
Germline POT1 Deregulation Can Predispose to Myeloid Malignancies in Childhood

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Identification of a Novel Germline POT1 Stop-Gain Mutation (p.Q199*) in a Child with AML
2.2. POT1 p.Q199* Leads to a Loss of POT1 Expression from the Respective Allele
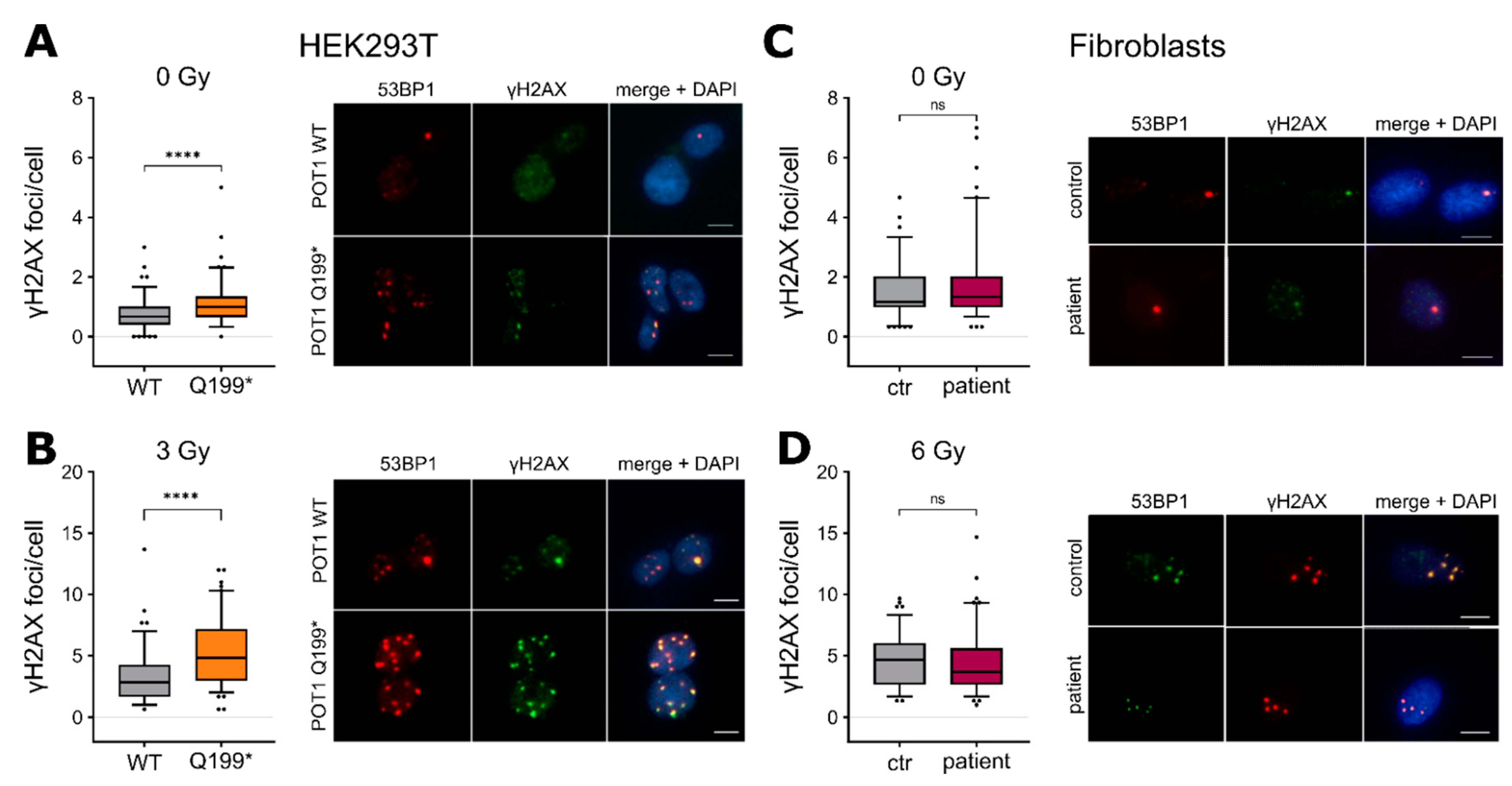
2.3. POT1 p.Q199* Overexpression Confers Increased DNA Damage Response Induction
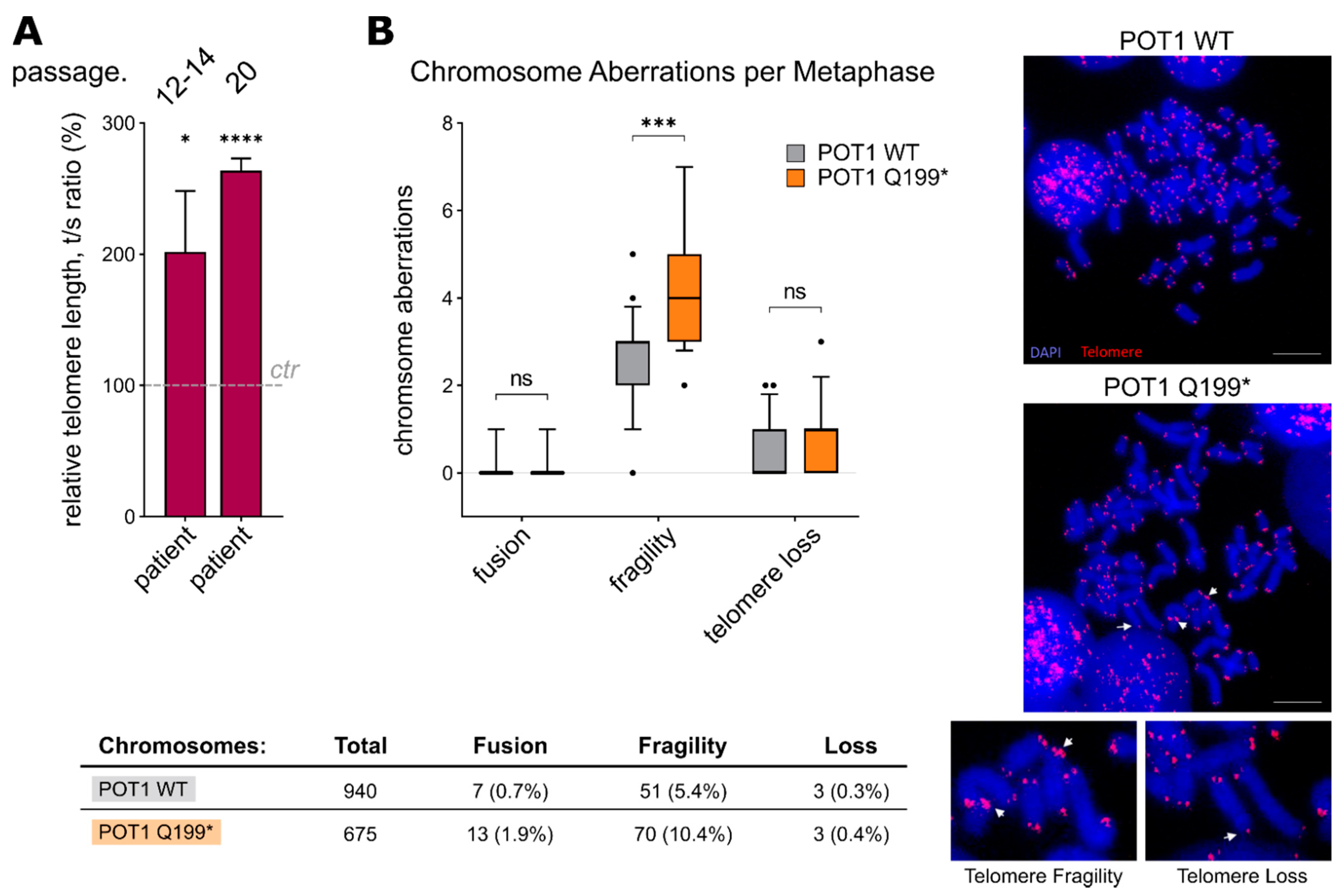
2.4. POT1 p.Q199* Leads to Telomere Elongation and Chromosomal Instability
2.5. Reduced POT1 Levels in Myeloid Cells Confer Telomere Elongation
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Whole Exome Sequencing (WES)
4.3. Sanger Sequencing Validation
4.4. POT1 Variation Analysis
4.5. Cloning
4.6. Cell Culture
4.7. HEK293T Cell Transfection
4.8. POT1 Knockdown in HL-60 Cells
4.9. Quantitative Real-Time (qRT)-PCR Analysis
4.10. Western Blot
4.11. QRT-PCR of the Telomere Length (TL)
4.12. Telomeric FISH on Metaphase Chromosomes
4.13. Immunofluorescence Staining
4.14. Single-Cell RNA Sequencing Analysis
4.15. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lim, C.J.; Cech, T.R. Shaping human telomeres: From shelterin and CST complexes to telomeric chromatin organization. Nat. Rev. Mol. Cell Biol. 2021, 22, 283–298. [Google Scholar] [CrossRef]
- de Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [Green Version]
- Barbaro, P.M.; Ziegler, D.S.; Reddel, R.R. The wide-ranging clinical implications of the short telomere syndromes. Intern. Med. J. 2016, 46, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P.; Cech, T.R. Pot1, the putative telomere end-binding protein in fission yeast and humans. Science 2001, 292, 1171–1175. [Google Scholar] [CrossRef] [Green Version]
- Loayza, D.; De Lange, T. POT1 as a terminal transducer of TRF1 telomere length control. Nature 2003, 423, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Podell, E.R.; Cech, T.R. Structure of human POT1 bound to telomeric single-stranded DNA provides a model for chromosome end-protection. Nat. Struct. Mol. Biol. 2004, 11, 1223–1229. [Google Scholar] [CrossRef]
- Houghtaling, B.R.; Cuttonaro, L.; Chang, W.; Smith, S. A dynamic molecular link between the telomere length regulator TRF1 and the chromosome end protector TRF2. Curr. Biol. 2004, 14, 1621–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Safari, A.; O’Connor, M.S.; Chan, D.W.; Laegeler, A.; Qin, J.; Songyang, Z. PTOP interacts with POT1 and regulates its localization to telomeres. Nat. Cell Biol. 2004, 6, 673–680. [Google Scholar] [CrossRef]
