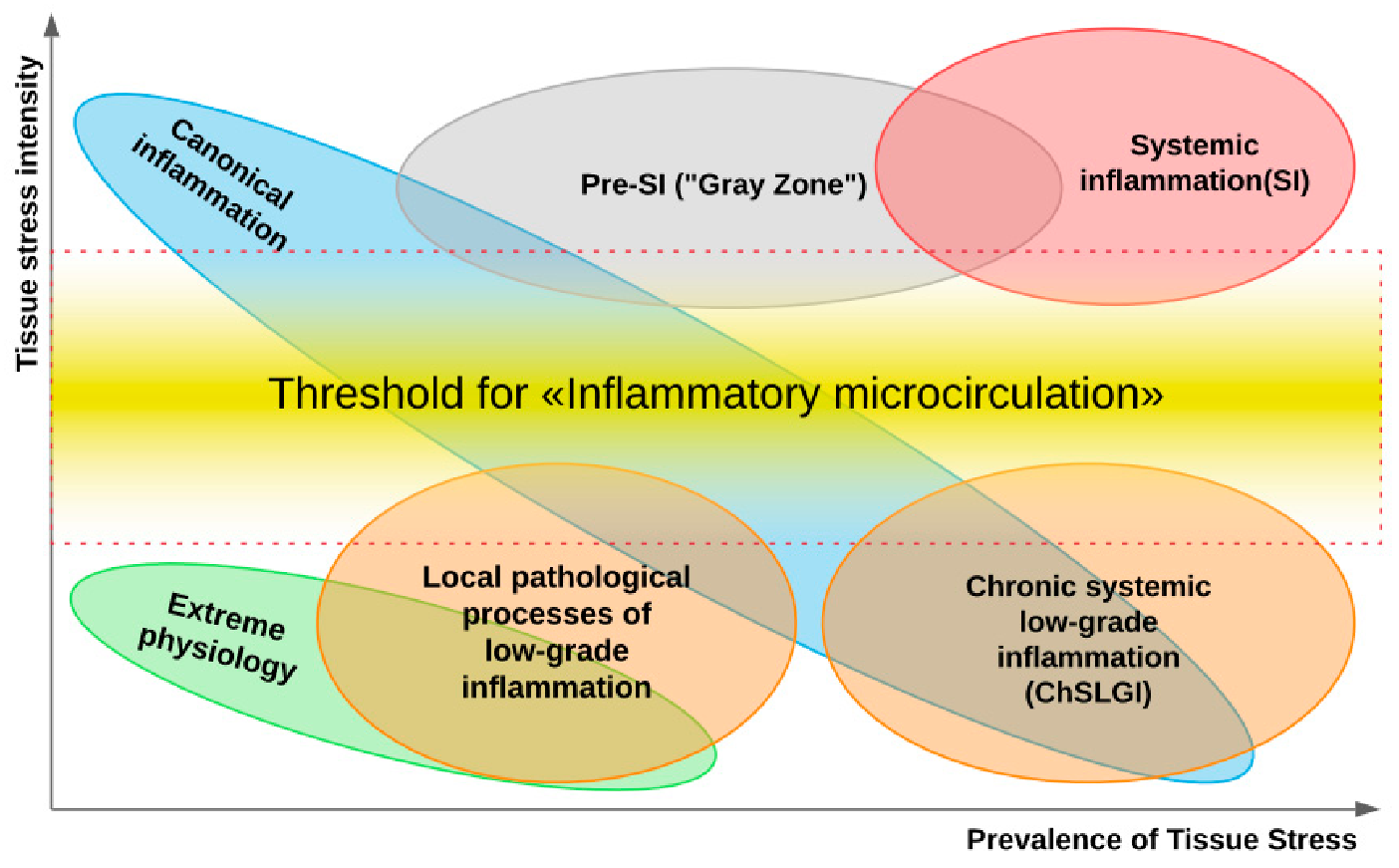
4.1. General Patterns of Development of Chronic Low-Grade Inflammation (ChLGI)
- ChLGI is tissue stress in response to local or systemic damage, which is insufficient for the development of classical or systemic inflammation, respectively.
- ChLGI is characterized by relatively low manifestations of SIR: an increase in C-reactive protein (CRP) in the blood, usually within the marginal zone between norm and pathology (3-10 mg/L), and an increase in key proinflammatory cytokines up to 2–4 times higher than the upper limit normal value.
- Signs of tissue decay and systemic coagulopathy are not typical; signs of organ dysfunction develop slowly; the accelerated development of atherosclerosis, hypertension, and tissue aging is characteristic; there is no connection of these changes with systemic manifestations of infections and autoimmune diseases, with pronounced signs of chronic classical inflammation.
- Key inducers of ChLGI are metabolic factors with a low ability to induce tissue changes, including modified proteins (denatured, oxidized, glycated), high concentrations of saturated free fatty acids (FFA), and oxidized low-density lipoproteins (oxLDL), homocysteine, and many other metabolites. The gradual accumulation of damage to the genome, proteome, and metabolome, as well as dysfunctions of organs during aging, contribute to an increase in the proinflammatory status of the organism.
- ChLGI involves a large number of parenchymal and stromal cells of various organs with relatively weak participation of “professional cells” of inflammation (leukocytes and their descendants, characteristic of the focus of inflammation). Consequently, ChLGI lacks barrier functions and many other signs of classical inflammation, including hyperemia, edema (exudation), and pronounced leukocyte infiltration, which determines the basic cellular composition of the inflammation focus [64,65].
- ChLGI can be defined as a para-inflammatory or quasi-inflammatory process. However, some of its features can be found in physiological processes. For example, in healthy people, the reaction of the acute phase of the liver can be detected situationally [66], and the physiological state of the intestinal mucosa is characterized by the presence of inflammasomes in the epithelial cells that perform a protective function against infection and tumors [67]. In athletes with pronounced work of skeletal muscles, there may be a significant increase in the blood levels of proinflammatory cytokines, especially IL-6, and other proinflammatory myokines [68].
- Pathological systemic manifestations of ChLGI can be directly associated with metabolic syndrome and, especially, with type 2 diabetes mellitus, as well as with neurodegeneration and chronic heart failure in the elderly [52]. Currently, it is very difficult to separate systemic ChLGI from SIR of classical inflammation in autoimmune, infectious, and many other chronic diseases due to the functional overlap of these processes (Figure 1).
4.2. Pathogenetic Significance of ChLGI in the Onset of ESRD
- proinflammatory cytokine of activated macrophages IL-18 (also accumulates in the urine);
- soluble forms of TNF receptors (sTNFR1 and sTNFR2);
- soluble forms of endothelial adhesion receptors (sICAM-1/CD54 and sVCAM-1/CD106);
- reactive oxygen species (ROS), oxLDL, AGE, and other biomarkers of systemic ChLGI.
- angiotensin II (Ang-II), ET-1, adenosine (ADO), ROS;
- transforming growth factor-β (TGF-β);
- growth factors of platelets (PDGF), connective tissue (CTGF), and fibroblasts-23 (FGF-23);
- chemokines: monocyte chemoattractant protein-1 (MCP-1, CCL2) and stromal cell factor-1 (SDF-1, CXCL12);
- cytokines: TNF-α, IL-6, IL-11, IL-18, and IL-20;
- kidney damage molecule-1 (KIM-1);
- direct intercellular contact interactions of Notch family receptors and Notch ligands involved in the epithelial–mesenchymal transition.
- oxidative stress;
- many signaling pathways of cellular stress, including Notch, MARK, PKC, PI3K/Akt/mTOR, and AMPK;
- activation of key factors of cellular stress transcription: NF-κB, AP-1, and HIF;
- the action of many regulatory miRNAs.
5. Typical Patterns of Classical Inflammation in CKD and ESRD
5.1. General Patterns of Classic Inflammation
- exudative–vascular catarrhal and serous inflammation (with more pronounced exudation in serous);
- productive (proliferative) inflammation, with the predominance of cellular infiltration of a particular composition depending on the nature of the damage factor and the stage of the process;
- exudative–destructive inflammation (purulent, curd, fibrinous, and gangrenous);
- mixed inflammation when it is impossible to clearly define its specific type.
5.2. Typical Manifestations of Classical Inflammation in Nephrites
- deposition of soluble immune complexes (post-streptococcal, lupus, and mesangiocapillary nephritis, Schönlein–Henoch disease, and IgA nephropathy);
- antibodies to the basement membrane of the renal glomeruli (Goodpasture’s disease, also known as anti-glomerular basement membrane disease);
- antineutrophil cytoplasmic antibody (ANCA-associated vasculitis).
