
Diversity of Lipid Function in Atherogenesis: A Focus on Endothelial Mechanobiology


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Current Understanding of Endothelial Function
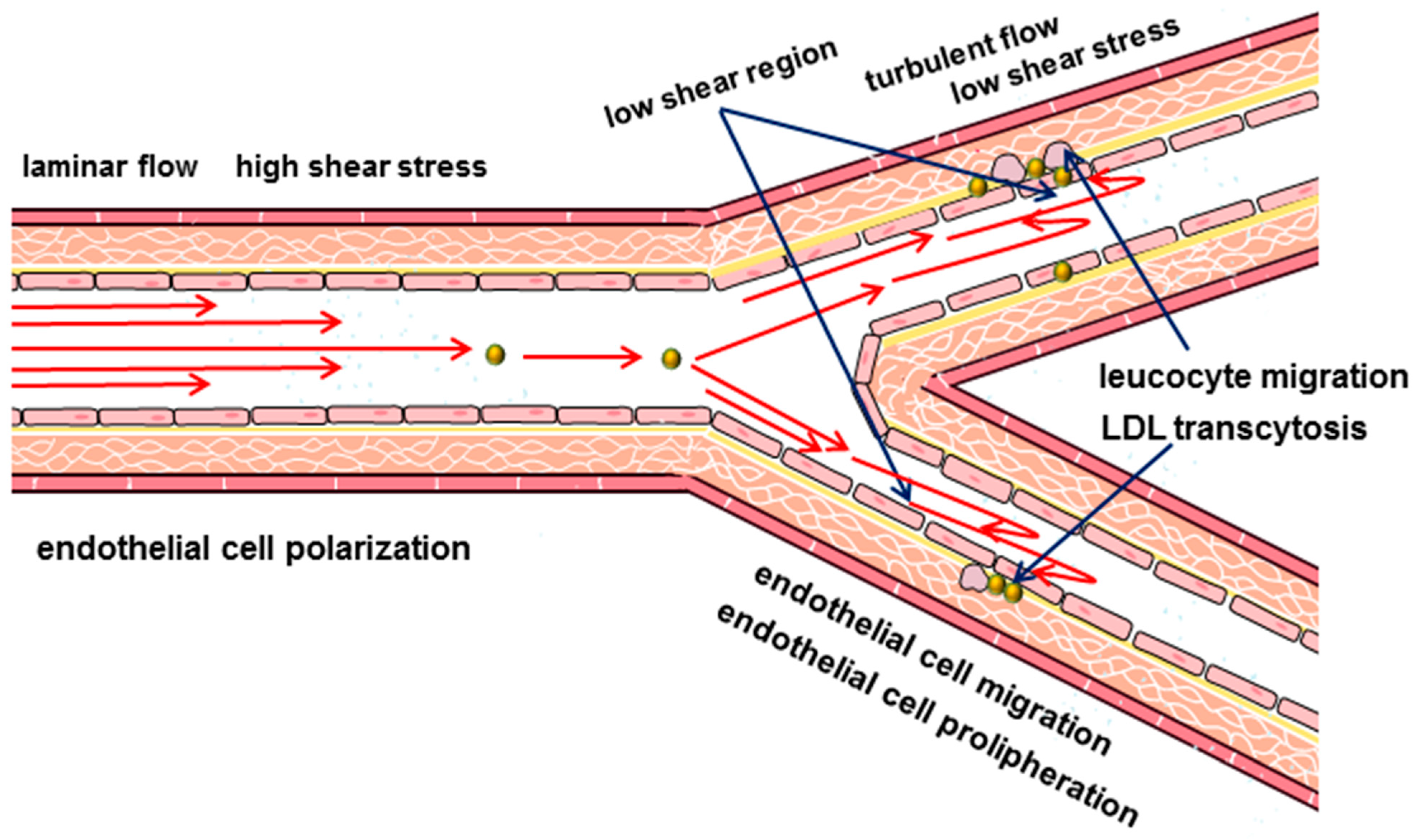
3. Hydrodynamic Characteristics of Blood Flow
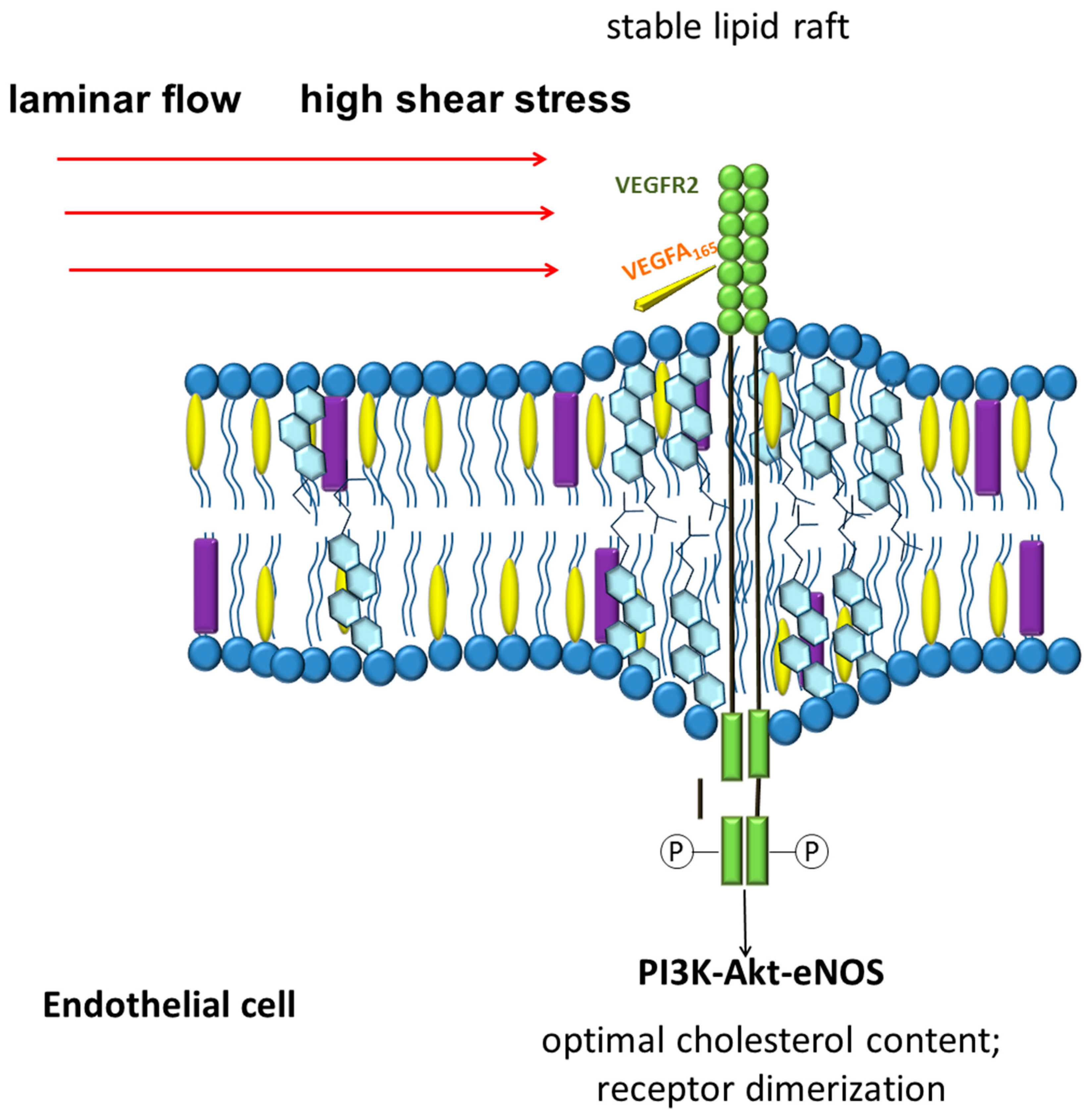
4. Lipids in Endothelial Mechanobiology
5. The Intersection of Endothelial Biomechanical Properties and Protein Function
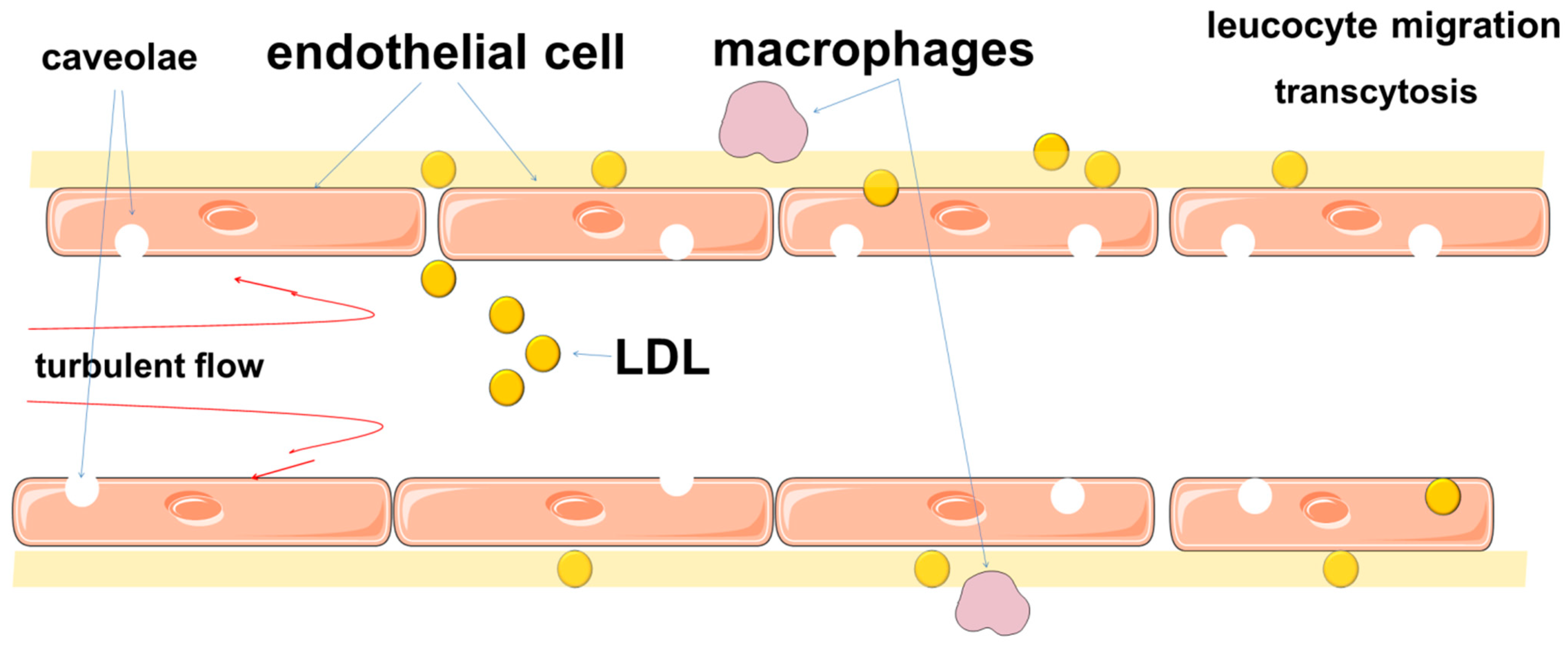
6. Regulation of Lipid Permeability and Its Disorders in Atherosclerosis
7. Endothelial Microparticles
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Song, P.; Fang, Z.; Wang, H.; Cai, Y.; Rahimi, K.; Zhu, Y.; Fowkes, F.G.R.; Fowkes, F.J.I.; Rudan, I. Global and regional prevalence, burden, and risk factors for carotid atherosclerosis: A systematic review, meta-analysis, and modelling study. Lancet Glob. Health 2020, 8, e721–e729. [Google Scholar] [CrossRef]
- Roquer, J.; Ois, A. Atherosclerotic Burden and Mortality. In Handbook of Disease Burdens and Quality of Life Measures; Preedy, V.R., Watson, R.R., Eds.; Springer: New York, NY, USA, 2010; pp. 899–918. [Google Scholar]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Noncommunicable Diseases. Available online: https://www.who.int/news-room/fact-sheets/detail/noncommunicable-diseases (accessed on 30 September 2021).
