
Epigenetic Dysregulations in Merkel Cell Polyomavirus-Driven Merkel Cell Carcinoma

 ,
,  ,
, 
 and
and
Abstract
:1. Introduction
1.1. Merkel Cell Polyomavirus: Genomic Organization and Oncogenic Activity
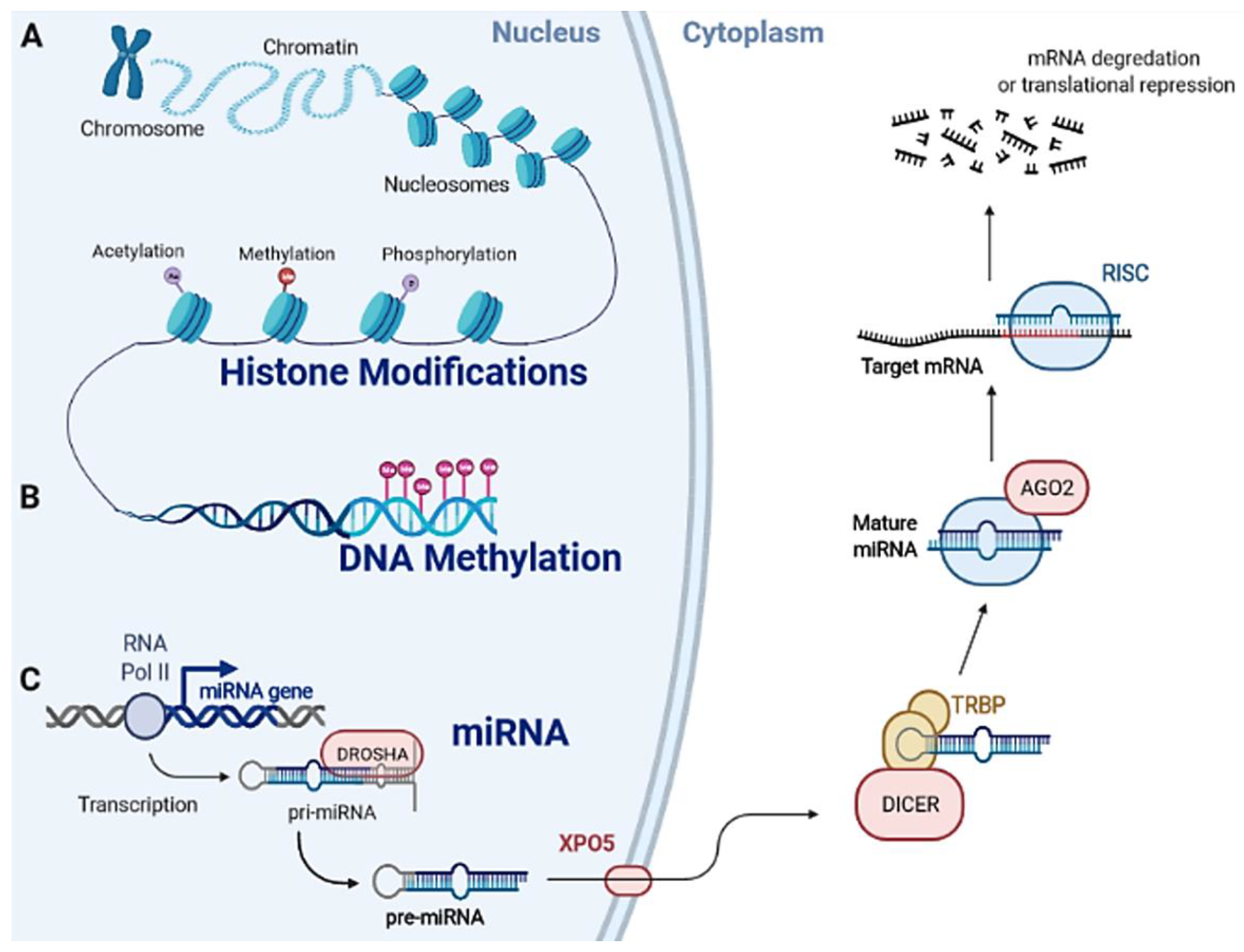
1.2. Epigenetic Machinery
2. Methods
3. Epigenetic Dysregulations in Merkel-Cell-Polyomavirus-Driven Merkel Cell Carcinoma
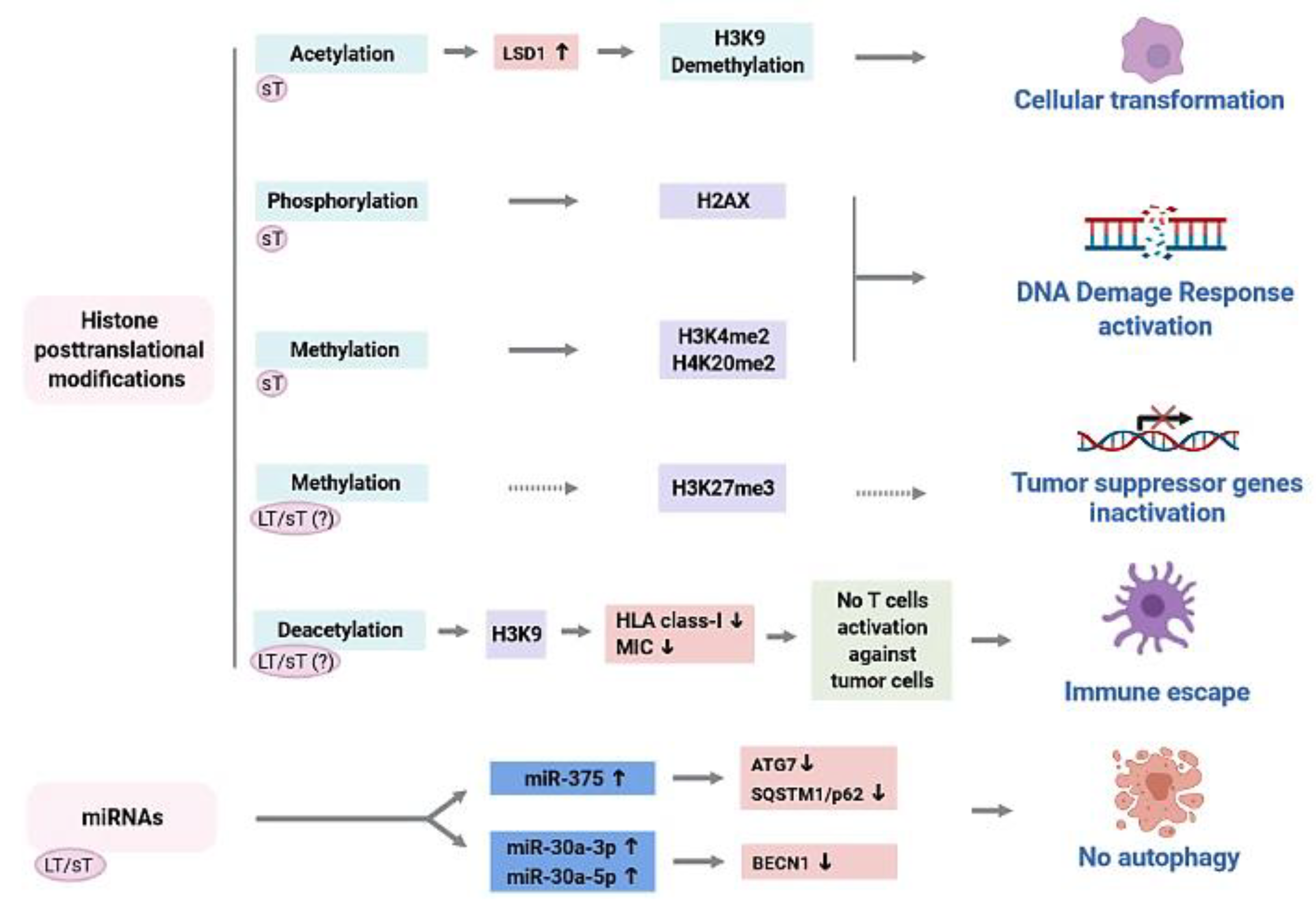
3.1. Aberrant Epigenetic Modifications in Merkel-Cell-Polyomavirus-Driven Merkel Cell Carcinoma
3.2. Role of Merkel Cell Polyomavirus (MCPyV) Oncoproteins in the Epigenetic Dysregulation of MCPyV-Driven Merkel Cell Carcinoma
3.3. Epigenetic Dysregulations as Diagnostic, Prognostic, and Therapy Target Tools in Merkel Cell Polyomavirus-Driven Merkel Cell Carcinoma
4. Discussion and Future Perspectives
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [Green Version]
- Rotondo, J.C.; Bononi, I.; Puozzo, A.; Govoni, M.; Foschi, V.; Lanza, G.; Gafa, R.; Gaboriaud, P.; Touzé, F.A.; Selvatici, R.; et al. Merkel cell carcinomas arising in autoimmune disease affected patients treated with biologic drugs including anti-TNF. Clin. Cancer Res. 2017, 23, 3929–3934. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.T.; Park, S.J.; Choi, E.K.; Kim, Y.S. The frequency of Merkel cell polyomavirus in whole blood from immunocompetent and immunosuppressed patients with kidney disease and healthy donors. Microb. Pathog. 2019, 131, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.W.; Qazi, J.; Hippe, D.S.; Lachance, K.; Thomas, H.; Cook, M.M.; Juhlin, I.; Singh, N.; Thuesmunn, Z.; Takagishi, S.R.; et al. Patterns of distant metastases in 215 Merkel cell carcinoma patients: Implications for prognosis and surveillance. Cancer Med. 2020, 9, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Zwijnenburg, E.M.; Lubeek, S.F.K.; Werner, J.E.M.; Amir, A.L.; Weijs, W.L.J.; Takes, R.P.; Pegge, S.A.H.; van Herpen, C.M.L.; Adema, G.J.; Kaanders, J.H.A.M. Merkel cell carcinoma: New trends. Cancers 2021, 13, 1614. [Google Scholar] [CrossRef]
- Pietropaolo, V.; Prezioso, C.; Moens, U. merkel cell polyomavirus and merkel cell carcinoma. Cancers 2020, 12, 1774. [Google Scholar] [CrossRef] [PubMed]
- Starrett, G.J.; Thakuria, M.; Chen, T.; Marcelus, C.; Cheng, J.; Nomburg, J.; Thorner, A.R.; Slevin, M.K.; Powers, W.; Burns, R.T.; et al. Clinical and molecular characterization of virus-positive and virus-negative Merkel cell carcinoma. Genome Med. 2020, 12, 30. [Google Scholar] [CrossRef] [Green Version]
- Leiendecker, L.; Jung, P.S.; Krecioch, I.; Neumann, T.; Schleiffer, A.; Mechtler, K.; Wiesner, T.; Obenauf, A.C. LSD 1 inhibition induces differentiation and cell death in Merkel cell carcinoma. EMBO Mol. Med. 2020, 12, e12525. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Diaz, J.; Tsang, S.H.; Buck, C.B.; You, J. Merkel Cell Polyomavirus Large T Antigen Disrupts Host Genomic Integrity and Inhibits Cellular Proliferation. J. Virol. 2013, 87, 9173–9188. [Google Scholar] [CrossRef] [Green Version]
- Del Marmol, V.; Lebbé, C. New perspectives in Merkel cell carcinoma. Curr. Opin. Oncol. 2019, 31, 72–83. [Google Scholar] [CrossRef]
- Kanwal, R.; Gupta, S. Epigenetic modifications in cancer. Clin. Genet. 2012, 81, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Kashyap, M.P.; Sinha, R.; Mukhtar, M.S.; Athar, M. Epigenetic regulation in the pathogenesis of non-melanoma skin cancer. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Li, F.-J.; Wu, Y.; Lv, Q.; Yang, X.-Y.; Jiang, M.-J.; Li, L.-M. Aberrant DNA Methylation in Cutaneous Squamous Cell Carcinoma. Int. J. Dermatol. Venereol. 2019, 2, 227–232. [Google Scholar] [CrossRef]
- Thuijs, N.B.; Berkhof, J.; Özer, M.; Duin, S.; van Splunter, A.P.; Snoek, B.C.; Heideman, D.A.M.; van Beurden, M.; Steenbergen, R.D.M.; Bleeker, M.C.G. DNA methylation markers for cancer risk prediction of vulvar intraepithelial neoplasia. Int. J. Cancer 2021, 148, 2481–2488. [Google Scholar] [CrossRef]
- Hsieh, I.-y.; He, J.; Wang, L.; Lin, B.; Liang, Z.; Lu, B.; Chen, W.; Lu, G.; Li, F.; Lv, W.; et al. H3K27me3 loss plays a vital role in CEMIP mediated carcinogenesis and progression of breast cancer with poor prognosis. Biomed. Pharmacother. 2020, 123, 109728. [Google Scholar] [CrossRef] [PubMed]
- Saki, J.; Sabaghan, M.; Arjmand, R.; Teimoori, A.; Rashno, M.; Saki, G.; Shojaee, S. Curcumin as an indirect methylation inhibitor modulates the effects of toxoplasma gondii on genes involved in male fertility. EXCLI J. 2020, 19, 1196–1207. [Google Scholar] [PubMed]
- Tsai, K.; Cullen, B.R. Epigenetic and epitranscriptomic regulation of viral replication. Nat. Rev. Microbiol. 2020, 18, 559–570. [Google Scholar] [CrossRef]
- Fischer, N. Infection-induced epigenetic changes and their impact on the pathogenesis of diseases. Semin. Immunopathol. 2020, 42, 127–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lourenço de Freitas, N.; Deberaldini, M.G.; Gomes, D.; Pavan, A.R.; Sousa, Â.; Dos Santos, J.L.; Soares, C.P. Histone Deacetylase Inhibitors as Therapeutic Interventions on Cervical Cancer Induced by Human Papillomavirus. Front. Cell Dev. Biol. 2021, 8, 592868. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Kurokawa, T.; Mima, M.; Imamoto, S.; Mizokami, H.; Kondo, S.; Okamoto, Y.; Misawa, K.; Hanazawa, T.; Kaneda, A. DNA methylation and hpv-associated head and neck cancer. Microorganisms 2021, 9, 801. [Google Scholar] [CrossRef]
- Emmett, S.E.; Stark, M.S.; Pandeya, N.; Panizza, B.; Whiteman, D.C.; Antonsson, A. MicroRNA expression is associated with human papillomavirus status and prognosis in mucosal head and neck squamous cell carcinomas. Oral Oncol. 2021, 113, 105136. [Google Scholar] [CrossRef]
