
The Role of Maternal Immune Activation in the Pathogenesis of Autism: A Review of the Evidence, Proposed Mechanisms and Implications for Treatment



Abstract
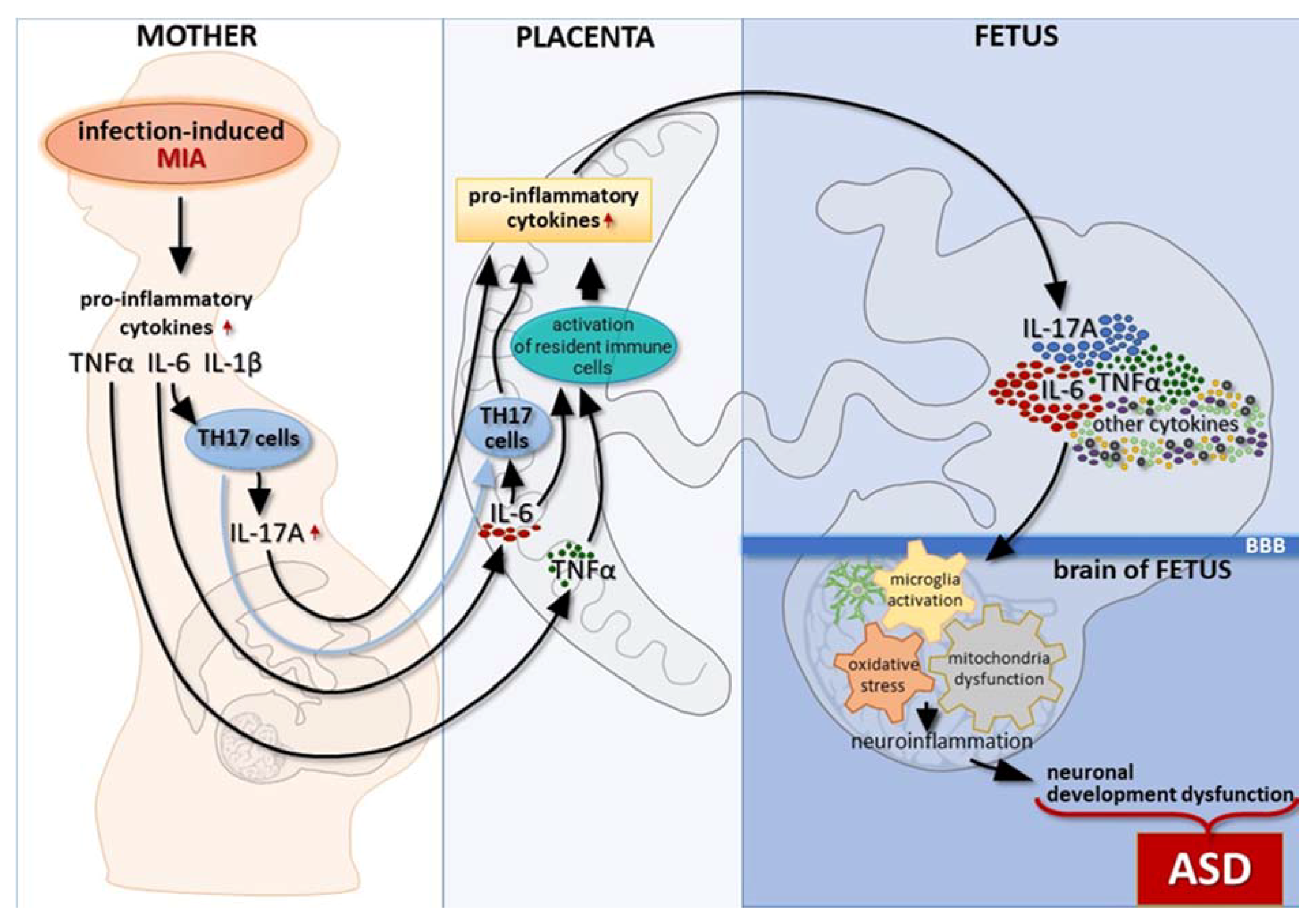
:1. Introduction
2. Proinflammatory Cytokines as the Link between Maternal Inflammation and Autism Development in Offspring
2.1. Interleukin-6
2.2. Interleukin-17
2.3. Interleukin-1β
3. Microglial Abnormalities in MIA-Induced Autism Spectrum Disorders
4. Oxidative Stress in ASD Individuals and MIA Models
5. Mitochondrial Dysfunction in MIA-Evoked Autism Spectrum Disorders
6. Potential Treatment Strategies Targeting Inflammatory Pathways in ASD
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Edition, F. Diagnostic and Statistical Manual of Mental Disorders; American Psychiatric Association: Washington, DC, USA, 2013; Volume 21. [Google Scholar]
- Rice, C.E.; Baio, J.; Van Naarden Braun, K.; Doernberg, N.; Meaney, F.J.; Kirby, R.S. A public health collaboration for the surveillance of autism spectrum disorders. Paediatr. Perinat. Epidemiol. 2007, 21, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Maenner, M.J.; Shaw, K.A.; Baio, J.; Washington, A.; Patrick, M.; DiRienzo, M.; Christensen, D.L.; Wiggins, L.D.; Pettygrove, S.; Andrews, J.G.; et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2016. MMWR Surveill. Summ. 2020, 69, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Young, H.; Oreve, M.J.; Speranza, M. Clinical characteristics and problems diagnosing autism spectrum disorder in girls. Arch. Pédiatr. 2018, 25, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Allely, C.S. Understanding and recognising the female phenotype of autism spectrum disorder and the “camouflage” hypothesis: A systematic PRISMA review. Adv. Autism 2019, 5, 14–37. [Google Scholar] [CrossRef]
- Chaste, P.; Leboyer, M. Autism risk factors: Genes, environment, and gene-environment interactions. Dialogues Clin. Neurosci. 2012, 14, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Tick, B.; Bolton, P.; Happé, F.; Rutter, M.; Rijsdijk, F. Heritability of autism spectrum disorders: A meta-analysis of twin studies. J. Child Psychol. Psychiatry Allied Discip. 2016, 57, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Vorstman, J.A.S.; Parr, J.R.; Moreno-De-Luca, D.; Anney, R.J.L.; Nurnberger, J.I., Jr.; Hallmayer, J.F. Autism genetics: Opportunities and challenges for clinical translation. Nat. Rev. Genet. 2017, 18, 362–376. [Google Scholar] [CrossRef]
- Gentile, S. Risks of neurobehavioral teratogenicity associated with prenatal exposure to valproate monotherapy: A systematic review with regulatory repercussions. CNS Spectr. 2014, 19, 305–315. [Google Scholar] [CrossRef]
- Lam, J.; Sutton, P.; Kalkbrenner, A.; Windham, G.; Halladay, A.; Koustas, E.; Lawler, C.; Davidson, L.; Daniels, N.; Newschaffer, C.; et al. A Systematic Review and Meta-Analysis of Multiple Airborne Pollutants and Autism Spectrum Disorder. PLoS ONE 2016, 11, e0161851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Wu, F.; Ding, Y.; Hou, J.; Bi, J.; Zhang, Z. Advanced parental age and autism risk in children: A systematic review and meta-analysis. Acta Psychiatr. Scand. 2017, 135, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Xiang, A.H.; Wang, X.; Martinez, M.P.; Page, K.; Buchanan, T.A.; Feldman, R.K. Maternal Type 1 Diabetes and Risk of Autism in Offspring. JAMA 2018, 320, 89–91. [Google Scholar] [CrossRef]
- Ornoy, A.; Weinstein-Fudim, L.; Ergaz, Z. Prenatal factors associated with autism spectrum disorder (ASD). Reprod. Toxicol. 2015, 56, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Chess, S. Follow-up report on autism in congenital rubella. J. Autism Child. Schizophr. 1977, 7, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Reisinger, S.; Khan, D.; Kong, E.; Berger, A.; Pollak, A.; Pollak, D.D. The poly(I:C)-induced maternal immune activation model in preclinical neuropsychiatric drug discovery. Pharmacol. Ther. 2015, 149, 213–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, Y.; Fujimoto, C.; Nakajima, E.; Isagai, T.; Matsuishi, T. Possible association between congenital cytomegalovirus infection and autistic disorder. J. Autism Dev. Disord. 2003, 33, 455–459. [Google Scholar] [CrossRef]
- Severance, E.G.; Xiao, J.; Jones-Brando, L.; Sabunciyan, S.; Li, Y.; Pletnikov, M.; Prandovszky, E.; Yolken, R. Toxoplasma gondii-A Gastrointestinal Pathogen Associated with Human Brain Diseases. Int. Rev. Neurobiol. 2016, 131, 143–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mawson, A.R.; Croft, A.M. Rubella Virus Infection, the Congenital Rubella Syndrome, and the Link to Autism. Int. J. Environ. Res. Public Health 2019, 16, 3543. [Google Scholar] [CrossRef] [Green Version]
- Boulanger-Bertolus, J.; Pancaro, C.; Mashour, G.A. Increasing Role of Maternal Immune Activation in Neurodevelopmental Disorders. Front. Behav. Neurosci. 2018, 12, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.Y.; Xu, L.L.; Shao, L.; Xia, R.M.; Yu, Z.H.; Ling, Z.X.; Yang, F.; Deng, M.; Ruan, B. Maternal infection during pregnancy and risk of autism spectrum disorders: A systematic review and meta-analysis. Brain Behav. Immun. 2016, 58, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Hornig, M.; Bresnahan, M.A.; Che, X.; Schultz, A.F.; Ukaigwe, J.E.; Eddy, M.L.; Hirtz, D.; Gunnes, N.; Lie, K.K.; Magnus, P.; et al. Prenatal fever and autism risk. Mol. Psychiatry 2018, 23, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Atladottir, H.O.; Henriksen, T.B.; Schendel, D.E.; Parner, E.T. Autism after infection, febrile episodes, and antibiotic use during pregnancy: An exploratory study. Pediatrics 2012, 130, e1447–e1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atladóttir, H.O.; Thorsen, P.; Østergaard, L.; Schendel, D.E.; Lemcke, S.; Abdallah, M.; Parner, E.T. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J. Autism Dev. Disord. 2010, 40, 1423–1430. [Google Scholar] [CrossRef]