- Hoshino, T.; Sissani, L.; Labreuche, J.; Ducrocq, G.; Lavallée, P.C.; Meseguer, E.; Guidoux, C.; Cabrejo, L.; Hobeanu, C.; Gongora-Rivera, F.; et al. Prevalence of Systemic Atherosclerosis Burdens and Overlapping Stroke Etiologies and Their Associations With Long-term Vascular Prognosis in Stroke With Intracranial Atherosclerotic Disease. JAMA Neurol. 2018, 75, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Bauersachs, R.; Zeymer, U.; Brière, J.-B.; Marre, C.; Bowrin, K.; Huelsebeck, M. Burden of Coronary Artery Disease and Peripheral Artery Disease: A Literature Review. Cardiovasc. Ther. 2019, 2019, 8295054. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.; Alarcão, J.; Araujo, F.; Ascenção, R.; Caldeira, D.; Fiorentino, F.; Gil, V.; Gouveia, M.; Lourenço, F.; Mello e Silva, A.; et al. The burden of atherosclerosis in Portugal. Eur. Heart J. Qual. Care Clin. Outcomes 2020, 7, 154–162. [Google Scholar] [CrossRef]
- Lambert, M.A.; Weir-McCall, J.R.; Salsano, M.; Gandy, S.J.; Levin, D.; Cavin, I.; Littleford, R.; MacFarlane, J.A.; Matthew, S.Z.; Nicholas, R.S.; et al. Prevalence and Distribution of Atherosclerosis in a Low- to Intermediate-Risk Population: Assessment with Whole-Body MR Angiography. Radiology 2018, 287, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.-M.; Capodanno, D.; et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice: Developed by the Task Force for cardiovascular disease prevention in clinical practice with representatives of the European Society of Cardiology and 12 medical societies With the special contribution of the European Association of Preventive Cardiology (EAPC). Eur. Heart J. 2021, 42, 3227–3337. [Google Scholar] [CrossRef] [PubMed]
- Konstantinov, I.E.; Mejevoi, N.; Anichkov, N.M.; Nikolai, N. Anichkov and his theory of atherosclerosis. Tex. Heart Inst. J. 2006, 33, 417–423. [Google Scholar]
- Wilkins, L.W. Classics in arteriosclerosis research: On experimental cholesterin steatosis and its significance in the origin of some pathological processes by N. Anitschkow and S. Chalatow, translated by Mary, Z. Pelias, 1913. Arteriosclerosis 1983, 3, 178–182. [Google Scholar]
- Meier, R.; Rachamin, Y.; Rosemann, T.; Markun, S. The Impact of the 2019 European Guideline for Cardiovascular Risk Management: A Cross-Sectional Study in General Practice. J. Clin. Med. 2020, 9, 2140. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Glagov, S.; Zarins, C.; Giddens, D.P.; Ku, D.N. Hemodynamics and atherosclerosis. Insights and perspectives gained from studies of human arteries. Arch. Pathol. Lab. Med. 1988, 112, 1018–1031. [Google Scholar]
- Morbiducci, U.; Kok, A.M.; Kwak, B.R.; Stone, P.H.; Steinman, D.A.; Wentzel, J.J. Atherosclerosis at arterial bifurcations: Evidence for the role of haemodynamics and geometry. Thromb. Haemost. 2016, 115, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Chen, Z.; Hippe, D.S.; Watase, H.; Sun, B.; Lin, R.; Yang, Z.; Xue, Y.; Zhao, X.; Yuan, C. Association Between Carotid Bifurcation Geometry and Atherosclerotic Plaque Vulnerability. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1383–1391. [Google Scholar] [CrossRef]
- Frangos, S.G.; Gahtan, V.; Sumpio, B. Localization of Atherosclerosis: Role of Hemodynamics. Arch. Surg. 1999, 134, 1142–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arjmandi-Tash, O.; Razavi, S.E.; Zanbouri, R. Possibility of atherosclerosis in an arterial bifurcation model. Bioimpacts 2011, 1, 225–228. [Google Scholar] [CrossRef]
- Antoniadis, A.P.; Chatzizisis, Y.S. Chapter 8—Local blood flow parameters and atherosclerosis in coronary artery bifurcations. In Biomechanics of Coronary Atherosclerotic Plaque; Ohayon, J., Finet, G., Pettigrew, R.I., Eds.; Academic Press: Cambridge, MA, USA, 2021; Volume 4, pp. 193–202. [Google Scholar]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Davignon, J.; Ganz, P. Role of Endothelial Dysfunction in Atherosclerosis. Circulation 2004, 109, III-27–III-32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodorou, K.; Boon, R.A. Endothelial Cell Metabolism in Atherosclerosis. Front. Cell Dev. Biol. 2018, 6, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galley, H.F.; Webster, N.R. Physiology of the endothelium. Br. J. Anaesth. 2004, 93, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Chiu, J.-J.; Chien, S. Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectives. Physiol. Rev. 2011, 91, 327–387. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Li, Y.-S.; Chien, S. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2191–2198. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Wang, M.; Ye, J.; Sun, G.; Sun, X. Mechanism overview and target mining of atherosclerosis: Endothelial cell injury in atherosclerosis is regulated by glycolysis (Review). Int. J. Mol. Med. 2021, 47, 65–76. [Google Scholar] [CrossRef]
- Mantovani, A.; Dejana, E. Cytokines as communication signals between leukocytes and endothelial cells. Immunol. Today 1989, 10, 370–375. [Google Scholar] [CrossRef]
- Davies, P.F. Flow-mediated endothelial mechanotransduction. Physiol. Rev. 1995, 75, 519–560. [Google Scholar] [CrossRef] [PubMed]
- Sandoo, A.; van Zanten, J.J.C.S.V.; Metsios, G.S.; Carroll, D.; Kitas, G.D. The endothelium and its role in regulating vascular tone. Open Cardiovasc. Med. J. 2010, 4, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Janaszak-Jasiecka, A.; Siekierzycka, A.; Płoska, A.; Dobrucki, I.T.; Kalinowski, L. Endothelial Dysfunction Driven by Hypoxia—The Influence of Oxygen Deficiency on NO Bioavailability. Biomolecules 2021, 11, 982. [Google Scholar] [CrossRef]
- O’Riordan, E.; Chen, J.; Brodsky, S.V.; Smirnova, I.; Li, H.; Goligorsky, M.S. Endothelial cell dysfunction: The syndrome in making. Kidney Int. 2005, 67, 1654–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, B.G.; Economides, C.; Mayeda, G.S.; Burstein, S.; Kloner, R.A. The endothelial cell in health and disease: Its function, dysfunction, measurement and therapy. Int. J. Impot. Res. 2010, 22, 77–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mestas, J.; Ley, K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc. Med. 2008, 18, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Qian, M.; Kyler, K.; Xu, J. Endothelial–Vascular Smooth Muscle Cells Interactions in Atherosclerosis. Front. Cardiovasc. Med. 2018, 5, 151. [Google Scholar] [CrossRef] [Green Version]
- Hunt, B.J.; Jurd, K.M. Endothelial cell activation. A central pathophysiological process. BMJ 1998, 316, 1328–1329. [Google Scholar] [CrossRef] [PubMed]
- Pober, J.S. Warner-Lambert/Parke-Davis award lecture. Cytokine-mediated activation of vascular endothelium. Physiology and pathology. Am. J. Pathol. 1988, 133, 426–433. [Google Scholar]
- Gimbrone, M.A., Jr.; Nagel, T.; Topper, J.N. Biomechanical activation: An emerging paradigm in endothelial adhesion biology. J. Clin. Investig. 1997, 99, 1809–1813. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Matsuto, T.; Miida, T.; Inano, K. Differences in the effects of cytokines on the expression of adhesion molecules in endothelial cells. Ann. Med. Interne 1997, 148, 125–129. [Google Scholar]
- De Caterina, R.; Libby, P.; Peng, H.B.; Thannickal, V.J.; Rajavashisth, T.B.; Gimbrone, M.A., Jr.; Shin, W.S.; Liao, J.K. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Investig. 1995, 96, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; DeFelice, A.F.; Hanig, J.P.; Colatsky, T. Biomarkers of Endothelial Cell Activation Serve as Potential Surrogate Markers for Drug-induced Vascular Injury. Toxicol. Pathol. 2010, 38, 856–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nourshargh, S.; Alon, R. Leukocyte Migration into Inflamed Tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef] [Green Version]
- De Caterina, R.; Liao, J.K.; Libby, P. Fatty acid modulation of endothelial activation. Am. J. Clin. Nutr. 2000, 71, 213S–223S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, J.K. Linking endothelial dysfunction with endothelial cell activation. J. Clin. Investig. 2013, 123, 540–541. [Google Scholar] [CrossRef] [PubMed]
- Jambusaria, A.; Hong, Z.; Zhang, L.; Srivastava, S.; Jana, A.; Toth, P.T.; Dai, Y.; Malik, A.B.; Rehman, J. Endothelial heterogeneity across distinct vascular beds during homeostasis and inflammation. eLife 2020, 9, e51413. [Google Scholar] [CrossRef]
- Potente, M.; Mäkinen, T. Vascular heterogeneity and specialization in development and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 477–494. [Google Scholar] [CrossRef]
- Aird, W.C. Phenotypic Heterogeneity of the Endothelium. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Monahan-Earley, R.; Dvorak, A.M.; Aird, W.C. Evolutionary origins of the blood vascular system and endothelium. J. Thromb. Haemost. 2013, 11, 46–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Chápuli, R.; Carmona, R.; Guadix, J.A.; Macías, D.; Pérez-Pomares, J.M. The origin of the endothelial cells: An evo-devo approach for the invertebrate/vertebrate transition of the circulatory system. Evol. Dev. 2005, 7, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Reiber, C.L.; McGaw, I.J. A Review of the “Open” and “Closed” Circulatory Systems: New Terminology for Complex Invertebrate Circulatory Systems in Light of Current Findings. Int. J. Zool. 2009, 2009, 301284. [Google Scholar] [CrossRef] [Green Version]
- Browning, J. Octopus microvasculature: Permeability to ferritin and carbon. Tissue Cell 1979, 11, 371–383. [Google Scholar] [CrossRef]
- Shigei, T.; Tsuru, H.; Ishikawa, N.; Yoshioka, K. Absence of endothelium in invertebrate blood vessels: Significance of endothelium and sympathetic nerve/medial smooth muscle in the vertebrate vascular system. Jpn. J. Pharmacol. 2001, 87, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Moller, P.C.; Philpott, C.W. The circulatory system of Amphioxus (Branchiostoma floridae). I. Morphology of the major vessels of the pharyngeal area. J. Morphol. 1973, 139, 389–406. [Google Scholar] [CrossRef]
- Moller, P.C.; Philpott, C.W. The circulatory system of amphioxus (Branchiostoma floridae). II. Uptake of exogenous proteins by endothelial cells. Z. Zellforsch Mikrosk. Anat. 1973, 143, 135–141. [Google Scholar] [CrossRef]
- Mai, J.; Virtue, A.; Shen, J.; Wang, H.; Yang, X.-F. An evolving new paradigm: Endothelial cells—Conditional innate immune cells. J. Hematol. Oncol. 2013, 6, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelis, U.R. Mechanisms of endothelial cell migration. Cell. Mol. Life Sci. 2014, 71, 4131–4148. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Saredy, J.; Yang, W.Y.; Sun, Y.; Lu, Y.; Saaoud, F.; Drummer, C.T.; Johnson, C.; Xu, K.; Jiang, X.; et al. Vascular Endothelial Cells and Innate Immunity. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e138–e152. [Google Scholar] [CrossRef]
- Fu, H.; Vadalia, N.; Xue, E.R.; Johnson, C.; Wang, L.; Yang, W.Y.; Sanchez, C.; Nelson, J.; Chen, Q.; Choi, E.T.; et al. Thrombus leukocytes exhibit more endothelial cell-specific angiogenic markers than peripheral blood leukocytes do in acute coronary syndrome patients, suggesting a possibility of trans-differentiation: A comprehensive database mining study. J. Hematol. Oncol. 2017, 10, 74. [Google Scholar] [CrossRef] [Green Version]
- Schulte, K.; Kunter, U.; Moeller, M.J. The evolution of blood pressure and the rise of mankind. Nephrol. Dial. Transplant. 2014, 30, 713–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc. Pathol. 2013, 22, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, P.F. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 2009, 6, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Eckmann, D.M.; Bowers, S.; Stecker, M.; Cheung, A.T. Hematocrit, volume expander, temperature, and shear rate effects on blood viscosity. Anesth. Analg. 2000, 91, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Santamaría, R.; González-Álvarez, M.; Delgado, R.; Esteban, S.; Arroyo, A.G. Remodeling of the Microvasculature: May the Blood Flow Be With You. Front. Physiol. 2020, 11, 586852. [Google Scholar] [CrossRef]