- Ye, J.Z.; Hockemeyer, D.; Krutchinsky, A.N.; Loayza, D.; Hooper, S.M.; Chait, B.T.; de Lange, T. POT1-interacting protein PIP1: A telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 2004, 18, 1649–1654. [Google Scholar] [CrossRef] [Green Version]
- Denchi, E.L.; de Lange, T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 2007, 448, 1068–1071. [Google Scholar] [CrossRef]
- Kratz, K.; de Lange, T. Protection of telomeres 1 proteins POT1a and POT1b can repress ATR signaling by RPA exclusion, but binding to CST limits ATR repression by POT1b. J. Biol. Chem. 2018, 293, 14384–14392. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Poulos, R.C.; Reddel, R.R. Role of POT1 in Human Cancer. Cancers 2020, 12, 2739. [Google Scholar] [CrossRef]
- Shen, E.; Xiu, J.; Lopez, G.Y.; Bentley, R.; Jalali, A.; Heimberger, A.B.; Bainbridge, M.N.; Bondy, M.L.; Walsh, K.M. POT1 mutation spectrum in tumour types commonly diagnosed among POT1-associated hereditary cancer syndrome families. J. Med. Genet. 2020, 57, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Henry, M.L.; Osborne, J.; Else, T. POT1 Tumor Predisposition. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; Seattle, WA, USA, 1993; Available online: https://europepmc.org/article/NBK/nbk563529 (accessed on 30 September 2021).
- Lim, T.L.; Lieberman, D.B.; Davis, A.R.; Loren, A.W.; Hausler, R.; Bigdeli, A.; Li, Y.; Powers, J.; Raper, A.; Regeneron Genetics, C.; et al. Germline POT1 variants can predispose to myeloid and lymphoid neoplasms. Leukemia 2021. [Google Scholar] [CrossRef]
- Wagener, R.; Taeubner, J.; Walter, C.; Yasin, L.; Alzoubi, D.; Bartenhagen, C.; Attarbaschi, A.; Classen, C.F.; Kontny, U.; Hauer, J.; et al. Comprehensive germline-genomic and clinical profiling in 160 unselected children and adolescents with cancer. Eur. J. Hum. Genet. 2021, 29, 1301–1311. [Google Scholar] [CrossRef]
- Chen, C.; Gu, P.L.; Wu, J.; Chen, X.Y.; Niu, S.S.; Sun, H.; Wu, L.J.; Li, N.; Peng, J.H.; Shi, S.H.; et al. Structural insights into POT1-TPP1 interaction and POT1 C-terminal mutations in human cancer. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aramburu, T.; Plucinsky, S.; Skordalakes, E. POT1-TPP1 telomere length regulation and disease. Comput. Struct. Biotechnol. J. 2020, 18, 1939–1946. [Google Scholar] [CrossRef]