- Arsenault, D.; St-Amour, I.; Cisbani, G.; Rousseau, L.S.; Cicchetti, F. The different effects of LPS and poly I:C prenatal immune challenges on the behavior, development and inflammatory responses in pregnant mice and their offspring. Brain Behav. Immun. 2014, 38, 77–90. [Google Scholar] [CrossRef]
- Gąssowska-Dobrowolska, M.; Cieślik, M.; Czapski, G.A.; Jęśko, H.; Frontczak-Baniewicz, M.; Gewartowska, M.; Dominiak, A.; Polowy, R.; Filipkowski, R.K.; Babiec, L.; et al. Prenatal Exposure to Valproic Acid Affects Microglia and Synaptic Ultrastructure in a Brain-Region-Specific Manner in Young-Adult Male Rats: Relevance to Autism Spectrum Disorders. Int. J. Mol. Sci. 2020, 21, 3576. [Google Scholar] [CrossRef] [PubMed]
- Boksa, P. Effects of prenatal infection on brain development and behavior: A review of findings from animal models. Brain Behav. Immun. 2010, 24, 881–897. [Google Scholar] [CrossRef] [PubMed]
- Cieślik, M.; Gąssowska-Dobrowolska, M.; Jęśko, H.; Czapski, G.A.; Wilkaniec, A.; Zawadzka, A.; Dominiak, A.; Polowy, R.; Filipkowski, R.K.; Boguszewski, P.M.; et al. Maternal Immune Activation Induces Neuroinflammation and Cortical Synaptic Deficits in the Adolescent Rat Offspring. Int. J. Mol. Sci. 2020, 21, 4097. [Google Scholar] [CrossRef] [PubMed]
- Cieślik, M.; Gassowska-Dobrowolska, M.; Zawadzka, A.; Frontczak-Baniewicz, M.; Gewartowska, M.; Dominiak, A.; Czapski, G.A.; Adamczyk, A. The Synaptic Dysregulation in Adolescent Rats Exposed to Maternal Immune Activation. Front. Mol. Neurosci. 2020, 13, 555290. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Sengupta, P. Defining pregnancy phases with cytokine shift. J. Pregnancy Reprod. 2017, 1, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Racicot, K.; Kwon, J.Y.; Aldo, P.; Silasi, M.; Mor, G. Understanding the complexity of the immune system during pregnancy. Am. J. Reprod. Immunol. 2014, 72, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zheng, Q.; Jin, L. Dynamic Function and Composition Changes of Immune Cells During Normal and Pathological Pregnancy at the Maternal-Fetal Interface. Front. Immunol. 2019, 10, 2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochoa-Bernal, M.A.; Fazleabas, A.T. Physiologic Events of Embryo Implantation and Decidualization in Human and Non-Human Primates. Int. J. Mol. Sci. 2020, 21, 1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, X.; Wu, L.; Hu, W. The regulation of leukemia inhibitory factor. Cancer Cell Microenviron. 2015, 2. [Google Scholar] [CrossRef]
- Yockey, L.J.; Iwasaki, A. Interferons and Proinflammatory Cytokines in Pregnancy and Fetal Development. Immunity 2018, 49, 397–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prins, J.R.; Gomez-Lopez, N.; Robertson, S.A. Interleukin-6 in pregnancy and gestational disorders. J. Reprod. Immunol. 2012, 95, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Equils, O.; Kellogg, C.; McGregor, J.; Gravett, M.; Neal-Perry, G.; Gabay, C. The role of the IL-1 system in pregnancy and the use of IL-1 system markers to identify women at risk for pregnancy complications. Biol. Reprod. 2020, 103, 684–694. [Google Scholar] [CrossRef]
- Zhao, B.; Schwartz, J.P. Involvement of cytokines in normal CNS development and neurological diseases: Recent progress and perspectives. J. Neurosci. Res. 1998, 52, 7–16. [Google Scholar] [CrossRef]
- Parker-Athill, E.C.; Tan, J. Maternal immune activation and autism spectrum disorder: Interleukin-6 signaling as a key mechanistic pathway. Neuro-Signals 2010, 18, 113–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackmon, R.; Pinnaduwage, L.; Zhang, J.; Lye, S.J.; Geraghty, D.E.; Dunk, C.E. Definitive class I human leukocyte antigen expression in gestational placentation: HLA-F, HLA-E, HLA-C, and HLA-G in extravillous trophoblast invasion on placentation, pregnancy, and parturition. Am. J. Reprod. Immunol. 2017, 77. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Zhou, Q.; Li, L.; Wang, S. HLA-C promotes proliferation and cell cycle progression in trophoblast cells. J. Matern. Fetal Neonatal Med. 2021, 34, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Ravaccia, D.; Ghafourian, T. Critical Role of the Maternal Immune System in the Pathogenesis of Autism Spectrum Disorder. Biomedicines 2020, 8, 557. [Google Scholar] [CrossRef]
- Estes, M.L.; McAllister, A.K. Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nat. Rev. Neurosci. 2015, 16, 469–486. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.; Hoeffer, C. Maternal IL-17A in autism. Exp. Neurol. 2018, 299, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, V.L.; Ellwanger, J.H.; Matte, M.C.C.; Savaris, R.F.; Vianna, P.; Chies, J.A.B. IL-17 blood levels increase in healthy pregnancy but not in spontaneous abortion. Mol. Biol. Rep. 2018, 45, 1565–1568. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.M.; Cowan, M.; Moonah, S.N.; Petri, W.A., Jr. The Impact of Systemic Inflammation on Neurodevelopment. Trends Mol. Med. 2018, 24, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Griffith, O.W.; Chavan, A.R.; Protopapas, S.; Maziarz, J.; Romero, R.; Wagner, G.P. Embryo implantation evolved from an ancestral inflammatory attachment reaction. Proc. Natl. Acad. Sci. USA 2017, 114, E6566–E6575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuelsson, A.M.; Jennische, E.; Hansson, H.A.; Holmäng, A. Prenatal exposure to interleukin-6 results in inflammatory neurodegeneration in hippocampus with NMDA/GABA(A) dysregulation and impaired spatial learning. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R1345–R1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2005, 57, 67–81. [Google Scholar] [CrossRef]