- Papaioannou, T.G.; Stefanadis, C. Vascular wall shear stress: Basic principles and methods. Hellenic J. Cardiol. 2005, 46, 9–15. [Google Scholar]
- Nigro, P.; Abe, J.-I.; Berk, B.C. Flow shear stress and atherosclerosis: A matter of site specificity. Antioxid. Redox Signal. 2011, 15, 1405–1414. [Google Scholar] [CrossRef]
- Ni, C.W.; Qiu, H.; Rezvan, A.; Kwon, K.; Nam, D.; Son, D.J.; Visvader, J.E.; Jo, H. Discovery of novel mechanosensitive genes in vivo using mouse carotid artery endothelium exposed to disturbed flow. Blood 2010, 116, e66–e73. [Google Scholar] [CrossRef] [PubMed]
- Malek, A.M.; Alper, S.L.; Izumo, S. Hemodynamic shear stress and its role in atherosclerosis. JAMA 1999, 282, 2035–2042. [Google Scholar] [CrossRef] [PubMed]
- Ku, D.N.; Giddens, D.P.; Zarins, C.K.; Glagov, S. Pulsatile flow and atherosclerosis in the human carotid bifurcation. Positive correlation between plaque location and low oscillating shear stress. Arteriosclerosis 1985, 5, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Cybulsky, M.I.; Marsden, P.A. Effect of Disturbed Blood Flow on Endothelial Cell Gene Expression. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1806–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barakat, A.I. Blood flow and arterial endothelial dysfunction: Mechanisms and implications. Comptes Rendus Phys. 2013, 14, 479–496. [Google Scholar] [CrossRef]
- Saqr, K.M.; Tupin, S.; Rashad, S.; Endo, T.; Niizuma, K.; Tominaga, T.; Ohta, M. Physiologic blood flow is turbulent. Sci. Rep. 2020, 10, 15492. [Google Scholar] [CrossRef]
- Butler, P.J. Mechanobiology of dynamic enzyme systems. APL Bioeng. 2020, 4, 010907. [Google Scholar] [CrossRef]
- Stepanchuk, A.P. Blood motion: Turbulent or laminar? Wiad. Lek. 2017, 70, 331–334. [Google Scholar] [PubMed]
- Dewey, C. Dynamics of arterial flow. Adv. Exp. Med. Biol. 1979, 115, 55–89. [Google Scholar]
- Giddens, D.; Zarins, C.; Glagov, S. Response of arteries to near-wall fluid dynamic behavior. Appl. Mech. Rev. 1990, 43, 98–102. [Google Scholar] [CrossRef]
- Nerem, R.M. Vascular fluid mechanics, the arterial wall, and atherosclerosis. J. Biomech. Eng. 1992, 114, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Dessalles, C.A.; Leclech, C.; Castagnino, A.; Barakat, A.I. Integration of substrate- and flow-derived stresses in endothelial cell mechanobiology. Commun. Biol. 2021, 4, 764. [Google Scholar] [CrossRef]
- Campinho, P.; Vilfan, A.; Vermot, J. Blood Flow Forces in Shaping the Vascular System: A Focus on Endothelial Cell Behavior. Front. Physiol. 2020, 11, 552. [Google Scholar] [CrossRef]
- Sriram, K.; Laughlin, J.G.; Rangamani, P.; Tartakovsky, D.M. Shear-Induced Nitric Oxide Production by Endothelial Cells. Biophys. J. 2016, 111, 208–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaunas, R. Good advice for endothelial cells: Get in line, relax tension, and go with the flow. APL Bioeng. 2020, 4, 010905. [Google Scholar] [CrossRef] [PubMed]
- Baeyens, N.; Bandyopadhyay, C.; Coon, B.G.; Yun, S.; Schwartz, M.A. Endothelial fluid shear stress sensing in vascular health and disease. J. Clin. Investig. 2016, 126, 821–828. [Google Scholar] [CrossRef]
- Joannides, R.; Haefeli, W.E.; Linder, L.; Richard, V.; Bakkali, E.H.; Thuillez, C.; Lüscher, T.F. Nitric oxide is responsible for flow-dependent dilatation of human peripheral conduit arteries in vivo. Circulation 1995, 91, 1314–1319. [Google Scholar] [CrossRef] [PubMed]
- Frangos, J.A.; Eskin, S.G.; McIntire, L.V.; Ives, C.L. Flow effects on prostacyclin production by cultured human endothelial cells. Science 1985, 227, 1477–1479. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Leyen, T.A.; Fontijn, R.D.; Fledderus, J.O.; Baggen, J.M.; Volger, O.L.; van Nieuw Amerongen, G.P.; Horrevoets, A.J. KLF2-induced actin shear fibers control both alignment to flow and JNK signaling in vascular endothelium. Blood 2010, 115, 2533–2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakache, M.; Gaub, H.E. Hydrodynamic hyperpolarization of endothelial cells. Proc. Natl. Acad. Sci. USA 1988, 85, 1841–1843. [Google Scholar] [CrossRef] [Green Version]
- Davies, P.F.; Reidy, M.A.; Goode, T.B.; Bowyer, D.E. Scanning electron microscopy in the evaluation of endothelial integrity of the fatty lesion in atherosclerosis. Atherosclerosis 1976, 25, 125–130. [Google Scholar] [CrossRef]
- Flaherty, J.T.; Pierce, J.E.; Ferrans, V.J.; Patel, D.J.; Tucker, W.K.; Fry, D.L. Endothelial nuclear patterns in the canine arterial tree with particular reference to hemodynamic events. Circ. Res. 1972, 30, 23–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goode, T.B.; Davies, P.F.; Reidy, M.A.; Bowyer, D.E. Aortic endothelial cell morphology observed in situ by scanning electron microscopy during atherogenesis in the rabbit. Atherosclerosis 1977, 27, 235–251. [Google Scholar] [CrossRef]
- Langille, B.L.; Adamson, S.L. Relationship between blood flow direction and endothelial cell orientation at arterial branch sites in rabbits and mice. Circ. Res. 1981, 48, 481–488. [Google Scholar] [CrossRef] [Green Version]
- Levesque, M.J.; Liepsch, D.; Moravec, S.; Nerem, R.M. Correlation of endothelial cell shape and wall shear stress in a stenosed dog aorta. Arteriosclerosis 1986, 6, 220–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levesque, M.J.; Nerem, R.M. The study of rheological effects on vascular endothelial cells in culture. Biorheology 1989, 26, 345–357. [Google Scholar] [CrossRef]
- Nerem, R.M.; Levesque, M.J.; Cornhill, J.F. Vascular endothelial morphology as an indicator of the pattern of blood flow. J. Biomech. Eng. 1981, 103, 172–176. [Google Scholar] [CrossRef]
- Okano, M.; Yoshida, Y. Influence of shear stress on endothelial cell shapes and junction complexes at flow dividers of aortic bifurcations in cholesterol-fed rabbits. Front. Med. Biol. Eng. 1993, 5, 95–120. [Google Scholar] [PubMed]
- Reidy, M.A.; Langille, B.L. The effect of local blood flow patterns on endothelial cell morphology. Exp. Mol. Pathol. 1980, 32, 276–289. [Google Scholar] [CrossRef]
- Dewey, C.F., Jr.; Bussolari, S.R.; Gimbrone, M.A., Jr.; Davies, P.F. The dynamic response of vascular endothelial cells to fluid shear stress. J. Biomech. Eng. 1981, 103, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Eskin, S.G.; Ives, C.L.; McIntire, L.V.; Navarro, L.T. Response of cultured endothelial cells to steady flow. Microvasc. Res. 1984, 28, 87–94. [Google Scholar] [CrossRef]
- Remuzzi, A.; Dewey, C.F., Jr.; Davies, P.F.; Gimbrone, M.A., Jr. Orientation of endothelial cells in shear fields in vitro. Biorheology 1984, 21, 617–630. [Google Scholar] [CrossRef]
- Franco, C.A.; Jones, M.L.; Bernabeu, M.O.; Vion, A.C.; Barbacena, P.; Fan, J.; Mathivet, T.; Fonseca, C.G.; Ragab, A.; Yamaguchi, T.P.; et al. Non-canonical Wnt signalling modulates the endothelial shear stress flow sensor in vascular remodelling. eLife 2016, 5, e07727. [Google Scholar] [CrossRef]
- Tkachenko, E.; Gutierrez, E.; Saikin, S.K.; Fogelstrand, P.; Kim, C.; Groisman, A.; Ginsberg, M.H. The nucleus of endothelial cell as a sensor of blood flow direction. Biol. Open 2013, 2, 1007–1012. [Google Scholar] [CrossRef] [Green Version]
- Rogers, K.A.; McKee, N.H.; Kalnins, V.I. Preferential orientation of centrioles toward the heart in endothelial cells of major blood vessels is reestablished after reversal of a segment. Proc. Natl. Acad. Sci. USA 1985, 82, 3272–3276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malek, A.M.; Izumo, S. Mechanism of endothelial cell shape change and cytoskeletal remodeling in response to fluid shear stress. J. Cell Sci. 1996, 109, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Hikita, T.; Mirzapourshafiyi, F.; Barbacena, P.; Riddell, M.; Pasha, A.; Li, M.; Kawamura, T.; Brandes, R.P.; Hirose, T.; Ohno, S.; et al. PAR-3 controls endothelial planar polarity and vascular inflammation under laminar flow. EMBO Rep. 2018, 19, e45253. [Google Scholar] [CrossRef]
- Coan, D.E.; Wechezak, A.R.; Viggers, R.F.; Sauvage, L.R. Effect of shear stress upon localization of the Golgi apparatus and microtubule organizing center in isolated cultured endothelial cells. J. Cell Sci. 1993, 104, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Franco, C.A.; Jones, M.L.; Bernabeu, M.O.; Geudens, I.; Mathivet, T.; Rosa, A.; Lopes, F.M.; Lima, A.P.; Ragab, A.; Collins, R.T.; et al. Dynamic endothelial cell rearrangements drive developmental vessel regression. PLoS Biol. 2015, 13, e1002125. [Google Scholar] [CrossRef]
- Gomes, E.R.; Jani, S.; Gundersen, G.G. Nuclear movement regulated by Cdc42, MRCK, myosin, and actin flow establishes MTOC polarization in migrating cells. Cell 2005, 121, 451–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gundersen, G.G.; Worman, H.J. Nuclear positioning. Cell 2013, 152, 1376–1389. [Google Scholar] [CrossRef] [Green Version]
- Morgan, J.T.; Pfeiffer, E.R.; Thirkill, T.L.; Kumar, P.; Peng, G.; Fridolfsson, H.N.; Douglas, G.C.; Starr, D.A.; Barakat, A.I. Nesprin-3 regulates endothelial cell morphology, perinuclear cytoskeletal architecture, and flow-induced polarization. Mol. Biol. Cell 2011, 22, 4324–4334. [Google Scholar] [CrossRef] [PubMed]
- Tzima, E.; Kiosses, W.B.; del Pozo, M.A.; Schwartz, M.A. Localized cdc42 activation, detected using a novel assay, mediates microtubule organizing center positioning in endothelial cells in response to fluid shear stress. J. Biol. Chem. 2003, 278, 31020–31023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, P.F.; Remuzzi, A.; Gordon, E.J.; Dewey, C.F., Jr.; Gimbrone, M.A., Jr. Turbulent fluid shear stress induces vascular endothelial cell turnover in vitro. Proc. Natl. Acad. Sci. USA 1986, 83, 2114–2117. [Google Scholar] [CrossRef] [Green Version]
- Hahn, C.; Schwartz, M.A. Mechanotransduction in vascular physiology and atherogenesis. Nat. Rev. Mol. Cell Biol. 2009, 10, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.B.; Wang, S.; Helker, C.S.; Rasouli, S.J.; Maischein, H.M.; Offermanns, S.; Herzog, W.; Stainier, D.Y. In vivo modulation of endothelial polarization by Apelin receptor signalling. Nat. Commun. 2016, 7, 11805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruter, D.L.; Liu, Z.; Ngo, K.M.; Shaka, X.; Marvin, A.; Buglak, D.B.; Kidder, E.J.; Bautch, V.L. SMAD6 transduces endothelial cell flow responses required for blood vessel homeostasis. Angiogenesis 2021, 24, 387–398. [Google Scholar] [CrossRef]
- Dieterich, P.; Odenthal-Schnittler, M.; Mrowietz, C.; Krämer, M.; Sasse, L.; Oberleithner, H.; Schnittler, H.J. Quantitative morphodynamics of endothelial cells within confluent cultures in response to fluid shear stress. Biophys. J. 2000, 79, 1285–1297. [Google Scholar] [CrossRef] [Green Version]
- McCue, S.; Dajnowiec, D.; Xu, F.; Zhang, M.; Jackson, M.R.; Langille, B.L. Shear stress regulates forward and reverse planar cell polarity of vascular endothelium in vivo and in vitro. Circ. Res. 2006, 98, 939–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noria, S.; Cowan, D.B.; Gotlieb, A.I.; Langille, B.L. Transient and steady-state effects of shear stress on endothelial cell adherens junctions. Circ. Res. 1999, 85, 504–514. [Google Scholar] [CrossRef] [Green Version]
- Schnittler, H.-J.; Püschel, B.; Drenckhahn, D. Role of cadherins and plakoglobin in interendothelial adhesion under resting conditions and shear stress. Am. J. Physiol. Heart Circ. Physiol. 1997, 273, H2396–H2405. [Google Scholar] [CrossRef]
- Shikata, Y.; Rios, A.; Kawkitinarong, K.; DePaola, N.; Garcia, J.G.; Birukov, K.G. Differential effects of shear stress and cyclic stretch on focal adhesion remodeling, site-specific FAK phosphorylation, and small GTPases in human lung endothelial cells. Exp. Cell Res. 2005, 304, 40–49. [Google Scholar] [CrossRef]