- Hockemeyer, D.; Daniels, J.P.; Takai, H.; de Lange, T. Recent expansion of the telomeric complex in rodents: Two distinct POT1 proteins protect mouse telomeres. Cell 2006, 126, 63–77. [Google Scholar] [CrossRef] [Green Version]
- Pinzaru, A.M.; Hom, R.A.; Beal, A.; Phillips, A.F.; Ni, E.; Cardozo, T.; Nair, N.; Choi, J.; Wuttke, D.S.; Sfeir, A.; et al. Telomere Replication Stress Induced by POT1 Inactivation Accelerates Tumorigenesis. Cell Rep. 2016, 15, 2170–2184. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.T.; Hennick, K.; Johnson, J.; Finnerty, B.; Choo, S.; Short, S.B.; Drubin, C.; Forster, R.; McMaster, M.L.; Hockemeyer, D. Cancer-associated POT1 mutations lead to telomere elongation without induction of a DNA damage response. EMBO J. 2021, 40, e107346. [Google Scholar] [CrossRef] [PubMed]
- Glousker, G.; Briod, A.S.; Quadroni, M.; Lingner, J. Human shelterin protein POT1 prevents severe telomere instability induced by homology-directed DNA repair. EMBO J. 2020, 39, e104500. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Zaug, A.J.; Podell, E.R.; Cech, T.R. Switching human telomerase on and off with hPOT1 protein in vitro. J. Biol. Chem. 2005, 280, 20449–20456. [Google Scholar] [CrossRef] [Green Version]
- Telomeres Mendelian Randomization, C.; Haycock, P.C.; Burgess, S.; Nounu, A.; Zheng, J.; Okoli, G.N.; Bowden, J.; Wade, K.H.; Timpson, N.J.; Evans, D.M.; et al. Association Between Telomere Length and Risk of Cancer and Non-Neoplastic Diseases: A Mendelian Randomization Study. JAMA Oncol. 2017, 3, 636–651. [Google Scholar] [CrossRef] [Green Version]
- Schratz, K.E.; Haley, L.; Danoff, S.K.; Blackford, A.L.; DeZern, A.E.; Gocke, C.D.; Duffield, A.S.; Armanios, M. Cancer spectrum and outcomes in the Mendelian short telomere syndromes. Blood 2020, 135, 1946–1956. [Google Scholar] [CrossRef]
- Ramsay, A.J.; Quesada, V.; Foronda, M.; Conde, L.; Martinez-Trillos, A.; Villamor, N.; Rodriguez, D.; Kwarciak, A.; Garabaya, C.; Gallardo, M.; et al. POT1 mutations cause telomere dysfunction in chronic lymphocytic leukemia. Nat. Genet. 2013, 45, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Robles-Espinoza, C.D.; Harland, M.; Ramsay, A.J.; Aoude, L.G.; Quesada, V.; Ding, Z.; Pooley, K.A.; Pritchard, A.L.; Tiffen, J.C.; Petljak, M.; et al. POT1 loss-of-function variants predispose to familial melanoma. Nat. Genet. 2014, 46, 478–481. [Google Scholar] [CrossRef] [Green Version]