- Ocadiz-Delgado, R.; Cruz-Colin, J.-L.; Alvarez-Rios, E.; Torres-Carrillo, A.; Hernandez-Mendoza, K.; Conde-Pérezprina, J.-C.; Dominguez-Gomez, G.-I.; Garcia-Villa, E.; Lambert, P.F.; Gariglio, P. Expression of miR-34a and miR-15b during the progression of cervical cancer in a murine model expressing the HPV16 E7 oncoprotein. J. Physiol. Biochem. 2021. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhao, H.; Zhang, Q.; Shi, Z.; Zhang, Y.; Zhao, L.; Ren, Y.; Ou, R.; Xu, Y. Human papillomavirus type 16 E7 oncoprotein-induced upregulation of lysine-specific demethylase 5A promotes cervical cancer progression by regulating the microRNA-424–5p/suppressor of zeste 12 pathway. Exp. Cell Res. 2020, 396, 112277. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos Bezerra, R.; Bitencourt, H.T.; Covas, D.T.; Kashima, S.; Slavov, S.N. Molecular evolution pattern of Merkel cell polyomavirus identified by viral metagenomics in plasma of high-risk blood donors from the Brazilian Amazon. Infect. Genet. Evol. 2020, 85, 104563. [Google Scholar] [CrossRef]
- Prezioso, C.; Bianchi, M.; Obregon, F.; Ciotti, M.; Sarmati, L.; Andreoni, M.; Palamara, A.T.; Pascarella, S.; Moens, U.; Pietropaolo, V. Structural analysis of merkel cell polyomavirus (MCPyV) viral capsid protein 1 (VP1) in HIV-1 infected individuals. Int. J. Mol. Sci. 2020, 21, 7998. [Google Scholar] [CrossRef] [PubMed]
- McIlroy, D.; Halary, F.; Bressollette-Bodin, C. Intra-patient viral evolution in polyomavirus-related diseases. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180301. [Google Scholar] [CrossRef] [Green Version]
- Decaprio, J.A. Molecular Pathogenesis of Merkel Cell Carcinoma. Annu. Rev. Pathol. Mech. Dis. 2021, 16, 69–91. [Google Scholar] [CrossRef]
- Gales, J.P.; Kubina, J.; Geldreich, A.; Dimitrova, M. Strength in Diversity: Nuclear Export of Viral RNAs. Viruses 2020, 12, 1014. [Google Scholar] [CrossRef]
- Wendzicki, J.A.; Moore, P.S.; Chang, Y. Large T and small T antigens of Merkel cell polyomavirus. Curr. Opin. Virol. 2015, 11, 38–43. [Google Scholar] [CrossRef] [Green Version]
- Carter, J.J.; Daugherty, M.D.; Qi, X.; Bheda-Malge, A.; Wipf, G.C.; Robinson, K.; Roman, A.; Malik, H.S.; Galloway, D.A. Identification of an overprinting gene in Merkel cell polyomavirus provides evolutionary insight into the birth of viral genes. Proc. Natl. Acad. Sci. USA 2013, 110, 12744–12749. [Google Scholar] [CrossRef] [Green Version]
- Coursaget, P.; Samimi, M.; Nicol, J.T.J.; Gardair, C.; Touzé, A. Human Merkel cell polyomavirus: Virological background and clinical implications. APMIS 2013, 121, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Konstantinell, A.; Coucheron, D.H.; Sveinbjørnsson, B.; Moens, U. MicroRNAs as Potential Biomarkers in Merkel Cell Carcinoma. Int. J. Mol. Sci. 2018, 19, 1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanotta, N.; Delbue, S.; Signorini, L.; Villani, S.; D’alessandro, S.; Campisciano, G.; Colli, C.; De Seta, F.; Ferrante, P.; Comar, M. Merkel cell polyomavirus is associated with anal infections in men who have sex with men. Microorganisms 2019, 7, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayer, N.J.; Januliene, D.; Zocher, G.; Stehle, T.; Moeller, A.; Blaum, B.S. Structure of Merkel Cell Polyomavirus Capsid and Interaction with Its Glycosaminoglycan Attachment Receptor. J. Virol. 2020, 94, e01664-19. [Google Scholar] [CrossRef]
- Liu, W.; You, J. Molecular Mechanisms of Merkel Cell Polyomavirus Transformation and Replication. Annu. Rev. Virol. 2020, 7, 289–307. [Google Scholar] [CrossRef]
- Csoboz, B.; Rasheed, K.; Sveinbjørnsson, B.; Moens, U. Merkel cell polyomavirus and non-Merkel cell carcinomas: Guilty or circumstantial evidence? APMIS 2020, 128, 104–120. [Google Scholar] [CrossRef] [Green Version]
- Hesbacher, S.; Pfitzer, L.; Wiedorfer, K.; Angermeyer, S.; Borst, A.; Haferkamp, S.; Scholz, C.J.; Wobser, M.; Schrama, D.; Houben, R. RB1 is the crucial target of the Merkel cell polyomavirus Large T antigen in Merkel cell carcinoma cells. Oncotarget 2016, 7, 32956–32968. [Google Scholar] [CrossRef]
- Houben, R.; Adam, C.; Baeurle, A.; Hesbacher, S.; Grimm, J.; Angermeyer, S.; Henzel, K.; Hauser, S.; Elling, R.; Bröcker, E.-B.; et al. An intact retinoblastoma protein-binding site in Merkel cell polyomavirus large T antigen is required for promoting growth of Merkel cell carcinoma cells. Int. J. Cancer 2012, 130, 847–856. [Google Scholar] [CrossRef]
- Angermeyer, S.; Hesbacher, S.; Becker, J.C.; Schrama, D.; Houben, R. Merkel cell polyomavirus-positive merkel cell carcinoma cells do not require expression of the viral small T antigen. J. Invest. Dermatol. 2013, 133, 2059–2064. [Google Scholar] [CrossRef] [Green Version]
- Shuda, M.; Guastafierro, A.; Geng, X.; Shuda, Y.; Ostrowski, S.M.; Lukianov, S.; Jenkins, F.J.; Honda, K.; Maricich, S.M.; Moore, P.S.; et al. Merkel Cell Polyomavirus Small T Antigen Induces Cancer and Embryonic Merkel Cell Proliferation in a Transgenic Mouse Model. PLoS ONE 2015, 10, e0142329. [Google Scholar] [CrossRef] [Green Version]
- Nwogu, N.; Ortiz, L.E.; Kwun, H.J. Surface charge of Merkel cell polyomavirus small T antigen determines cell transformation through allosteric FBW7 WD40 domain targeting. Oncogenesis 2020, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Rotondo, J.C.; Oton-Gonzalez, L.; Mazziotta, C.; Lanzillotti, C.; Iaquinta, M.R.; Tognon, M.; Martini, F. Simultaneous detection and viral DNA load quantification of different human papillomavirus types in clinical specimens by the high analytical droplet digital PCR method. Front. Microbiol. 2020, 11, 591452. [Google Scholar] [CrossRef]