- Lin, K.; Hsu, P.P.; Chen, B.P.; Yuan, S.; Usami, S.; Shyy, J.Y.; Li, Y.S.; Chien, S. Molecular mechanism of endothelial growth arrest by laminar shear stress. Proc. Natl. Acad. Sci. USA 2000, 97, 9385–9389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akimoto, S.; Mitsumata, M.; Sasaguri, T.; Yoshida, Y. Laminar shear stress inhibits vascular endothelial cell proliferation by inducing cyclin-dependent kinase inhibitor p21(Sdi1/Cip1/Waf1). Circ. Res. 2000, 86, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Ostrowski, M.A.; Huang, N.F.; Walker, T.W.; Verwijlen, T.; Poplawski, C.; Khoo, A.S.; Cooke, J.P.; Fuller, G.G.; Dunn, A.R. Microvascular endothelial cells migrate upstream and align against the shear stress field created by impinging flow. Biophys. J. 2014, 106, 366–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wechezak, A.R.; Viggers, R.F.; Sauvage, L.R. Fibronectin and F-actin redistribution in cultured endothelial cells exposed to shear stress. Lab. Investig. 1985, 53, 639–647. [Google Scholar]
- White, G.E.; Fujiwara, K. Expression and intracellular distribution of stress fibers in aortic endothelium. J. Cell Biol. 1986, 103, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langille, B.L.; Graham, J.J.; Kim, D.; Gotlieb, A.I. Dynamics of shear-induced redistribution of F-actin in endothelial cells in vivo. Arterioscler. Thromb. 1991, 11, 1814–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojciak-Stothard, B.; Ridley, A.J. Shear stress-induced endothelial cell polarization is mediated by Rho and Rac but not Cdc42 or PI 3-kinases. J. Cell Biol. 2003, 161, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Noria, S.; Xu, F.; McCue, S.; Jones, M.; Gotlieb, A.I.; Langille, B.L. Assembly and reorientation of stress fibers drives morphological changes to endothelial cells exposed to shear stress. Am. J. Pathol. 2004, 164, 1211–1223. [Google Scholar] [CrossRef] [Green Version]
- DePaola, N.; Davies, P.F.; Pritchard, W.F., Jr.; Florez, L.; Harbeck, N.; Polacek, D.C. Spatial and temporal regulation of gap junction connexin43 in vascular endothelial cells exposed to controlled disturbed flows in vitro. Proc. Natl. Acad. Sci. USA 1999, 96, 3154–3159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franke, R.P.; Gräfe, M.; Schnittler, H.; Seiffge, D.; Mittermayer, C.; Drenckhahn, D. Induction of human vascular endothelial stress fibres by fluid shear stress. Nature 1984, 307, 648–649. [Google Scholar] [CrossRef] [PubMed]
- Helmke, B.P.; Goldman, R.D.; Davies, P.F. Rapid displacement of vimentin intermediate filaments in living endothelial cells exposed to flow. Circ. Res. 2000, 86, 745–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.W.; Langille, B.L.; Wong, M.K.; Gotlieb, A.I. Patterns of endothelial microfilament distribution in the rabbit aorta in situ. Circ. Res. 1989, 64, 21–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levesque, M.J.; Nerem, R.M. The elongation and orientation of cultured endothelial cells in response to shear stress. J. Biomech. Eng. 1985, 107, 341–347. [Google Scholar] [CrossRef]
- Kim, D.W.; Gotlieb, A.I.; Langille, B.L. In vivo modulation of endothelial F-actin microfilaments by experimental alterations in shear stress. Arteriosclerosis 1989, 9, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Vyalov, S.; Langille, B.L.; Gotlieb, A.I. Decreased blood flow rate disrupts endothelial repair in vivo. Am. J. Pathol. 1996, 149, 2107–2118. [Google Scholar]
- Masuda, H.; Shozawa, T.; Hosoda, S.; Kanda, M.; Kamiya, A. Cytoplasmic microfilaments in endothelial cells of flow loaded canine carotid arteries. Heart Vessel. 1985, 1, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Ookawa, K.; Sato, M.; Ohshima, N. Changes in the microstructure of cultured porcine aortic endothelial cells in the early stage after applying a fluid-imposed shear stress. J. Biomech. 1992, 25, 1321–1328. [Google Scholar] [CrossRef]
- Wong, A.J.; Pollard, T.D.; Herman, I.M. Actin filament stress fibers in vascular endothelial cells in vivo. Science 1983, 219, 867–869. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Butler, J.P.; Ingber, D.E. Mechanotransduction across the cell surface and through the cytoskeleton. Science 1993, 260, 1124–1127. [Google Scholar] [CrossRef]
- Li, Y.S.; Haga, J.H.; Chien, S. Molecular basis of the effects of shear stress on vascular endothelial cells. J. Biomech. 2005, 38, 1949–1971. [Google Scholar] [CrossRef]
- Peiffer, V.; Sherwin, S.J.; Weinberg, P.D. Does low and oscillatory wall shear stress correlate spatially with early atherosclerosis? A systematic review. Cardiovasc. Res. 2013, 99, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Ando, J.; Yamamoto, K. Effects of shear stress and stretch on endothelial function. Antioxid. Redox Signal. 2011, 15, 1389–1403. [Google Scholar] [CrossRef]
- Le Roux, A.-L.; Quiroga, X.; Walani, N.; Arroyo, M.; Roca-Cusachs, P. The plasma membrane as a mechanochemical transducer. Philos. Trans. R. Soc. Biol. Sci. 2019, 374, 20180221. [Google Scholar] [CrossRef] [PubMed]
- White, C.R.; Frangos, J.A. The shear stress of it all: The cell membrane and mechanochemical transduction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2007, 362, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Sachs, F. Mechanical transduction in biological systems. Crit. Rev. Biomed. Eng. 1988, 16, 141–169. [Google Scholar]
- Lombard, J. Once upon a time the cell membranes: 175 years of cell boundary research. Biol. Direct 2014, 9, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardino de la Serna, J.; Schütz, G.J.; Eggeling, C.; Cebecauer, M. There Is No Simple Model of the Plasma Membrane Organization. Front. Cell Dev. Biol. 2016, 4, 106. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira Andrade, L. Understanding the role of cholesterol in cellular biomechanics and regulation of vesicular trafficking: The power of imaging. Biomed. Spectrosc. Imaging 2016, 5, S101–S117. [Google Scholar] [CrossRef] [Green Version]
- Steck, T.L.; Lange, Y. Transverse distribution of plasma membrane bilayer cholesterol: Picking sides. Traffic 2018, 19, 750–760. [Google Scholar] [CrossRef] [Green Version]
- Subczynski, W.K.; Pasenkiewicz-Gierula, M.; Widomska, J.; Mainali, L.; Raguz, M. High Cholesterol/Low Cholesterol: Effects in Biological Membranes: A Review. Cell Biochem. Biophys. 2017, 75, 369–385. [Google Scholar] [CrossRef]
- Fantini, J.; Barrantes, F.J. How cholesterol interacts with membrane proteins: An exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front. Physiol. 2013, 4, 31. [Google Scholar] [CrossRef] [Green Version]
- Fantini, J.; Barrantes, F.J. Sphingolipid/cholesterol regulation of neurotransmitter receptor conformation and function. Biochim. Biophys. Acta 2009, 1788, 2345–2361. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.E.; Homann, U. Cell surface area regulation and membrane tension. J. Membr. Biol. 2001, 179, 79–102. [Google Scholar] [CrossRef]
- Biswas, A.; Kashyap, P.; Datta, S.; Sengupta, T.; Sinha, B. Cholesterol Depletion by MβCD Enhances Cell Membrane Tension and Its Variations-Reducing Integrity. Biophys. J. 2019, 116, 1456–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosmalska, A.J.; Casares, L.; Elosegui-Artola, A.; Thottacherry, J.J.; Moreno-Vicente, R.; González-Tarragó, V.; Del Pozo, M.; Mayor, S.; Arroyo, M.; Navajas, D.; et al. Physical principles of membrane remodelling during cell mechanoadaptation. Nat. Commun. 2015, 6, 7292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, E.; Needham, D. Physical properties of surfactant bilayer membranes: Thermal transitions, elasticity, rigidity, cohesion and colloidal interactions. J. Phys. Chem. 1987, 91, 4219–4228. [Google Scholar] [CrossRef]
- Needham, D.; Nunn, R.S. Elastic deformation and failure of lipid bilayer membranes containing cholesterol. Biophys. J. 1990, 58, 997–1009. [Google Scholar] [CrossRef] [Green Version]
- Karatekin, E.; Sandre, O.; Guitouni, H.; Borghi, N.; Puech, P.H.; Brochard-Wyart, F. Cascades of transient pores in giant vesicles: Line tension and transport. Biophys. J. 2003, 84, 1734–1749. [Google Scholar] [CrossRef] [Green Version]
- Subczynski, W.K.; Antholine, W.E.; Hyde, J.S.; Kusumi, A. Microimmiscibility and three-dimensional dynamic structures of phosphatidylcholine-cholesterol membranes: Translational diffusion of a copper complex in the membrane. Biochemistry 1990, 29, 7936–7945. [Google Scholar] [CrossRef] [PubMed]
- Subczynski, W.K.; Wisniewska, A.; Hyde, J.S.; Kusumi, A. Three-dimensional dynamic structure of the liquid-ordered domain in lipid membranes as examined by pulse-EPR oxygen probing. Biophys. J. 2007, 92, 1573–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subczynski, W.K.; Hyde, J.S.; Kusumi, A. Effect of alkyl chain unsaturation and cholesterol intercalation on oxygen transport in membranes: A pulse ESR spin labeling study. Biochemistry 1991, 30, 8578–8590. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, D.; Kučerka, N.; Wassall, S.R.; Harroun, T.A.; Katsaras, J. Cholesterol’s location in lipid bilayers. Chem. Phys. Lipids 2016, 199, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Golani, G.; Ariotti, N.; Parton, R.G.; Kozlov, M.M. Membrane Curvature and Tension Control the Formation and Collapse of Caveolar Superstructures. Dev. Cell 2019, 48, 523–538.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthaeus, C.; Taraska, J.W. Energy and Dynamics of Caveolae Trafficking. Front. Cell Dev. Biol. 2021, 8, 614472. [Google Scholar] [CrossRef]
- Echarri, A.; Pavón, D.M.; Sánchez, S.; García-García, M.; Calvo, E.; Huerta-López, C.; Velázquez-Carreras, D.; Viaris de Lesegno, C.; Ariotti, N.; Lázaro-Carrillo, A.; et al. An Abl-FBP17 mechanosensing system couples local plasma membrane curvature and stress fiber remodeling during mechanoadaptation. Nat. Commun. 2019, 10, 5828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilch, P.F.; Liu, L. Fat caves: Caveolae, lipid trafficking and lipid metabolism in adipocytes. Trends Endocrinol. Metab. 2011, 22, 318–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilch, P.F.; Meshulam, T.; Ding, S.; Liu, L. Caveolae and lipid trafficking in adipocytes. Clin. Lipidol. 2011, 6, 49–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, P.G.; Pavlides, S.; Lisanti, M.P. Caveolae and transcytosis in endothelial cells: Role in atherosclerosis. Cell Tissue Res. 2009, 335, 41–47. [Google Scholar] [CrossRef]
- Cheng, J.P.X.; Nichols, B.J. Caveolae: One Function or Many? Trends Cell Biol. 2016, 26, 177–189. [Google Scholar] [CrossRef]
- Okamoto, T.; Schlegel, A.; Scherer, P.E.; Lisanti, M.P. Caveolins, a family of scaffolding proteins for organizing “preassembled signaling complexes” at the plasma membrane. J. Biol. Chem. 1998, 273, 5419–5422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, N.L.; Park, H.; Yi, H.; Boo, Y.C.; Sorescu, G.P.; Sykes, M.; Jo, H. Chronic shear induces caveolae formation and alters ERK and Akt responses in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1113–H1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, X.; Matthaeus, C.; Kaßmann, M.; Daumke, O.; Gollasch, M. Pathophysiological Role of Caveolae in Hypertension. Front. Med. 2019, 6, 153. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaul, P.W. Endothelial nitric oxide synthase, caveolae and the development of atherosclerosis. J. Physiol. 2003, 547, 21–33. [Google Scholar] [CrossRef]
- Tran, J.; Magenau, A.; Rodriguez, M.; Rentero, C.; Royo, T.; Enrich, C.; Thomas, S.R.; Grewal, T.; Gaus, K. Activation of Endothelial Nitric Oxide (eNOS) Occurs through Different Membrane Domains in Endothelial Cells. PLoS ONE 2016, 11, e0151556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, X.-L.; Yang, X.-Y.; Li, J.-Y.; Ye, L.; Jia, X.; Xiong, Z.-F.; Wang, Y.-M.; Jin, S. Cavin-1 regulates caveolae-mediated LDL transcytosis: Crosstalk in an AMPK/eNOS/ NF-κB/Sp1 loop. Oncotarget 2017, 8, 103985–103995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briand, N.; Dugail, I.; Le Lay, S. Cavin proteins: New players in the caveolae field. Biochimie 2011, 93, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Deng, X.; Li, J.; Wang, M.; Li, Q.; An, W.; Deli, A.; Cong, Y.S. Regulation of cellular senescence by the essential caveolar component PTRF/Cavin-1. Cell Res. 2011, 21, 1088–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayer, A.; Stoeber, M.; Bissig, C.; Helenius, A. Biogenesis of caveolae: Stepwise assembly of large caveolin and cavin complexes. Traffic 2010, 11, 361–382. [Google Scholar] [CrossRef]