- Wei, H.; Zou, H.; Sheikh, A.M.; Malik, M.; Dobkin, C.; Brown, W.T.; Li, X. IL-6 is increased in the cerebellum of autistic brain and alters neural cell adhesion, migration and synaptic formation. J. Neuroinflamm. 2011, 8, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuillermot, S.; Luan, W.; Meyer, U.; Eyles, D. Vitamin D treatment during pregnancy prevents autism-related phenotypes in a mouse model of maternal immune activation. Mol. Autism 2017, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Simões, L.R.; Sangiogo, G.; Tashiro, M.H.; Generoso, J.S.; Faller, C.J.; Dominguini, D.; Mastella, G.A.; Scaini, G.; Giridharan, V.V.; Michels, M.; et al. Maternal immune activation induced by lipopolysaccharide triggers immune response in pregnant mother and fetus, and induces behavioral impairment in adult rats. J. Psychiatr. Res. 2018, 100, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, J.; Zhang, H.; Yu, J.; Yao, Z. Oral probiotic administration during pregnancy prevents autism-related behaviors in offspring induced by maternal immune activation via anti-inflammation in mice. Autism Res. 2019, 12, 576–588. [Google Scholar] [CrossRef]
- Hsiao, E.Y.; Patterson, P.H. Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav. Immun. 2011, 25, 604–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, N.; Li, X.; Zhong, Y. Inflammatory cytokines: Potential biomarkers of immunologic dysfunction in autism spectrum disorders. Mediat. Inflamm. 2015, 2015, 531518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baran, P.; Hansen, S.; Waetzig, G.H.; Akbarzadeh, M.; Lamertz, L.; Huber, H.J.; Ahmadian, M.R.; Moll, J.M.; Scheller, J. The balance of interleukin (IL)-6, IL-6·soluble IL-6 receptor (sIL-6R), and IL-6·sIL-6R·sgp130 complexes allows simultaneous classic and trans-signaling. J. Biol. Chem. 2018, 293, 6762–6775. [Google Scholar] [CrossRef] [Green Version]
- Greenhill, C.J.; Rose-John, S.; Lissilaa, R.; Ferlin, W.; Ernst, M.; Hertzog, P.J.; Mansell, A.; Jenkins, B.J. IL-6 trans-signaling modulates TLR4-dependent inflammatory responses via STAT3. J. Immunol. 2011, 186, 1199–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linker, S.B.; Mendes, A.P.D.; Marchetto, M.C. IGF-1 treatment causes unique transcriptional response in neurons from individuals with idiopathic autism. Mol. Autism 2020, 11, 55. [Google Scholar] [CrossRef]
- Wu, W.L.; Hsiao, E.Y.; Yan, Z.; Mazmanian, S.K.; Patterson, P.H. The placental interleukin-6 signaling controls fetal brain development and behavior. Brain Behav. Immun. 2017, 62, 11–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaretsky, M.V.; Alexander, J.M.; Byrd, W.; Bawdon, R.E. Transfer of inflammatory cytokines across the placenta. Obstet. Gynecol. 2004, 103, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, J.; Samuelsson, A.M.; Jansson, T.; Holmäng, A. Interleukin-6 in the maternal circulation reaches the rat fetus in mid-gestation. Pediatric Res. 2006, 60, 147–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conroy, S.M.; Nguyen, V.; Quina, L.A.; Blakely-Gonzales, P.; Ur, C.; Netzeband, J.G.; Prieto, A.L.; Gruol, D.L. Interleukin-6 produces neuronal loss in developing cerebellar granule neuron cultures. J. Neuroimmunol. 2004, 155, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Alberts, I.; Li, X. Brain IL-6 and autism. Neuroscience 2013, 252, 320–325. [Google Scholar] [CrossRef]
- Smith, S.E.; Li, J.; Garbett, K.; Mirnics, K.; Patterson, P.H. Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci. 2007, 27, 10695–10702. [Google Scholar] [CrossRef] [Green Version]
- Choi, G.B.; Yim, Y.S.; Wong, H.; Kim, S.; Kim, H.; Kim, S.V.; Hoeffer, C.A.; Littman, D.R.; Huh, J.R. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 2016, 351, 933–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, W.; Dong, C. IL-17 cytokines in immunity and inflammation. Emerg. Microbes Infect. 2013, 2, e60. [Google Scholar] [CrossRef] [PubMed]
- Al-Ayadhi, L.Y.; Mostafa, G.A. Elevated serum levels of interleukin-17A in children with autism. J. Neuroinflamm. 2012, 9, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadeem, A.; Ahmad, S.F.; Attia, S.M.; Al-Ayadhi, L.Y.; Bakheet, S.A.; Al-Harbi, N.O. Oxidative and inflammatory mediators are upregulated in neutrophils of autistic children: Role of IL-17A receptor signaling. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 90, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Wienecke, J.; Hebel, K.; Hegel, K.J.; Pierau, M.; Brune, T.; Reinhold, D.; Pethe, A.; Brunner-Weinzierl, M.C. Pro-inflammatory effector Th cells transmigrate through anti-inflammatory environments into the murine fetus. Placenta 2012, 33, 39–46. [Google Scholar] [CrossRef]
- Toy, D.; Kugler, D.; Wolfson, M.; Vanden Bos, T.; Gurgel, J.; Derry, J.; Tocker, J.; Peschon, J. Cutting edge: Interleukin 17 signals through a heteromeric receptor complex. J. Immunol. 2006, 177, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zepp, J.; Li, X. Function of Act1 in IL-17 family signaling and autoimmunity. Adv. Exp. Med. Biol. 2012, 946, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Commane, M.; Nie, H.; Hua, X.; Chatterjee-Kishore, M.; Wald, D.; Haag, M.; Stark, G.R. Act1, an NF-kappa B-activating protein. Proc. Natl. Acad. Sci. USA 2000, 97, 10489–10493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohgakiuchi, Y.; Saino, Y.; Muromoto, R.; Komori, Y.; Sato, A.; Hirashima, K.; Kitai, Y.; Kashiwakura, J.I.; Oritani, K.; Matsuda, T. Dimethyl fumarate dampens IL-17-ACT1-TBK1 axis-mediated phosphorylation of Regnase-1 and suppresses IL-17-induced IκB-ζ expression. Biochem. Biophys. Res. Commun. 2020, 521, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Amatya, N.; Garg, A.V.; Gaffen, S.L. IL-17 Signaling: The Yin and the Yang. Trends Immunol. 2017, 38, 310–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, H.; Hale, V.A.; Dolcet, X.; Davies, A. NF-kappaB signalling regulates the growth of neural processes in the developing PNS and CNS. Development 2005, 132, 1713–1726. [Google Scholar] [CrossRef] [Green Version]
- Liao, X.; Li, Y. Nuclear Factor Kappa B in Autism Spectrum Disorder: A Systematic Review. Pharmacol. Res. 2020, 159, 104918. [Google Scholar] [CrossRef]