- Preti, M.; Rotondo, J.C.; Holzinger, D.; Micheletti, L.; Gallio, N.; Robitaille, A.; Mckay-Chopin, S.; Carreira, C.; Privitera, S.S.; Watanabe, R.; et al. Role of human papillomavirus infection in the etiology of vulvar cancer in Italian women. Infect. Agents Cancer 2020, 15, e2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tognon, M.; Tagliapietra, A.; Magagnoli, F.; Mazziotta, C.; Oton-Gonzalez, L.; Lanzillotti, C.; Vesce, F.; Contini, C.; Rotondo, J.C.; Martini, F.; et al. Investigation on Spontaneous Abortion and Human Papillomavirus Infection. Vaccines 2020, 8, 473. [Google Scholar] [CrossRef]
- Waldvogel-Abramowski, S.; Taleb, S.; Alessandrini, M.; Preynat-Seauve, O. Viral Metagenomics of Blood Donors and Blood-Derived Products Using Next-Generation Sequencing. Transfus. Med. Hemother. 2019, 46, 87–93. [Google Scholar] [CrossRef] [PubMed]
- L’Huillier, A.G.; Brito, F.; Wagner, N.; Cordey, S.; Zdobnov, E.; Posfay-Barbe, K.M.; Kaiser, L. Identification of Viral Signatures Using High-Throughput Sequencing on Blood of Patients With Kawasaki Disease. Front. Pediatr. 2019, 7, 524. [Google Scholar] [CrossRef] [PubMed]
- Motavalli Khiavi, F.; Nasimi, M.; Rahimi, H. Merkel Cell Polyomavirus Gene Expression and Mutational Analysis of Large Tumor Antigen in Non-Merkel Cell Carcinoma Tumors of Iranian Patients. Public Health Genom. 2021, 23, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Schrama, D.; Sarosi, E.M.; Adam, C.; Ritter, C.; Kaemmerer, U.; Klopocki, E.; König, E.M.; Utikal, J.; Becker, J.C.; Houben, R. Characterization of six Merkel cell polyomavirus-positive Merkel cell carcinoma cell lines: Integration pattern suggest that large T antigen truncating events occur before or during integration. Int. J. Cancer 2019, 145, 1020–1032. [Google Scholar] [CrossRef]
- Qin, J.; Wen, B.; Liang, Y.; Yu, W.; Li, H. Histone Modifications and their Role in Colorectal Cancer (Review). Pathol. Oncol. Res. 2020, 26, 2023–2033. [Google Scholar] [CrossRef] [Green Version]
- Marchione, D.M.; Lisby, A.; Viaene, A.N.; Santi, M.; Nasrallah, M.L.; Wang, L.P.; Williams, E.A.; Larque, A.B.; Chebib, I.; Garcia, B.A.; et al. Histone H3K27 dimethyl loss is highly specific for malignant peripheral nerve sheath tumor and distinguishes true PRC2 loss from isolated H3K27 trimethyl loss. Mod. Pathol. 2019, 32, 1434–1444. [Google Scholar] [CrossRef]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Chen, P.; Guo, Z.; Chen, C.; Tian, S.; Bai, X.; Zhai, G.; Ma, Z.; Wu, H.; Zhang, K. Identification of dual histone modification-binding protein interaction by combining mass spectrometry and isothermal titration calorimetric analysis. J. Adv. Res. 2020, 22, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Nishida, Y.; Tsutsumida, H.; Hamada, T.; Goto, M.; Higashi, M.; Nomoto, M.; Yonezawa, S. MUC1 expression is regulated by DNA methylation and histone H3 lysine 9 modification in cancer cells. Cancer Res. 2008, 68, 2708–2716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Sharma, K.L.; Bansal, C.; Kumar, A. Updates on “Cancer Genomics and Epigenomics”. World J. Clin. Oncol. 2020, 11, 890–897. [Google Scholar] [CrossRef] [PubMed]
- McCool, K.W.; Xu, X.; Singer, D.B.; Murdoch, F.E.; Fritsch, M.K. The role of histone acetylation in regulating early gene expression patterns during early embryonic stem cell differentiation. J. Biol. Chem. 2007, 282, 6696–6706. [Google Scholar] [CrossRef] [Green Version]
- Milon, B.C.; Cheng, H.; Tselebrovsky, M.V.; Lavrov, S.A.; Nenasheva, V.V.; Mikhaleva, E.A.; Shevelyov, Y.Y.; Nurminsky, D.I. Role of Histone Deacetylases in Gene Regulation at Nuclear Lamina. PLoS ONE 2012, 7, e49692. [Google Scholar] [CrossRef]
- Pelzel, H.R.; Schlamp, C.L.; Nickells, R.W. Histone H4 deacetylation plays a critical role in early gene silencing during neuronal apoptosis. BMC Neurosci. 2010, 11, 62. [Google Scholar] [CrossRef] [Green Version]
- Goyama, S.; Nitta, E.; Yoshino, T.; Kako, S.; Watanabe-Okochi, N.; Shimabe, M.; Imai, Y.; Takahashi, K.; Kurokawa, M. EVI-1 interacts with histone methyltransferases SUV39H1 and G9a for transcriptional repression and bone marrow immortalization. Leukemia 2010, 24, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Ma, S.; Song, N.; Li, X.; Liu, L.; Yang, S.; Ding, X.; Shan, L.; Zhou, X.; Su, D.; et al. Stabilization of histone demethylase PHF8 by USP7 promotes breast carcinogenesis. J. Clin. Invest. 2016, 126, 2205–2220. [Google Scholar] [CrossRef] [Green Version]
- Sheng, W.; LaFleur, M.W.; Nguyen, T.H.; Chen, S.; Chakravarthy, A.; Conway, J.R.; Li, Y.; Chen, H.; Yang, H.; Hsu, P.H.; et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 2018, 174, 549–563. [Google Scholar] [CrossRef] [Green Version]
- Carnesecchi, J.; Forcet, C.; Zhang, L.; Tribollet, V.; Barenton, C.; Boudra, R.; Cerutti, C.; Billas, I.M.L.; Sérandour, A.A.; Carroll, J.S.; et al. ERRα induces H3K9 demethylation by LSD1 to promote cell invasion. Proc. Natl. Acad. Sci. USA 2017, 114, 3909–3914. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeld, J.A.; Wang, Z.; Schones, D.E.; Zhao, K.; DeSalle, R.; Zhang, M.Q. Determination of enriched histone modifications in non-genic portions of the human genome. BMC Genomics 2009, 10, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Rosa, H.; Schneider, R.; Bernstein, B.E.; Karabetsou, N.; Morillon, A.; Weise, C.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Methylation of histone H3 K4 mediates association of the Isw1p ATPase with chromatin. Mol. Cell 2003, 12, 1325–1332. [Google Scholar] [CrossRef]
- Fernandez-Capetillo, O.; Allis, C.D.; Nussenzweig, A. Phosphorylation of histone H2B at DNA double-strand breaks. J. Exp. Med. 2004, 199, 1671–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzger, E.; Yin, N.; Wissmann, M.; Kunowska, N.; Fischer, K.; Friedrichs, N.; Patnaik, D.; Higgins, J.M.G.; Potier, N.; Scheidtmann, K.H.; et al. Phosphorylation of histone H3 at threonine 11 establishes a novel chromatin mark for transcriptional regulation. Nat. Cell Biol. 2008, 10, 53–60. [Google Scholar] [CrossRef]