- Krishna, A.; Sengupta, D. Interplay between Membrane Curvature and Cholesterol: Role of Palmitoylated Caveolin-1. Biophys. J. 2019, 116, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Aravamudan, B.; VanOosten, S.K.; Meuchel, L.W.; Vohra, P.; Thompson, M.; Sieck, G.C.; Prakash, Y.S.; Pabelick, C.M. Caveolin-1 knockout mice exhibit airway hyperreactivity. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L669–L681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayer, A.; Stoeber, M.; Ritz, D.; Engel, S.; Meyer, H.H.; Helenius, A. Caveolin-1 is ubiquitinated and targeted to intralumenal vesicles in endolysosomes for degradation. J. Cell Biol. 2010, 191, 615–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.M.; Bastiani, M.; Luetterforst, R.; Kirkham, M.; Kirkham, A.; Nixon, S.J.; Walser, P.; Abankwa, D.; Oorschot, V.M.; Martin, S.; et al. PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell 2008, 132, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Brown, D.; McKee, M.; Lebrasseur, N.K.; Yang, D.; Albrecht, K.H.; Ravid, K.; Pilch, P.F. Deletion of Cavin/PTRF causes global loss of caveolae, dyslipidemia, and glucose intolerance. Cell Metab. 2008, 8, 310–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drab, M.; Verkade, P.; Elger, M.; Kasper, M.; Lohn, M.; Lauterbach, B.; Menne, J.; Lindschau, C.; Mende, F.; Luft, F.C.; et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 2001, 293, 2449–2452. [Google Scholar] [CrossRef] [Green Version]
- Vogel, E.R.; Manlove, L.J.; Kuipers, I.; Thompson, M.A.; Fang, Y.H.; Freeman, M.R.; Britt, R.D., Jr.; Faksh, A.; Yang, B.; Prakash, Y.S.; et al. Caveolin-1 scaffolding domain peptide prevents hyperoxia-induced airway remodeling in a neonatal mouse model. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 317, L99–L108. [Google Scholar] [CrossRef] [PubMed]
- Filippini, A.; D’Alessio, A. Caveolae and Lipid Rafts in Endothelium: Valuable Organelles for Multiple Functions. Biomolecules 2020, 10, 1218. [Google Scholar] [CrossRef]
- Razani, B.; Engelman, J.A.; Wang, X.B.; Schubert, W.; Zhang, X.L.; Marks, C.B.; Macaluso, F.; Russell, R.G.; Li, M.; Pestell, R.G. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J. Biol. Chem. 2001, 276, 38121–38138. [Google Scholar] [CrossRef] [PubMed]
- Bucci, M.; Gratton, J.P.; Rudic, R.D.; Acevedo, L.; Roviezzo, F.; Cirino, G.; Sessa, W.C. In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat. Med. 2000, 6, 1362–1367. [Google Scholar] [CrossRef] [PubMed]
- Goligorsky, M.S.; Li, H.; Brodsky, S.; Chen, J. Relationships between caveolae and eNOS: Everything in proximity and the proximity of everything. Am. J. Physiol. Ren. Physiol. 2002, 283, F1–F10. [Google Scholar] [CrossRef] [Green Version]
- Dudzinski, D.M.; Michel, T. Life history of eNOS: Partners and pathways. Cardiovasc. Res. 2007, 75, 247–260. [Google Scholar] [CrossRef]
- García-Cardeña, G.; Martasek, P.; Masters, B.S.; Skidd, P.M.; Couet, J.; Li, S.; Lisanti, M.P.; Sessa, W.C. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J. Biol. Chem. 1997, 272, 25437–25440. [Google Scholar] [CrossRef] [Green Version]
- Mineo, C.; Shaul, P.W. Regulation of eNOS in caveolae. Adv. Exp. Med. Biol. 2012, 729, 51–62. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, Q.; Wang, M.; Zhao, S.; Ma, J.; Luo, N.; Li, N.; Li, Y.; Xu, G.; Li, J. Eicosapentaenoic acid modifies lipid composition in caveolae and induces translocation of endothelial nitric oxide synthase. Biochimie 2007, 89, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Engel, D.; Beckers, L.; Wijnands, E.; Seijkens, T.; Lievens, D.; Drechsler, M.; Gerdes, N.; Soehnlein, O.; Daemen, M.J.A.P.; Stan, R.V.; et al. Caveolin-1 deficiency decreases atherosclerosis by hampering leukocyte influx into the arterial wall and generating a regulatory T-cell response. FASEB J. 2011, 25, 3838–3848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Feo, J.A.; Hellings, W.E.; Moll, F.L.; De Vries, J.-P.P.; van Middelaar, B.J.; Algra, A.; Sluijter, J.; Velema, E.; Van Der Broek, T.; Sessa, W.C. Caveolin-1 influences vascular protease activity and is a potential stabilizing factor in human atherosclerotic disease. PLoS ONE 2008, 3, e2612. [Google Scholar] [CrossRef] [PubMed]
- Zulli, A.; Buxton, B.F.; Black, M.J.; Ming, Z.; Cameron, A.; Hare, D.L. The immunoquantification of caveolin-1 and eNOS in human and rabbit diseased blood vessels. J. Histochem. Cytochem. 2006, 54, 151–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwencke, C.; Schmeisser, A.; Walter, C.; Wachter, R.; Pannach, S.; Weck, B.; Braun-Dullaeus, R.C.; Kasper, M.; Strasser, R.H. Decreased caveolin-1 in atheroma: Loss of antiproliferative control of vascular smooth muscle cells in atherosclerosis. Cardiovasc. Res. 2005, 68, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.W.; Lin, Y.C.; Chang, T.Y.; Tsai, S.H.; Ho, H.C.; Chen, Y.T.; Yang, V.C. Caveolin-1 expression is associated with plaque formation in hypercholesterolemic rabbits. J. Histochem. Cytochem. 2006, 54, 897–904. [Google Scholar] [CrossRef]
- Frank, P.G.; Lee, H.; Park, D.S.; Tandon, N.N.; Scherer, P.E.; Lisanti, M.P. Genetic ablation of caveolin-1 confers protection against atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 98–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Hernando, C.; Yu, J.; Suárez, Y.; Rahner, C.; Dávalos, A.; Lasunción, M.A.; Sessa, W.C. Genetic evidence supporting a critical role of endothelial caveolin-1 during the progression of atherosclerosis. Cell Metab. 2009, 10, 48–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Hernando, C.; Yu, J.; Dávalos, A.; Prendergast, J.; Sessa, W.C. Endothelial-specific overexpression of caveolin-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Am. J. Pathol. 2010, 177, 998–1003. [Google Scholar] [CrossRef]
- Sinha, B.; Köster, D.; Ruez, R.; Gonnord, P.; Bastiani, M.; Abankwa, D.; Stan, R.V.; Butler-Browne, G.; Vedie, B.; Johannes, L.; et al. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell 2011, 144, 402–413. [Google Scholar] [CrossRef] [Green Version]
- Keren, K. Cell motility: The integrating role of the plasma membrane. Eur. Biophys. J. 2011, 40, 1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michell, D.L.; Shihata, W.A.; Andrews, K.L.; Abidin, N.A.Z.; Jefferis, A.-M.; Sampson, A.K.; Lumsden, N.G.; Huet, O.; Parat, M.-O.; Jennings, G.L.; et al. High intraluminal pressure promotes vascular inflammation via caveolin-1. Sci. Rep. 2021, 11, 5894. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.P.; Mendoza-Topaz, C.; Howard, G.; Chadwick, J.; Shvets, E.; Cowburn, A.S.; Dunmore, B.J.; Crosby, A.; Morrell, N.W.; Nichols, B.J. Caveolae protect endothelial cells from membrane rupture during increased cardiac output. J. Cell Biol. 2015, 211, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Elliott, M.H.; Ashpole, N.E.; Gu, X.; Herrnberger, L.; McClellan, M.E.; Griffith, G.L.; Reagan, A.M.; Boyce, T.M.; Tanito, M.; Tamm, E.R.; et al. Caveolin-1 modulates intraocular pressure: Implications for caveolae mechanoprotection in glaucoma. Sci. Rep. 2016, 6, 37127. [Google Scholar] [CrossRef] [Green Version]
- Haidekker, M.A.; L’Heureux, N.; Frangos, J.A. Fluid shear stress increases membrane fluidity in endothelial cells: A study with DCVJ fluorescence. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1401–H1406. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Nogimori, Y.; Imamura, H.; Ando, J. Shear stress activates mitochondrial oxidative phosphorylation by reducing plasma membrane cholesterol in vascular endothelial cells. Proc. Natl. Acad. Sci. USA 2020, 117, 33660–33667. [Google Scholar] [CrossRef] [PubMed]
- Butler, P.J.; Norwich, G.; Weinbaum, S.; Chien, S. Shear stress induces a time—And position-dependent increase in endothelial cell membrane fluidity. Am. J. Physiol. Cell Physiol. 2001, 280, C962–C969. [Google Scholar] [CrossRef] [PubMed]
- Tabouillot, T.; Muddana, H.S.; Butler, P.J. Endothelial Cell Membrane Sensitivity to Shear Stress is Lipid Domain Dependent. Cell Mol. Bioeng. 2011, 4, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Ando, J. Endothelial cell and model membranes respond to shear stress by rapidly decreasing the order of their lipid phases. J. Cell Sci. 2013, 126, 1227–1234. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Ando, J. Emerging Role of Plasma Membranes in Vascular Endothelial Mechanosensing. Circ. J. 2018, 82, 2691–2698. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Ando, J. Vascular endothelial cell membranes differentiate between stretch and shear stress through transitions in their lipid phases. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1178–H1185. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Northup, N.; Marga, F.; Huber, T.; Byfield, F.J.; Levitan, I.; Forgacs, G. The effect of cellular cholesterol on membrane-cytoskeleton adhesion. J. Cell Sci. 2007, 120, 2223–2231. [Google Scholar] [CrossRef] [Green Version]
- Byfield, F.J.; Aranda-Espinoza, H.; Romanenko, V.G.; Rothblat, G.H.; Levitan, I. Cholesterol depletion increases membrane stiffness of aortic endothelial cells. Biophys. J. 2004, 87, 3336–3343. [Google Scholar] [CrossRef] [Green Version]
- Kowalsky, G.B.; Byfield, F.J.; Levitan, I. oxLDL facilitates flow-induced realignment of aortic endothelial cells. Am. J. Physiol. Cell Physiol. 2008, 295, C332–C340. [Google Scholar] [CrossRef] [Green Version]
- Khatibzadeh, N.; Gupta, S.; Farrell, B.; Brownell, W.E.; Anvari, B. Effects of cholesterol on nano-mechanical properties of the living cell plasma membrane. Soft Matter 2012, 8, 8350–8360. [Google Scholar] [CrossRef] [PubMed]
- Le Master, E.; Ahn, S.J.; Levitan, I. Mechanisms of endothelial stiffening in dyslipidemia and aging: Oxidized lipids and shear stress. Curr. Top. Membr. 2020, 86, 185–215. [Google Scholar] [CrossRef] [PubMed]
- Parat, M.-O.; Anand-Apte, B.; Fox, P.L. Differential caveolin-1 polarization in endothelial cells during migration in two and three dimensions. Mol. Biol. Cell 2003, 14, 3156–3168. [Google Scholar] [CrossRef] [PubMed]
- Czarny, M.; Liu, J.; Oh, P.; Schnitzer, J.E. Transient Mechanoactivation of Neutral Sphingomyelinase in Caveolae to Generate Ceramide. J. Biol. Chem. 2003, 278, 4424–4430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, T.; Yamamoto, K.; Ikeda, K.; Arita, M. Functional lipidomics of vascular endothelial cells in response to laminar shear stress. FASEB J. 2021, 35, e21301. [Google Scholar] [CrossRef]
- Czarny, M.; Schnitzer, J.E. Neutral sphingomyelinase inhibitor scyphostatin prevents and ceramide mimics mechanotransduction in vascular endothelium. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1344–H1352. [Google Scholar] [CrossRef]
- Espinosa, G.; López-Montero, I.; Monroy, F.; Langevin, D. Shear rheology of lipid monolayers and insights on membrane fluidity. Proc. Natl. Acad. Sci. USA 2011, 108, 6008–6013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Montero, I.; Monroy, F.; Vélez, M.; Devaux, P.F. Ceramide: From lateral segregation to mechanical stress. Biochim. Biophys. Acta (BBA) Biomembr. 2010, 1798, 1348–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolesnick, R.; Hannun, Y.A. Ceramide and apoptosis. Trends Biochem. Sci. 1999, 24, 224–225. [Google Scholar] [CrossRef]
- Freed, J.K.; Durand, M.J.; Hoffmann, B.R.; Densmore, J.C.; Greene, A.S.; Gutterman, D.D. Mitochondria-regulated formation of endothelium-derived extracellular vesicles shifts the mediator of flow-induced vasodilation. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H1096–H1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Huang, S.; Duan, W.; Liu, Q.; Lei, M. Inhibition of acid sphingomyelinase activity ameliorates endothelial dysfunction in db/db mice. Biosci. Rep. 2019, 39, 4. [Google Scholar] [CrossRef] [Green Version]