- Samuels, I.S.; Saitta, S.C.; Landreth, G.E. MAP’ing CNS development and cognition: An ERKsome process. Neuron 2009, 61, 160–167. [Google Scholar] [CrossRef] [Green Version]
- Kalkman, H.O. Potential opposite roles of the extracellular signal-regulated kinase (ERK) pathway in autism spectrum and bipolar disorders. Neurosci. Biobehav. Rev. 2012, 36, 2206–2213. [Google Scholar] [CrossRef]
- Rosenzweig, J.M.; Lei, J.; Burd, I. Interleukin-1 receptor blockade in perinatal brain injury. Front. Pediatr. 2014, 2, 108. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Castejon, G.; Brough, D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 2011, 22, 189–195. [Google Scholar] [CrossRef]
- Saghazadeh, A.; Ataeinia, B.; Keynejad, K.; Abdolalizadeh, A.; Hirbod-Mobarakeh, A.; Rezaei, N. A meta-analysis of pro-inflammatory cytokines in autism spectrum disorders: Effects of age, gender, and latitude. J. Psychiatr. Res. 2019, 115, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Enstrom, A.M.; Onore, C.E.; Van de Water, J.A.; Ashwood, P. Differential monocyte responses to TLR ligands in children with autism spectrum disorders. Brain Behav. Immun. 2010, 24, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Sadowska, G.B.; Chen, X.; Zhang, J.; Lim, Y.P.; Cummings, E.E.; Makeyev, O.; Besio, W.G.; Gaitanis, J.; Padbury, J.F.; Banks, W.A.; et al. Interleukin-1β transfer across the blood-brain barrier in the ovine fetus. J. Cereb. Blood Flow Metab. 2015, 35, 1388–1395. [Google Scholar] [CrossRef]
- Jovanovic, D.V.; Di Battista, J.A.; Martel-Pelletier, J.; Jolicoeur, F.C.; He, Y.; Zhang, M.; Mineau, F.; Pelletier, J.P. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J. Immunol. 1998, 160, 3513–3521. [Google Scholar] [PubMed]
- Chudnovets, A.; Lei, J.; Na, Q.; Dong, J.; Narasimhan, H.; Klein, S.L.; Burd, I. Dose-dependent structural and immunological changes in the placenta and fetal brain in response to systemic inflammation during pregnancy. Am. J. Reprod. Immunol. 2020, 84, e13248. [Google Scholar] [CrossRef]
- Crampton, S.J.; Collins, L.M.; Toulouse, A.; Nolan, Y.M.; O’Keeffe, G.W. Exposure of foetal neural progenitor cells to IL-1β impairs their proliferation and alters their differentiation—A role for maternal inflammation? J. Neurochem. 2012, 120, 964–973. [Google Scholar] [CrossRef]
- Scholz, C.C.; Cavadas, M.A.; Tambuwala, M.M.; Hams, E.; Rodríguez, J.; von Kriegsheim, A.; Cotter, P.; Bruning, U.; Fallon, P.G.; Cheong, A.; et al. Regulation of IL-1β-induced NF-κB by hydroxylases links key hypoxic and inflammatory signaling pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 18490–18495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girard, S.; Tremblay, L.; Lepage, M.; Sébire, G. IL-1 receptor antagonist protects against placental and neurodevelopmental defects induced by maternal inflammation. J. Immunol. 2010, 184, 3997–4005. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef]
- Norris, G.T.; Kipnis, J. Immune cells and CNS physiology: Microglia and beyond. J. Exp. Med. 2019, 216, 60–70. [Google Scholar] [CrossRef]
- Aarum, J.; Sandberg, K.; Haeberlein, S.L.; Persson, M.A. Migration and differentiation of neural precursor cells can be directed by microglia. Proc. Natl. Acad. Sci. USA 2003, 100, 15983–15988. [Google Scholar] [CrossRef] [Green Version]
- Cowan, M.; Petri, W.A., Jr. Microglia: Immune Regulators of Neurodevelopment. Front. Immunol. 2018, 9, 2576. [Google Scholar] [CrossRef] [Green Version]
- Smolders, S.; Notter, T.; Smolders, S.M.T.; Rigo, J.M.; Brône, B. Controversies and prospects about microglia in maternal immune activation models for neurodevelopmental disorders. Brain Behav. Immun. 2018, 73, 51–65. [Google Scholar] [CrossRef]
- Kierdorf, K.; Prinz, M. Factors regulating microglia activation. Front. Cell. Neurosci. 2013, 7, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Sugihara, G.; Ouchi, Y.; Nakamura, K.; Futatsubashi, M.; Takebayashi, K.; Yoshihara, Y.; Omata, K.; Matsumoto, K.; Tsuchiya, K.J.; et al. Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry 2013, 70, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.T.; Chana, G.; Pardo, C.A.; Achim, C.; Semendeferi, K.; Buckwalter, J.; Courchesne, E.; Everall, I.P. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol. Psychiatry 2010, 68, 368–376. [Google Scholar] [CrossRef]
- Tetreault, N.A.; Hakeem, A.Y.; Jiang, S.; Williams, B.A.; Allman, E.; Wold, B.J.; Allman, J.M. Microglia in the cerebral cortex in autism. J. Autism Dev. Disord. 2012, 42, 2569–2584. [Google Scholar] [CrossRef]
- Li, W.Y.; Chang, Y.C.; Lee, L.J.; Lee, L.J. Prenatal infection affects the neuronal architecture and cognitive function in adult mice. Dev. Neurosci. 2014, 36, 359–370. [Google Scholar] [CrossRef]
- Gumusoglu, S.B.; Fine, R.S.; Murray, S.J.; Bittle, J.L.; Stevens, H.E. The role of IL-6 in neurodevelopment after prenatal stress. Brain Behav. Immun. 2017, 65, 274–283. [Google Scholar] [CrossRef] [PubMed]
- O’Loughlin, E.; Pakan, J.M.P.; Yilmazer-Hanke, D.; McDermott, K.W. Acute in utero exposure to lipopolysaccharide induces inflammation in the pre- and postnatal brain and alters the glial cytoarchitecture in the developing amygdala. J. Neuroinflamm. 2017, 14, 212. [Google Scholar] [CrossRef] [PubMed]