- Basnet, H.; Su, X.B.; Tan, Y.; Meisenhelder, J.; Merkurjev, D.; Ohgi, K.A.; Hunter, T.; Pillus, L.; Rosenfeld, M.G. Tyrosine phosphorylation of histone H2A by CK2 regulates transcriptional elongation. Nature 2014, 516, 267–271. [Google Scholar] [CrossRef]
- Polioudaki, H.; Markaki, Y.; Kourmouli, N.; Dialynas, G.; Theodoropoulos, P.A.; Singh, P.B.; Georgatos, S.D. Mitotic phosphorylation of histone H3 at threonine 3. FEBS Lett. 2004, 560, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Cook, P.J.; Ju, B.G.; Telese, F.; Wang, X.; Glass, C.K.; Rosenfeld, M.G. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 2009, 458, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, M.; Iwasaki, T.; Wardhani, L.O.; Kuwamoto, S.; Nonaka, D.; Nagata, K.; Kato, M.; Kitamura, Y.; Hayashi, K. Decreased H3K27me3 expression is associated with merkel cell polyomavirus-negative merkel cell carcinoma, especially combined with cutaneous squamous cell carcinoma. Anticancer Res. 2019, 39, 5573–5579. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Stomper, J.; Rotondo, J.C.; Greve, G.; Lübbert, M. Hypomethylating agents (HMA) for the treatment of acute myeloid leukemia and myelodysplastic syndromes: Mechanisms of resistance and novel HMA-based therapies. Leukemia 2021, 35, 1873–1889. [Google Scholar] [CrossRef]
- Rotondo, J.C.; Oton-Gonzalez, L.; Selvatici, R.; Rizzo, P.; Pavasini, R.; Campo, G.C.; Lanzillotti, C.; Mazziotta, C.; De Mattei, M.; Tognon, M.; et al. SERPINA1 Gene Promoter Is Differentially Methylated in Peripheral Blood Mononuclear Cells of Pregnant Women. Front. Cell Dev. Biol. 2020, 8, 5505. [Google Scholar] [CrossRef] [PubMed]
- Rotondo, J.C.; Lanzillotti, C.; Mazziotta, C.; Tognon, M.; Martini, F. Epigenetics of male infertility: The role of DNA methylation. Front. Cell Dev. Biol. 2021, 9, 689624. [Google Scholar] [CrossRef]
- Rotondo, J.C.; Bosi, S.; Bazzan, E.; Di Domenico, M.; De Mattei, M.; Selvatici, R.; Patella, A.; Marci, R.; Tognon, M.; Martini, F. Methylenetetrahydrofolate reductase gene promoter hypermethylation in semen samples of infertile couples correlates with recurrent spontaneous abortion. Hum. Reprod. 2012, 27, 3632–3638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theka, I.; Sottile, F.; Cammisa, M.; Bonnin, S.; Sanchez-Delgado, M.; Di Vicino, U.; Neguembor, M.V.; Arumugam, K.; Aulicino, F.; Monk, D.; et al. Wnt/β-catenin signaling pathway safeguards epigenetic stability and homeostasis of mouse embryonic stem cells. Sci. Rep. 2019, 9, 948. [Google Scholar] [CrossRef]
- Fathima, T.; Arumugam, P.; AS, S.G.; Priyadharsini, J.V. Decoding the Genetic Alterations in Genes of DNMT Family (DNA Methyl-Transferase) and their Association with Head and Neck Squamous Cell Carcinoma. Asian Pac. J. Cancer Prev. 2020, 21, 3605–3612. [Google Scholar] [CrossRef]
- Barau, J.; Teissandier, A.; Zamudio, N.; Roy, S.; Nalesso, V.; Hérault, Y.; Guillou, F.; Bourc’his, D. The DNA methyltransferase DNMT3C protects male germ cells from transposon activity. Science 2016, 354, 909–912. [Google Scholar] [CrossRef]
- Lopomo, A.; Ricciardi, R.; Maestri, M.; Rosa, A.; Melfi, F.; Lucchi, M.; Mussi, A.; Coppedè, F.; Migliore, L. Gene-specific methylation analysis in thymomas of patients with myasthenia gravis. Int. J. Mol. Sci. 2016, 17, 2121. [Google Scholar] [CrossRef] [Green Version]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome-biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Valinluck, V.; Sowers, L.C. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007, 67, 946–950. [Google Scholar] [CrossRef] [Green Version]
- Melamed, P.; Yosefzon, Y.; David, C.; Tsukerman, A.; Pnueli, L. Tet enzymes, variants, and differential effects on function. Front. Cell Dev. Biol. 2018, 6, 22. [Google Scholar] [CrossRef] [Green Version]
- Jin, C.; Qin, T.; Barton, M.C.; Jelinek, J.; Issa, J.P.J. Minimal role of base excision repair in TET-induced global DNA demethylation in HEK293T cells. Epigenetics 2015, 10, 1006–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotondo, J.C.; Selvatici, R.; Di Domenico, M.; Marci, R.; Vesce, F.; Tognon, M.; Martini, F. Methylation loss at H19 imprinted gene correlates with methylenetetrahydrofolate reductase gene promoter hypermethylation in semen samples from infertile males. Epigenetics 2013, 8, 990–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trietsch, M.D.; Nooij, L.S.; Gaarenstroom, K.N.; Van Poelgeest, M.I.E. Genetic and epigenetic changes in vulvar squamous cell carcinoma and its precursor lesions: A review of the current literature. Gynecol. Oncol. 2015, 136, 143–157. [Google Scholar] [CrossRef]
- Rotondo, J.C.; Giari, L.; Guerranti, C.; Tognon, M.; Castaldelli, G.; Fano, E.A.; Martini, F. Environmental doses of perfluorooctanoic acid change the expression of genes in target tissues of common carp. Environ. Toxicol. Chem. 2018, 37, 942–948. [Google Scholar] [CrossRef]
- Khambata, K.; Raut, S.; Deshpande, S.; Mohan, S.; Sonawane, S.; Gaonkar, R.; Ansari, Z.; Datar, M.; Bansal, V.; Patil, A.; et al. DNA methylation defects in spermatozoa of male partners from couples experiencing recurrent pregnancy loss. Hum. Reprod. 2021, 36, 48–60. [Google Scholar]
- Shaker, M.M.; Shalabi, T.A.; Amr, K.S. Correlation of methylation status in MTHFR promoter region with recurrent pregnancy loss. J. Genet. Eng. Biotechnol. 2021, 19, 44. [Google Scholar] [CrossRef]