- Kornhuber, J.; Tripal, P.; Reichel, M.; Mühle, C.; Rhein, C.; Muehlbacher, M.; Groemer, T.W.; Gulbins, E. Functional Inhibitors of Acid Sphingomyelinase (FIASMAs): A Novel Pharmacological Group of Drugs with Broad Clinical Applications. Cell. Physiol. Biochem. 2010, 26, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.-Y.; Hupe, D.C.; Otto, G.P.; Sprenger, M.; Bunck, A.C.; Dorer, M.J.; Bockmeyer, C.L.; Deigner, H.-P.; Gräler, M.H.; Claus, R.A. Acid Sphingomyelinase Promotes Endothelial Stress Response in Systemic Inflammation and Sepsis. Mol. Med. 2016, 22, 412–423. [Google Scholar] [CrossRef]
- Beckmann, N.; Becker, K.A. Ceramide and Related Molecules in Viral Infections. Int. J. Mol. Sci. 2021, 22, 5676. [Google Scholar] [CrossRef] [PubMed]
- DeVerse, J.S.; Sandhu, A.S.; Mendoza, N.; Edwards, C.M.; Sun, C.; Simon, S.I.; Passerini, A.G. Shear stress modulates VCAM-1 expression in response to TNF-α and dietary lipids via interferon regulatory factor-1 in cultured endothelium. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1149–H1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, X.; Tian, J.; Zhang, P.; Fan, Y.; Chen, L.; Guan, Y.; Fu, Y.; Zhu, Y.; Chien, S.; Wang, N. Laminar shear stress up-regulates the expression of stearoyl-CoA desaturase-1 in vascular endothelial cells. Cardiovasc. Res. 2007, 74, 506–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byfield, F.J.; Tikku, S.; Rothblat, G.H.; Gooch, K.J.; Levitan, I. OxLDL increases endothelial stiffness, force generation, and network formation. J. Lipid Res. 2006, 47, 715–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayee, M.A.; Levitan, I. Paradoxical impact of cholesterol on lipid packing and cell stiffness. Front. Biosci. Landmark 2016, 21, 1245–1259. [Google Scholar] [CrossRef] [Green Version]
- Shentu, T.P.; Titushkin, I.; Singh, D.K.; Gooch, K.J.; Subbaiah, P.V.; Cho, M.; Levitan, I. oxLDL-induced decrease in lipid order of membrane domains is inversely correlated with endothelial stiffness and network formation. Am. J. Physiol. Cell. Physiol. 2010, 299, C218–C229. [Google Scholar] [CrossRef] [Green Version]
- Levitan, I.; Shentu, T.-P. Impact of oxLDL on Cholesterol-Rich Membrane Rafts. J. Lipids 2011, 2011, 730209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, A.; Shaul, P.W.; Yuhanna, I.S.; Conrad, P.A.; Smart, E.J. Oxidized Low Density Lipoprotein Displaces Endothelial Nitric-oxide Synthase (eNOS) from Plasmalemmal Caveolae and Impairs eNOS Activation. J. Biol. Chem. 1999, 274, 32512–32519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S. CD36 as a therapeutic target for endothelial dysfunction in stroke. Curr. Pharm. Des. 2012, 18, 3721–3730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uittenbogaard, A.; Shaul, P.W.; Yuhanna, I.S.; Blair, A.; Smart, E.J. High density lipoprotein prevents oxidized low density lipoprotein-induced inhibition of endothelial nitric-oxide synthase localization and activation in caveolae. J. Biol. Chem. 2000, 275, 11278–11283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kincer, J.F.; Uittenbogaard, A.; Dressman, J.; Guerin, T.M.; Febbraio, M.; Guo, L.; Smart, E.J. Hypercholesterolemia promotes a CD36-dependent and endothelial nitric-oxide synthase-mediated vascular dysfunction. J. Biol. Chem. 2002, 277, 23525–23533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essler, M.; Retzer, M.; Bauer, M.; Heemskerk, J.W.; Aepfelbacher, M.; Siess, W. Mildly oxidized low density lipoprotein induces contraction of human endothelial cells through activation of Rho/Rho kinase and inhibition of myosin light chain phosphatase. J. Biol. Chem. 1999, 274, 30361–30364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couto, N.F.; Rezende, L.; Fernandes-Braga, W.; Alves, A.P.; Agero, U.; Alvarez-Leite, J.; Damasceno, N.R.T.; Castro-Gomes, T.; Andrade, L.O. OxLDL alterations in endothelial cell membrane dynamics leads to changes in vesicle trafficking and increases cell susceptibility to injury. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183139. [Google Scholar] [CrossRef] [PubMed]
- Augé, N.; Andrieu, N.; Nègre-Salvayre, A.; Thiers, J.-C.; Levade, T.; Salvayre, R. The Sphingomyelin-Ceramide Signaling Pathway Is Involved in Oxidized Low Density Lipoprotein-induced Cell Proliferation. J. Biol. Chem. 1996, 271, 19251–19255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escargueil-Blanc, I.; Andrieu-Abadie, N.; Caspar-Bauguil, S.; Brossmer, R.; Levade, T.; Nègre-Salvayre, A.; Salvayre, R. Apoptosis and Activation of the Sphingomyelin-Ceramide Pathway Induced by Oxidized Low Density Lipoproteins Are Not Causally Related in ECV-304 Endothelial Cells. J. Biol. Chem. 1998, 273, 27389–27395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada-Shiba, M.; Kinoshita, M.; Kamido, H.; Shimokado, K. Oxidized Low Density Lipoprotein Induces Apoptosis in Cultured Human Umbilical Vein Endothelial Cells by Common and Unique Mechanisms. J. Biol. Chem. 1998, 273, 9681–9687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peretó, J.; López-García, P.; Moreira, D. Ancestral lipid biosynthesis and early membrane evolution. Trends Biochem. Sci. 2004, 29, 469–477. [Google Scholar] [CrossRef]
- Lombard, J.; López-García, P.; Moreira, D. The early evolution of lipid membranes and the three domains of life. Nat. Rev. Microbiol. 2012, 10, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Corradi, V.; Sejdiu, B.I.; Mesa-Galloso, H.; Abdizadeh, H.; Noskov, S.Y.; Marrink, S.J.; Tieleman, D.P. Emerging Diversity in Lipid-Protein Interactions. Chem. Rev. 2019, 119, 5775–5848. [Google Scholar] [CrossRef] [Green Version]
- Axelrod, D. Lateral motion of membrane proteins and biological function. J. Membr. Biol. 1983, 75, 1–10. [Google Scholar] [CrossRef]
- Butler, P.J.; Tsou, T.-C.; Li, J.Y.-S.; Usami, S.; Chien, S. Rate sensitivity of shear-induced changes in the lateral diffusion of endothelial cell membrane lipids: A role for membrane perturbation in shear-induced MAPK activation. FASEB J. 2002, 16, 1–18. [Google Scholar] [CrossRef]
- Grouleff, J.; Irudayam, S.J.; Skeby, K.K.; Schiøtt, B. The influence of cholesterol on membrane protein structure, function, and dynamics studied by molecular dynamics simulations. Biochim. Biophys. Acta 2015, 1848, 1783–1795. [Google Scholar] [CrossRef] [Green Version]
- Sprong, H.; van der Sluijs, P.; van Meer, G. How proteins move lipids and lipids move proteins. Nat. Rev. Mol. Cell Biol. 2001, 2, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R.; Ursell, T.; Wiggins, P.; Sens, P. Emerging roles for lipids in shaping membrane-protein function. Nature 2009, 459, 379–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marguet, D.; Lenne, P.F.; Rigneault, H.; He, H.T. Dynamics in the plasma membrane: How to combine fluidity and order. EMBO J. 2006, 25, 3446–3457. [Google Scholar] [CrossRef] [Green Version]
- Zabroski, I.O.; Nugent, M.A. Lipid Raft Association Stabilizes VEGF Receptor 2 in Endothelial Cells. Int. J. Mol. Sci. 2021, 22, 798. [Google Scholar] [CrossRef]
- Zhu, L.; Gu, Q.; Fang, L. Cholesterol-mediated regulation of angiogenesis: An emerging paradigm. Cardiol. Plus 2019, 4, 1. [Google Scholar] [CrossRef]
- Vion, A.-C.; Perovic, T.; Petit, C.; Hollfinger, I.; Bartels-Klein, E.; Frampton, E.; Gordon, E.; Claesson-Welsh, L.; Gerhardt, H. Endothelial Cell Orientation and Polarity Are Controlled by Shear Stress and VEGF Through Distinct Signaling Pathways. Front. Physiol. 2021, 11, 1743. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.-G.; Ueba, H.; Tanimoto, T.; Lungu, A.O.; Frame, M.D.; Berk, B.C. Ligand-Independent Activation of Vascular Endothelial Growth Factor Receptor 2 by Fluid Shear Stress Regulates Activation of Endothelial Nitric Oxide Synthase. Circ. Res. 2003, 93, 354–363. [Google Scholar] [CrossRef] [Green Version]
- dela Paz, N.G.; Melchior, B.; Frangos, J.A. Early VEGFR2 activation in response to flow is VEGF-dependent and mediated by MMP activity. Biochem. Biophys. Res. Commun. 2013, 434, 641–646. [Google Scholar] [CrossRef] [Green Version]
- Tzima, E.; Irani-Tehrani, M.; Kiosses, W.B.; Dejana, E.; Schultz, D.A.; Engelhardt, B.; Cao, G.; DeLisser, H.; Schwartz, M.A. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 2005, 437, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, W.; Weth, A.; Gutberlet, J.; Harms, G.; Groschner, K. Localization of VE-cadherin in plasmalemmal cholesterol rich microdomains and the effects of cholesterol depletion on VE-cadherin mediated cell-cell adhesion. Biochim. Biophys. Acta 2014, 1841, 1725–1732. [Google Scholar] [CrossRef] [PubMed]
- Mani, A.M.; Chattopadhyay, R.; Singh, N.K.; Rao, G.N. Cholesterol crystals increase vascular permeability by inactivating SHP2 and disrupting adherens junctions. Free Radic. Biol. Med. 2018, 123, 72–84. [Google Scholar] [CrossRef]
- Baumer, Y.; McCurdy, S.; Weatherby, T.M.; Mehta, N.N.; Halbherr, S.; Halbherr, P.; Yamazaki, N.; Boisvert, W.A. Hyperlipidemia-induced cholesterol crystal production by endothelial cells promotes atherogenesis. Nat. Commun. 2017, 8, 1129. [Google Scholar] [CrossRef] [PubMed]
- Villar, V.A.M.; Cuevas, S.; Zheng, X.; Jose, P.A. Chapter 1—Localization and signaling of GPCRs in lipid rafts. In Methods in Cell Biology; Shukla, A.K., Ed.; Academic Press: Cambridge, MA, USA, 2016; Volume 132, pp. 3–23. [Google Scholar]
- Barnett-Norris, J.; Lynch, D.; Reggio, P.H. Lipids, lipid rafts and caveolae: Their importance for GPCR signaling and their centrality to the endocannabinoid system. Life Sci. 2005, 77, 1625–1639. [Google Scholar] [CrossRef]
- Tilghman, R.W.; Hoover, R.L. E-selectin and ICAM-1 are incorporated into detergent-insoluble membrane domains following clustering in endothelial cells. FEBS Lett. 2002, 525, 83–87. [Google Scholar] [CrossRef] [Green Version]
- Marin, E.P.; Derakhshan, B.; Lam, T.T.; Davalos, A.; Sessa, W.C. Endothelial cell palmitoylproteomic identifies novel lipid-modified targets and potential substrates for protein acyl transferases. Circ. Res. 2012, 110, 1336–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.J.; Fan, Y.; Boehning, D. Regulation of Dynamic Protein S-Acylation. Front. Mol. Biosci. 2021, 8, 656440. [Google Scholar] [CrossRef] [PubMed]
- Main, A.; Fuller, W. Protein S-Palmitoylation: Advances and challenges in studying a therapeutically important lipid modification. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Yeh, D.C.; Duncan, J.A.; Yamashita, S.; Michel, T. Depalmitoylation of endothelial nitric-oxide synthase by acyl-protein thioesterase 1 is potentiated by Ca(2+)-calmodulin. J. Biol. Chem. 1999, 274, 33148–33154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahinian, S.; Silvius, J.R. Doubly-lipid-modified protein sequence motifs exhibit long-lived anchorage to lipid bilayer membranes. Biochemistry 1995, 34, 3813–3822. [Google Scholar] [CrossRef]
- Greaves, J.; Prescott, G.R.; Gorleku, O.A.; Chamberlain, L.H. The fat controller: Roles of palmitoylation in intracellular protein trafficking and targeting to membrane microdomains (Review). Mol. Membr. Biol. 2009, 26, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; García-Cardeña, G.; Sessa, W.C. Biosynthesis and palmitoylation of endothelial nitric oxide synthase: Mutagenesis of palmitoylation sites, cysteines-15 and/or -26, argues against depalmitoylation-induced translocation of the enzyme. Biochemistry 1995, 34, 12333–12340. [Google Scholar] [CrossRef] [PubMed]
- García-Cardeña, G.; Oh, P.; Liu, J.; Schnitzer, J.E.; Sessa, W.C. Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: Implications for nitric oxide signaling. Proc. Natl. Acad. Sci. USA 1996, 93, 6448–6453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafikov, R.; Fonseca, F.V.; Kumar, S.; Pardo, D.; Darragh, C.; Elms, S.; Fulton, D.; Black, S.M. eNOS activation and NO function: Structural motifs responsible for the posttranslational control of endothelial nitric oxide synthase activity. J. Endocrinol. 2011, 210, 271–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaskovic, S.; Blanc, M.; van der Goot, F.G. What does S-palmitoylation do to membrane proteins? FEBS J. 2013, 280, 2766–2774. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; García-Cardeña, G.; Sessa, W.C. Palmitoylation of endothelial nitric oxide synthase is necessary for optimal stimulated release of nitric oxide: Implications for caveolae localization. Biochemistry 1996, 35, 13277–13281. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, M.; Barrett, T.J.; Fisher, E.A. HDL and Reverse Cholesterol Transport. Circ. Res. 2019, 124, 1505–1518. [Google Scholar] [CrossRef]
