
The Initial Human Atherosclerotic Lesion and Lipoprotein Modification—A Deep Connection

Abstract
:1. Introduction
2. The Evolution of Classification Systems
3. The Matter of Insudation
4. Extra- or Intracellular Lipoproteins?
5. The Fate of Tissue-Stranded LDL
6. Atherogenesis in Infants and Children?
7. Lesion Progression Due to…?
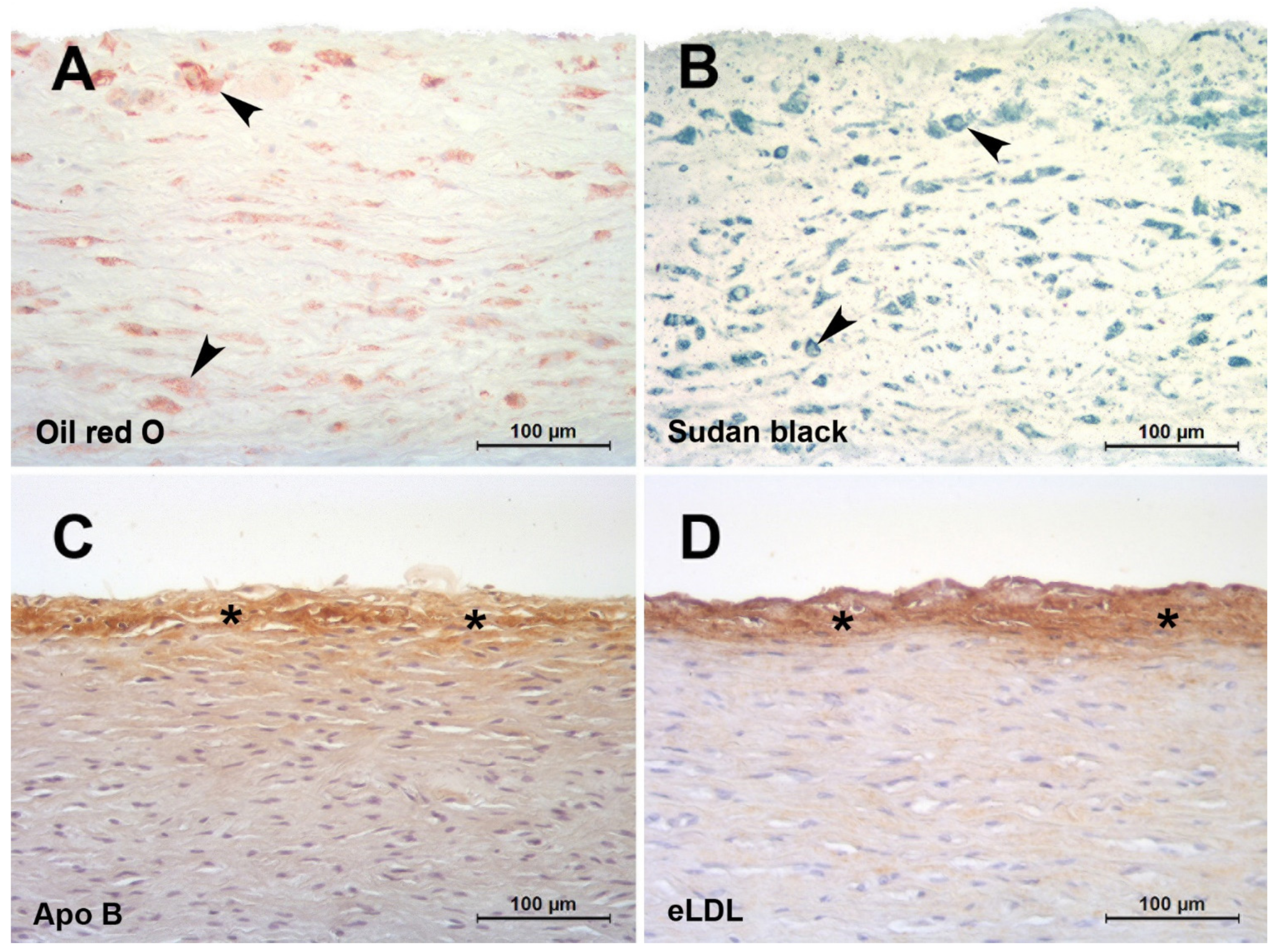
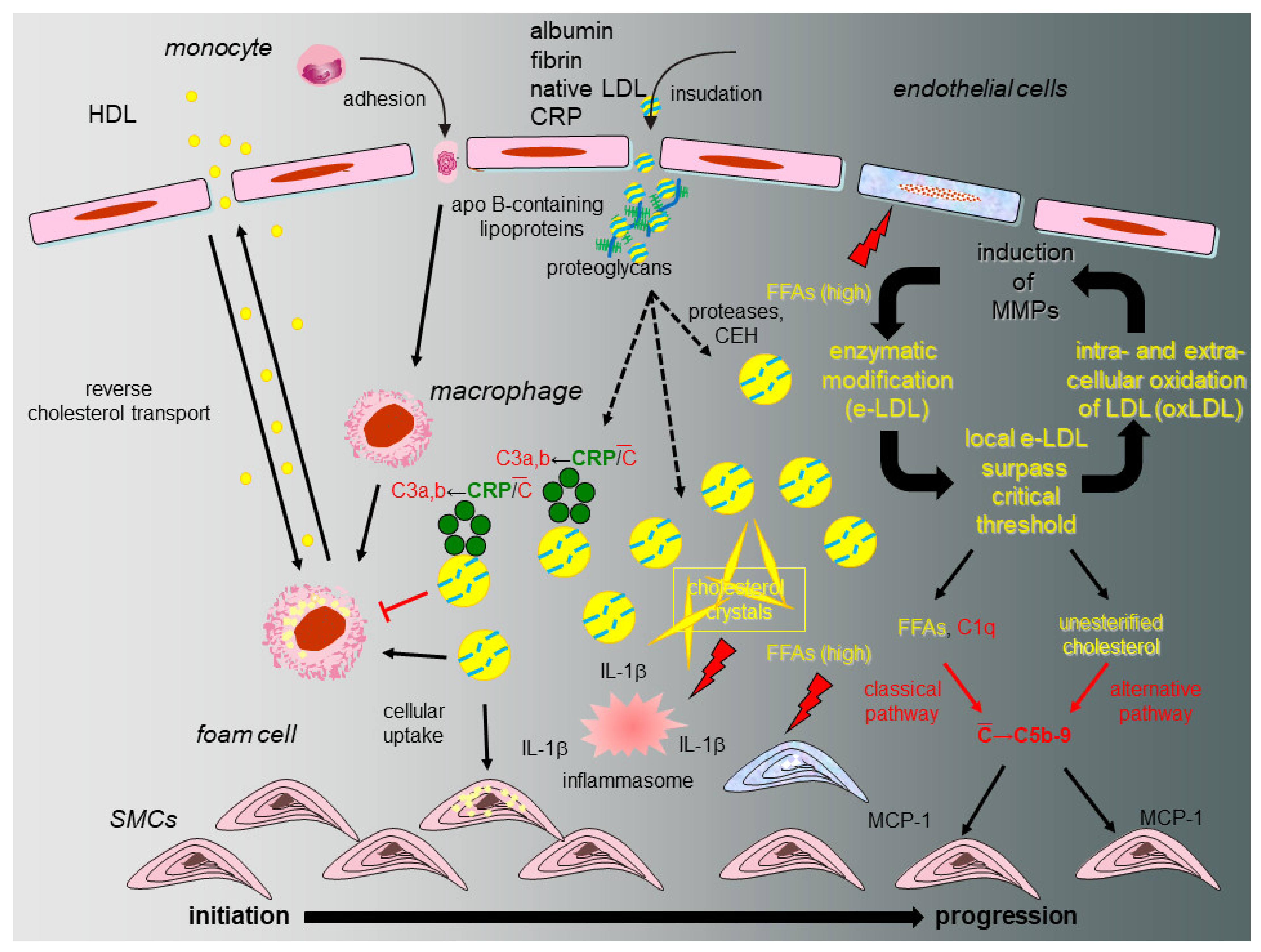
8. The eLDL Hypothesis (Figure 2)
9. Alternative Hypotheses on Atherosclerotic Lesion Initiation and Progression
9.1. Vascular-Associated Lymphoid Tissue (VALT)
9.2. Lysosomal Storage Disease
9.3. Inflammasome and Mitochondria
9.4. Micro RNAs
10. Conclusions
Funding
Conflicts of Interest
Abbreviations
| ACAT | acetyl-CoA acetyltransferase |
| AHA | American Heart Association |
| CRP | C-reactive protein |
| DAMPs | damage-associated molecular patterns |
| DIT | diffuse intimal thickening |
| eLDL | enzymatically modified LDL |
| FFA | free fatty acid |
| GMA | glycol methacrylate) |
| HEMA | 2-hydroxyethylmethacrylate |
| IL-1 β | interleukin-1 β |
| IL-6 | interleukin-6 |
| LDL | Low Density Lipoproteins |
| MCP-1 | monocyte chemotactic protein-1 |
| mtDNA | mitochondral DNA |
| miRNAs | micro RNAs |
| MMP | matrix metalloproteinase |
| NLRP3 | NOD-, LRR- and pyrin-domain containing 3 |
| oxLDL | oxidized LDL |
| PIT | pathological intimal thickening |
| SMC | smooth muscle cell |
| VALT | vascular-associated lymphoid tissue |
References
- Klotz, O.; Mannig, M.F. Fatty streaks in the intima of arteries. J. Path. Bact. 1911, 16, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Holman, R.L.; Mc, G.H., Jr.; Strong, J.P.; Geer, J.C. The natural history of atherosclerosis: The early aortic lesions as seen in New Orleans in the middle of the of the 20th century. Am. J. Pathol. 1958, 34, 209–235. [Google Scholar]
- Stary, H.C. Natural history and histological classification of atherosclerotic lesions: An update. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1177–1178. [Google Scholar] [CrossRef] [Green Version]
- Stary, H.C. Lipid and macrophage accumulations in arteries of children and the development of atherosclerosis. Am. J. Clin. Nutr. 2000, 72, 1297S–1306S. [Google Scholar] [CrossRef]
- Virmani, R.; Kolodgie, F.D.; Burke, A.P.; Farb, A.; Schwartz, S.M. Lessons from sudden coronary death: A comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1262–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolodgie, F.D.; Burke, A.P.; Nakazawa, G.; Cheng, Q.; Xu, X.; Virmani, R. Free cholesterol in atherosclerotic plaques: Where does it come from? Curr. Opin. Lipidol. 2007, 18, 500–507. [Google Scholar] [CrossRef]