- Rotondo, J.C.; Borghi, A.; Selvatici, R.; Magri, E.; Bianchini, E.; Montinari, E.; Corazza, M.; Virgili, A.; Tognon, M.; Martini, F. Hypermethylation-induced inactivation of the IRF6 gene as a possible early event in progression of vulvar squamous cell carcinoma associated with lichen sclerosus. JAMA Dermatol. 2016, 152, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Haag, T.; Richter, A.M.; Schneider, M.B.; Jiménez, A.P.; Dammann, R.H. The dual specificity phosphatase 2 gene is hypermethylated in human cancer and regulated by epigenetic mechanisms. BMC Cancer 2016, 16, 49. [Google Scholar] [CrossRef] [Green Version]
- Yanatatsaneejit, P.; Chalertpet, K.; Sukbhattee, J.; Nuchcharoen, I.; Phumcharoen, P.; Mutirangura, A. Promoter methylation of tumor suppressor genes induced by human papillomavirus in cervical cancer. Oncol. Lett. 2020, 20, 955–961. [Google Scholar] [CrossRef]
- Park, E.; Gong, E.Y.; Romanelli, M.G.; Lee, K. Suppression of estrogen receptor-alpha transactivation by thyroid transcription factor-2 in breast cancer cells. Biochem. Biophys. Res. Commun. 2012, 421, 532–537. [Google Scholar] [CrossRef]
- Sheaffer, K.L.; Elliott, E.N.; Kaestner, K.H. DNA hypomethylation contributes to genomic instability and intestinal cancer initiation. Cancer Prev. Res. 2016, 9, 534–546. [Google Scholar] [CrossRef] [Green Version]
- Lanzillotti, C.; De Mattei, M.; Mazziotta, C.; Taraballi, F.; Rotondo, J.C.; Tognon, M.; Martini, F. Long Non-coding RNAs and MicroRNAs Interplay in Osteogenic Differentiation of Mesenchymal Stem Cells. Front. Cell Dev. Biol. 2021, 9, 646032. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, J.P.; Bowman, M.J.; Azodi, C.B.; Sowers, R.P.; Moghe, G.D.; Childs, K.L.; Shiu, S.H. Evolutionary characteristics of intergenic transcribed regions indicate rare novel genes and widespread noisy transcription in the Poaceae. Sci. Rep. 2019, 9, 12122. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef]
- Cai, X.; Hagedorn, C.H.; Cullen, B.R. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 2004, 10, 1957–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dexheime, P.; Cochella, L. MicroRNAs: From Mechanism to Organism. Front. Cell Dev. Biol. 2020, 8, 409. [Google Scholar] [CrossRef]
- Hayes, J.; Peruzzi, P.P.; Lawler, S. MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol. Med. 2014, 20, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Liu, B.; Qu, S.; Liang, G.; Luo, W.; Gong, C. MicroRNAs and cancer: Key paradigms in molecular therapy (Review). Oncol. Lett. 2018, 15, 2735–2742. [Google Scholar] [CrossRef] [Green Version]
- Mazziotta, C.; Rotondo, J.C.; Lanzillotti, C.; Campione, G.; Martini, F.; Tognon, M. Cancer biology and molecular genetics of A3 adenosine receptor. Oncogene 2021, in press. [Google Scholar]
- Zhang, B.; Pan, X.; Cobb, G.P.; Anderson, T.A. microRNAs as oncogenes and tumor suppressors. Dev. Biol. 2007, 302, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juan, A.H.; Wang, S.; Ko, K.D.; Zare, H.; Tsai, P.F.; Feng, X.; Vivanco, K.O.; Ascoli, A.M.; Gutierrez-Cruz, G.; Krebs, J.; et al. Roles of H3K27me2 and H3K27me3 Examined during Fate Specification of Embryonic Stem Cells. Cell Rep. 2016, 17, 1369–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böhm, J.; Muenzner, J.K.; Caliskan, A.; Ndreshkjana, B.; Erlenbach-Wünsch, K.; Merkel, S.; Croner, R.; Rau, T.T.; Geppert, C.I.; Hartmann, A.; et al. Loss of enhancer of zeste homologue 2 (EZH2) at tumor invasion front is correlated with higher aggressiveness in colorectal cancer cells. J. Cancer Res. Clin. Oncol. 2019, 145, 2227–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harms, K.L.; Chubb, H.; Zhao, L.; Fullen, D.R.; Bichakjian, C.K.; Johnson, T.M.; Carskadon, S.; Palanisamy, N.; Harms, P.W. Increased expression of EZH2 in Merkel cell carcinoma is associated with disease progression and poorer prognosis. Hum. Pathol. 2017, 67, 78–84. [Google Scholar] [CrossRef]
- Veija, T.; Koljonen, V.; Bohling, T.; Kero, M.; Knuutila, S.; Sarhadi, V.K. Aberrant expression of ALK and EZH2 in Merkel cell carcinoma. BMC Cancer 2017, 17, 236. [Google Scholar] [CrossRef] [Green Version]
- Ritter, C.; Fan, K.; Paulson, K.G.; Nghiem, P.; Schrama, D.; Becker, J.C. Reversal of epigenetic silencing of MHC class i chain-related protein A and B improves immune recognition of Merkel cell carcinoma. Sci. Rep. 2016, 23, 21678. [Google Scholar] [CrossRef] [Green Version]
- Ritter, C.; Fan, K.; Paschen, A.; Hardrup, S.R.; Ferrone, S.; Nghiem, P.; Ugurel, S.; Schrama, D.; Becker, J.C. Epigenetic priming restores the HLA class-I antigen processing machinery expression in Merkel cell carcinoma. Sci. Rep. 2017, 7, 2290. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Park, D.E.; Berrios, C.; White, E.A.; Arora, R.; Yoon, R.; Branigan, T.; Xiao, T.; Westerling, T.; Federation, A.; et al. Merkel cell polyomavirus recruits MYCL to the EP400 complex to promote oncogenesis. PLoS Pathog. 2017, 13, e1006668. [Google Scholar] [CrossRef]
- Park, D.E.; Cheng, J.; McGrath, J.P.; Lim, M.Y.; Cushman, C.; Swanson, S.K.; Tillgren, M.L.; Paulo, J.A.; Gokhale, P.C.; Florens, L.; et al. Merkel cell polyomavirus activates LSD1-mediated blockade of non-canonical BAF to regulate transformation and tumorigenesis. Nat. Cell Biol. 2020, 22, 603–615. [Google Scholar] [CrossRef]
- Busam, K.J.; Pulitzer, M.P.; Coit, D.C.; Arcila, M.; Leng, D.; Jungbluth, A.A.; Wiesner, T. Reduced H3K27me3 expression in Merkel cell polyoma virus-positive tumors. Mod. Pathol. 2017, 30, 877–883. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.H.; Narayanan, D.; Limmer, A.L.; Simonette, R.A.; Rady, P.L.; Tyring, S.K. Merkel Cell Polyomavirus Small T Antigen Induces DNA Damage Response. Intervirology 2019, 62, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Kotowski, U.; Erović, B.M.; Schnöll, J.; Stanek, V.; Janik, S.; Steurer, M.; Mitulović, G. Quantitative proteome analysis of Merkel cell carcinoma cell lines using SILAC. Clin. Proteom. 2019, 16, 42. [Google Scholar] [CrossRef] [PubMed]