- Corradini, I.; Focchi, E.; Rasile, M.; Morini, R.; Desiato, G.; Tomasoni, R.; Lizier, M.; Ghirardini, E.; Fesce, R.; Morone, D.; et al. Maternal Immune Activation Delays Excitatory-to-Inhibitory Gamma-Aminobutyric Acid Switch in Offspring. Biol. Psychiatry 2018, 83, 680–691. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, K.; Kato, D.; Ikegami, A.; Hashimoto, A.; Sugio, S.; Guo, Z.; Shibushita, M.; Tatematsu, T.; Haruwaka, K.; Moorhouse, A.J.; et al. Maternal immune activation induces sustained changes in fetal microglia motility. Sci. Rep. 2020, 10, 21378. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, E.Y.; McBride, S.W.; Chow, J.; Mazmanian, S.K.; Patterson, P.H. Modeling an autism risk factor in mice leads to permanent immune dysregulation. Proc. Natl. Acad. Sci. USA 2012, 109, 12776–12781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garay, P.A.; Hsiao, E.Y.; Patterson, P.H.; McAllister, A.K. Maternal immune activation causes age- and region-specific changes in brain cytokines in offspring throughout development. Brain Behav. Immun. 2013, 31, 54–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovanoli, S.; Notter, T.; Richetto, J.; Labouesse, M.A.; Vuillermot, S.; Riva, M.A.; Meyer, U. Late prenatal immune activation causes hippocampal deficits in the absence of persistent inflammation across aging. J. Neuroinflamm. 2015, 12, 221. [Google Scholar] [CrossRef] [Green Version]
- Mouihate, A. Prenatal Activation of Toll-Like Receptor-4 Dampens Adult Hippocampal Neurogenesis in An IL-6 Dependent Manner. Front. Cell. Neurosci. 2016, 10, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaafsma, W.; Basterra, L.B.; Jacobs, S.; Brouwer, N.; Meerlo, P.; Schaafsma, A.; Boddeke, E.W.G.M.; Eggen, B.J.L. Maternal inflammation induces immune activation of fetal microglia and leads to disrupted microglia immune responses, behavior, and learning performance in adulthood. Neurobiol. Dis. 2017, 106, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Limatola, C.; Ransohoff, R.M. Modulating neurotoxicity through CX3CL1/CX3CR1 signaling. Front. Cell. Neurosci. 2014, 8, 229. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, P.; Ziemka-Nalecz, M.; Sypecka, J.; Zalewska, T. The Impact of the CX3CL1/CX3CR1 Axis in Neurological Disorders. Cells 2020, 9, 2277. [Google Scholar] [CrossRef] [PubMed]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Microglia and Aging: The Role of the TREM2-DAP12 and CX3CL1-CX3CR1 Axes. Int. J. Mol. Sci. 2018, 19, 318. [Google Scholar] [CrossRef] [Green Version]
- Lauro, C.; Chece, G.; Monaco, L.; Antonangeli, F.; Peruzzi, G.; Rinaldo, S.; Paone, A.; Cutruzzolà, F.; Limatola, C. Fractalkine Modulates Microglia Metabolism in Brain Ischemia. Front. Cell. Neurosci. 2019, 13, 414. [Google Scholar] [CrossRef] [PubMed]
- Ngwa, C.; Liu, F. CD200-CD200R signaling and diseases: A potential therapeutic target? Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 297–309. [Google Scholar] [PubMed]
- Shrivastava, K.; Gonzalez, P.; Acarin, L. The immune inhibitory complex CD200/CD200R is developmentally regulated in the mouse brain. J. Comp. Neurol. 2012, 520, 2657–2675. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Fonken, L.K.; Annis, J.L.; Watkins, L.R.; Maier, S.F. Stress disinhibits microglia via down-regulation of CD200R: A mechanism of neuroinflammatory priming. Brain Behav. Immun. 2018, 69, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Chamera, K.; Kotarska, K.; Szuster-Głuszczak, M.; Trojan, E.; Skórkowska, A.; Pomierny, B.; Krzyżanowska, W.; Bryniarska, N.; Basta-Kaim, A. The prenatal challenge with lipopolysaccharide and polyinosinic:polycytidylic acid disrupts CX3CL1-CX3CR1 and CD200-CD200R signalling in the brains of male rat offspring: A link to schizophrenia-like behaviours. J. Neuroinflamm. 2020, 17, 247. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Bazhin, A.V.; Werner, J.; Karakhanova, S. Reactive oxygen species in the immune system. Int. Rev. Immunol. 2013, 32, 249–270. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Manivasagam, T.; Arunadevi, S.; Essa, M.M.; SaravanaBabu, C.; Borah, A.; Thenmozhi, A.J.; Qoronfleh, M.W. Role of Oxidative Stress and Antioxidants in Autism. Adv. Neurobiol. 2020, 24, 193–206. [Google Scholar] [CrossRef]
- Pangrazzi, L.; Balasco, L.; Bozzi, Y. Oxidative Stress and Immune System Dysfunction in Autism Spectrum Disorders. Int. J. Mol. Sci. 2020, 21, 3293. [Google Scholar] [CrossRef]
- Rose, S.; Melnyk, S.; Pavliv, O.; Bai, S.; Nick, T.G.; Frye, R.E.; James, S.J. Evidence of oxidative damage and inflammation associated with low glutathione redox status in the autism brain. Transl. Psychiatry 2012, 2, e134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Shi, X.-J.; Liu, H.; Mao, X.; Gui, L.-N.; Wang, H.; Cheng, Y. Oxidative stress marker aberrations in children with autism spectrum disorder: A systematic review and meta-analysis of 87 studies (N = 9109). Transl. Psychiatry 2021, 11, 15. [Google Scholar] [CrossRef]
- Abruzzo, P.M.; Matté, A.; Bolotta, A.; Federti, E.; Ghezzo, A.; Guarnieri, T.; Marini, M.; Posar, A.; Siciliano, A.; De Franceschi, L.; et al. Plasma peroxiredoxin changes and inflammatory cytokines support the involvement of neuro-inflammation and oxidative stress in Autism Spectrum Disorder. J. Transl. Med. 2019, 17, 332. [Google Scholar] [CrossRef] [PubMed]
- Lanté, F.; Meunier, J.; Guiramand, J.; Maurice, T.; Cavalier, M.; de Jesus Ferreira, M.C.; Aimar, R.; Cohen-Solal, C.; Vignes, M.; Barbanel, G. Neurodevelopmental damage after prenatal infection: Role of oxidative stress in the fetal brain. Free. Radic. Biol. Med. 2007, 42, 1231–1245. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxidative Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.P.; Hooker, B.S.; Herbert, M.R. Bridging from Cells to Cognition in Autism Pathophysiology: Biological Pathways to Defective Brain Function and Plasticity. Am. J. Biochem. Biotechnol. 2008, 4, 167–176. [Google Scholar] [CrossRef] [Green Version]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frye, R.E. Mitochondrial Dysfunction in Autism Spectrum Disorder: Unique Abnormalities and Targeted Treatments. Semin. Pediatr. Neurol. 2020, 35, 100829. [Google Scholar] [CrossRef]