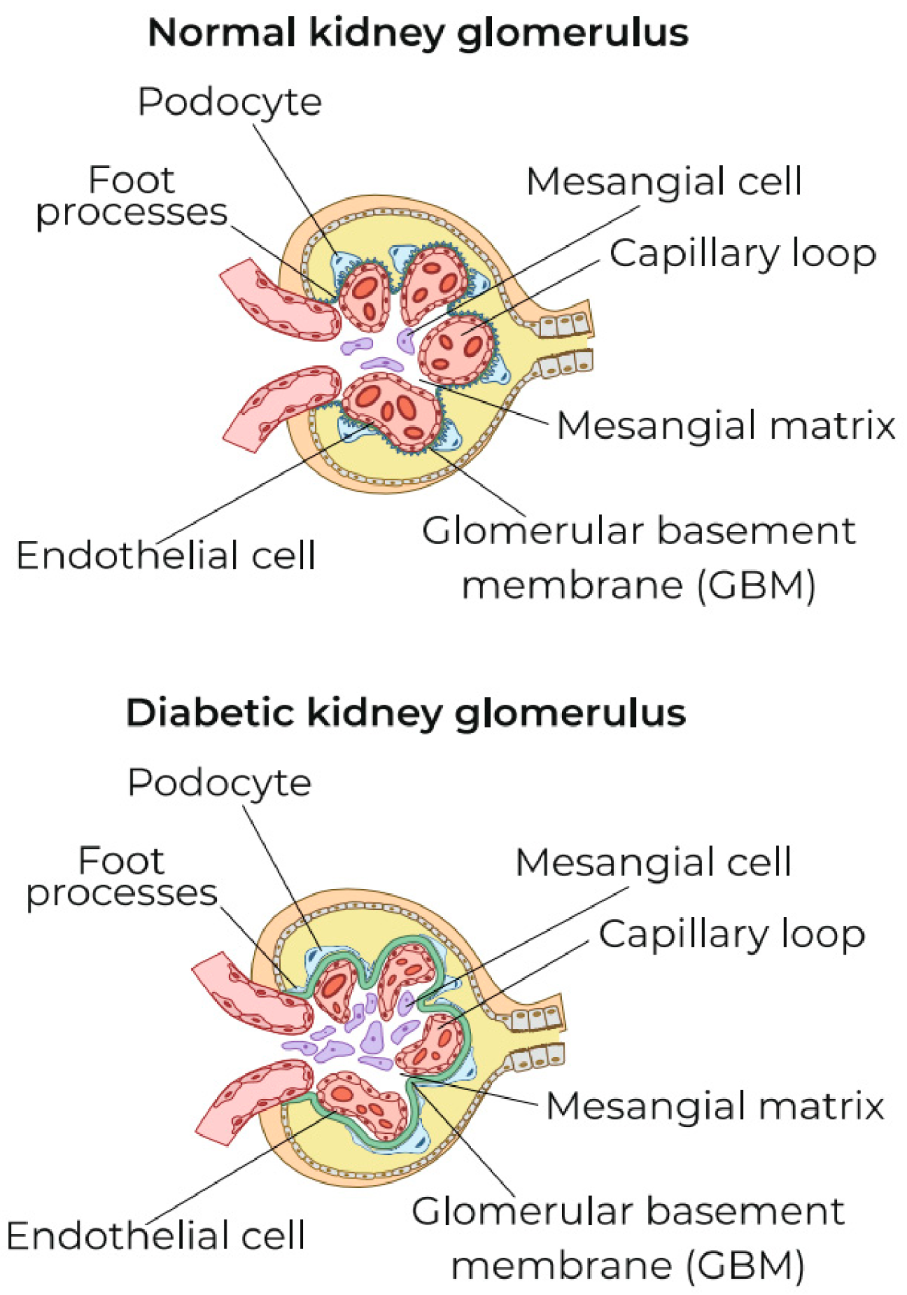
5.3. Possible Causes of the Transformation of Local ChLGI into Classic Inflammation in Diabetic Kidney Disease
- intercellular contact receptors Notch-1;
- membrane GTP-binding proteins;
- stress protein kinases MAPK (mitogen-activated protein kinase) and mTORC1 (mammalian target of rapamycin);
- many stress miRNAs and long non-coding (regulatory) RNAs;
- heat-shock proteins, especially HSP70;
- various proapoptotic factors;
5.4. Typical Features of Inflammation in Dysfunction and Rejection of the Renal Allograft
6. Systemic Inflammatory Processes in ESRD
6.1. General Characteristics of Systemic Inflammation
6.2. Features of SIR Development in ESRD
- various ROS;
- proinflammatory cytokines: TNF-α, IL-6, and IL-18;
- chemokines: IL-8 (CXCL8), IL-34, SDF1α (CXCL12), MCP-1 (CCL2), and MIP-1β (CCL4);
- growth factors: GM-CSF (granulocyte-macrophage colony-stimulating factor), FGF-23, and HGF (hepatocyte growth factor);
- soluble forms of receptors: sTNFR1 and sTNFR2, sCD40L, and sCD163 (SR-I3);
- cyclophilin A as an inducer of proinflammatory cytokines [186].
- multiple increases in average and median values of TNF-α, IL-8 and the soluble form of the IL-2 receptor (sCD25);
- less significant (approximately two-fold from the upper level of the norm) changes in C-reactive protein (CRP), IL-6, and TGF-β1;
- at the same time, the concentration of anti-inflammatory IL-10 did not differ significantly from its level in the blood of conventionally healthy people.
6.3. Systemic Inflammatory Phenomena Specific to ESRD
6.4. Use of the Integral Criterion of Systemic Inflammation in ChSI
- RL-0 characterizes the reference values of the norm;
- RL-1 is typical of ChLGI and SIR manifestations in classic acute and chronic inflammation;
- RL-2 is typical of severe acute purulent-inflammatory processes of the classical type;
- RL-3 is the overlap zone SIR of classical acute purulent inflammation and systemic inflammation;
- RL-4 and RL-5 are levels typical of the hyperergic variant of acute systemic inflammation;
- RL-3–5 in chronic processes, in our opinion, a priori confirm ChSI, while verification of ChSI at lower SIR values (RL-1–2) requires additional criteria.
- systemic autoimmune diseases (systemic lupus erythematosus, rheumatoid and reactive arthritis, and primary antiphospholipid syndrome);
- critical atherosclerotic ischemia of the lower extremities, complicated by gangrene of the toes;
- programmed hemodialysis (glomerulonephritis, diabetes mellitus, and chronic pyelonephritis);
- chronic allograft dysfunction regardless of the morphological variant of Banff classification based on biopsy data (taking into account the presence of large-scale anti-inflammatory and immunosuppressive therapy).

{kind=link}
{kind=link}
{kind=link}
| Group | n | RL | D-d | ChSI | |||||
|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | ||||
| Healthy people, age <55 years old | 50 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Conditionally healthy people, age >65 years old | 18 | 88.9 | 11.1 | 0 | 0 | 0 | 0 | 0 | 0 |
| PID | 16 | 75 | 25 | 0 | 0 | 0 | 0 | 0 | 0 |
| Chronic phlegmons | 42 | 19 | 78.6 | 2.4 | 0 | 0 | 0 | 9.5 | 7.1 |
| Hypertension, PMS | 16 | 93.7 | 6.3 | 0 | 0 | 0 | 0 | 0 | 0 |
| Elderly patients (>65 years old) with CHF and encephalopathy | 49 | 53.1 | 36.7 | 10.2 | 0 | 0 | 0 | 32.7 | 2.0 |
| Atherosclerotic stenosis of CFA with gangrene | 38 | 5.3 | 31.6 | 52.6 | 10.5 | 0 | 0 | 47.4 | 57.9 |
| AIT | 29 | 79.3 | 20.7 | 0 | 0 | 0 | 0 | 0 | 0 |
| PsA | 12 | 33.3 | 50 | 16.7 | 0 | 0 | 0 | 8.3 | 8.3 |
| Ankylosing spondylitis | 27 | 44.5 | 33.3 | 22.2 | 0 | 0 | 0 | 11.1 | 11.1 |
| Valvular heart diseases | 15 | 53.5 | 33.3 | 13.3 | 0 | 0 | 0 | 13.3 | 13.3 |
| ReA | 30 | 46.7 | 33.3 | 20 | 0 | 0 | 0 | 23.3 | 20 |
| Rheumatoid arthritis | 42 | 31 | 47.6 | 19 | 2.4 | 0 | 0 | 54.8 | 38.5 |
| SLE | 49 | 8.2 | 4.1 | 16.3 | 32.6 | 34.7 | 4.1 | 40.8 | 75.5 |
| PAPS 1 | 5 | 0 | 0 | 20 | 80 | 0 | 0 | 100 | 100 |
| End-stage renal disease (program hemodialysis) 2 | 42 | 4.8 | 16.6 | 54.8 | 21.4 | 2.4 | 0 | 38.1 | 88.1 |
| CAD 3 | 23 | 8.7 | 69.6 | 17.4 | 4.3 | 0 | 0 | 21.7 | 43.5 |
| Normal function of renal allograft | 24 | 58.3 | 25 | 16.7 | 0 | 0 | 0 | 4.2 | 0 |
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ammirati, A.L. Chronic Kidney Disease. Rev. Assoc. Med. Bras. 2020, 66, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Faria, M.; de Pinho, M.N. Challenges of Reducing Protein-Bound Uremic Toxin Levels in Chronic Kidney Disease and End Stage Renal Disease. Transl. Res. 2021, 229, 115–134. [Google Scholar] [CrossRef] [PubMed]
- Roufosse, C.; Simmonds, N.; Clahsen-van Groningen, M.; Haas, M.; Henriksen, K.J.; Horsfield, C.; Loupy, A.; Mengel, M.; Perkowska-Ptasińska, A.; Rabant, M.; et al. A 2018 Reference Guide to the Banff Classification of Renal Allograft Pathology. Transplantation 2018, 102, 1795–1814. [Google Scholar] [CrossRef] [PubMed]
- Saran, R.; Robinson, B.; Abbott, K.C.; Bragg-Gresham, J.; Chen, X.; Gipson, D.; Gu, H.; Hirth, R.A.; Hutton, D.; Jin, Y.; et al. US Renal Data System 2019 Annual Data Report: Epidemiology of Kidney Disease in the United States. Am. J. Kidney Dis. 2020, 75, A6–A7. [Google Scholar] [CrossRef]
- Benjamin, O.; Lappin, S.L. End-Stage Renal Disease. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Girndt, M.; Fiedler, R.; Martus, P.; Pawlak, M.; Storr, M.; Bohler, T.; Glomb, M.A.; Liehr, K.; Henning, C.; Templin, M.; et al. High Cut-off Dialysis in Chronic Haemodialysis Patients. Eur. J. Clin. Investig. 2015, 45, 1333–1340. [Google Scholar] [CrossRef]
- Lai, S.; Bagordo, D.; Perrotta, A.M.; Gigante, A.; Gasperini, M.L.; Muscaritoli, M.; Mazzaferro, S.; Cianci, R. Autonomic Dysfunction in Kidney Diseases. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 8458–8468. [Google Scholar] [CrossRef]
- Politano, S.A.; Colbert, G.B.; Hamiduzzaman, N. Nephrotic Syndrome. Prim. Care 2020, 47, 597–613. [Google Scholar] [CrossRef]
- Whitney, D.G.; Schmidt, M.; Bell, S.; Morgenstern, H.; Hirth, R.A. Incidence Rate of Advanced Chronic Kidney Disease Among Privately Insured Adults with Neurodevelopmental Disabilities. Clin. Epidemiol. 2020, 12, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Zemaitis, M.R.; Foris, L.A.; Katta, S.; Bashir, K. Uremia. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Murdeshwar, H.N.; Anjum, F. Hemodialysis. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Panocchia, N.; Tazza, L.; Di Stasio, E.; Liberatori, M.; Vulpio, C.; Giungi, S.; Lucani, G.; Antocicco, M.; Bossola, M. Mortality in Hospitalized Chronic Kidney Disease Patients Starting Unplanned Urgent Haemodialysis. Nephrology 2016, 21, 62–67. [Google Scholar] [CrossRef]
- Pazos, F. Range of Adiposity and Cardiorenal Syndrome. World J. Diabetes 2020, 11, 322–350. [Google Scholar] [CrossRef]
- Daehn, I.S. Glomerular Endothelial Cell Stress and Cross-Talk with Podocytes in Early [Corrected] Diabetic Kidney Disease. Front. Med. 2018, 5, 76. [Google Scholar] [CrossRef]
- Finnigan, N.A.; Leslie, S.W. Polycystic Kidney Disease in Adults. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Gasparotto, M.; Gatto, M.; Binda, V.; Doria, A.; Moroni, G. Lupus Nephritis: Clinical Presentations and Outcomes in the 21st Century. Rheumatology 2020, 59, v39–v51. [Google Scholar] [CrossRef]
- Ademola, B.L.; Atanda, A.T.; Aji, S.A.; Abdu, A. Clinical, Morphologic and Histological Features of Chronic Pyelonephritis: An 8-Year Review. Niger. Postgrad. Med. J. 2020, 27, 37–41. [Google Scholar] [CrossRef]
- Devuyst, O.; Olinger, E.; Weber, S.; Eckardt, K.-U.; Kmoch, S.; Rampoldi, L.; Bleyer, A.J. Autosomal Dominant Tubulointerstitial Kidney Disease. Nat. Rev. Dis. Primers 2019, 5, 60. [Google Scholar] [CrossRef] [Green Version]
- Padmanabhan, A.; Gohil, S.; Gadgil, N.M.; Sachdeva, P. Chronic Renal Failure: An Autopsy Study. Saudi J. Kidney Dis. Transplant. 2017, 28, 545–551. [Google Scholar] [CrossRef]
- Apel, M.; Maia, V.P.L.; Zeidan, M.; Schinkoethe, C.; Wolf, G.; Reinhart, K.; Sakr, Y. End-Stage Renal Disease and Outcome in a Surgical Intensive Care Unit. Crit. Care 2013, 17, R298. [Google Scholar] [CrossRef] [Green Version]