- Otsuka, F.; Kramer, M.C.; Woudstra, P.; Yahagi, K.; Ladich, E.; Finn, A.V.; de Winter, R.J.; Kolodgie, F.D.; Wight, T.N.; Davis, H.R.; et al. Natural progression of atherosclerosis from pathologic intimal thickening to late fibroatheroma in human coronary arteries: A pathology study. Atherosclerosis 2015, 241, 772–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, Y.; Fujii, H.; Sumiyoshi, S.; Wight, T.N.; Sueishi, K. Early human atherosclerosis: Accumulation of lipid and proteoglycans in intimal thickenings followed by macrophage infiltration. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1159–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Cliff, W.J.; Schoefl, G.I.; Higgins, G. Plasma protein insudation as an index of early coronary atherogenesis. Am. J. Pathol. 1993, 143, 496–506. [Google Scholar]
- Duguid, J.B. Thrombosis as a factor in the pathogenesis of aortic atherosclerosis. J. Pathol. Bacteriol. 1948, 60, 57–61. [Google Scholar] [CrossRef]
- Smith, E.B. Molecular interactions in human atherosclerotic plaques. Am. J. Pathol. 1977, 86, 665–674. [Google Scholar] [PubMed]
- Wolinsky, H. A proposal linking clearance of circulating lipoproteins to tissue metabolic activity as a basis for understanding atherogenesis. Circ. Res. 1980, 47, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Eliska, O.; Eliskova, M.; Miller, A.J. The absence of lymphatics in normal and atherosclerotic coronary arteries in man: A morphologic study. Lymphology 2006, 39, 76–83. [Google Scholar] [PubMed]
- Tabas, I.; Williams, K.J.; Borén, J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: Update and therapeutic implications. Circulation 2007, 116, 1832–1844. [Google Scholar] [CrossRef] [PubMed]
- Torzewski, M.; Bhakdi, S. Complement and atherosclerosis-united to the point of no return? Clin. Biochem. 2013, 46, 20–25. [Google Scholar] [CrossRef]
- Torzewski, M.; Navarro, B.; Cheng, F.; Canisius, A.; Schmidt, T.; Bhakdi, S.; Urban, R.; Lackner, K.J. Investigation of Sudan IV staining areas in aortas of infants and children: Possible prelesional stages of atherogenesis. Atherosclerosis 2009, 206, 159–167. [Google Scholar] [CrossRef]
- Bhakdi, S.; Lackner, K.J.; Han, S.R.; Torzewski, M.; Husmann, M. Beyond cholesterol: The enigma of atherosclerosis revisited. Thromb. Haemost. 2004, 91, 639–645. [Google Scholar]
- Torzewski, M. Enzymatically modified LDL, atherosclerosis and beyond: Paving the way to acceptance. Front. Biosci. Landmark 2018, 23, 257–271. [Google Scholar] [CrossRef] [Green Version]
- Klouche, M.; Gottschling, S.; Gerl, V.; Hell, W.; Husmann, M.; Dorweiler, B.; Messner, M.; Bhakdi, S. Atherogenic properties of enzymatically degraded LDL: Selective induction of MCP-1 and cytotoxic effects on human macrophages. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1376–1385. [Google Scholar] [CrossRef] [Green Version]
- Bhakdi, S.; Dorweiler, B.; Kirchmann, R.; Torzewski, J.; Weise, E.; Tranum-Jensen, J.; Walev, I.; Wieland, E. On the pathogenesis of atherosclerosis: Enzymatic transformation of human low density lipoprotein to an atherogenic moiety. J. Exp. Med. 1995, 182, 1959–1971. [Google Scholar] [CrossRef]