- Anitha, A.; Nakamura, K.; Thanseem, I.; Matsuzaki, H.; Miyachi, T.; Tsujii, M.; Iwata, Y.; Suzuki, K.; Sugiyama, T.; Mori, N. Downregulation of the expression of mitochondrial electron transport complex genes in autism brains. Brain Pathol. 2013, 23, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Gu, F.; Essa, M.M.; Wegiel, J.; Kaur, K.; Brown, W.T.; Chauhan, V. Brain region-specific deficit in mitochondrial electron transport chain complexes in children with autism. J. Neurochem. 2011, 117, 209–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, G.; Gutierrez Rios, P.; Kuo, S.-H.; Akman, H.O.; Rosoklija, G.; Tanji, K.; Dwork, A.; Schon, E.A.; DiMauro, S.; Goldman, J.; et al. Mitochondrial abnormalities in temporal lobe of autistic brain. Neurobiol. Dis. 2013, 54, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Gu, F.; Chauhan, V.; Kaur, K.; Brown, W.T.; LaFauci, G.; Wegiel, J.; Chauhan, A. Alterations in mitochondrial DNA copy number and the activities of electron transport chain complexes and pyruvate dehydrogenase in the frontal cortex from subjects with autism. Transl. Psychiatry 2013, 3, e299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anitha, A.; Nakamura, K.; Thanseem, I.; Yamada, K.; Iwayama, Y.; Toyota, T.; Matsuzaki, H.; Miyachi, T.; Yamada, S.; Tsujii, M.; et al. Brain region-specific altered expression and association of mitochondria-related genes in autism. Mol. Autism 2012, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Rose, S.; Frye, R.E.; Slattery, J.; Wynne, R.; Tippett, M.; Pavliv, O.; Melnyk, S.; James, S.J. Oxidative stress induces mitochondrial dysfunction in a subset of autism lymphoblastoid cell lines in a well-matched case control cohort. PLoS ONE 2014, 9, e85436. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Bennuri, S.C.; Wynne, R.; Melnyk, S.; James, S.J.; Frye, R.E. Mitochondrial and redox abnormalities in autism lymphoblastoid cells: A sibling control study. FASEB J. 2017, 31, 904–909. [Google Scholar] [CrossRef] [Green Version]
- Pecorelli, A.; Ferrara, F.; Messano, N.; Cordone, V.; Schiavone, M.L.; Cervellati, F.; Woodby, B.; Cervellati, C.; Hayek, J.; Valacchi, G. Alterations of mitochondrial bioenergetics, dynamics, and morphology support the theory of oxidative damage involvement in autism spectrum disorder. FASEB J. 2020, 34, 6521–6538. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Singh, I.N.; Diggins, E.; Connors, S.L.; Karim, M.A.; Lee, D.; Zimmerman, A.W.; Frye, R.E. Developmental regression and mitochondrial function in children with autism. Ann. Clin. Transl. Neurol. 2020, 7, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Giulivi, C.; Napoli, E.; Schwartzer, J.; Careaga, M.; Ashwood, P. Gestational exposure to a viral mimetic poly(i:C) results in long-lasting changes in mitochondrial function by leucocytes in the adult offspring. Mediat. Inflamm. 2013, 2013, 609602. [Google Scholar] [CrossRef] [Green Version]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef]
- Morris, G.; Maes, M. Mitochondrial dysfunctions in myalgic encephalomyelitis/chronic fatigue syndrome explained by activated immuno-inflammatory, oxidative and nitrosative stress pathways. Metab. Brain Dis. 2014, 29, 19–36. [Google Scholar] [CrossRef]
- Yang, D.; Elner, S.G.; Bian, Z.M.; Till, G.O.; Petty, H.R.; Elner, V.M. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp. Eye Res. 2007, 85, 462–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Xu, M.; Xo, R.; Mates, A.; Wilson, G.L.; Pearsall, A.W.; Grishko, V. Mitochondrial DNA damage is involved in apoptosis caused by pro-inflammatory cytokines in human OA chondrocytes. Osteoarthr. Cartil. 2010, 18, 424–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.; Berk, M. The many roads to mitochondrial dysfunction in neuroimmune and neuropsychiatric disorders. BMC Med. 2015, 13, 68. [Google Scholar] [CrossRef] [Green Version]
- Pineda, E.; Shin, D.; You, S.J.; Auvin, S.; Sankar, R.; Mazarati, A. Maternal immune activation promotes hippocampal kindling epileptogenesis in mice. Ann. Neurol. 2013, 74, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Coiro, P.; Padmashri, R.; Suresh, A.; Spartz, E.; Pendyala, G.; Chou, S.; Jung, Y.; Meays, B.; Roy, S.; Gautam, N.; et al. Impaired synaptic development in a maternal immune activation mouse model of neurodevelopmental disorders. Brain Behav. Immun. 2015, 50, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Ruskin, D.N.; Murphy, M.I.; Slade, S.L.; Masino, S.A. Ketogenic diet improves behaviors in a maternal immune activation model of autism spectrum disorder. PLoS ONE 2017, 12, e0171643. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef] [Green Version]
- King, B.H.; Wright, D.M.; Handen, B.L.; Sikich, L.; Zimmerman, A.W.; McMahon, W.; Cantwell, E.; Davanzo, P.A.; Dourish, C.T.; Dykens, E.M.; et al. Double-blind, placebo-controlled study of amantadine hydrochloride in the treatment of children with autistic disorder. J. Am. Acad. Child Adolesc. Psychiatry 2001, 40, 658–665. [Google Scholar] [CrossRef]
- Mohammadi, M.R.; Yadegari, N.; Hassanzadeh, E.; Farokhnia, M.; Yekehtaz, H.; Mirshafiee, O.; Akhondzadeh, S. Double-blind, placebo-controlled trial of risperidone plus amantadine in children with autism: A 10-week randomized study. Clin. Neuropharmacol. 2013, 36, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Asadabadi, M.; Mohammadi, M.R.; Ghanizadeh, A.; Modabbernia, A.; Ashrafi, M.; Hassanzadeh, E.; Forghani, S.; Akhondzadeh, S. Celecoxib as adjunctive treatment to risperidone in children with autistic disorder: A randomized, double-blind, placebo-controlled trial. Psychopharmacology 2013, 225, 51–59. [Google Scholar] [CrossRef]
- Stefanatos, G.A.; Grover, W.; Geller, E. Case study: Corticosteroid treatment of language regression in pervasive developmental disorder. J. Am. Acad. Child Adolesc. Psychiatry 1995, 34, 1107–1111. [Google Scholar] [CrossRef]