- Tuegel, C.; Katz, R.; Alam, M.; Bhat, Z.; Bellovich, K.; de Boer, I.; Brosius, F.; Gadegbeku, C.; Gipson, D.; Hawkins, J.; et al. GDF-15, Galectin 3, Soluble ST2, and Risk of Mortality and Cardiovascular Events in CKD. Am. J. Kidney Dis. 2018, 72, 519–528. [Google Scholar] [CrossRef]
- Onuigbo, M.A.C. End-Stage Renal Disease Risk in Different Glomerulonephropathies. Mayo Clin. Proc. 2018, 93, 958–959. [Google Scholar] [CrossRef] [Green Version]
- Sim, J.J.; Bhandari, S.K.; Batech, M.; Hever, A.; Harrison, T.N.; Shu, Y.-H.; Kujubu, D.A.; Jonelis, T.Y.; Kanter, M.H.; Jacobsen, S.J. End-Stage Renal Disease and Mortality Outcomes Across Different Glomerulonephropathies in a Large Diverse US Population. Mayo Clin. Proc. 2018, 93, 167–178. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Z.; Zhuo, L.; Chen, D.-P.; Li, W.-G. The Spectrum of Biopsy-Proven Glomerular Disease in China: A Systematic Review. Chin. Med. J. 2018, 131, 731–735. [Google Scholar] [CrossRef]
- Lin, J.; Jiang, Z.; Liu, C.; Zhou, D.; Song, J.; Liao, Y.; Chen, J. Emerging Roles of Long Non-Coding RNAs in Renal Fibrosis. Life 2020, 10, 131. [Google Scholar] [CrossRef]
- Miesen, L.; Eymael, J.; Sharma, S.; Loeven, M.A.; Willemsen, B.; Bakker-van Bebber, M.; Mooren, F.; Meyer-Schwesinger, C.; Dijkman, H.; Wetzels, J.F.M.; et al. Inhibition of MTOR Delayed but Could Not Prevent Experimental Collapsing Focal Segmental Glomerulosclerosis. Sci. Rep. 2020, 10, 8580. [Google Scholar] [CrossRef]
- Hesp, A.C.; Schaub, J.A.; Prasad, P.V.; Vallon, V.; Laverman, G.D.; Bjornstad, P.; van Raalte, D.H. The Role of Renal Hypoxia in the Pathogenesis of Diabetic Kidney Disease: A Promising Target for Newer Renoprotective Agents Including SGLT2 Inhibitors? Kidney Int. 2020, 98, 579–589. [Google Scholar] [CrossRef]
- Hamza, E.; Metzinger, L.; Metzinger-Le Meuth, V. Uremic Toxins Affect Erythropoiesis during the Course of Chronic Kidney Disease: A Review. Cells 2020, 9, 2039. [Google Scholar] [CrossRef]
- Masuda, T.; Nagata, D. Recent Advances in the Management of Secondary Hypertension: Chronic Kidney Disease. Hypertens. Res. 2020, 43, 869–875. [Google Scholar] [CrossRef]
- Saritas, T.; Floege, J. Cardiovascular Disease in Patients with Chronic Kidney Disease. Herz 2020, 45, 122–128. [Google Scholar] [CrossRef]
- Coronado Daza, J.; Martí-Carvajal, A.J.; Ariza García, A.; Rodelo Ceballos, J.; Yomayusa González, N.; Páez-Canro, C.; Loza Munárriz, C.; Urrútia, G. Early versus Delayed Erythropoietin for the Anaemia of End-Stage Kidney Disease. Cochrane Database Syst. Rev. 2015, 2015, CD011122. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Ganz, T.; Goodnough, L.T. Anemia of Inflammation. Blood 2019, 133, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, G. Immune Dysfunction in Uremia 2020. Toxins 2020, 12, 439. [Google Scholar] [CrossRef] [PubMed]
- Betjes, M.G. Uremia-Associated Ageing of the Thymus and Adaptive Immune Responses. Toxins 2020, 12, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, Y.-L.; Tsai, W.-C.; Hung, R.-W.; Chen, I.-Y.; Shu, K.-H.; Pan, S.-Y.; Yang, F.-J.; Ting, T.-T.; Jiang, J.-Y.; Peng, Y.-S.; et al. Emergence of T Cell Immunosenescence in Diabetic Chronic Kidney Disease. Immun. Ageing 2020, 17, 31. [Google Scholar] [CrossRef]
- Ebert, T.; Pawelzik, S.-C.; Witasp, A.; Arefin, S.; Hobson, S.; Kublickiene, K.; Shiels, P.G.; Bäck, M.; Stenvinkel, P. Inflammation and Premature Ageing in Chronic Kidney Disease. Toxins 2020, 12, 227. [Google Scholar] [CrossRef] [Green Version]
- Querfeld, U.; Mak, R.H.; Pries, A.R. Microvascular Disease in Chronic Kidney Disease: The Base of the Iceberg in Cardiovascular Comorbidity. Clin. Sci. 2020, 134, 1333–1356. [Google Scholar] [CrossRef]
- Eroglu, E.; Kocyigit, I.; Lindholm, B. The Endothelin System as Target for Therapeutic Interventions in Cardiovascular and Renal Disease. Clin. Chim. Acta 2020, 506, 92–106. [Google Scholar] [CrossRef]
- Jung, J.; Bae, G.H.; Kang, M.; Kim, S.W.; Lee, D.H. Statins and All-Cause Mortality in Patients Undergoing Hemodialysis. J. Am. Heart Assoc. 2020, 9, e014840. [Google Scholar] [CrossRef]
- Deferrari, G.; Cipriani, A.; La Porta, E. Renal Dysfunction in Cardiovascular Diseases and Its Consequences. J. Nephrol. 2021, 34, 137–153. [Google Scholar] [CrossRef]
- Joseph, M.S.; Palardy, M.; Bhave, N.M. Management of Heart Failure in Patients with End-Stage Kidney Disease on Maintenance Dialysis: A Practical Guide. Rev. Cardiovasc. Med. 2020, 21, 31–39. [Google Scholar] [CrossRef]
- Raina, R.; Nair, N.; Chakraborty, R.; Nemer, L.; Dasgupta, R.; Varian, K. An Update on the Pathophysiology and Treatment of Cardiorenal Syndrome. Cardiol. Res 2020, 11, 76–88. [Google Scholar] [CrossRef]
- Triposkiadis, F.; Starling, R.C.; Boudoulas, H.; Giamouzis, G.; Butler, J. The Cardiorenal Syndrome in Heart Failure: Cardiac? Renal? Syndrome? Heart Fail. Rev. 2012, 17, 355–366. [Google Scholar] [CrossRef]
- Sharma, A.; Sharma, A.; Gahlot, S.; Prasher, P.K. A Study of Pulmonary Function in End-Stage Renal Disease Patients on Hemodialysis: A Cross-Sectional Study. Sao Paulo Med. J. 2017, 135, 568–572. [Google Scholar] [CrossRef] [Green Version]
- Yılmaz, S.; Yildirim, Y.; Yilmaz, Z.; Kara, A.V.; Taylan, M.; Demir, M.; Coskunsel, M.; Kadiroglu, A.K.; Yilmaz, M.E. Pulmonary Function in Patients with End-Stage Renal Disease: Effects of Hemodialysis and Fluid Overload. Med. Sci. Monit. 2016, 22, 2779–2784. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.J.; Wen, J.; Ni, L.; Zhong, J.; Liang, X.; Zheng, G.; Lu, G.M. Predominant Gray Matter Volume Loss in Patients with End-Stage Renal Disease: A Voxel-Based Morphometry Study. Metab. Brain Dis. 2013, 28, 647–654. [Google Scholar] [CrossRef]
- Chiu, Y.-L.; Tsai, H.-H.; Lai, Y.-J.; Tseng, H.-Y.; Wu, Y.-W.; Peng, Y.-S.; Chiu, C.-M.; Chuang, Y.-F. Cognitive Impairment in Patients with End-Stage Renal Disease: Accelerated Brain Aging? J. Formos. Med. Assoc. 2019, 118, 867–875. [Google Scholar] [CrossRef]
- Mindikoglu, A.L.; Pappas, S.C. New Developments in Hepatorenal Syndrome. Clin. Gastroenterol. Hepatol. 2018, 16, 162–177.e1. [Google Scholar] [CrossRef] [Green Version]
- Simonetto, D.A.; Gines, P.; Kamath, P.S. Hepatorenal Syndrome: Pathophysiology, Diagnosis, and Management. BMJ 2020, 370, m2687. [Google Scholar] [CrossRef]
- Francoz, C.; Durand, F.; Kahn, J.A.; Genyk, Y.S.; Nadim, M.K. Hepatorenal Syndrome. Clin. J. Am. Soc. Nephrol. 2019, 14, 774–781. [Google Scholar] [CrossRef]
- Zhou, Z.-F.; Jiang, L.; Zhao, Q.; Wang, Y.; Zhou, J.; Chen, Q.-K.; Lv, J.-L. Roles of Pattern Recognition Receptors in Diabetic Nephropathy. J. Zhejiang Univ.-Sci. B 2020, 21, 192–203. [Google Scholar] [CrossRef]
- Gusev, E.Y.; Zotova, N.V. Cellular Stress and General Pathological Processes. Curr. Pharm. Des. 2019, 25, 251–297. [Google Scholar] [CrossRef]
- Kielstein, J.T.; Pontremoli, R.; Burnier, M. Management of Hyperuricemia in Patients with Chronic Kidney Disease: A Focus on Renal Protection. Curr. Hypertens. Rep. 2020, 22, 102. [Google Scholar] [CrossRef]
- Sabatino, A.; Regolisti, G.; Cosola, C.; Gesualdo, L.; Fiaccadori, E. Intestinal Microbiota in Type 2 Diabetes and Chronic Kidney Disease. Curr. Diabetes Rep. 2017, 17, 16. [Google Scholar] [CrossRef]
- Terpstra, M.L.; Sinnige, M.J.; Hugenholtz, F.; Peters-Sengers, H.; Remmerswaal, E.B.; Geerlings, S.E.; Bemelman, F.J. Butyrate Production in Patients with End-Stage Renal Disease. Int. J. Nephrol. Renovasc. Dis. 2019, 12, 87–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhang, W.; Nie, J. Gut Microbiota and Renal Injury. Adv. Exp. Med. Biol. 2020, 1238, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular Stress Responses: Cell Survival and Cell Death. Int. J. Cell. Biol. 2010, 2010, 214074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardwell, L.; Zou, X.; Nie, Q.; Komarova, N.L. Mathematical Models of Specificity in Cell Signaling. Biophys. J. 2007, 92, 3425–3441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Tsuchiya, R.; Hozumi, Y.; Nakano, T.; Okada, M.; Goto, K. Reciprocal Regulation of P53 and NF-ΚB by Diacylglycerol Kinase ζ. Adv. Biol. Regul. 2016, 60, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Webster, G.A.; Perkins, N.D. Transcriptional Cross Talk between NF-κB and P53. Mol. Cell. Biol. 1999, 19, 3485–3495. [Google Scholar] [CrossRef] [Green Version]