- Helmbold, P.; Lahtz, C.; Enk, A.; Herrmann-Trost, P.; Marsch, W.C.; Kutzner, H.; Dammann, R.H.; Herpel, E.; Schnabel, P.A.; Dammann, R.H. Frequent occurrence of RASSF1A promoter hypermethylation and Merkel cell polyomavirus in Merkel cell carcinoma. Eur. J. Cancer 2009, 45, 2207–2211. [Google Scholar] [CrossRef]
- Sahi, H.; Savola, S.; Sihto, H.; Koljonen, V.; Bohling, T.; Knuutila, S. RB1 gene in Merkel cell carcinoma: Hypermethylation in all tumors and concurrent heterozygous deletions in the polyomavirus-negative subgroup. APMIS 2014, 122, 1157–1166. [Google Scholar] [CrossRef]
- Higaki-Mori, H.; Kuwamoto, S.; Iwasaki, T.; Kato, M.; Murakami, I.; Nagata, K.; Sano, H.; Horie, Y.; Yoshida, Y.; Yamamoto, O.; et al. Association of Merkel cell polyomavirus infection with clinicopathological differences in Merkel cell carcinoma. Hum. Pathol. 2012, 43, 2282–2291. [Google Scholar] [CrossRef]
- Richter, A.; Haag, T.; Walesch, S.; Herrmann-Trost, P.; Marsch, W.; Kutzner, H.; Helmbold, P.; Dammann, R. Aberrant Promoter Hypermethylation of RASSF Family Members in Merkel Cell Carcinoma. Cancers 2013, 5, 1566–1576. [Google Scholar] [CrossRef]
- Improta, G.; Ritter, C.; Pettinato, A.; Vasta, V.; Schrama, D.; Fraggetta, F.; Becker, J.C. MGMT promoter methylation status in Merkel cell carcinoma: In vitro versus invivo. J. Cancer Res. Clin. Oncol. 2017, 143, 1489–1497. [Google Scholar] [CrossRef]
- Gambichler, T.; Dreißigacker, M.; Kasakovski, D.; Skrygan, M.; Wieland, U.; Silling, S.; Gravemeyer, J.; Melior, A.; Cherouny, A.; Stücker, M.; et al. Patched 1 expression in Merkel cell carcinoma. J. Dermatol. 2021, 48, 64–74. [Google Scholar] [CrossRef]
- Ricci, C.; Morandi, L.; Righi, A.; Gibertoni, D.; Maletta, F.; Ambrosi, F.; Agostinelli, C.; Uccella, S.; Asioli, S.; Sessa, F.; et al. PD-1 (PDCD1) promoter methylation in Merkel cell carcinoma: Prognostic relevance and relationship with clinico-pathological parameters. Mod. Pathol. 2019, 32, 1359–1372. [Google Scholar] [CrossRef] [PubMed]
- Chteinberg, E.; Vogt, J.; Kolarova, J.; Bormann, F.; van den Oord, J.; Speel, E.J.; Winnepenninckx, V.; Kurz, A.K.; Zenke, M.; Siebert, R.; et al. The curious case of Merkel cell carcinoma: Epigenetic youth and lack of pluripotency. Epigenetics 2020, 15, 1319–1324. [Google Scholar] [CrossRef]
- Gujar, H.; Mehta, A.; Li, H.; Tsai, Y.; Qiu, X.; Weisenberger, D.; Jasiulionis, M.; In, G.; Liang, G. Characterizing DNA methylation signatures and their potential functional roles in Merkel cell carcinoma. Genome Med. 2021, 13, 130. [Google Scholar] [CrossRef]
- Neagu, M.; Constantin, C.; Cretoiu, S.M.; Zurac, S. miRNAs in the Diagnosis and Prognosis of Skin Cancer. Front. Cell Dev. Biol. 2020, 8, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, K.; Gravemeyer, J.; Ritter, C.; Rasheed, K.; Gambichler, T.; Moens, U.; Shuda, M.; Schrama, D.; Becker, J.C. MCPyV Large T Antigen-Induced Atonal Homolog 1 Is a Lineage-Dependency Oncogene in Merkel Cell Carcinoma. J. Invest. Dermatol. 2020, 140, 56–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Xie, H.; Shi, H.; Gao, J.; Juhlin, C.C.; Björnhagen, V.; Höög, A.; Lee, L.; Larsson, C.; Lui, W.O. Merkel cell polyomavirus oncoproteins induce microRNAs that suppress multiple autophagy genes. Int. J. Cancer 2020, 146, 1652–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, K.; Ritter, C.; Nghiem, P.; Blom, A.; Verhaegen, M.E.; Dlugosz, A.; Dum, N.; Woetmann, A.; Tothill, R.W.; Hicks, R.J.; et al. Circulating cell-free miR-375 as surrogate marker of tumor burden in Merkel cell carcinoma. Clin. Cancer Res. 2018, 24, 5873–5882. [Google Scholar] [CrossRef] [Green Version]
- Renwick, N.; Cekan, P.; Masry, P.A.; McGeary, S.E.; Miller, J.B.; Hafner, M.; Li, Z.; Mihailovic, A.; Morozov, P.; Brown, M.; et al. Multicolor microRNA FISH effectively differentiates tumor types. J. Clin. Investig. 2013, 123, 2694–2702. [Google Scholar] [CrossRef]
- Abraham, K.J.; Zhang, X.; Vidal, R.; Paré, G.C.; Feilotter, H.E.; Tron, V.A. Roles for miR-375 in neuroendocrine differentiation and tumor suppression via notch pathway suppression in merkel cell Carcinoma. Am. J. Pathol. 2016, 186, 1025–1035. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Lee, L.; Caramuta, S.; Höög, A.; Browaldh, N.; Björnhagen, V.; Larsson, C.; Lui, W.O. MicroRNA expression patterns related to merkel cell polyomavirus infection in human Merkel cell carcinoma. J. Invest. Dermatol. 2014, 134, 507–517. [Google Scholar] [CrossRef] [Green Version]
- Costa-Pinheiro, P.; Ramalho-Carvalho, J.; Vieira, F.Q.; Torres-Ferreira, J.; Oliveira, J.; Gonçalves, C.S.; Costa, B.M.; Henrique, R.; Jerónimo, C. MicroRNA-375 plays a dual role in prostate carcinogenesis. Clin. Epigenetics 2015, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Hong, Z.; Gao, F.; Feng, W. Upregulation of microRNA-375 is associated with poor prognosis in pediatric acute myeloid leukemia. Mol. Cell. Biochem. 2013, 383, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Rocha Simonini, P.D.S.; Breiling, A.; Gupta, N.; Malekpour, M.; Youns, M.; Omranipour, R.; Malekpour, F.; Volinia, S.; Croce, C.M.; Najmabadi, H.; et al. Epigenetically deregulated microRNA-375 is involved in a positive feedback loop with estrogen receptor α in breast cancer cells. Cancer Res. 2010, 70, 9175–9184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.; Lin, J.; Tsung, A. Manipulation of autophagy by MIR375 generates antitumor effects in liver cancer. Autophagy 2012, 8, 1833–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Z.; Chu, X.; Wu, Y.; Wu, J.; Lu, C.; Lü, R.; Ding, M.; Mao, N. MicroRNA-375 functions as a tumor suppressor in osteosarcoma by targeting PIK3CA. Tumor Biol. 2015, 36, 8579–8584. [Google Scholar] [CrossRef]