- Shenoy, S.; Arnold, S.; Chatila, T. Response to steroid therapy in autism secondary to autoimmune lymphoproliferative syndrome. J. Pediatr. 2000, 136, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, N.; Pacini, S.; Ruggiero, M. Clinical Experience of Integrative Autism treatment with a Novel type of Immunotherapy. J. Vaccines 2019, 3, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Chez, M.; Low, R.; Parise, C.; Donnel, T. Safety and observations in a pilot study of lenalidomide for treatment in autism. Autism Res. Treat. 2012, 2012, 291601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taliou, A.; Zintzaras, E.; Lykouras, L.; Francis, K. An open-label pilot study of a formulation containing the anti-inflammatory flavonoid luteolin and its effects on behavior in children with autism spectrum disorders. Clin. Ther. 2013, 35, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Tsilioni, I.; Taliou, A.; Francis, K.; Theoharides, T.C. Children with autism spectrum disorders, who improved with a luteolin-containing dietary formulation, show reduced serum levels of TNF and IL-6. Transl. Psychiatry 2015, 5, e647. [Google Scholar] [CrossRef] [Green Version]
- Theoharides, T.C.; Asadi, S.; Panagiotidou, S. A case series of a luteolin formulation (NeuroProtek®) in children with autism spectrum disorders. Int. J. Immunopathol. Pharmacol. 2012, 25, 317–323. [Google Scholar] [CrossRef] [Green Version]
- Ghaleiha, A.; Asadabadi, M.; Mohammadi, M.R.; Shahei, M.; Tabrizi, M.; Hajiaghaee, R.; Hassanzadeh, E.; Akhondzadeh, S. Memantine as adjunctive treatment to risperidone in children with autistic disorder: A randomized, double-blind, placebo-controlled trial. Int. J. Neuropsychopharmacol. 2013, 16, 783–789. [Google Scholar] [CrossRef] [Green Version]
- Joshi, G.; Wozniak, J.; Faraone, S.V.; Fried, R.; Chan, J.; Furtak, S.; Grimsley, E.; Conroy, K.; Kilcullen, J.R.; Woodworth, K.Y.; et al. A Prospective Open-Label Trial of Memantine Hydrochloride for the Treatment of Social Deficits in Intellectually Capable Adults with Autism Spectrum Disorder. J. Clin. Psychopharmacol. 2016, 36, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Pardo, C.A.; Buckley, A.; Thurm, A.; Lee, L.C.; Azhagiri, A.; Neville, D.M.; Swedo, S.E. A pilot open-label trial of minocycline in patients with autism and regressive features. J. Neurodev. Disord. 2013, 5, 9. [Google Scholar] [CrossRef] [Green Version]
- Ghaleiha, A.; Alikhani, R.; Kazemi, M.R.; Mohammadi, M.R.; Mohammadinejad, P.; Zeinoddini, A.; Hamedi, M.; Shahriari, M.; Keshavarzi, Z.; Akhondzadeh, S. Minocycline as Adjunctive Treatment to Risperidone in Children with Autistic Disorder: A Randomized, Double-Blind Placebo-Controlled Trial. J. Child Adolesc. Psychopharmacol. 2016, 26, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Buitelaar, J.K.; van Engeland, H.; van Ree, J.M.; de Wied, D. Behavioral effects of Org 2766, a synthetic analog of the adrenocorticotrophic hormone (4-9), in 14 outpatient autistic children. J. Autism Dev. Disord. 1990, 20, 467–478. [Google Scholar] [CrossRef]
- Buitelaar, J.K.; van Engeland, H.; de Kogel, K.H.; de Vries, H.; van Hooff, J.A.; van Ree, J.M. The use of adrenocorticotrophic hormone (4-9) analog ORG 2766 in autistic children: Effects on the organization of behavior. Biol. Psychiatry 1992, 31, 1119–1129. [Google Scholar] [CrossRef]
- Bradstreet, J.J.; Smith, S.; Granpeesheh, D.; El-Dahr, J.M.; Rossignol, D. Spironolactone might be a desirable immunologic and hormonal intervention in autism spectrum disorders. Med. Hypotheses 2007, 68, 979–987. [Google Scholar] [CrossRef]
- Hafizi, S.; Tabatabaei, D.; Lai, M.C. Review of Clinical Studies Targeting Inflammatory Pathways for Individuals with Autism. Front. Psychiatry 2019, 10, 849. [Google Scholar] [CrossRef] [PubMed]
- Ossola, B.; Schendzielorz, N.; Chen, S.H.; Bird, G.S.; Tuominen, R.K.; Männistö, P.T.; Hong, J.S. Amantadine protects dopamine neurons by a dual action: Reducing activation of microglia and inducing expression of GDNF in astroglia. Neuropharmacology 2011, 61, 574–582. [Google Scholar] [CrossRef] [Green Version]
- Kubera, M.; Maes, M.; Budziszewska, B.; Basta-Kaim, A.; Leśkiewicz, M.; Grygier, B.; Rogóz, Z.; Lasoń, W. Inhibitory effects of amantadine on the production of pro-inflammatory cytokines by stimulated in vitro human blood. Pharmacol. Rep. 2009, 61, 1105–1112. [Google Scholar] [CrossRef]
- Ma, H.M.; Zafonte, R.D. Amantadine and memantine: A comprehensive review for acquired brain injury. Brain Inj. 2020, 34, 299–315. [Google Scholar] [CrossRef]
- Maleki, S.J.; Crespo, J.F.; Cabanillas, B. Anti-inflammatory effects of flavonoids. Food Chem. 2019, 299, 125124. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.F.; Braidy, N.; Gortzi, O.; Sobarzo-Sanchez, E.; Daglia, M.; Skalicka-Woźniak, K.; Nabavi, S.M. Luteolin as an anti-inflammatory and neuroprotective agent: A brief review. Brain Res. Bull. 2015, 119, 1–11. [Google Scholar] [CrossRef]


{kind=link}
| Animal Model | Applied Treatment | Results | Reference |
|---|---|---|---|
| Poly(I:C) mice model; 2.5 mg/kg i.p. from GD 12 to 16 | Anti-IL-6 or anti-IL-1β or anti-IL-6 plus anti-IL-1β antibody co-administered with poly(I:C) from GD 12 to 16 | Anti-IL-6 antibody reversed the effect of MIA on impaired social interaction | [149] |
| Poly(I:C) mice model; 20 mg/kg i.p. on GD 12.5 | IL-17A antibody administered at GD 14.5 | IL-17A antibody and IL-17Ra KO prevented elevation of cytokine levels and reversed effects of MIA on impaired ultrasonic vocalizations (USVs), sociability, and repetitive/compulsive-like behaviors | [64] |
| Poly(I:C) mice model; 20 mg/kg i.p. or IL-6, IFN-γ, 5 µg/kg i.p. on GD 12.5 and IL-6 KO | Anti-IL-6, IFN-γ, or IL-1β antibodies co-administered with poly(I:C) or IL-6 on GD 12.5 | anti-IL-6, IFN-γ, or IL-1β antibodies reversed the effect of MIA on pre-pulse inhibition (PPI). Anti-IL-6 antibodies prevented the exploratory, anxiety, and social deficits provoked by MIA | [63] |
| Poly(I:C) mice model; 20 mg/kg i.p. on GD 12.5 | Ibudilast (anti-inflammatory drug) administered to lactating females i.p. for 2 weeks (starting 24 h post-parturition) | Ibudilast prevented increased repetitive/compulsive-like behaviors | [150] |