- Torzewski, M.; Torzewski, J.; Bowyer, D.E.; Waltenberger, J.; Fitzsimmons, C.; Hombach, V.; Gabbert, H.E. Immunohistochemical colocalization of the terminal complex of human complement and smooth muscle cell alpha-actin in early atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2448–2452. [Google Scholar] [CrossRef]
- Torzewski, J.; Oldroyd, R.; Lachmann, P.; Fitzsimmons, C.; Proudfoot, D.; Bowyer, D. Complement-induced release of monocyte chemotactic protein-1 from human smooth muscle cells. A possible initiating event in atherosclerotic lesion formation. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 673–677. [Google Scholar] [CrossRef]
- Torzewski, M.; Klouche, M.; Hock, J.; Messner, M.; Dorweiler, B.; Torzewski, J.; Gabbert, H.E.; Bhakdi, S. Immunohistochemical demonstration of enzymatically modified human LDL and its colocalization with the terminal complement complex in the early atherosclerotic lesion. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Bhakdi, S.; Torzewski, M.; Klouche, M.; Hemmes, M. Complement and atherogenesis: Binding of CRP to degraded, nonoxidized LDL enhances complement activation. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2348–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torzewski, M.; Rist, C.; Mortensen, R.F.; Zwaka, T.P.; Bienek, M.; Waltenberger, J.; Koenig, W.; Schmitz, G.; Hombach, V.; Torzewski, J. C-reactive protein in the arterial intima: Role of C-reactive protein receptor-dependent monocyte recruitment in atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2094–2099. [Google Scholar] [CrossRef] [Green Version]
- Geer, J.C.; Mc, G.H., Jr.; Strong, J.P. The fine structure of human atherosclerotic lesions. Am. J. Pathol. 1961, 38, 263–287. [Google Scholar] [PubMed]
- Khalil, M.F.; Wagner, W.D.; Goldberg, I.J. Molecular interactions leading to lipoprotein retention and the initiation of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2211–2218. [Google Scholar] [CrossRef] [PubMed]
- Chao, F.F.; Amende, L.M.; Blanchette-Mackie, E.J.; Skarlatos, S.I.; Gamble, W.; Resau, J.H.; Mergner, W.T.; Kruth, H.S. Unesterified cholesterol-rich lipid particles in atherosclerotic lesions of human and rabbit aortas. Am. J. Pathol. 1988, 131, 73–83. [Google Scholar]
- Napoli, C.; D‘Armiento, F.P.; Mancini, F.P.; Postiglione, A.; Witztum, J.L.; Palumbo, G.; Palinski, W. Fatty streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia. Intimal accumulation of low density lipoprotein and its oxidation precede monocyte recruitment into early atherosclerotic lesions. J. Clin. Investig. 1997, 100, 2680–2690. [Google Scholar] [CrossRef]
- Guyton, J.R.; Klemp, K.F. Transitional features in human atherosclerosis. Intimal thickening, cholesterol clefts, and cell loss in human aortic fatty streaks. Am. J. Pathol. 1993, 143, 1444–1457. [Google Scholar]
- Millonig, G.; Schwentner, C.; Mueller, P.; Mayerl, C.; Wick, G. The vascular-associated lymphoid tissue: A new site of local immunity. Curr. Opin. Lipidol. 2001, 12, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Millonig, G.; Malcom, G.T.; Wick, G. Early inflammatory-immunological lesions in juvenile atherosclerosis from the Pathobiological Determinants of Atherosclerosis in Youth (PDAY)-study. Atherosclerosis 2002, 160, 441. [Google Scholar] [CrossRef]