- Osako, Y.; Seki, N.; Kita, Y.; Yonemori, K.; Koshizuka, K.; Kurozumi, A.; Omoto, I.; Sasaki, K.; Uchikado, Y.; Kurahara, H.; et al. Regulation of MMP13 by antitumor microRNA-375 markedly inhibits cancer cell migration and invasion in esophageal squamous cell carcinoma. Int. J. Oncol. 2016, 49, 2255–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veija, T.; Sahi, H.; Koljonen, V.; Bohling, T.; Knuutila, S.; Mosakhani, N. miRNA-34a underexpressed in Merkel cell polyomavirus-negative Merkel cell carcinoma. Virchows Arch. 2015, 466, 289–295. [Google Scholar] [CrossRef]
- Gravemeyer, J.; Lange, A.; Ritter, C.; Spassova, I.; Song, L.; Picard, D.; Remke, M.; Horny, K.; Sriram, A.; Gambichler, T.; et al. Classical and Variant Merkel Cell Carcinoma Cell Lines Display Different Degrees of Neuroendocrine Differentiation and Epithelial-Mesenchymal Transition. J. Invest. Dermatol. 2021, 141, 1675–1686.e4. [Google Scholar] [CrossRef]
- Theiss, J.M.; Günther, T.; Alawi, M.; Neumann, F.; Tessmer, U.; Fischer, N.; Grundhoff, A. A Comprehensive Analysis of Replicating Merkel Cell Polyomavirus Genomes Delineates the Viral Transcription Program and Suggests a Role for mcv-miR-M1 in Episomal Persistence. PLoS Pathog. 2015, 7, e1004974. [Google Scholar] [CrossRef]
- Seo, G.J.; Chen, C.J.; Sullivan, C.S. Merkel cell polyomavirus encodes a microRNA with the ability to autoregulate viral gene expression. Virology 2009, 383, 183–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Paulson, K.G.; Murchison, E.P.; Afanasiev, O.K.; Alkan, C.; Leonard, J.H.; Byrd, D.R.; Hannon, G.J.; Nghiem, P. Identification and validation of a novel mature microRNA encoded by the Merkel cell polyomavirus in human Merkel cell carcinomas. J. Clin. Virol. 2011, 52, 272–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhbari, P.; Tobin, D.; Poterlowicz, K.; Roberts, W.; Boyne, J.R. MCV-miR-M1 Targets the Host-Cell Immune Response Resulting in the Attenuation of Neutrophil Chemotaxis. J. Investig. Dermatol. 2018, 138, 2343–2354. [Google Scholar] [CrossRef] [Green Version]
- Mauri, F.; Blanpain, C. Targeting the epigenetic addiction of Merkel cell carcinoma. EMBO Mol. Med. 2020, 12, e13347. [Google Scholar] [CrossRef] [PubMed]
- Harms, P.W.; Harms, K.L.; Moore, P.S.; DeCaprio, J.A.; Nghiem, P.; Wong, M.K.K.; Brownell, I. The biology and treatment of Merkel cell carcinoma: Current understanding and research priorities. Nat. Rev. Clin. Oncol. 2018, 15, 763–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Xie, H.; Scicluna, P.; Lee, L.; Björnhagen, V.; Höög, A.; Larsson, C.; Lui, W.O. MiR-375 regulation of LDHB plays distinct roles in polyomavirus-positive and-negative merkel cell carcinoma. Cancers 2018, 14, 443. [Google Scholar] [CrossRef] [Green Version]
- Fan, K.; Zebisch, A.; Horny, K.; Schrama, D.; Becker, J.C. Highly expressed MiR-375 is not an intracellular oncogene in merkel cell polyomavirus-associated merkel cell carcinoma. Cancers 2020, 12, 529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Wang, Q.J.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–476. [Google Scholar] [CrossRef]
- Paulson, K.G.; Tegeder, A.; Willmes, C.; Iyer, J.G.; Afanasiev, O.K.; Schrama, D.; Koba, S.; Thibodeau, R.; Nagase, K.; Simonson, W.T.; et al. Downregulation of MHC-I expression is prevalent but reversible in Merkel cell carcinoma. Cancer Immunol. Res. 2014, 2, 1071–1079. [Google Scholar] [CrossRef] [Green Version]
- Schadendorf, D.; Nghiem, P.; Bhatia, S.; Hauschild, A.; Saiag, P.; Mahnke, L.; Hariharan, S.; Kaufman, H.L. Immune evasion mechanisms and immune checkpoint inhibition in advanced merkel cell carcinoma. Oncoimmunology 2017, 6, e1338237. [Google Scholar] [CrossRef]
- Song, L.; Bretz, A.C.; Gravemeyer, J.; Spassova, I.; Muminova, S.; Gambichler, T.; Sriram, A.; Ferrone, S.; Becker, J.C. The HDAC Inhibitor Domatinostat Promotes Cell-Cycle Arrest, Induces Apoptosis, and Increases Immunogenicity of Merkel Cell Carcinoma Cells. J. Invest. Dermatol. 2020, 141, 903–912.e4. [Google Scholar] [CrossRef]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.U.; Rashoff, A.Q.; Krabbenhoft, S.D.; Hoelper, D.; Do, T.J.; Gibson, T.J.; Lundgren, S.M.; Bondra, E.R.; Deshmukh, S.; Harutyunyan, A.S.; et al. H3 K27M and EZHIP Impede H3K27-Methylation Spreading by Inhibiting Allosterically Stimulated PRC2. Mol. Cell 2020, 80, 726–735.e7. [Google Scholar] [CrossRef]
- Chung, C.; Sweha, S.R.; Pratt, D.; Tamrazi, B.; Panwalkar, P.; Banda, A.; Bayliss, J.; Hawes, D.; Yang, F.; Lee, H.J.; et al. Integrated Metabolic and Epigenomic Reprograming by H3K27M Mutations in Diffuse Intrinsic Pontine Gliomas. Cancer Cell 2020, 38, 334–349.e9. [Google Scholar] [CrossRef]
- Lassacher, A.; Heitzer, E.; Kerl, H.; Wolf, P. p14ARF hypermethylation is common but INK4a-ARF locus or p53 mutations are rare in Merkel cell carcinoma. J. Invest. Dermatol. 2008, 128, 1788–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenke, R.; Reßing, N.; Hansen, F.K.; Aigner, A.; Büch, T. Anticancer Therapy with HDAC Inhibitors: Mechanism-Based Combination Strategies and Future Perspectives. Cancers 2021, 13, 634. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, M.; Yao, Y.; Yu, B.; Liu, H. Targeting LSD1 for acute myeloid leukemia (AML) treatment. Pharmacol. Res. 2021, 164, 105335. [Google Scholar] [CrossRef] [PubMed]
- Nikolouzakis, T.K.; Falzone, L.; Lasithiotakis, K.; Krüger-Krasagakis, S.; Kalogeraki, A.; Sifaki, M.; Spandidos, D.A.; Chrysos, E.; Tsatsakis, A.; Tsiaoussis, J. Current and Future Trends in Molecular Biomarkers for Diagnostic, Prognostic, and Predictive Purposes in Non-Melanoma Skin Cancer. J. Clin. Med. 2020, 9, 2868. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.A.; Tetzlaff, M.T.; Pattanaprichakul, P.; Fox, P.; Torres-Cabala, C.A.; Bassett, R.L.; Prieto, V.G.; Richards, H.W.; Curry, J.L. Detection of mitotic figures and G2+ tumor nuclei with histone markers correlates with worse overall survival in patients with Merkel cell carcinoma. J. Cutan. Pathol. 2014, 41, 846–852. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; You, J. Merkel Cell Polyomavirus and Human Merkel Cell Carcinoma. Recent Results Cancer Res. 2021, 217, 303–323. [Google Scholar] [PubMed]