| Poly(I:C) mice model; 5 mg/kg i.p. on GD 10.5, 12.5, and 14.5 | A ketogenic diet (KD) at 5 weeks of offspring age for 3–4 weeks | A KD reversed the effect of MIA on impaired sociability and communication as well as repetitive behaviors | [151] |
| Poly(I:C) mice model; 20 mg/kg i.p. on GD 12.5 | Probiotics: Bacteroides fragilis (ATCC 9343) administered to offspring every other day for 6 days post-weaning (1 × 1010 CFU) | B. fragilis regulated serum metabolomic profiles and prevented anxiety-like behaviors | [152] |
| Poly(I:C) mice model; 20 mg/kg i.p. on GD 12.5 | Probiotics sachet children’s formula: Bifidobacteria (B. bifidum & B. infantis), Lactobacillus helveticus, and fructooligosaccharides administered to mother from GD 0.5 to PD 21 | Oral probiotics prevented increases in the IL-6 and IL-17A levels in both maternal serum and fetal brains, and it prevented the MIA-induced social deficits, repetitive/stereotyped behaviors, and anxiety in adult offspring | [52] |
| Applied Drug | Type of Trial | Participants | Results | Reference |
|---|---|---|---|---|
| Amantadine | Double-blind placebo-controlled trial | 39 patients, age: 5–19 years | Significant improvement in absolute score of hyperactivity and inappropriate speech, as compared to placebo group | [153] |
| Amantadine plus risperidone | Double-blind placebo-controlled trial | 39 patients, age: 4–12 years | Significant improvement in hyperactivity and on CGI | [154] |
| Celecoxib (a nonsteroidal anti-inflammatory drug, selective inhibitor of cyclooxygenase-2 (COX-2) enzyme) plus risperidone | Double-blind placebo-controlled trial | 40 patients, age: 4–12 years | Significantly improved scores in irritability, social withdrawal/lethargy, and stereotypic behavior | [155] |
| Corticosteroid therapy | Case study | 6-year-old patient with autoimmune condition plus ASD | Significantly improved spontaneous speech, responsiveness to verbal communications, social relatedness, and receptive and expressive vocabulary Decreased stereotypical utterances | [156] |
| Corticosteroid therapy (low dose) | Case study | 18-month-old patient | Increased social interaction and improvements in speech, gesturing, nonverbal communication, language expression, and comprehensive subjective improvement | [157] |
| Imuno® (supplement containing low-molecular-weight chondroitin sulfate, phosphatidylcholine and vitamin D3) | Open-label study | 3 patients | Improvement in behavioral symptoms | [158] |
| Lenalidomide (derivative of thalidomide) | Pilot, open-label study | 7 patients, age: 6–12 years | Some improvement of behavioral symptoms | [159] |
| Luteolin plus quercetin | Pilot, open-label study | 40 patients, age: 4–10 years | Improvement in Vineland Adaptive Behavior Scale (VABS) scores (10 patients), correlated with a significant decrease in serum IL-6 and TNF levels | [160] |
| Luteolin, quercetin, plus rutin (NeuroProtek® supplement) | Open-label study | 40 patients | Significant improvement in bowel color, form, and habits, as well as in eye contact/attention to directions (50%), retention of learned tasks, social interactions, and speaking skills | [161,162] |
| Memantine plus Risperidone | Double-blind placebo-controlled study | 40 patients, age: 4–12 years | Significant improvement in irritability, stereotypic behavior, and hyperactivity (ABC) | [163] |
| Memantine | Open-label treatment trial | 18 patients, age: 18–50 years | Significant improvement in the severity of core features of autism based on SRS and CGI, and improvement in impaired reading and nonverbal communication | [164] |
| Minocycline | Pilot pen-label study | 10 patients, age: 3–13 years | Changes in profiles of BDNF in CSF and blood, HGF in CSF, and CXCL8 (IL-8) in serum. However, the group of children was too small to observe clinical improvement | [165] |
| Minocycline plus Risperidone | A randomized double-blind placebo-controlled study | 46 patients, age: 3–14 years | Significant improvement of irritability and hyperactivity/noncompliance | [166] |
| ORG 2766 (analog of adrenocorticotropic hormone—(ACTH) | Placebo-controlled double-blind cross-over trial | 14 patients, age: 5–13 years | Significant improvement in irritability, stereotypic behaviors, hyperactivity, and excessive speech | [167] |
| ORG 2766 | Double-blind placebo-controlled cross-over trial | 20 patients, age: 5–15 years | Significant improvement in stereotypic behaviors and social interaction (play behavior); adverse effects were minimal | [168] |
| Spironolactone (an aldosterone antagonist and potassium-sparing diuretic) | Case study | 12-year-old boy | Significant improvement in irritability, social withdrawal, stereotypy, hyperactivity, inappropriate speech, and receptive language | [169] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zawadzka, A.; Cieślik, M.; Adamczyk, A. The Role of Maternal Immune Activation in the Pathogenesis of Autism: A Review of the Evidence, Proposed Mechanisms and Implications for Treatment. Int. J. Mol. Sci. 2021, 22, 11516. https://doi.org/10.3390/ijms222111516
Zawadzka A, Cieślik M, Adamczyk A. The Role of Maternal Immune Activation in the Pathogenesis of Autism: A Review of the Evidence, Proposed Mechanisms and Implications for Treatment. International Journal of Molecular Sciences. 2021; 22(21):11516. https://doi.org/10.3390/ijms222111516
Chicago/Turabian StyleZawadzka, Aleksandra, Magdalena Cieślik, and Agata Adamczyk. 2021. "The Role of Maternal Immune Activation in the Pathogenesis of Autism: A Review of the Evidence, Proposed Mechanisms and Implications for Treatment" International Journal of Molecular Sciences 22, no. 21: 11516. https://doi.org/10.3390/ijms222111516
APA StyleZawadzka, A., Cieślik, M., & Adamczyk, A. (2021). The Role of Maternal Immune Activation in the Pathogenesis of Autism: A Review of the Evidence, Proposed Mechanisms and Implications for Treatment. International Journal of Molecular Sciences, 22(21), 11516. https://doi.org/10.3390/ijms222111516