- Haley, N.J.; Fowler, S.; de Duve, C. Lysosomal acid cholesteryl esterase activity in normal and lipid-laden aortic cells. J. Lipid Res. 1980, 21, 961–969. [Google Scholar] [CrossRef]
- Bobryshev, Y.V.; Shchelkunova, T.A.; Morozov, I.A.; Rubtsov, P.M.; Sobenin, I.A.; Orekhov, A.N.; Smirnov, A.N. Changes of lysosomes in the earliest stages of the development of atherosclerosis. J. Cell. Mol. Med. 2013, 17, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Kruth, H.S. Subendothelial accumulation of unesterified cholesterol. An early event in atherosclerotic lesion development. Atherosclerosis 1985, 57, 337–341. [Google Scholar] [CrossRef]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef] [PubMed]
- Bezsonov, E.E.; Sobenin, I.A.; Orekhov, A.N. Immunopathology of Atherosclerosis and Related Diseases: Focus on Molecular Biology. Int. J. Mol. Sci. 2021, 22, 4080. [Google Scholar] [CrossRef]
- Andreou, I.; Sun, X.; Stone, P.H.; Edelman, E.R.; Feinberg, M.W. miRNAs in atherosclerotic plaque initiation, progression, and rupture. Trends Mol. Med. 2015, 21, 307–318. [Google Scholar] [CrossRef]
- Hartmann, P.; Zhou, Z.; Natarelli, L.; Wei, Y.; Nazari-Jahantigh, M.; Zhu, M.; Grommes, J.; Steffens, S.; Weber, C.; Schober, A. Endothelial Dicer promotes atherosclerosis and vascular inflammation by miRNA-103-mediated suppression of KLF4. Nat. Commun. 2016, 7, 11907. [Google Scholar] [CrossRef]
- Li, X.; He, X.; Wang, J.; Wang, D.; Cong, P.; Zhu, A.; Chen, W. The Regulation of Exosome-Derived miRNA on Heterogeneity of Macrophages in Atherosclerotic Plaques. Front. Immunol. 2020, 11, 2175. [Google Scholar] [CrossRef]
- Woo, C.C.; Liu, W.; Lin, X.Y.; Dorajoo, R.; Lee, K.W.; Richards, A.M.; Lee, C.N.; Wongsurawat, T.; Nookaew, I.; Sorokin, V. The interaction between 30b-5p miRNA and MBNL1 mRNA is involved in vascular smooth muscle cell differentiation in patients with coronary atherosclerosis. Int. J. Mol. Sci. 2019, 21, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stary, H.C.; Blankenhorn, D.H.; Chandler, A.B.; Glagov, S.; Insull, W., Jr.; Richardson, M.; Rosenfeld, M.E.; Schaffer, S.A.; Schwartz, C.J.; Wagner, W.D. A definition of the intima of human arteries and of its atherosclerosis-prone regions. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1992, 85, 391–405. [Google Scholar] [CrossRef] [Green Version]
- Stary, H.C.; Chandler, A.B.; Glagov, S.; Guyton, J.R.; Insull, W., Jr.; Rosenfeld, M.E.; Schaffer, S.A.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler. Thromb. 1994, 14, 840–856. [Google Scholar] [CrossRef] [Green Version]
- Stary, H.C.; Chandler, A.B.; Dinsmore, R.E.; Fuster, V.; Glagov, S.; Insull, W., Jr.; Rosenfeld, M.E.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1512–1531. [Google Scholar] [CrossRef]