- Mitra, A.; Mishra, L.; Li, S. Technologies for deriving primary tumor cells for use in personalized cancer therapy. Trends Biotechnol. 2013, 31, 347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotondo, J.C.; Bosi, S.; Bassi, C.; Ferracin, M.; Lanza, G.; Gafà, R.; Magri, E.; Selvatici, R.; Torresani, S.; Marci, R.; et al. Gene expression changes in progression of cervical neoplasia revealed by microarray analysis of cervical neoplastic keratinocytes. J. Cell. Physiol. 2015, 230, 806–812. [Google Scholar] [CrossRef]
- Wang, J.; Liu, S.; Shi, J. The Role of miRNA in the Diagnosis, Prognosis, and Treatment of Osteosarcoma. Cancer Biother. Radiopharm. 2019, 34, 605–613. [Google Scholar] [CrossRef]
- Rotondo, J.C.; Martini, F.; Maritati, M.; Mazziotta, C.; Di Mauro, G.; Lanzillotti, C.; Barp, N.; Gallerani, A.; Tognon, M.; Contini, C. SARS-CoV-2 Infection: New Molecular, Phylogenetic, and Pathogenetic Insights. Efficacy of Current Vaccines and the Potential Risk of Variants. Viruses 2021, 13, 1687. [Google Scholar] [CrossRef] [PubMed]
- Oton-Gonzalez, L.; Rotondo, J.C.; Cerritelli, L.; Malagutti, N.; Lanzillotti, C.; Bononi, I.; Ciorba, A.; Bianchini, C.; Mazziotta, C.; De Mattei, M.; et al. Association between oncogenic human papillomavirus type 16 and Killian polyp. Infect. Agents Cancer 2021, 16, 3. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Modification | Histone | Site | Experimental Model | Enzyme | Reference |
|---|---|---|---|---|---|
| Deacetylation | H3 | K9 | MCPyV-positive MCC cell lines | - | [106] |
| Deacetylation | H3 | K9 | MCPyV-positive MCC cell lines and mouse models | - | [107] |
| Acetylation | - | - | MCPyV-positive/-negative MCC cell lines | MYCL and EP400 complex | [108] |
| Demethylation | - | - | MCPyV-positive MCC cell lines | LSD1 | [109] |
| Demethylation | - | - | MCPyV-positive MCC cell lines | LSD1 | [8] |
| Methylation (me3) | H3 | K27 | MCPyV-positive/-negative MCC tissues | - | [69] |
| Methylation (me1-2-3) | - | - | MCPyV-positive/-negative MCC tissues | EZH2 | [105] |
| Low methylation (me3) | H3 | K27 | MCPyV-positive/-negative MCC tissues | - | [110] |
| Methylation (me2) | H3 | K4 | No-MCC cell lines expressing MCPyV sT antigen | - | [111] |
| Methylation (me2) | H4 | K20 | No-MCC cell lines expressing MCPyV sT antigen | - | |
| Phosphorylation | H2AX | S139 | No-MCC cell lines expressing MCPyV sT antigen | - |
| Gene | Function | Promoter Methylation | Experimental Model | Reference |
|---|---|---|---|---|
| P14ARF | Tumor suppressor protein | Hypermethylated | MCPyV-positive/-negative MCC tissues | [115] |
| CDKN2A | Tumor suppressor protein | Hypermethylated | MCPyV-positive/-negative MCC tissues | [113] |
| RASSF1A | Tumor suppressor protein | Hypermethylated | ||
| RASSF2 | Tumor suppressor protein | Hypermethylated | MCPyV-positive/-negative MCC tissues | [116] |
| RASSF5C | Tumor suppressor protein | Hypermethylated | ||
| RASSF10 | Embryonic neurogenesis | Hypermethylated | ||
| RB1 | Tumor suppressor protein | Hypermethylated | MCPyV-positive/-negative MCC tissues | [114] |
| MGMT | DNA repair and apoptosis | Hyper-/Hypomethylated | MCPyV-positive MCC cell lines | [117] |
| Hypomethylated | MCC tissues * | |||
| PTCH1 | HH receptor | Hypomethylated | MCPyV-positive/-negative MCC tissues | [118] |
| PD-1 | Immune-inhibitory receptor | Hypomethylated | MCPyV-positive/-negative MCC tissues | [119] |
| Multiple genes | Hyper-/Hypomethylated | MCPyV-positive/-negative MCC tissues/cell lines | [120] | |
| Multiple genes | Hyper-/Hypomethylated | MCPyV-positive/-negative MCC tissues/cell lines | [121] | |
| KDM6B | H3K27 demethylation | Hypomethylated | MCPyV-positive tissues |
| miRNA↑ | miRNA↓ | Experimental Model | Reference |
|---|---|---|---|
| miR-375 | → | MCPyV-positive/-negative MCC vs. non-MCC tissues and cells lines * | [126] |
| miR-375 | MCPyV-positive vs. MCPyV-negative MCC cell lines | [127] | |
| miR-200c-141 miR-183-96-182 | MCPyV-positive vs. MCPyV-negative MCC cell lines | [136] | |
| miR-30a-3p miR-30a-5p miR-375 miR-34a miR-769-5p | miR-203 | MCPyV-positive vs. MCPyV-negative MCC tissues and cell lines | [128] |
| miR-30a miR-34a miR-142-3p miR-1539 | MCPyV-positive vs. MCPyV-negative MCC tissues | [135] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rotondo, J.C.; Mazziotta, C.; Lanzillotti, C.; Tognon, M.; Martini, F. Epigenetic Dysregulations in Merkel Cell Polyomavirus-Driven Merkel Cell Carcinoma. Int. J. Mol. Sci. 2021, 22, 11464. https://doi.org/10.3390/ijms222111464
Rotondo JC, Mazziotta C, Lanzillotti C, Tognon M, Martini F. Epigenetic Dysregulations in Merkel Cell Polyomavirus-Driven Merkel Cell Carcinoma. International Journal of Molecular Sciences. 2021; 22(21):11464. https://doi.org/10.3390/ijms222111464
Chicago/Turabian StyleRotondo, John Charles, Chiara Mazziotta, Carmen Lanzillotti, Mauro Tognon, and Fernanda Martini. 2021. "Epigenetic Dysregulations in Merkel Cell Polyomavirus-Driven Merkel Cell Carcinoma" International Journal of Molecular Sciences 22, no. 21: 11464. https://doi.org/10.3390/ijms222111464
APA StyleRotondo, J. C., Mazziotta, C., Lanzillotti, C., Tognon, M., & Martini, F. (2021). Epigenetic Dysregulations in Merkel Cell Polyomavirus-Driven Merkel Cell Carcinoma. International Journal of Molecular Sciences, 22(21), 11464. https://doi.org/10.3390/ijms222111464