- Kotlyarov, S.; Kotlyarova, A. The Role of ABC Transporters in Lipid Metabolism and the Comorbid Course of Chronic Obstructive Pulmonary Disease and Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 6711. [Google Scholar] [CrossRef] [PubMed]
- Terasaka, N.; Yu, S.; Yvan-Charvet, L.; Wang, N.; Mzhavia, N.; Langlois, R.; Pagler, T.; Li, R.; Welch, C.L.; Goldberg, I.J.; et al. ABCG1 and HDL protect against endothelial dysfunction in mice fed a high-cholesterol diet. J. Clin. Investig. 2008, 118, 3701–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erbilgin, A.; Siemers, N.; Kayne, P.; Yang, W.-P.; Berliner, J.; Lusis, A.J. Gene expression analyses of mouse aortic endothelium in response to atherogenic stimuli. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2509–2517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaisman, B.L.; Demosky, S.J.; Stonik, J.A.; Ghias, M.; Knapper, C.L.; Sampson, M.L.; Dai, C.; Levine, S.J.; Remaley, A.T. Endothelial expression of human ABCA1 in mice increases plasma HDL cholesterol and reduces diet-induced atherosclerosis. J. Lipid Res. 2012, 53, 158–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münch, G.; Bültmann, A.; Li, Z.; Holthoff, H.P.; Ullrich, J.; Wagner, S.; Ungerer, M. Overexpression of ABCG1 protein attenuates arteriosclerosis and endothelial dysfunction in atherosclerotic rabbits. Heart Int. 2012, 7, e12. [Google Scholar] [CrossRef]
- Qin, L.; Zhu, N.; Ao, B.-X.; Liu, C.; Shi, Y.-N.; Du, K.; Chen, J.-X.; Zheng, X.-L.; Liao, D.-F. Caveolae and Caveolin-1 Integrate Reverse Cholesterol Transport and Inflammation in Atherosclerosis. Int. J. Mol. Sci. 2016, 17, 429. [Google Scholar] [CrossRef] [Green Version]
- Chao, W.T.; Tsai, S.H.; Lin, Y.C.; Lin, W.W.; Yang, V.C. Cellular localization and interaction of ABCA1 and caveolin-1 in aortic endothelial cells after HDL incubation. Biochem. Biophys. Res. Commun. 2005, 332, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-C.; Ma, C.; Hsu, W.-C.; Lo, H.-F.; Yang, V.C. Molecular interaction between caveolin-1 and ABCA1 on high-density lipoprotein-mediated cholesterol efflux in aortic endothelial cells. Cardiovasc. Res. 2007, 75, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Orsó, E.; Broccardo, C.; Kaminski, W.E.; Böttcher, A.; Liebisch, G.; Drobnik, W.; Götz, A.; Chambenoit, O.; Diederich, W.; Langmann, T.; et al. Transport of lipids from golgi to plasma membrane is defective in tangier disease patients and Abc1-deficient mice. Nat. Genet. 2000, 24, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Hong, C.; Rong, X.; Zhu, X.; Tarling, E.J.; Hedde, P.N.; Gratton, E.; Parks, J.; Tontonoz, P. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. eLife 2015, 4, e08009. [Google Scholar] [CrossRef]
- Chawla, A.; Boisvert, W.A.; Lee, C.-H.; Laffitte, B.A.; Barak, Y.; Joseph, S.B.; Liao, D.; Nagy, L.; Edwards, P.A.; Curtiss, L.K.; et al. A PPARγ-LXR-ABCA1 Pathway in Macrophages Is Involved in Cholesterol Efflux and Atherogenesis. Mol. Cell 2001, 7, 161–171. [Google Scholar] [CrossRef]
- Wang, N.; Ranalletta, M.; Matsuura, F.; Peng, F.; Tall, A.R. LXR-Induced Redistribution of ABCG1 to Plasma Membrane in Macrophages Enhances Cholesterol Mass Efflux to HDL. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1310–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, H.; Langmann, T.; Schmitz, G.; Zhu, Y. Native LDL upregulation of ATP-binding cassette transporter-1 in human vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 127–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Liao, H.; Xie, X.; Yuan, Y.; Lee, T.S.; Wang, N.; Wang, X.; Shyy, J.Y.; Stemerman, M.B. Oxidized LDL downregulates ATP-binding cassette transporter-1 in human vascular endothelial cells via inhibiting liver X receptor (LXR). Cardiovasc. Res. 2005, 68, 425–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-L.; Chen, C.-T.; Tung, C.-S.; Tsai, M.-C. Laminar shear stress upregulates the expression of PPARs in vascular endothelial cells under high free fatty acid-induced stress. Exp. Ther. Med. 2021, 21, 438. [Google Scholar] [CrossRef]
- Xu, P.; Zhai, Y.; Wang, J. The Role of PPAR and Its Cross-Talk with CAR and LXR in Obesity and Atherosclerosis. Int. J. Mol. Sci. 2018, 19, 1260. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Fu, Y.; Hou, Y.; Wang, N.; Guan, Y.; Tang, C.; Shyy, J.Y.; Zhu, Y. Laminar shear stress regulates liver X receptor in vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 527–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westerterp, M.; Tsuchiya, K.; Tattersall, I.W.; Fotakis, P.; Bochem, A.E.; Molusky, M.M.; Ntonga, V.; Abramowicz, S.; Parks, J.S.; Welch, C.L.; et al. Deficiency of ATP-Binding Cassette Transporters A1 and G1 in Endothelial Cells Accelerates Atherosclerosis in Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1328–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatikos, A.; Dronadula, N.; Ng, P.; Palmer, D.; Knight, E.; Wacker, B.K.; Tang, C.; Kim, F.; Dichek, D.A. ABCA1 Overexpression in Endothelial Cells In Vitro Enhances ApoAI-Mediated Cholesterol Efflux and Decreases Inflammation. Hum. Gene Ther. 2019, 30, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Young, S.G.; Parthasarathy, S. Why are low-density lipoproteins atherogenic? West J. Med. 1994, 160, 153–164. [Google Scholar]
- Jialal, I.; Devaraj, S. The role of oxidized low density lipoprotein in atherogenesis. J. Nutr. 1996, 126, 1053s–1057s. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Nikiforov, N.G.; Markin, A.M.; Kashirskikh, D.A.; Myasoedova, V.A.; Gerasimova, E.V.; Orekhov, A.N. Overview of OxLDL and Its Impact on Cardiovascular Health: Focus on Atherosclerosis. Front. Pharmacol. 2021, 11, 2248. [Google Scholar] [CrossRef] [PubMed]
- Summerhill, V.I.; Grechko, A.V.; Yet, S.-F.; Sobenin, I.A.; Orekhov, A.N. The Atherogenic Role of Circulating Modified Lipids in Atherosclerosis. Int. J. Mol. Sci. 2019, 20, 3561. [Google Scholar] [CrossRef] [Green Version]
- Medina-Leyte, D.J.; Zepeda-García, O.; Domínguez-Pérez, M.; González-Garrido, A.; Villarreal-Molina, T.; Jacobo-Albavera, L. Endothelial Dysfunction, Inflammation and Coronary Artery Disease: Potential Biomarkers and Promising Therapeutical Approaches. Int. J. Mol. Sci. 2021, 22, 3850. [Google Scholar] [CrossRef] [PubMed]
- Mundi, S.; Massaro, M.; Scoditti, E.; Carluccio, M.A.; Van Hinsbergh, V.W.; Iruela-Arispe, M.L.; De Caterina, R. Endothelial permeability, LDL deposition, and cardiovascular risk factors—A review. Cardiovasc. Res. 2018, 114, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Lampugnani, M.G. Endothelial cell-to-cell junctions: Adhesion and signaling in physiology and pathology. Cold Spring Harb. Perspect. Med. 2012, 2, a006528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, K.Y.Y.; Fairn, G.D.; Lee, W.L. Transcellular vesicular transport in epithelial and endothelial cells: Challenges and opportunities. Traffic 2018, 19, 5–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Yang, X.; Xing, S.; Bian, F.; Yao, W.; Bai, X.; Zheng, T.; Wu, G.; Jin, S. Endogenous ceramide contributes to the transcytosis of oxLDL across endothelial cells and promotes its subendothelial retention in vascular wall. Oxid. Med. Cell. Longev. 2014, 2014, 823071. [Google Scholar] [CrossRef] [Green Version]
- Krauss, R.M.; Burke, D.J. Identification of multiple subclasses of plasma low density lipoproteins in normal humans. J. Lipid Res. 1982, 23, 97–104. [Google Scholar] [CrossRef]
- Simionescu, N.; Simionescu, M.; Palade, G.E. Open junctions in the endothelium of the postcapillary venules of the diaphragm. J. Cell Biol. 1978, 79, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Jang, E.; Robert, J.; Rohrer, L.; von Eckardstein, A.; Lee, W.L. Transendothelial transport of lipoproteins. Atherosclerosis 2020, 315, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Rippe, B.; Haraldsson, B. Transport of macromolecules across microvascular walls: The two-pore theory. Physiol. Rev. 1994, 74, 163–219. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.C.; Curry, F.E. Microvascular permeability. Physiol. Rev. 1999, 79, 703–761. [Google Scholar] [CrossRef]
- Pappenheimer, J.R.; Renkin, E.M.; Borrero, L.M. Filtration, diffusion and molecular sieving through peripheral capillary membranes; a contribution to the pore theory of capillary permeability. Am. J. Physiol. 1951, 167, 13–46. [Google Scholar] [CrossRef] [PubMed]
- Rippe, B.; Rosengren, B.I.; Carlsson, O.; Venturoli, D. Transendothelial transport: The vesicle controversy. J. Vasc. Res. 2002, 39, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, S.M.; Sugiyama, M.G.; Fung, K.Y.; Gao, Y.; Wang, C.; Levy, A.S.; Azizi, P.; Roufaiel, M.; Zhu, S.N.; Neculai, D.; et al. A novel assay uncovers an unexpected role for SR-BI in LDL transcytosis. Cardiovasc. Res. 2015, 108, 268–277. [Google Scholar] [CrossRef] [Green Version]
- Borén, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef] [PubMed]
- Frank, P.G.; Lisanti, M.P. Caveolin-1 and caveolae in atherosclerosis: Differential roles in fatty streak formation and neointimal hyperplasia. Curr. Opin. Lipidol. 2004, 15, 523–529. [Google Scholar] [CrossRef]
- Kraehling, J.R.; Chidlow, J.H.; Rajagopal, C.; Sugiyama, M.G.; Fowler, J.W.; Lee, M.Y.; Zhang, X.; Ramírez, C.M.; Park, E.J.; Tao, B.; et al. Genome-wide RNAi screen reveals ALK1 mediates LDL uptake and transcytosis in endothelial cells. Nat. Commun. 2016, 7, 13516. [Google Scholar] [CrossRef]
- Zhang, X.; Sessa, W.C.; Fernández-Hernando, C. Endothelial Transcytosis of Lipoproteins in Atherosclerosis. Front. Cardiovasc. Med. 2018, 5, 130. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Chambliss, K.L.; Gao, X.; Yuhanna, I.S.; Behling-Kelly, E.; Bergaya, S.; Ahmed, M.; Michaely, P.; Luby-Phelps, K.; Darehshouri, A.; et al. SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature 2019, 569, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Zhou, F.; Katirai, F.; Li, P.L. Lipid raft redox signaling: Molecular mechanisms in health and disease. Antioxid. Redox Signal. 2011, 15, 1043–1083. [Google Scholar] [CrossRef] [PubMed]
- Vasile, E.; Simionescu, M.; Simionescu, N. Visualization of the binding, endocytosis, and transcytosis of low-density lipoprotein in the arterial endothelium in situ. J. Cell Biol. 1983, 96, 1677–1689. [Google Scholar] [CrossRef]
- Sun, S.W.; Zu, X.Y.; Tuo, Q.H.; Chen, L.X.; Lei, X.Y.; Li, K.; Tang, C.K.; Liao, D.F. Caveolae and caveolin-1 mediate endocytosis and transcytosis of oxidized low density lipoprotein in endothelial cells. Acta Pharmacol. Sin. 2010, 31, 1336–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramírez, C.M.; Zhang, X.; Bandyopadhyay, C.; Rotllan, N.; Sugiyama, M.G.; Aryal, B.; Liu, X.; He, S.; Kraehling, J.R.; Ulrich, V.; et al. Caveolin-1 Regulates Atherogenesis by Attenuating Low-Density Lipoprotein Transcytosis and Vascular Inflammation Independently of Endothelial Nitric Oxide Synthase Activation. Circulation 2019, 140, 225–239. [Google Scholar] [CrossRef]
- Cogolludo, A.; Villamor, E.; Perez-Vizcaino, F.; Moreno, L. Ceramide and Regulation of Vascular Tone. Int. J. Mol. Sci. 2019, 20, 411. [Google Scholar] [CrossRef] [Green Version]
- Schweitzer, K.S.; Hatoum, H.; Brown, M.B.; Gupta, M.; Justice, M.J.; Beteck, B.; Van Demark, M.; Gu, Y.; Presson, R.G., Jr.; Hubbard, W.C.; et al. Mechanisms of lung endothelial barrier disruption induced by cigarette smoke: Role of oxidative stress and ceramides. Am. J Physiol. Lung Cell Mol. Physiol. 2011, 301, L836–L846. [Google Scholar] [CrossRef] [PubMed]
- Simmons, S.; Erfinanda, L.; Bartz, C.; Kuebler, W.M. Novel mechanisms regulating endothelial barrier function in the pulmonary microcirculation. J. Physiol. 2019, 597, 997–1021. [Google Scholar] [CrossRef]