- Nakashima, Y.; Chen, Y.X.; Kinukawa, N.; Sueishi, K. Distributions of diffuse intimal thickening in human arteries: Preferential expression in atherosclerosis-prone arteries from an early age. Virchows Arch. 2002, 441, 279–288. [Google Scholar] [CrossRef]
- Stender, S.; Zilversmit, D.B. Transfer of plasma lipoprotein components and of plasma proteins into aortas of cholesterol-fed rabbits. Molecular size as a determinant of plasma lipoprotein influx. Arteriosclerosis 1981, 1, 38–49. [Google Scholar] [CrossRef]
- Camejo, G. The interaction of lipids and lipoproteins with the intercellular matrix of arterial tissue: Its possible role in atherogenesis. Adv. Lipid Res. 1982, 19, 1–53. [Google Scholar] [PubMed]
- Williams, K.J.; Tabas, I. The response-to-retention hypothesis of atherogenesis reinforced. Curr. Opin. Lipidol. 1998, 9, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Öörni, K.; Kovanen, P.T. Aggregation Susceptibility of Low-Density Lipoproteins—A Novel Modifiable Biomarker of Cardiovascular Risk. J. Clin. Med. 2021, 10, 1769. [Google Scholar] [CrossRef]
- Kruth, H.S. Localization of unesterified cholesterol in human atherosclerotic lesions. Demonstration of filipin-positive, oil-red-O-negative particles. Am. J. Pathol. 1984, 114, 201–208. [Google Scholar]
- Skalen, K.; Gustafsson, M.; Rydberg, E.K.; Hulten, L.M.; Wiklund, O.; Innerarity, T.L.; Boren, J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 2002, 417, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, H.; Gerrits, P.O.; Grond, J. The application of lipid-soluble stains in plastic embedded sections. Histochemistry 1986, 85, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Meyer, W.; Schmidt, J.; Busche, R.; Jacob, R.; Naim, H.Y. Demonstration of free fatty acids in the integument of semi-aquatic and aquatic mammals. Acta Histochem. 2012, 114, 145–150. [Google Scholar] [CrossRef]
- Steinberg, D. Low density lipoprotein oxidation and its pathobiological significance. J. Biol. Chem. 1997, 272, 20963–20966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torzewski, M.; Suriyaphol, P.; Paprotka, K.; Spath, L.; Ochsenhirt, V.; Schmitt, A.; Han, S.R.; Husmann, M.; Gerl, V.B.; Bhakdi, S.; et al. Enzymatic modification of low-density lipoprotein in the arterial wall: A new role for plasmin and matrix metalloproteinases in atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2130–2136. [Google Scholar] [CrossRef] [Green Version]
- Mezentsev, A.; Bezsonov, E.; Kashirskikh, D.; Baig, M.S.; Eid, A.H.; Orekhov, A. Proatherogenic Sialidases and Desialylated Lipoproteins: 35 Years of Research and Current State from Bench to Bedside. Biomedicines 2021, 9, 600. [Google Scholar] [CrossRef]
- Lehti, S.; Nguyen, S.D.; Belevich, I.; Vihinen, H.; Heikkilä, H.M.; Soliymani, R.; Käkelä, R.; Saksi, J.; Jauhiainen, M.; Grabowski, G.A.; et al. Extracellular Lipids Accumulate in Human Carotid Arteries as Distinct Three-Dimensional Structures and Have Proinflammatory Properties. Am. J. Pathol. 2018, 188, 525–538. [Google Scholar] [CrossRef] [Green Version]