- Chittiboyina, S.; Bai, Y.; Lelièvre, S.A. Microenvironment-Cell Nucleus Relationship in the Context of Oxidative Stress. Front. Cell Dev. Biol. 2018, 6, 23. [Google Scholar] [CrossRef]
- Zhou, D.; Huang, C.; Lin, Z.; Zhan, S.; Kong, L.; Fang, C.; Li, J. Macrophage Polarization and Function with Emphasis on the Evolving Roles of Coordinated Regulation of Cellular Signaling Pathways. Cell. Signal. 2014, 26, 192–197. [Google Scholar] [CrossRef]
- Movafagh, S.; Crook, S.; Vo, K. Regulation of Hypoxia-Inducible Factor-1a by Reactive Oxygen Species: New Developments in an Old Debate. J. Cell. Biochem. 2015, 116, 696–703. [Google Scholar] [CrossRef]
- Granger, D.N.; Senchenkova, E. Inflammation and the Microcirculation. Colloq. Ser. Integr. Syst. Physiol.: Mol. Funct. 2010, 2, 1–87. [Google Scholar] [CrossRef]
- Majno, G.; Joris, I. Cells, Tissues, and Disease: Principles of General Pathology, 2nd ed.; Oxford University Press: New York, NY, USA, 2004; 1005p. [Google Scholar]
- Pietzner, M.; Kaul, A.; Henning, A.-K.; Kastenmüller, G.; Artati, A.; Lerch, M.M.; Adamski, J.; Nauck, M.; Friedrich, N. Comprehensive Metabolic Profiling of Chronic Low-Grade Inflammation among Generally Healthy Individuals. BMC Med. 2017, 15, 210. [Google Scholar] [CrossRef] [Green Version]
- Zmora, N.; Levy, M.; Pevsner-Fishcer, M.; Elinav, E. Inflammasomes and Intestinal Inflammation. Mucosal. Immunol. 2017, 10, 865–883. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Febbraio, M.A. Muscle as an Endocrine Organ: Focus on Muscle-Derived Interleukin-6. Physiol. Rev. 2008, 88, 1379–1406. [Google Scholar] [CrossRef] [Green Version]
- Yeung, Y.T.; Aziz, F.; Guerrero-Castilla, A.; Arguelles, S. Signaling Pathways in Inflammation and Anti-Inflammatory Therapies. Curr. Pharm. Des. 2018, 24, 1449–1484. [Google Scholar] [CrossRef]
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J. Immunol. 2017, 198, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.; Holman, G.D. Emerging Role for AS160/TBC1D4 and TBC1D1 in the Regulation of GLUT4 Traffic. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E29–E37. [Google Scholar] [CrossRef]
- Avanoǧlu Güler, A.; Rossi, F.W.; Bellando-Randone, S.; Prevete, N.; Tufan, A.; Manetti, M.; de Paulis, A.; Matucci-Cerinic, M. The Role of Endogenous Eicosapentaenoic Acid and Docosahexaenoic Acid-Derived Resolvins in Systemic Sclerosis. Front. Immunol. 2020, 11, 1249. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-Resolving Lipid Mediators Are Leads for Resolution Physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Park, I.; Xun, P.; Tsinovoi, C.L.; Klemmer, P.; Liu, K.; He, K. Intakes of Long-Chain Omega-3 Polyunsaturated Fatty Acids and Non-Fried Fish in Relation to Incidence of Chronic Kidney Disease in Young Adults: A 25-Year Follow-Up. Eur. J. Nutr. 2020, 59, 399–407. [Google Scholar] [CrossRef]
- Senthilkumar, G.P.; Anithalekshmi, M.S.; Yasir, M.; Parameswaran, S.; Packirisamy, R.M.; Bobby, Z. Role of Omentin 1 and IL-6 in Type 2 Diabetes Mellitus Patients with Diabetic Nephropathy. Diabetes Metab. Syndr. 2018, 12, 23–26. [Google Scholar] [CrossRef]
- Gusev, E.Y.; Zotova, N.V.; Zhuravleva, Y.A.; Chereshnev, V.A. Physiological and pathogenic role of scavenger receptors in humans. Med. Immunol. 2020, 22, 7–48. [Google Scholar] [CrossRef]
- Kumar Pasupulati, A.; Chitra, P.S.; Reddy, G.B. Advanced Glycation End Products Mediated Cellular and Molecular Events in the Pathology of Diabetic Nephropathy. Biomol. Concepts 2016, 7, 293–309. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-C.; Chen, J.-S.; Lin, S.-H.; Chu, P.; Lin, Y.-F.; Lin, S.-M.; Liao, T.-N. Aberrant Activation of the TNF-Alpha System and Production of Fas and Scavenger Receptors on Monocytes in Patients with End-Stage Renal Disease. Artif. Organs 2005, 29, 701–707. [Google Scholar] [CrossRef]
- Yang, X.; Okamura, D.M.; Lu, X.; Chen, Y.; Moorhead, J.; Varghese, Z.; Ruan, X.Z. CD36 in Chronic Kidney Disease: Novel Insights and Therapeutic Opportunities. Nat. Rev. Nephrol. 2017, 13, 769–781. [Google Scholar] [CrossRef]
- McEwen, B.S.; Wingfield, J.C. The Concept of Allostasis in Biology and Biomedicine. Horm. Behav. 2003, 43, 2–15. [Google Scholar] [CrossRef]
- Smykiewicz, P.; Segiet, A.; Keag, M.; Żera, T. Proinflammatory Cytokines and Ageing of the Cardiovascular-Renal System. Mech. Ageing Dev. 2018, 175, 35–45. [Google Scholar] [CrossRef]
- Hernández-Saavedra, D.; Stanford, K.I. The Regulation of Lipokines by Environmental Factors. Nutrients 2019, 11, 2422. [Google Scholar] [CrossRef] [Green Version]
- Sorop, O.; Olver, T.D.; van de Wouw, J.; Heinonen, I.; van Duin, R.W.; Duncker, D.J.; Merkus, D. The Microcirculation: A Key Player in Obesity-Associated Cardiovascular Disease. Cardiovasc. Res. 2017, 113, 1035–1045. [Google Scholar] [CrossRef]
- Yoshida, T.; Delafontaine, P. Mechanisms of Cachexia in Chronic Disease States. Am. J. Med. Sci. 2015, 350, 250–256. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.C.; Brownlee, M.; Susztak, K.; Sharma, K.; Jandeleit-Dahm, K.A.M.; Zoungas, S.; Rossing, P.; Groop, P.-H.; Cooper, M.E. Diabetic Kidney Disease. Nat. Rev. Dis. Primers 2015, 1, 15018. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Qiao, F.; Zhao, Y.; Wang, Y.; Liu, G. HMGB1 Is Activated in Type 2 Diabetes Mellitus Patients and in Mesangial Cells in Response to High Glucose. Int. J. Clin. Exp. Pathol. 2015, 8, 6683–6691. [Google Scholar] [PubMed]
- Hojs, R.; Ekart, R.; Bevc, S.; Hojs, N. Biomarkers of Renal Disease and Progression in Patients with Diabetes. J. Clin. Med. 2015, 4, 1010–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraenkel, P.G. Understanding Anemia of Chronic Disease. Hematol. Am. Soc. Hematol. Educ. Program 2015, 2015, 14–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alnaggar, A.R.L.R.; Sayed, M.; El-Deena, K.E.; Gomaa, M.; Hamed, Y. Evaluation of Serum Adiponectin Levels in Diabetic Nephropathy. Diabetes Metab. Syndr. 2019, 13, 128–131. [Google Scholar] [CrossRef]
- Marouga, A.; Dalamaga, M.; Kastania, A.N.; Kroupis, C.; Lagiou, M.; Saounatsou, K.; Dimas, K.; Vlahakos, D.V. Circulating Resistin Is a Significant Predictor of Mortality Independently from Cardiovascular Comorbidities in Elderly, Non-Diabetic Subjects with Chronic Kidney Disease. Biomarkers 2016, 21, 73–79. [Google Scholar] [CrossRef]
- Tong, X.; Yu, Q.; Ankawi, G.; Pang, B.; Yang, B.; Yang, H. Insights into the Role of Renal Biopsy in Patients with T2DM: A Literature Review of Global Renal Biopsy Results. Diabetes Ther. 2020, 11, 1983–1999. [Google Scholar] [CrossRef]
- Tesch, G.H. Diabetic Nephropathy—Is this an immune disorder? Clin. Sci. 2017, 131, 2183–2199. [Google Scholar] [CrossRef]
- Buraczynska, M.; Swatowski, A.; Buraczynska, K.; Dragan, M.; Ksiazek, A. Heat-Shock Protein Gene Polymorphisms and the Risk of Nephropathy in Patients with Type 2 Diabetes. Clin. Sci. 2009, 116, 81–86. [Google Scholar] [CrossRef] [Green Version]
- Yamanouchi, M.; Mori, M.; Hoshino, J.; Kinowaki, K.; Fujii, T.; Ohashi, K.; Furuichi, K.; Wada, T.; Ubara, Y. Retinopathy Progression and the Risk of End-Stage Kidney Disease: Results from a Longitudinal Japanese Cohort of 232 Patients with Type 2 Diabetes and Biopsy-Proven Diabetic Kidney Disease. BMJ Open Diabetes Res. Care 2019, 7, e000726. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.-Y.; Zhou, X.-J.; Sun, P.-P.; Yu, X.-J.; Wang, S.-X.; Qu, L.; Zhang, F.; Ma, Y.-Y.; Lv, J.-C.; Liu, G.; et al. Interstitial Eosinophilic Infiltration in Diabetic Nephropathy Is Indicative of Poor Prognosis, with No Therapy Benefit from Steroid. J. Diabetes 2020, 12, 881–894. [Google Scholar] [CrossRef]
- Zeng, L.-F.; Xiao, Y.; Sun, L. A Glimpse of the Mechanisms Related to Renal Fibrosis in Diabetic Nephropathy. Adv. Exp. Med. Biol. 2019, 1165, 49–79. [Google Scholar] [CrossRef]
- Yang, X.; Mou, S. Role of Immune Cells in Diabetic Kidney Disease. Curr. Gene Ther. 2017, 17, 424–433. [Google Scholar] [CrossRef]