- Chakravarti, D.; LaBella, K.A.; DePinho, R.A. Telomeres: History, health, and hallmarks of aging. Cell 2021, 184, 306–322. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, B.S.; Quinlan, A.R. Who’s Who? Detecting and Resolving Sample Anomalies in Human DNA Sequencing Studies with Peddy. Am. J. Hum. Genet. 2017, 100, 406–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakken, S.; Saveliev, V.; Hofmann, O.; Møller, P.; Myklebost, O.; Hovig, E. Cancer Predisposition Sequencing Reporter (CPSR): A Flexible Variant Report Engine for Germline Screening in Cancer. Bioinformatics 2019. [Google Scholar] [CrossRef] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, S.B.; Ferchen, K.; Chetal, K.; Grimes, H.L.; Salomonis, N. The Human Cell Atlas bone marrow single-cell interactive web portal. Exp. Hematol. 2018, 68, 51–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michler, P.; Schedel, A.; Witschas, M.; Friedrich, U.A.; Wagener, R.; Mehtonen, J.; Brozou, T.; Menzel, M.; Walter, C.; Nabi, D.; et al. Germline POT1 Deregulation Can Predispose to Myeloid Malignancies in Childhood. Int. J. Mol. Sci. 2021, 22, 11572. https://doi.org/10.3390/ijms222111572
Michler P, Schedel A, Witschas M, Friedrich UA, Wagener R, Mehtonen J, Brozou T, Menzel M, Walter C, Nabi D, et al. Germline POT1 Deregulation Can Predispose to Myeloid Malignancies in Childhood. International Journal of Molecular Sciences. 2021; 22(21):11572. https://doi.org/10.3390/ijms222111572
Chicago/Turabian StyleMichler, Pia, Anne Schedel, Martha Witschas, Ulrike Anne Friedrich, Rabea Wagener, Juha Mehtonen, Triantafyllia Brozou, Maria Menzel, Carolin Walter, Dalileh Nabi, and et al. 2021. "Germline POT1 Deregulation Can Predispose to Myeloid Malignancies in Childhood" International Journal of Molecular Sciences 22, no. 21: 11572. https://doi.org/10.3390/ijms222111572
APA StyleMichler, P., Schedel, A., Witschas, M., Friedrich, U. A., Wagener, R., Mehtonen, J., Brozou, T., Menzel, M., Walter, C., Nabi, D., Pearce, G., Erlacher, M., Göhring, G., Dugas, M., Heinäniemi, M., Borkhardt, A., Stölzel, F., Hauer, J., & Auer, F. (2021). Germline POT1 Deregulation Can Predispose to Myeloid Malignancies in Childhood. International Journal of Molecular Sciences, 22(21), 11572. https://doi.org/10.3390/ijms222111572