- Simionescu, N.; Vasile, E.; Lupu, F.; Popescu, G.; Simionescu, M. Prelesional events in atherogenesis. Accumulation of extracellular cholesterol-rich liposomes in the arterial intima and cardiac valves of the hyperlipidemic rabbit. Am. J. Pathol. 1986, 123, 109–125. [Google Scholar]
- Kühn, H.; Heydeck, D.; Hugou, I.; Gniwotta, C. In vivo action of 15-lipoxygenase in early stages of human atherogenesis. J. Clin. Investig. 1997, 99, 888–893. [Google Scholar] [CrossRef]
- Calara, F.; Dimayuga, P.; Niemann, A.; Thyberg, J.; Diczfalusy, U.; Witztum, J.L.; Palinski, W.; Shah, P.K.; Cercek, B.; Nilsson, J.; et al. An animal model to study local oxidation of LDL and its biological effects in the arterial wall. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 884–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinserling, W.D. Untersuchungen über Atherosklerose. I. Über die Aortaverfettung bei Kindern. Virchows Arch. Path. Anat. 1925, 255, 677–705. [Google Scholar] [CrossRef]
- Zeek, P. Juvenile arteriosclerosis. Arch. Path. 1930, 10, 417–446. [Google Scholar]
- Albert, Z. Die Veränderungen der Aorta bei Kindern und ihr Verhältnis zur Atherosklerose. Virchows Arch. Path. Anat. 1939, 303, 265–279. [Google Scholar] [CrossRef]
- Velican, C.; Velican, D. Intimal histolysis as an early stage of atherosclerotic involvement. Observations on the human coronary arteries. Med. Interne 1976, 14, 185–190. [Google Scholar] [PubMed]
- Sakurai, I.; Tosaka, A.; Yamada, T.; Kuwahara, T.; Masubuchi, K. Childhood coronary sclerosis. Acta Pathol. Jpn. 1978, 28, 41–52. [Google Scholar] [CrossRef]
- Bland, J.; Skordalaki, A.; Emery, J.L. Early intimal lesions in the common carotid artery. Cardiovasc. Res. 1986, 20, 863–868. [Google Scholar] [CrossRef]
- Stary, H.C. Macrophages, macrophage foam cells, and eccentric intimal thickening in the coronary arteries of young children. Atherosclerosis 1987, 64, 91–108. [Google Scholar] [CrossRef]
- Napoli, C.; Glass, C.K.; Witztum, J.L.; Deutsch, R.; D‘Armiento, F.P.; Palinski, W. Influence of maternal hypercholesterolaemia during pregnancy on progression of early atherosclerotic lesions in childhood: Fate of Early Lesions in Children (FELIC) study. Lancet 1999, 354, 1234–1241. [Google Scholar] [CrossRef]
- Stary, H.C. Evolution and progression of atherosclerotic lesions in coronary arteries of children and young adults. Arteriosclerosis 1989, 9, I19–I32. [Google Scholar]
- Stary, H.C. Composition and classification of human atherosclerotic lesions. Virchows Arch. A Pathol. Anat. Histopathol. 1992, 421, 277–290. [Google Scholar] [CrossRef]
- Weber, C.; Zernecke, A.; Libby, P. The multifaceted contributions of leukocyte subsets to atherosclerosis: Lessons from mouse models. Nat. Rev. Immunol. 2008, 8, 802–815. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [Green Version]