- Fan, J.; Xie, K.; Wang, L.; Zheng, N.; Yu, X. Roles of Inflammasomes in Inflammatory Kidney Diseases. Mediat. Inflamm. 2019, 2019, 2923072. [Google Scholar] [CrossRef] [Green Version]
- Duni, A.; Liakopoulos, V.; Roumeliotis, S.; Peschos, D.; Dounousi, E. Oxidative Stress in the Pathogenesis and Evolution of Chronic Kidney Disease: Untangling Ariadne’s Thread. Int. J. Mol. Sci. 2019, 20, 3711. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.-S.; Hsu, Y.-H. The Role of IL-20 in Chronic Kidney Disease and Diabetic Nephropathy: Pathogenic and Therapeutic Implications. J. Leukoc. Biol. 2018, 104, 919–923. [Google Scholar] [CrossRef]
- Corden, B.; Adami, E.; Sweeney, M.; Schafer, S.; Cook, S.A. IL-11 in Cardiac and Renal Fibrosis: Late to the Party but a Central Player. Br. J. Pharmacol. 2020, 177, 1695–1708. [Google Scholar] [CrossRef]
- Klimczak-Tomaniak, D.; Pilecki, T.; Żochowska, D.; Sieńko, D.; Janiszewski, M.; Pączek, L.; Kuch, M. CXCL12 in Patients with Chronic Kidney Disease and Healthy Controls: Relationships to Ambulatory 24-Hour Blood Pressure and Echocardiographic Measures. Cardiorenal Med. 2018, 8, 249–258. [Google Scholar] [CrossRef]
- Liu, M.; Ning, X.; Li, R.; Yang, Z.; Yang, X.; Sun, S.; Qian, Q. Signalling Pathways Involved in Hypoxia-Induced Renal Fibrosis. J. Cell. Mol. Med. 2017, 21, 1248–1259. [Google Scholar] [CrossRef]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory Processes in Renal Fibrosis. Nat. Rev. Nephrol. 2014, 10, 493–503. [Google Scholar] [CrossRef]
- Prakoura, N.; Hadchouel, J.; Chatziantoniou, C. Novel Targets for Therapy of Renal Fibrosis. J. Histochem. Cytochem. 2019, 67, 701–715. [Google Scholar] [CrossRef]
- Isakova, T. Fibroblast Growth Factor 23 and Adverse Clinical Outcomes in Chronic Kidney Disease. Curr. Opin. Nephrol. Hypertens. 2012, 21, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.A.I.; Larsson, A.; Melhus, H.; Lind, L.; Larsson, T.E. Serum Intact FGF23 Associate with Left Ventricular Mass, Hypertrophy and Geometry in an Elderly Population. Atherosclerosis 2009, 207, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Solari, V.; Unemoto, K.; Piaseczna Piotrowska, A.; Puri, P. Increased Expression of Mast Cells in Reflux Nephropathy. Pediatric Nephrol. 2004, 19, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Tóth, T.; Tóth-Jakatics, R.; Jimi, S.; Takebayashi, S. Increased Density of Interstitial Mast Cells in Amyloid A Renal Amyloidosis. Mod. Pathol. 2000, 13, 1020–1028. [Google Scholar] [CrossRef] [Green Version]
- Karihaloo, A. Role of Inflammation in Polycystic Kidney Disease. In Polycystic Kidney Disease; Li, X., Ed.; Codon Publications: Brisbane, Australia, 2015; pp. 335–374. [Google Scholar]
- Ruan, X.; Guan, Y. Metabolic Syndrome and Chronic Kidney Disease. J. Diabetes 2009, 1, 236–245. [Google Scholar] [CrossRef]
- Dai, L.; Golembiewska, E.; Lindholm, B.; Stenvinkel, P. End-Stage Renal Disease, Inflammation and Cardiovascular Outcomes. Contrib. Nephrol. 2017, 191, 32–43. [Google Scholar] [CrossRef]
- Canaud, B.; Kooman, J.P.; Selby, N.M.; Taal, M.W.; Francis, S.; Maierhofer, A.; Kopperschmidt, P.; Collins, A.; Kotanko, P. Dialysis-Induced Cardiovascular and Multiorgan Morbidity. Kidney Int. Rep. 2020, 5, 1856–1869. [Google Scholar] [CrossRef]
- Ahmadmehrabi, S.; Tang, W.H.W. Hemodialysis-induced cardiovascular disease. Semin. Dial. 2018, 31, 258–267. [Google Scholar] [CrossRef]
- Kohlová, M.; Amorim, C.G.; Araújo, A.; Santos-Silva, A.; Solich, P.; Montenegro, M.C.B.S.M. The Biocompatibility and Bioactivity of Hemodialysis Membranes: Their Impact in End-Stage Renal Disease. J. Artif. Organs 2019, 22, 14–28. [Google Scholar] [CrossRef]
- Martinez Cantarin, M.P.; Whitaker-Menezes, D.; Lin, Z.; Falkner, B. Uremia Induces Adipose Tissue Inflammation and Muscle Mitochondrial Dysfunction. Nephrol. Dial. Transplant. 2017, 32, 943–951. [Google Scholar] [CrossRef]
- Gusev, E.Y.; Zhuravleva, Y.A.; Zotova, N.V. Correlation of the Evolution of Immunity and Inflammation in Vertebrates. Biol. Bull. Rev. 2019, 9, 358–372. (In Russian) [Google Scholar] [CrossRef]
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Cellular and Molecular Immunology, 9th ed.; Elsevier: Philadelphia, PA, USA, 2018; 608p. [Google Scholar]
- Yamaguchi, T.; Takizawa, F.; Fischer, U.; Dijkstra, J.M. Along the Axis between Type 1 and Type 2 Immunity; Principles Conserved in Evolution from Fish to Mammals. Biology 2015, 4, 814–859. [Google Scholar] [CrossRef] [Green Version]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of Monocytes, Macrophages, and Dendritic Cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Hirahara, K.; Nakayama, T. CD4+ T-Cell Subsets in Inflammatory Diseases: Beyond the Th1/Th2 Paradigm. Int. Immunol. 2016, 28, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.K.; Turner, H.; Maynard, C.L.; Oliver, J.R.; Chen, D.; Elson, C.O.; Weaver, C.T. Late Developmental Plasticity in the T Helper 17 Lineage. Immunity 2009, 30, 92–107. [Google Scholar] [CrossRef] [Green Version]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4+T Cells: Differentiation and Functions. Clin. Dev. Immunol. 2012, 2012, 925135. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Souabni, A.; Flavell, R.A.; Wan, Y.Y. An Intrinsic Mechanism Predisposes Foxp3-Expressing Regulatory T Cells to Th2 Conversion in Vivo. J. Immunol. 2010, 185, 5983–5992. [Google Scholar] [CrossRef] [Green Version]
- Rose, N.; Mackay, I. (Eds.) The Autoimmune Diseases, 6th ed.; Academic Press: Cambridge, MA, USA, 2019; p. 1532. [Google Scholar]
- Tuttle, K.S.L.; Vargas, S.O.; Callahan, M.J.; Bae, D.S.; Nigrovic, P.A. Enthesitis as a Component of Dactylitis in Psoriatic Juvenile Idiopathic Arthritis: Histology of an Established Clinical Entity. Pediatr. Rheumatol. Online J. 2015, 13, 7. [Google Scholar] [CrossRef] [Green Version]
- Costanza, M. Type 2 Inflammatory Responses in Autoimmune Demyelination of the Central Nervous System: Recent Advances. J. Immunol. Res. 2019, 2019, 4204512. [Google Scholar] [CrossRef] [Green Version]
- Binda, V.; Moroni, G.; Messa, P. ANCA-Associated Vasculitis with Renal Involvement. J. Nephrol. 2018, 31, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, T.; Joh, K.; Okonogi, H.; Koike, K.; Utsunomiya, Y.; Miyazaki, Y.; Matsushima, M.; Yoshimura, M.; Horikoshi, S.; Suzuki, Y.; et al. A Histologic Classification of IgA Nephropathy for Predicting Long-Term Prognosis: Emphasis on End-Stage Renal Disease. J. Nephrol. 2013, 26, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Tortajada, A.; Gutierrez, E.; Pickering, M.C.; Praga Terente, M.; Medjeral-Thomas, N. The Role of Complement in IgA Nephropathy. Mol. Immunol. 2019, 114, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-F.; Chen, M. Complement Activation in Progression of Chronic Kidney Disease. Adv. Exp. Med. Biol. 2019, 1165, 423–441. [Google Scholar] [CrossRef] [PubMed]
- Maixnerova, D.; Reily, C.; Bian, Q.; Neprasova, M.; Novak, J.; Tesar, V. Markers for the Progression of IgA Nephropathy. J. Nephrol. 2016, 29, 535–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villacorta, J.; Diaz-Crespo, F.; Acevedo, M.; Guerrero, C.; Campos-Martin, Y.; García-Díaz, E.; Mollejo, M.; Fernandez-Juarez, G. Glomerular C3d as a Novel Prognostic Marker for Renal Vasculitis. Hum. Pathol. 2016, 56, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Appel, G.B.; Cook, H.T.; Hageman, G.; Jennette, J.C.; Kashgarian, M.; Kirschfink, M.; Lambris, J.D.; Lanning, L.; Lutz, H.U.; Meri, S.; et al. Membranoproliferative Glomerulonephritis Type II (Dense Deposit Disease): An Update. J. Am. Soc. Nephrol. 2005, 16, 1392–1403. [Google Scholar] [CrossRef] [Green Version]
- Poppelaars, F.; Faria, B.; Gaya da Costa, M.; Franssen, C.F.M.; van Son, W.J.; Berger, S.P.; Daha, M.R.; Seelen, M.A. The Complement System in Dialysis: A Forgotten Story? Front. Immunol. 2018, 9, 71. [Google Scholar] [CrossRef] [Green Version]
- Azadegan-Dehkordi, F.; Bagheri, N.; Shirzad, H.; Rafieian-Kopaei, M. The Role of Th1 and Th17 Cells in Glomerulonephritis. J. Nephropathol. 2015, 4, 32–37. [Google Scholar] [CrossRef]