- Göggel, R.; Winoto-Morbach, S.; Vielhaber, G.; Imai, Y.; Lindner, K.; Brade, L.; Brade, H.; Ehlers, S.; Slutsky, A.S.; Schütze, S. PAF-mediated pulmonary edema: A new role for acid sphingomyelinase and ceramide. Nat. Med. 2004, 10, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Petrache, I.; Medler, T.R.; Richter, A.T.; Kamocki, K.; Chukwueke, U.; Zhen, L.; Gu, Y.; Adamowicz, J.; Schweitzer, K.S.; Hubbard, W.C.; et al. Superoxide dismutase protects against apoptosis and alveolar enlargement induced by ceramide. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L44–L53. [Google Scholar] [CrossRef] [Green Version]
- Medler, T.R.; Petrusca, D.N.; Lee, P.J.; Hubbard, W.C.; Berdyshev, E.V.; Skirball, J.; Kamocki, K.; Schuchman, E.; Tuder, R.M.; Petrache, I. Apoptotic sphingolipid signaling by ceramides in lung endothelial cells. Am. J. Respir. Cell Mol. Biol. 2008, 38, 639–646. [Google Scholar] [CrossRef] [Green Version]
- Petrache, I.; Natarajan, V.; Zhen, L.; Medler, T.R.; Richter, A.T.; Cho, C.; Hubbard, W.C.; Berdyshev, E.V.; Tuder, R.M. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat. Med. 2005, 11, 491–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindner, K.; Uhlig, U.; Uhlig, S. Ceramide alters endothelial cell permeability by a nonapoptotic mechanism. Br. J. Pharmacol. 2005, 145, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Bowler, R.P.; Jacobson, S.; Cruickshank, C.; Hughes, G.J.; Siska, C.; Ory, D.S.; Petrache, I.; Schaffer, J.E.; Reisdorph, N.; Kechris, K. Plasma sphingolipids associated with chronic obstructive pulmonary disease phenotypes. Am. J. Respir. Crit. Care Med. 2015, 191, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Thomashow, M.A.; Shimbo, D.; Parikh, M.A.; Hoffman, E.A.; Vogel-Claussen, J.; Hueper, K.; Fu, J.; Liu, C.Y.; Bluemke, D.A.; Ventetuolo, C.E.; et al. Endothelial microparticles in mild chronic obstructive pulmonary disease and emphysema. The Multi-Ethnic Study of Atherosclerosis Chronic Obstructive Pulmonary Disease study. Am. J. Respir. Crit. Care Med. 2013, 188, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Serban, K.A.; Rezania, S.; Petrusca, D.N.; Poirier, C.; Cao, D.; Justice, M.J.; Patel, M.; Tsvetkova, I.; Kamocki, K.; Mikosz, A.; et al. Structural and functional characterization of endothelial microparticles released by cigarette smoke. Sci. Rep. 2016, 6, 31596. [Google Scholar] [CrossRef] [PubMed]
- Cavelier, C.; Rohrer, L.; von Eckardstein, A. ATP-Binding cassette transporter A1 modulates apolipoprotein A-I transcytosis through aortic endothelial cells. Circ. Res. 2006, 99, 1060–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrer, L.; Ohnsorg, P.M.; Lehner, M.; Landolt, F.; Rinninger, F.; von Eckardstein, A. High-density lipoprotein transport through aortic endothelial cells involves scavenger receptor BI and ATP-binding cassette transporter G1. Circ. Res. 2009, 104, 1142–1150. [Google Scholar] [CrossRef] [Green Version]
- VanWijk, M.J.; VanBavel, E.; Sturk, A.; Nieuwland, R. Microparticles in cardiovascular diseases. Cardiovasc. Res. 2003, 59, 277–287. [Google Scholar] [CrossRef]
- Boulanger, C.M.; Leroyer, A.S.; Amabile, N.; Tedgui, A. Circulating endothelial microparticles: A new marker of vascular injury. Ann. Cardiol. Angeiol. 2008, 57, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Shantsila, E. Endothelial microparticles: A universal marker of vascular health? J. Hum. Hypertens. 2009, 23, 359–361. [Google Scholar] [CrossRef]
- Chironi, G.N.; Boulanger, C.M.; Simon, A.; Dignat-George, F.; Freyssinet, J.M.; Tedgui, A. Endothelial microparticles in diseases. Cell Tissue Res. 2009, 335, 143–151. [Google Scholar] [CrossRef]
- Sabatier, F.; Camoin-Jau, L.; Anfosso, F.; Sampol, J.; Dignat-George, F. Circulating endothelial cells, microparticles and progenitors: Key players towards the definition of vascular competence. J. Cell. Mol. Med. 2009, 13, 454–471. [Google Scholar] [CrossRef]
- Wills, T.B.; Heaney, A.M.; Jane Wardrop, K.; Haldorson, G.J. Immunomagnetic isolation of canine circulating endothelial and endothelial progenitor cells. Vet. Clin. Pathol. 2009, 38, 437–442. [Google Scholar] [CrossRef]
- Berezin, A.; Zulli, A.; Kerrigan, S.; Petrovic, D.; Kruzliak, P. Predictive role of circulating endothelial-derived microparticles in cardiovascular diseases. Clin. Biochem. 2015, 48, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Paudel, K.R.; Panth, N.; Kim, D.-W. Circulating Endothelial Microparticles: A Key Hallmark of Atherosclerosis Progression. Scientifica 2016, 2016, 8514056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- França, C.N.; Izar, M.C.; Amaral, J.B.; Tegani, D.M.; Fonseca, F.A. Microparticles as potential biomarkers of cardiovascular disease. Arq. Bras. Cardiol. 2015, 104, 169–174. [Google Scholar] [CrossRef]
- Angelillo-Scherrer, A. Leukocyte-derived microparticles in vascular homeostasis. Circ. Res. 2012, 110, 356–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faure, V.; Dou, L.; Sabatier, F.; Cerini, C.; Sampol, J.; Berland, Y.; Brunet, P.; Dignat-George, F. Elevation of circulating endothelial microparticles in patients with chronic renal failure. J. Thromb. Haemost. 2006, 4, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, C.M.; Scoazec, A.; Ebrahimian, T.; Henry, P.; Mathieu, E.; Tedgui, A.; Mallat, Z. Circulating microparticles from patients with myocardial infarction cause endothelial dysfunction. Circulation 2001, 104, 2649–2652. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Hugel, B.; Ohan, J.; Lesèche, G.; Freyssinet, J.M.; Tedgui, A. Shed membrane microparticles with procoagulant potential in human atherosclerotic plaques: A role for apoptosis in plaque thrombogenicity. Circulation 1999, 99, 348–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berezin, A.E.; Kremzer, A.A.; Martovitskaya, Y.V.; Samura, T.A.; Berezina, T.A. The predictive role of circulating microparticles in patients with chronic heart failure. BBA Clin. 2015, 3, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Gu, S.; Mao, X.; Tan, Y.; Wang, H.; Li, S.; Zhou, Y. Endothelial Cell Morphology Regulates Inflammatory Cells Through MicroRNA Transferred by Extracellular Vesicles. Front. Bioeng. Biotechnol. 2020, 8, 369. [Google Scholar] [CrossRef] [PubMed]
- Markiewicz, M.; Richard, E.; Marks, N.; Ludwicka-Bradley, A. Impact of Endothelial Microparticles on Coagulation, Inflammation, and Angiogenesis in Age-Related Vascular Diseases. J. Aging Res. 2013, 2013, 734509. [Google Scholar] [CrossRef] [Green Version]
- Mause, S.F.; Weber, C. Microparticles: Protagonists of a novel communication network for intercellular information exchange. Circ. Res. 2010, 107, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Leroyer, A.S.; Ebrahimian, T.G.; Cochain, C.; Récalde, A.; Blanc-Brude, O.; Mees, B.; Vilar, J.; Tedgui, A.; Levy, B.I.; Chimini, G.; et al. Microparticles from ischemic muscle promotes postnatal vasculogenesis. Circulation 2009, 119, 2808–2817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmoud, A.M.; Wilkinson, F.L.; McCarthy, E.M.; Moreno-Martinez, D.; Langford-Smith, A.; Romero, M.; Duarte, J.; Alexander, M.Y. Endothelial microparticles prevent lipid-induced endothelial damage via Akt/eNOS signaling and reduced oxidative stress. FASEB J. 2017, 31, 4636–4648. [Google Scholar] [CrossRef] [Green Version]
- Jansen, F.; Yang, X.; Hoelscher, M.; Cattelan, A.; Schmitz, T.; Proebsting, S.; Wenzel, D.; Vosen, S.; Franklin, B.S.; Fleischmann, B.K.; et al. Endothelial microparticle-mediated transfer of MicroRNA-126 promotes vascular endothelial cell repair via SPRED1 and is abrogated in glucose-damaged endothelial microparticles. Circulation 2013, 128, 2026–2038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, F.; Yang, X.; Baumann, K.; Przybilla, D.; Schmitz, T.; Flender, A.; Paul, K.; Alhusseiny, A.; Nickenig, G.; Werner, N. Endothelial microparticles reduce ICAM-1 expression in a microRNA-222-dependent mechanism. J. Cell. Mol. Med. 2015, 19, 2202–2214. [Google Scholar] [CrossRef]
- Jansen, F.; Stumpf, T.; Proebsting, S.; Franklin, B.S.; Wenzel, D.; Pfeifer, P.; Flender, A.; Schmitz, T.; Yang, X.; Fleischmann, B.K.; et al. Intercellular transfer of miR-126-3p by endothelial microparticles reduces vascular smooth muscle cell proliferation and limits neointima formation by inhibiting LRP6. J. Mol. Cell. Cardiol. 2017, 104, 43–52. [Google Scholar] [CrossRef]
- Alexy, T.; Rooney, K.; Weber, M.; Gray, W.D.; Searles, C.D. TNF-α alters the release and transfer of microparticle-encapsulated miRNAs from endothelial cells. Physiol. Genom. 2014, 46, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Jansen, F.; Yang, X.; Hoyer, F.F.; Paul, K.; Heiermann, N.; Becher, M.U.; Abu Hussein, N.; Kebschull, M.; Bedorf, J.; Franklin, B.S.; et al. Endothelial microparticle uptake in target cells is annexin I/phosphatidylserine receptor dependent and prevents apoptosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1925–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vion, A.-C.; Ramkhelawon, B.; Loyer, X.; Chironi, G.; Devue, C.; Loirand, G.; Tedgui, A.; Lehoux, S.; Boulanger, C.M. Shear Stress Regulates Endothelial Microparticle Release. Circ. Res. 2013, 112, 1323–1333. [Google Scholar] [CrossRef]
- Hafiane, A.; Genest, J. ATP binding cassette A1 (ABCA1) mediates microparticle formation during high-density lipoprotein (HDL) biogenesis. Atherosclerosis 2017, 257, 90–99. [Google Scholar] [CrossRef]
- Chatterjee, S. Endothelial Mechanotransduction, Redox Signaling and the Regulation of Vascular Inflammatory Pathways. Front. Physiol. 2018, 9, 524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Fisher, A.B. Mechanotransduction in the endothelium: Role of membrane proteins and reactive oxygen species in sensing, transduction, and transmission of the signal with altered blood flow. Antioxid. Redox Signal. 2014, 20, 899–913. [Google Scholar] [CrossRef] [PubMed]
- Virchow, R. Cellular Pathology as Based Upon Physiological and Pathological Histology. Twenty Lectures Delivered in the Pathological Institute of Berlin During the Months of February, March, and April, 1858; John Churchill: London, UK, 1860. [Google Scholar]
- Ross, R. Rous-Whipple Award Lecture. Atherosclerosis: A defense mechanism gone awry. Am. J. Pathol. 1993, 143, 987–1002. [Google Scholar]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.R.; Kinnun, J.J.; Leng, X.; Williams, J.A.; Wassall, S.R. How polyunsaturated fatty acids modify molecular organization in membranes: Insight from NMR studies of model systems. Biochim. Biophys. Acta (BBA) Biomembr. 2015, 1848, 211–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, R.P.; Jacob, R.F.; Shrivastava, S.; Sherratt, S.C.R.; Chattopadhyay, A. Eicosapentaenoic acid reduces membrane fluidity, inhibits cholesterol domain formation, and normalizes bilayer width in atherosclerotic-like model membranes. Biochim. Biophys. Acta 2016, 1858, 3131–3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, M.; Hossain, S.; Yamasaki, H.; Yazawa, K.; Masumura, S. Effects of eicosapentaenoic acid and docosahexaenoic acid on plasma membrane fluidity of aortic endothelial cells. Lipids 1999, 34, 1297–1304. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotlyarov, S. Diversity of Lipid Function in Atherogenesis: A Focus on Endothelial Mechanobiology. Int. J. Mol. Sci. 2021, 22, 11545. https://doi.org/10.3390/ijms222111545
Kotlyarov S. Diversity of Lipid Function in Atherogenesis: A Focus on Endothelial Mechanobiology. International Journal of Molecular Sciences. 2021; 22(21):11545. https://doi.org/10.3390/ijms222111545
Chicago/Turabian StyleKotlyarov, Stanislav. 2021. "Diversity of Lipid Function in Atherogenesis: A Focus on Endothelial Mechanobiology" International Journal of Molecular Sciences 22, no. 21: 11545. https://doi.org/10.3390/ijms222111545
APA StyleKotlyarov, S. (2021). Diversity of Lipid Function in Atherogenesis: A Focus on Endothelial Mechanobiology. International Journal of Molecular Sciences, 22(21), 11545. https://doi.org/10.3390/ijms222111545