- Vedder, V.L.; Aherrahrou, Z.; Erdmann, J. Dare to Compare. Development of Atherosclerotic Lesions in Human, Mouse, and Zebrafish. Front. Cardiovasc. Med. 2020, 7, 109. [Google Scholar] [CrossRef]
- Lu, X.; Kakkar, V. Inflammasome and atherogenesis. Curr. Pharm. Des. 2014, 20, 108–124. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Tuzcu, E.M.; Brewer, H.B.; Sipahi, I.; Nicholls, S.J.; Ganz, P.; Schoenhagen, P.; Waters, D.D.; Pepine, C.J.; Crowe, T.D.; et al. Effect of ACAT inhibition on the progression of coronary atherosclerosis. N. Engl. J. Med. 2006, 354, 1253–1263. [Google Scholar] [CrossRef]
- Meuwese, M.C.; de Groot, E.; Duivenvoorden, R.; Trip, M.D.; Ose, L.; Maritz, F.J.; Basart, D.C.; Kastelein, J.J.; Habib, R.; Davidson, M.H.; et al. ACAT inhibition and progression of carotid atherosclerosis in patients with familial hypercholesterolemia: The CAPTIVATE randomized trial. JAMA 2009, 301, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Fichtlscherer, S.; De Rosa, S.; Fox, H.; Schwietz, T.; Fischer, A.; Liebetrau, C.; Weber, M.; Hamm, C.W.; Röxe, T.; Müller-Ardogan, M.; et al. Circulating microRNAs in patients with coronary artery disease. Circ. Res. 2010, 107, 677. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Denomination | Features | Lesion Progression Due to … | Ref. |
|---|---|---|---|
| fatty streak, minimal sudanophilic intimal deposit | both intra- and extracellular “globules” of lipid, slight increase in interstitial mucinous material | conversion into fibrous plaques | [2] |
| type I (initial) lesion | isolated macrophage foam cells | small pools of lipid droplets and dead cell remnants as a source of extracellular lipid in addition to macrophage foam cells (preatheroma) | [3,4] |
| intimal xanthoma | isolated macrophage foam cells | extracellular lipid accumulation (lipid pools) that are rich in extracellular matrix proteoglycans (pathologic intimal thickening (PIT)) | [5,6,7] |
| grade of lipid deposition 1 | fatty streaks with extracellular lipids colocalizing with biglycan and decorin in the outer layer of the intima | n/a | [8] |
| early lesion | plasma albumin and apolipoprotein B insudation | n/a | [9] |
| early lesion | interstitial lipid deposits resulting from the encrustation or imbibition of fibrin onto or into the intima | n/a | [10] |
| gelatinous lesion | balances of intact LDL/“deposited” cholesterol and of fibrinogen/fibrin | loss of steady state concentrations reflecting rates of egress of macromolecules depending on molecular sieving (immobilization of LDL by fibrin) | [11] |
| n/a | n/a | influx-efflux imbalance in the cell and blood vessel wall | [12] |
| epicardial coronary atherosclerosis | impairment of lymphatic drainage from the coronary arteries (absence of a potential system for removing protein, fluid and lipids from the arterial wall) | impairment of lymphatic drainage from the coronary arteries (absence of a potential system for removing protein, fluid and lipids from the arterial wall) | [13] |