- Kurts, C.; Heymann, F.; Lukacs-Kornek, V.; Boor, P.; Floege, J. Role of T Cells and Dendritic Cells in Glomerular Immunopathology. Semin. Immunopathol. 2007, 29, 317–335. [Google Scholar] [CrossRef]
- Tang, Y.; He, H.; Hu, P.; Xu, X. T Lymphocytes in IgA Nephropathy. Exp. Ther. Med. 2020, 20, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Odobasic, D.; Gan, P.-Y.; Summers, S.A.; Semple, T.J.; Muljadi, R.C.M.; Iwakura, Y.; Kitching, A.R.; Holdsworth, S.R. Interleukin-17A Promotes Early but Attenuates Established Disease in Crescentic Glomerulonephritis in Mice. Am. J. Pathol. 2011, 179, 1188–1198. [Google Scholar] [CrossRef]
- Araújo, L.S.; Torquato, B.G.S.; da Silva, C.A.; Dos Reis Monteiro, M.L.G.; Dos Santos Martins, A.L.M.; da Silva, M.V.; Dos Reis, M.A.; Machado, J.R. Renal Expression of Cytokines and Chemokines in Diabetic Nephropathy. BMC Nephrol. 2020, 21, 308. [Google Scholar] [CrossRef]
- Calle, P.; Hotter, G. Macrophage Phenotype and Fibrosis in Diabetic Nephropathy. Int. J. Mol. Sci. 2020, 21, E2806. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.-J. Current Opinion for Hypertension in Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 37–47. [Google Scholar] [CrossRef]
- Ishii, H.; Kaneko, S.; Yanai, K.; Aomatsu, A.; Hirai, K.; Ookawara, S.; Ishibashi, K.; Morishita, Y. MicroRNAs in Podocyte Injury in Diabetic Nephropathy. Front. Genet. 2020, 11, 993. [Google Scholar] [CrossRef]
- Lassén, E.; Daehn, I.S. Molecular Mechanisms in Early Diabetic Kidney Disease: Glomerular Endothelial Cell Dysfunction. Int. J. Mol. Sci. 2020, 21, 9456. [Google Scholar] [CrossRef]
- Eftekhari, A.; Vahed, S.Z.; Kavetskyy, T.; Rameshrad, M.; Jafari, S.; Chodari, L.; Hosseiniyan, S.M.; Derakhshankhah, H.; Ahmadian, E.; Ardalan, M. Cell Junction Proteins: Crossing the Glomerular Filtration Barrier in Diabetic Nephropathy. Int. J. Biol. Macromol. 2020, 148, 475–482. [Google Scholar] [CrossRef]
- Roumeliotis, S.; Mallamaci, F.; Zoccali, C. Endothelial Dysfunction in Chronic Kidney Disease, from Biology to Clinical Outcomes: A 2020 Update. J. Clin. Med. 2020, 9, 2359. [Google Scholar] [CrossRef]
- Hohenstein, B.; Hugo, C.P.M.; Hausknecht, B.; Boehmer, K.P.; Riess, R.H.; Schmieder, R.E. Analysis of NO-Synthase Expression and Clinical Risk Factors in Human Diabetic Nephropathy. Nephrol. Dial. Transplant. 2008, 23, 1346–1354. [Google Scholar] [CrossRef] [Green Version]
- Ji, L.; Chen, Y.; Wang, H.; Zhang, W.; He, L.; Wu, J.; Liu, Y. Overexpression of Sirt6 Promotes M2 Macrophage Transformation, Alleviating Renal Injury in Diabetic Nephropathy. Int. J. Oncol. 2019, 55, 103–115. [Google Scholar] [CrossRef]
- Elmarakby, A.A.; Sullivan, J.C. Relationship between Oxidative Stress and Inflammatory Cytokines in Diabetic Nephropathy. Cardiovasc. Ther. 2012, 30, 49–59. [Google Scholar] [CrossRef]
- Landis, R.C.; Quimby, K.R.; Greenidge, A.R. M1/M2 Macrophages in Diabetic Nephropathy: Nrf2/HO-1 as Therapeutic Targets. Curr. Pharm. Des. 2018, 24, 2241–2249. [Google Scholar] [CrossRef]
- Zhao, L.; Zou, Y.; Liu, F. Transforming Growth Factor-Beta1 in Diabetic Kidney Disease. Front. Cell Dev. Biol. 2020, 8, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kume, S.; Maegawa, H. Lipotoxicity, Nutrient-Sensing Signals, and Autophagy in Diabetic Nephropathy. JMA J. 2020, 3, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Bai, X.; Chen, X. Autophagy and Diabetic Nephropathy. Adv. Exp. Med. Biol. 2020, 1207, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Ye, D.; Gao, C.; Huang, Q.; Gui, D. Mechanism of Progression of Diabetic Kidney Disease Mediated by Podocyte Mitochondrial Injury. Mol. Biol. Rep. 2020, 47, 8023–8035. [Google Scholar] [CrossRef] [PubMed]
- Console, L.; Scalise, M.; Giangregorio, N.; Tonazzi, A.; Barile, M.; Indiveri, C. The Link Between the Mitochondrial Fatty Acid Oxidation Derangement and Kidney Injury. Front. Physiol. 2020, 11, 794. [Google Scholar] [CrossRef]
- Wu, C.-C.; Sytwu, H.-K.; Lu, K.-C.; Lin, Y.-F. Role of T Cells in Type 2 Diabetic Nephropathy. Exp. Diabetes Res. 2011, 2011, 514738. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-C.; Sytwu, H.-K.; Lin, Y.-F. Cytokines in Diabetic Nephropathy. Adv. Clin. Chem. 2012, 56, 55–74. [Google Scholar] [CrossRef]
- Zhang, C.; Xiao, C.; Wang, P.; Xu, W.; Zhang, A.; Li, Q.; Xu, X. The Alteration of Th1/Th2/Th17/Treg Paradigm in Patients with Type 2 Diabetes Mellitus: Relationship with Diabetic Nephropathy. Hum. Immunol. 2014, 75, 289–296. [Google Scholar] [CrossRef]
- Hassanein, M.; Augustine, J.J. Chronic Kidney Transplant Rejection. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021; Available online: https://www.statpearls.com/articlelibrary/viewarticle/19539/ (accessed on 1 September 2021).
- Gusev, E.Y.; Solomatina, L.V.; Panshina, E.V.; Zhiravleva, J.A.; Zubova, T.E. Systemic Inflammation in Chronic Renal Allograft Dysfunction. Nephrol. Dyalysis 2011, 13, 82–88. (In Russian) [Google Scholar]
- Carron, C.; Pais de Barros, J.-P.; Gaiffe, E.; Deckert, V.; Adda-Rezig, H.; Roubiou, C.; Laheurte, C.; Masson, D.; Simula-Faivre, D.; Louvat, P.; et al. End-Stage Renal Disease-Associated Gut Bacterial Translocation: Evolution and Impact on Chronic Inflammation and Acute Rejection After Renal Transplantation. Front. Immunol. 2019, 10, 1630. [Google Scholar] [CrossRef] [Green Version]
- Loupy, A.; Haas, M.; Roufosse, C.; Naesens, M.; Adam, B.; Afrouzian, M.; Akalin, E.; Alachkar, N.; Bagnasco, S.; Becker, J.U.; et al. The Banff 2019 Kidney Meeting Report (I): Updates on and clarification of criteria for T cell- and antibody-mediated rejection. Am. J. Transplant. 2020, 20, 2318–2331. [Google Scholar] [CrossRef]
- Infante, B.; Rossini, M.; Di Lorenzo, A.; Coviello, N.; Giuseppe, C.; Gesualdo, L.; Giuseppe, G.; Stallone, G. Recurrence of Immunoglobulin A Nephropathy after Kidney Transplantation: A Narrative Review of the Incidence, Risk Factors, Pathophysiology and Management of Immunosuppressive Therapy. Clin. Kidney J. 2020, 13, 758–767. [Google Scholar] [CrossRef]
- Bruneau, S.; Berre, L.L.; Hervé, C.; Valanciuté, A.; Kamal, M.; Naulet, J.; Tesson, L.; Foucher, Y.; Soulillou, J.-P.; Sahali, D.; et al. Potential Role of Soluble ST2 Protein in Idiopathic Nephrotic Syndrome Recurrence Following Kidney Transplantation. Am. J. Kidney Dis. 2009, 54, 522–532. [Google Scholar] [CrossRef] [Green Version]
- Cohen, E.; Korah, M.; Callender, G.; Belfort de Aguiar, R.; Haakinson, D. Metabolic Disorders with Kidney Transplant. Clin. J. Am. Soc. Nephrol 2020, 15, 732–742. [Google Scholar] [CrossRef]
- Goto, E.; Honjo, S.; Yamashita, H.; Shomori, K.; Adachi, H.; Ito, H. Mast Cells in Human Allografted Kidney: Correlation with Interstitial Fibrosis. Clin. Transplant. 2002, 16, 7–11. [Google Scholar] [CrossRef]
- Bhatti, A.B.; Usman, M. Chronic Renal Transplant Rejection and Possible Anti-Proliferative Drug Targets. Cureus 2015, 7, e376. [Google Scholar] [CrossRef] [Green Version]
- Jager, N.M.; Poppelaars, F.; Daha, M.R.; Seelen, M.A. Complement in Renal Transplantation: The Road to Translation. Mol. Immunol. 2017, 89, 22–35. [Google Scholar] [CrossRef]
- Karahan, H.I.; Soyöz, M.; Pehlivan, M.; Tatar, E.; Uslu, A.; Çerçi Gürbüz, B.; Pirim, I.; Kiliçaslan Ayna, T. Assessment of Interleukin 2 Cytokine Expression Levels After Renal Transplantation. Transplant. Proc. 2019, 51, 1074–1077. [Google Scholar] [CrossRef]
- Lion, J.; Taflin, C.; Cross, A.R.; Robledo-Sarmiento, M.; Mariotto, E.; Savenay, A.; Carmagnat, M.; Suberbielle, C.; Charron, D.; Haziot, A.; et al. HLA Class II Antibody Activation of Endothelial Cells Promotes Th17 and Disrupts Regulatory T Lymphocyte Expansion. Am. J. Transplant. 2016, 16, 1408–1420. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Zhang, H.; Hu, K.; Lv, G.; Fu, Y.; Ayana, D.A.; Zhao, P.; Jiang, Y. The Imbalance between Tregs, Th17 Cells and Inflammatory Cytokines among Renal Transplant Recipients. BMC Immunol. 2015, 16, 56. [Google Scholar] [CrossRef] [Green Version]
- Tse, G.H.; Johnston, C.J.C.; Kluth, D.; Gray, M.; Gray, D.; Hughes, J.; Marson, L.P. Intrarenal B Cell Cytokines Promote Transplant Fibrosis and Tubular Atrophy. Am. J. Transplant. 2015, 15, 3067–3080. [Google Scholar] [CrossRef]
- Loverre, A.; Divella, C.; Castellano, G.; Tataranni, T.; Zaza, G.; Rossini, M.; Ditonno, P.; Battaglia, M.; Palazzo, S.; Gigante, M.; et al. T Helper 1, 2 and 17 Cell Subsets in Renal Transplant Patients with Delayed Graft Function. Transpl. Int. 2011, 24, 233–242. [Google Scholar] [CrossRef]
- Zotova, N.V.; Chereshnev, V.A.; Gusev, E.Y. Systemic Inflammation: Methodological Approaches to Identification of the Common Pathological Process. PLoS ONE 2016, 11, e0155138. [Google Scholar] [CrossRef]
- Zotova, N.V.; Zhuravleva, Y.V.; Zubova, T.E.; Gusev, E.Y. Integral Estimation of Systemic Inflammatory Response under Sepsis. Gen Physiol Biophys 2020, 39, 13–26. [Google Scholar] [CrossRef]
- Batko, K.; Krzanowski, M.; Gajda, M.; Dumnicka, P.; Fedak, D.; Woziwodzka, K.; Sułowicz, W.; Kuźniewski, M.; Litwin, J.A.; Krzanowska, K. Endothelial Injury Is Closely Related to Osteopontin and TNF Receptor-Mediated Inflammation in End-Stage Renal Disease. Cytokine 2019, 121, 154729. [Google Scholar] [CrossRef]
- Bi, X.; Chu, M.; Ai, H.; Hu, C.; Ding, W. Association of Serum IL-18 with Protein-Energy Wasting in End-Stage Renal Disease Patients on Haemodialysis. Int. Urol. Nephrol. 2019, 51, 1271–1278. [Google Scholar] [CrossRef]
- Gohda, T.; Maruyama, S.; Kamei, N.; Yamaguchi, S.; Shibata, T.; Murakoshi, M.; Horikoshi, S.; Tomino, Y.; Ohsawa, I.; Gotoh, H.; et al. Circulating TNF Receptors 1 and 2 Predict Mortality in Patients with End-Stage Renal Disease Undergoing Dialysis. Sci. Rep. 2017, 7, 43520. [Google Scholar] [CrossRef]
- Hartzell, S.; Bin, S.; Cantarelli, C.; Haverly, M.; Manrique, J.; Angeletti, A.; Manna, G.L.; Murphy, B.; Zhang, W.; Levitsky, J.; et al. Kidney Failure Associates with T Cell Exhaustion and Imbalanced Follicular Helper T Cells. Front. Immunol. 2020, 11, 583702. [Google Scholar] [CrossRef] [PubMed]
- Hojs, R.; Ekart, R.; Bevc, S.; Hojs, N. Markers of Inflammation and Oxidative Stress in the Development and Progression of Renal Disease in Diabetic Patients. Nephron 2016, 133, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Mak, R.H.; Cheung, W.; Cone, R.D.; Marks, D.L. Mechanisms of Disease: Cytokine and Adipokine Signaling in Uremic Cachexia. Nat. Clin. Pract. Nephrol. 2006, 2, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Oncel, M.; Akbulut, S.; Toka Ozer, T.; Kiyici, A.; Keles, M.; Baltaci, B.; Turk, S. Cytokines, Adipocytokines and Inflammatory Markers in Patients on Continuous Ambulatory Peritoneal Dialysis and Hemodialysis. Ren. Fail. 2016, 38, 1071–1075. [Google Scholar] [CrossRef] [Green Version]
- Rusu, C.C.; Racasan, S.; Kacso, I.M.; Ghervan, L.; Moldovan, D.; Potra, A.; Patiu, I.M.; Bondor, C.; Caprioara, M.G. The Association of High SCD163/STWEAK Ratio with Cardiovascular Disease in Hemodialysis Patients. Int. Urol. Nephrol. 2015, 47, 2023–2030. [Google Scholar] [CrossRef]
- Tao, R.; Fan, Q.; Zhang, H.; Xie, H.; Lu, L.; Gu, G.; Wang, F.; Xi, R.; Hu, J.; Chen, Q.; et al. Prognostic Significance of Interleukin-34 (IL-34) in Patients with Chronic Heart Failure with or Without Renal Insufficiency. J. Am. Heart Assoc. 2017, 6, e004911. [Google Scholar] [CrossRef]
- Jin, K.; Vaziri, N.D. Elevated Plasma Cyclophillin A in Hemodialysis and Peritoneal Dialysis Patients: A Novel Link to Systemic Inflammation. Iran. J. Kidney Dis. 2017, 11, 44–49. [Google Scholar]
- Bessa, J.; Albino-Teixeira, A.; Reina-Couto, M.; Sousa, T. Endocan: A Novel Biomarker for Risk Stratification, Prognosis and Therapeutic Monitoring in Human Cardiovascular and Renal Diseases. Clin. Chim. Acta 2020, 509, 310–335. [Google Scholar] [CrossRef]
- Desjardins, M.-P.; Thorin-Trescases, N.; Sidibé, A.; Fortier, C.; De Serres, S.A.; Larivière, R.; Thorin, E.; Agharazii, M. Levels of Angiopoietin-Like-2 Are Positively Associated with Aortic Stiffness and Mortality After Kidney Transplantation. Am. J. Hypertens. 2017, 30, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Erturk, I.; Yesildal, F.; Acar, R.; Ozgurtas, T.; Saglam, K. Vascular Endothelial Growth Factor and Soluble Vascular Endothelial Growth Factor Receptor-1 in Patients with End-Stage Renal Disease. Associations with Laboratory Findings, Comorbidities, and Medications. Saudi Med. J. 2018, 39, 586–591. [Google Scholar] [CrossRef] [Green Version]
- Gusev, E.Y.; Solomatina, L.V.; Zhuravleva, Y.A.; Zubova, T.E. Systemic Inflammatory Reaction in ESRD Patients. Nephrol. Dial. 2008, 10, 248–253. (In Russian) [Google Scholar]
- Gusev, E.Y.; Solomatina, L.V.; Zhuravleva, J.A.; Zubova, T.E. Comparative Analysis of Markers of Systemic Inflammatory Reaction in End-Stage Renal Disease (ESRD)Patients. Nephrol. Dial. 2009, 11, 123–128. (In Russian) [Google Scholar]
- Solomatina, L.V. The Role of Chronic Systemic Inflammation in the Pathogenesis of End-Stage Renal Failure in Patients Receiving Programmed Hemodialysis Replacement Therapy. Ph.D. Thesis, Institute of Immunology and Physiology of Ural Branch of the Russian Academy of Science, Ekaterinburg, Russia, 30 March 2012. (In Russian). [Google Scholar]
- Li, X.-Q.; Lerman, L.O.; Meng, Y. Potential Role of Extracellular Vesicles in the Pathophysiology of Glomerular Diseases. Clin. Sci. 2020, 134, 2741–2754. [Google Scholar] [CrossRef]
- Zhang, W.; Zhou, X.; Zhang, H.; Yao, Q.; Liu, Y.; Dong, Z. Extracellular Vesicles in Diagnosis and Therapy of Kidney Diseases. Am. J. Physiol.-Ren. Physiol. 2016, 311, F844–F851. [Google Scholar] [CrossRef] [Green Version]
- Lutz, J.; Menke, J.; Sollinger, D.; Schinzel, H.; Thürmel, K. Haemostasis in Chronic Kidney Disease. Nephrol. Dial. Transplant. 2014, 29, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Haller, C.; Zehelein, J.; Remppis, A.; Müller-Bardorff, M.; Katus, H.A. Cardiac Troponin T in Patients with End-Stage Renal Disease: Absence of Expression in Truncal Skeletal Muscle. Clin. Chem. 1998, 44, 930–938. [Google Scholar] [CrossRef] [Green Version]
- Contreras, A.M.; Ruiz, I.; Polanco-Cruz, G.; Monteón, F.J.; Celis, A.; Vázquez, G.; Gómez-Herrera, E.; García-Correa, J.E.; Male-Velázquez, R.; Ruelas-Hernández, S. End-Stage Renal Disease and Hepatitis C Infection: Comparison of Alanine Aminotransferase Levels and Liver Histology in Patients with and without Renal Damage. Ann. Hepatol. 2007, 6, 48–54. [Google Scholar] [CrossRef]
- Graul, A.I.; Stringer, M.; Sorbera, L. Cachexia. Drugs Today 2016, 52, 519–529. [Google Scholar] [CrossRef]
- Sabatino, A.; Cuppari, L.; Stenvinkel, P.; Lindholm, B.; Avesani, C.M. Sarcopenia in Chronic Kidney Disease: What Have We Learned so Far? J. Nephrol. 2021, 34, 1347–1372. [Google Scholar] [CrossRef]
- Arimura, T.; Shiba, T.; Takahashi, M.; Kumashiro, S.; Osamura, H.; Matsumoto, T.; Sakai, K.; Hori, Y. Assessment of Ocular Microcirculation in Patients with End-Stage Kidney Disease. Graefe’s Arch. Clin. Exp. Ophthalmol. 2018, 256, 2335–2340. [Google Scholar] [CrossRef]
- Nelson, A.J.; Dundon, B.K.; Worthley, S.G.; Richardson, J.D.; Puri, R.; Wong, D.T.L.; Coates, P.T.; Faull, R.J.; Worthley, M.I. End-Stage Renal Failure Is Associated with Impaired Coronary Microvascular Function. Coron. Artery Dis. 2019, 30, 520–527. [Google Scholar] [CrossRef]
- Smogorzewski, M.J. Skin Blood Flow and Vascular Endothelium Function in Uremia. J. Ren. Nutr. 2017, 27, 465–469. [Google Scholar] [CrossRef]
- Houben, A.J.H.M.; Martens, R.J.H.; Stehouwer, C.D.A. Assessing Microvascular Function in Humans from a Chronic Disease Perspective. J. Am. Soc. Nephrol. 2017, 28, 3461–3472. [Google Scholar] [CrossRef] [Green Version]
- Meinders, A.-J.; Nieuwenhuis, L.; Ince, C.; Bos, W.-J.; Elbers, P.W.G. Haemodialysis Impairs the Human Microcirculation Independent from Macrohemodynamic Parameters. Blood Purif. 2015, 40, 38–44. [Google Scholar] [CrossRef]
- Yeh, Y.C.; Chao, A.; Lee, C.-Y.; Lee, C.-T.; Yeh, C.-C.; Liu, C.-M.; Tsai, M.-K. An Observational Study of Microcirculation in Dialysis Patients and Kidney Transplant Recipients. Eur. J. Clin. Investig. 2017, 47, 630–637. [Google Scholar] [CrossRef]
- Popova, J.A.; Yadrihinskaya, V.N.; Krylova, M.I.; Sleptsova, S.S.; Borisova, N.V. Comparison of Clinical and Laboratory Parameters in Patients with End-Stage Renal Failure in the Outcome of Chronic Glomerulonephritis and Patients with End-Stage Renal Failure in the Outcome of Other Diseases. Wiad Lek. 2016, 69, 739–741. [Google Scholar]