| prelesional stage | ‘inert’ lipoprotein insudation without monocyte/ macrophage infiltration, lipoprotein modification and complement activation | overload of the cholesterol removal machinery, enzymatic modification of LDL, complement activation, persisting macrophages secreting a variety of molecules accelerating lipoprotein retention, plaque instability, and clotting on rupture | [14,15,16,17,18,19,20,21,22,23,24,25] |
| early fatty streak | intracellular lipid accumulation in SMCs | degeneration of lipid-containing cells with extravasation of lipid particles into the extracellular space | [26] |
| early lesion | ionic interaction of positively charged regions of apolipoprotein B with matrix proteins, including proteoglycans, collagen, and fibronectin | n/a | [27] |
| initial lipid deposition | unesterified cholesterol-rich lipid particles | n/a | [28] |
| fatty streak | LDL accumulation and oxidation preceding intimal accumulation of monocytes | n/a | [29] |
| n/a | n/a | cholesterol crystals or clefts in the musculoelastic (deep) layer of the intima or in the tunica media | [30] |
| fatty streak | accumulation of mononuclear cells | n/a | [31,32] |
| type I (initial) lesion | alteration in electron density of the matrix of lysosomal bodies as well as the formation of lamellar bodies in lysosomes | substantial structural changes of lysosomes in the ‘normal intima-initial lesion-fatty streak’ sequence | [33,34] |
| early lesion | unesterified, crystalline cholesterol | n/a | [35,36] |
| initial lesion | miRNAs mediating cellular regulation in endothelial activation and inflammation, differentiation of macrophages and their polarization, having important functional properties in lipoprotein homeostasis and playing a central role in the mechanisms determining SMC phenotype | miRNAs mediating cellular regulation in endothelial activation and inflammation, differentiation of macrophages and their polarization, having important functional properties in lipoprotein homeostasis and playing a central role in the mechanisms determining SMC phenotype | [37,38,39,40,41] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torzewski, M. The Initial Human Atherosclerotic Lesion and Lipoprotein Modification—A Deep Connection. Int. J. Mol. Sci. 2021, 22, 11488. https://doi.org/10.3390/ijms222111488
Torzewski M. The Initial Human Atherosclerotic Lesion and Lipoprotein Modification—A Deep Connection. International Journal of Molecular Sciences. 2021; 22(21):11488. https://doi.org/10.3390/ijms222111488
Chicago/Turabian StyleTorzewski, Michael. 2021. "The Initial Human Atherosclerotic Lesion and Lipoprotein Modification—A Deep Connection" International Journal of Molecular Sciences 22, no. 21: 11488. https://doi.org/10.3390/ijms222111488
APA StyleTorzewski, M. (2021). The Initial Human Atherosclerotic Lesion and Lipoprotein Modification—A Deep Connection. International Journal of Molecular Sciences, 22(21), 11488. https://doi.org/10.3390/ijms222111488