- Rios, D.R.A.; Carvalho, M.d.G.; Lwaleed, B.A.; Simões e Silva, A.C.; Borges, K.B.G.; Dusse, L.M.S. Hemostatic Changes in Patients with End Stage Renal Disease Undergoing Hemodialysis. Clin. Chim. Acta 2010, 411, 135–139. [Google Scholar] [CrossRef]
- Jalal, D.I.; Chonchol, M.; Targher, G. Disorders of Hemostasis Associated with Chronic Kidney Disease. Semin. Thromb. Hemost. 2010, 36, 34–40. [Google Scholar] [CrossRef]
- Hakim, R.M. Clinical Sequelae of Complement Activation in Hemodialysis. Clin. Nephrol. 1986, 26, S9–S12. [Google Scholar]
- Kubatiev, A.; Rudko, I.; Ermolenko, V. Complement Activation and Neutrophil Aggregation Changes during Haemodialysis. Int. J. Clin. Pharmacol. Res. 1993, 13, 293–299. [Google Scholar]
- Robertson, L.M.; Frith, J.A.; Tcheurekdjian, H.; Hostoffer, R.W. Possible Mast Cell Activation Syndrome in a Patient undergoing Long-Term Hemodialysis. Ann. Allergy Asthma Immunol. 2016, 116, 576–577. [Google Scholar] [CrossRef] [PubMed]
- Narita, I.; Iguchi, S.; Omori, K.; Gejyo, F. Uremic Pruritus in Chronic Hemodialysis Patients. J. Nephrol. 2008, 21, 161–165. [Google Scholar] [PubMed]
- Leong, S.O.; Tan, C.C.; Lye, W.C.; Lee, E.J.; Chan, H.L. Dermal Mast Cell Density and Pruritus in End-Stage Renal Failure. Ann. Acad. Med. Singap. 1994, 23, 327–329. [Google Scholar] [PubMed]
- Oweis, A.O.; Al-Qarqaz, F.; Bodoor, K.; Heis, L.; Alfaqih, M.A.; Almomani, R.; Obeidat, M.A.; Alshelleh, S.A. Elevated Interleukin 31 Serum Levels in Hemodialysis Patients Are Associated with Uremic Pruritus. Cytokine 2021, 138, 155369. [Google Scholar] [CrossRef]
- Ellis, H.A.; Peart, K.M.; Pierides, A.M. Effect of Renal Transplantation on Marrow Mast Cell Hyperplasia of Chronic Renal Failure. J. Clin. Pathol. 1977, 30, 960–965. [Google Scholar] [CrossRef] [Green Version]
- Dugas-Breit, S.; Schöpf, P.; Dugas, M.; Schiffl, H.; Ruëff, F.; Przybilla, B. Baseline Serum Levels of Mast Cell Tryptase Are Raised in Hemodialysis Patients and Associated with Severity of Pruritus. J. Dtsch. Dermatol. Ges. 2005, 3, 343–347. [Google Scholar] [CrossRef]
- Wasse, H.; Rivera, A.A.; Huang, R.; Martinson, D.E.; Long, Q.; McKinnon, W.; Naqvi, N.; Husain, A. Increased Plasma Chymase Concentration and Mast Cell Chymase Expression in Venous Neointimal Lesions of Patients with CKD and ESRD. Semin. Dial. 2011, 24, 688–693. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wu, J.-H.; Otto, J.C.; Gurley, S.B.; Hauser, E.R.; Shenoy, S.K.; Nagi, K.; Brian, L.; Wertman, V.; Mattocks, N.; et al. Interleukin-9 Mediates Chronic Kidney Disease-Dependent Vein Graft Disease: A Role for Mast Cells. Cardiovasc. Res. 2017, 113, 1551–1559. [Google Scholar] [CrossRef] [Green Version]
- Ekdahl, K.N.; Soveri, I.; Hilborn, J.; Fellström, B.; Nilsson, B. Cardiovascular Disease in Haemodialysis: Role of the Intravascular Innate Immune System. Nat. Rev. Nephrol. 2017, 13, 285–296. [Google Scholar] [CrossRef]
- Laudański, K.; Nowak, Z. Aberrant Function and Differentiation of Monocytes in End Stage Renal Disease. Arch. Immunol. Ther. Exp. 2012, 60, 453–459. [Google Scholar] [CrossRef]
- Bronze-da-Rocha, E.; Santos-Silva, A. Neutrophil Elastase Inhibitors and Chronic Kidney Disease. Int. J. Biol. Sci. 2018, 14, 1343–1360. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-K.; Lee, H.W.; Joo, N.; Lee, H.S.; Song, Y.R.; Kim, H.J.; Kim, S.G. Prognostic Role of Circulating Neutrophil Extracellular Traps Levels for Long-Term Mortality in New End-Stage Renal Disease Patients. Clin. Immunol. 2020, 210, 108263. [Google Scholar] [CrossRef]
- Fukushi, T.; Yamamoto, T.; Yoshida, M.; Fujikura, E.; Miyazaki, M.; Nakayama, M. Enhanced Neutrophil Apoptosis Accompanying Myeloperoxidase Release during Hemodialysis. Sci. Rep. 2020, 10, 21747. [Google Scholar] [CrossRef]
- Kim, J.-K.; Hong, C.-W.; Park, M.J.; Song, Y.R.; Kim, H.J.; Kim, S.G. Increased Neutrophil Extracellular Trap Formation in Uremia Is Associated with Chronic Inflammation and Prevalent Coronary Artery Disease. J. Immunol. Res. 2017, 2017, 8415179. [Google Scholar] [CrossRef]
- Hällgren, R.; Grefberg, N.; Venge, P. Elevated Circulating Levels of Eosinophil Cationic Protein in Uremia as Signs of Abnormal Eosinophil Homeostasis. Nephron 1984, 36, 10–14. [Google Scholar] [CrossRef]
- Ferraris, J.R.; Ramirez, J.A.; Goldberg, V.; Rivarola, M.A. Glucocorticoids and Adrenal Androgens in Children with End Stage Renal Disease. Acta Endocrinol. 1991, 124, 245–250. [Google Scholar] [CrossRef]
- Raff, H.; Trivedi, H. Circadian Rhythm of Salivary Cortisol, Plasma Cortisol, and Plasma ACTH in End-Stage Renal Disease. Endocr. Connect. 2013, 2, 23–31. [Google Scholar] [CrossRef]
- Arregger, A.L.; Cardoso, E.M.L.; Zucchini, A.; Aguirre, E.C.; Elbert, A.; Contreras, L.N. Adrenocortical Function in Hypotensive Patients with End Stage Renal Disease. Steroids 2014, 84, 57–63. [Google Scholar] [CrossRef]
- Kocyigit, I.; Unal, A.; Tanriverdi, F.; Hayri Sipahioglu, M.; Tokgoz, B.; Oymak, O.; Utas, C. Misdiagnosis of Addison’s Disease in a Patient with End-Stage Renal Disease. Ren. Fail. 2011, 33, 88–91. [Google Scholar] [CrossRef]
- Gusev, E.Y.; Chereshnev, V.A.; Zhuravleva, Y.A.; Solomatina, L.V.; Zubova, T.E. Progression Variants of Chronic Systemic Inflammation. Med. Immunol. 2009, 11, 131–140. (In Russian) [Google Scholar] [CrossRef] [Green Version]
- Arriens, C.; Chen, S.; Karp, D.R.; Saxena, R.; Sambandam, K.; Chakravarty, E.; James, J.A.; Merrill, J.T. Prognostic Significance of Repeat Biopsy in Lupus Nephritis: Histopathologic Worsening and a Short Time between Biopsies Is Associated with Significantly Increased Risk for End Stage Renal Disease and Death. Clin. Immunol. 2017, 185, 3–9. [Google Scholar] [CrossRef]
- Gusev, E.; Sarapultsev, A.; Hu, D.; Chereshnev, V. Problems of Pathogenesis and Pathogenetic Therapy of COVID-19 from the Perspective of the General Theory of Pathological Systems (General Pathological Processes). Int. J. Mol. Sci. 2021, 22, 7582. [Google Scholar] [CrossRef]
- Sasso, F.C.; Pafundi, P.C.; Simeon, V.; De Nicola, L.; Chiodini, P.; Galiero, R.; Rinaldi, L.; Nevola, R.; Salvatore, T.; Sardu, C.; et al. Efficacy and durability of multifactorial intervention on mortality and MACEs: A randomized clinical trial in type-2 diabetic kidney disease. Cardiovasc. Diabetol. 2021, 20, 145. [Google Scholar] [CrossRef] [PubMed]



| ChSI Phenomena | Partial ChSI Criteria | Unit | Norm | ChSI Scale Points |
|---|---|---|---|---|
| Systemic inflammatory response | RL scale | Points (0 to 5) | 0 | 1 RL point = 1 ChSI scale point |
| Microthrombus formation | D-dimers >500 | ng/ml | <250 | 1 point |
| Systemic alteration | Myoglobin >60 | ng/ml | <25 | 1 point |
| Troponin I >0.2 | ng/ml | <0.2 | ||
| Distress reaction of the hypothalamic pituitary adrenal system | Cortisol >690 | nmol/L | 138–690 | 1 point |
| Cortisol <100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gusev, E.; Solomatina, L.; Zhuravleva, Y.; Sarapultsev, A. The Pathogenesis of End-Stage Renal Disease from the Standpoint of the Theory of General Pathological Processes of Inflammation. Int. J. Mol. Sci. 2021, 22, 11453. https://doi.org/10.3390/ijms222111453
Gusev E, Solomatina L, Zhuravleva Y, Sarapultsev A. The Pathogenesis of End-Stage Renal Disease from the Standpoint of the Theory of General Pathological Processes of Inflammation. International Journal of Molecular Sciences. 2021; 22(21):11453. https://doi.org/10.3390/ijms222111453
Chicago/Turabian StyleGusev, Evgenii, Liliya Solomatina, Yulia Zhuravleva, and Alexey Sarapultsev. 2021. "The Pathogenesis of End-Stage Renal Disease from the Standpoint of the Theory of General Pathological Processes of Inflammation" International Journal of Molecular Sciences 22, no. 21: 11453. https://doi.org/10.3390/ijms222111453
APA StyleGusev, E., Solomatina, L., Zhuravleva, Y., & Sarapultsev, A. (2021). The Pathogenesis of End-Stage Renal Disease from the Standpoint of the Theory of General Pathological Processes of Inflammation. International Journal of Molecular Sciences, 22(21), 11453. https://doi.org/10.3390/ijms222111453