
Clustered Regularly Interspaced Short Palindromic Repeat Analysis of Clonal Complex 17 Serotype III Group B Streptococcus Strains Causing Neonatal Invasive Diseases

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
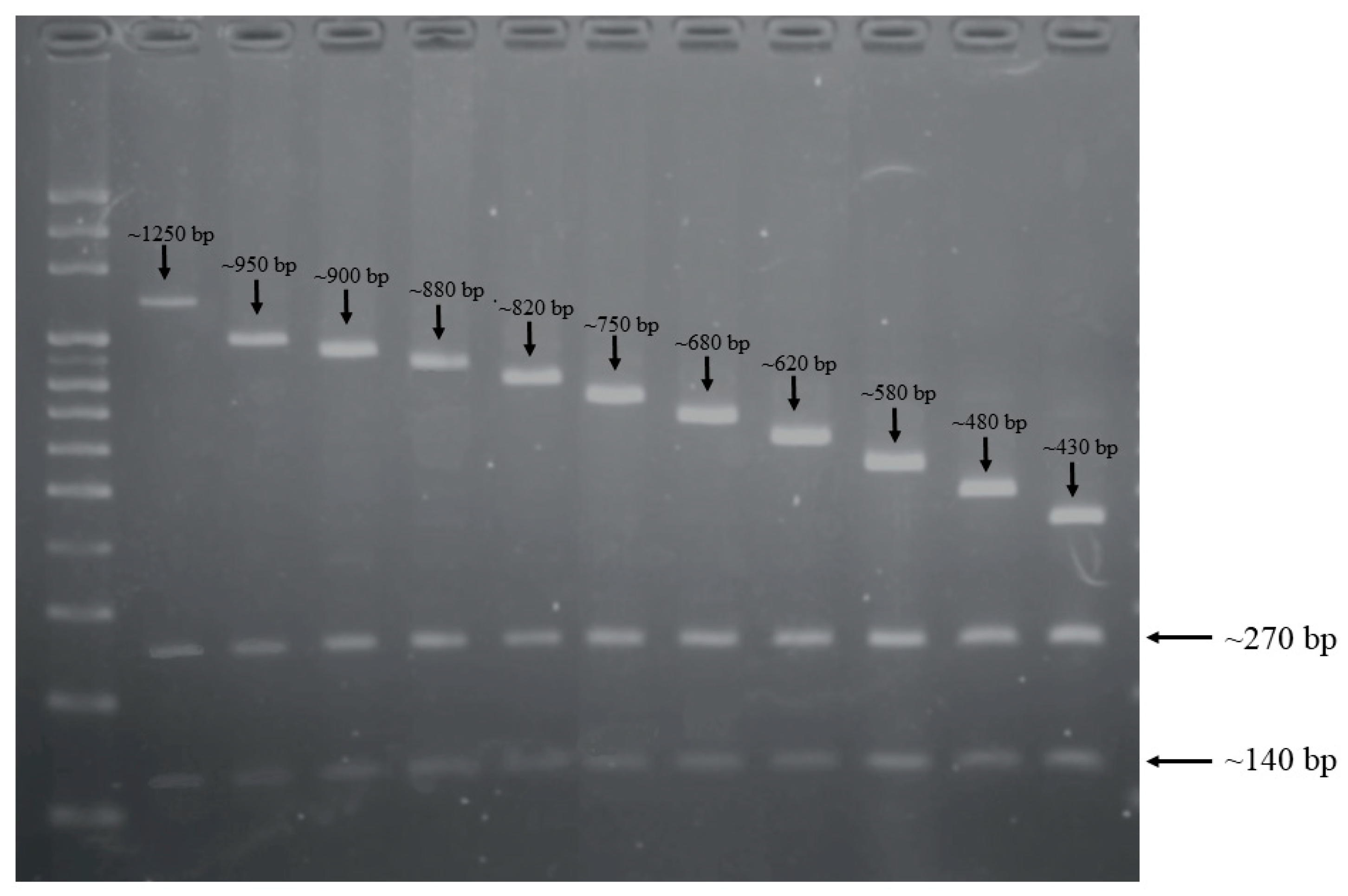
2.1. CRISPR-RFLP and CRISPR Direct Repeat Analysis
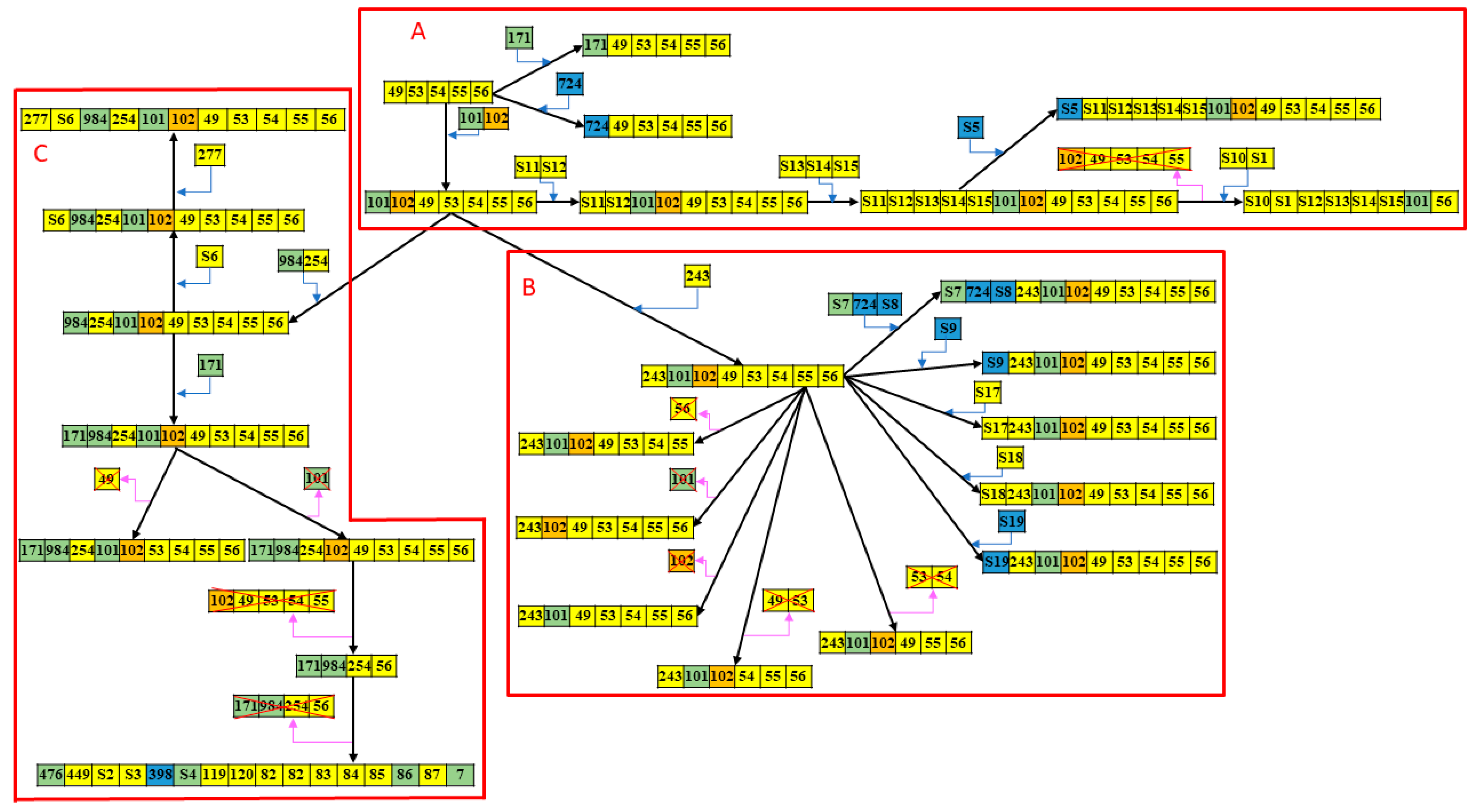
2.2. CRISPR1-Based Subgroups of CC17/III GBS Isolates and Spacer Analysis
2.3. WGS Analyses of CC17/III GBS Isolates with Different CRISPR-RFLP Patterns
3. Discussion
4. Materials and Methods
4.1. Bacterial Isolates
4.2. DNA Extraction
4.3. CRISPR1 Locus Amplification
4.4. Restriction Fragment Length Polymorphism (RFLP) Analysis
4.5. CRISPR1 Locus Sequencing and Analysis
4.6. MLST and Capsule Genotyping
4.7. Antimicrobial Susceptibility Testing
4.8. Whole-Genome Sequencing (WGS)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Joubrel, C.; Tazi, A.; Six, A.; Dmytruk, N.; Touak, G.; Bidet, P.; Raymond, J.; Trieu-Cuot, P.; Fouet, A.; Kernéis, S.; et al. Group B streptococcus neonatal invasive infections, France 2007–2012. Clin. Microbiol. Infect. 2015, 21, 910–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; Chu, S.M.; Wang, H.C.; Yang, P.H.; Huang, H.R.; Chiang, M.C.; Fu, R.H.; Tsai, M.H.; Hsu, J.F. Complicated Streptococcus agalactiae sepsis with/without meningitis in young infants and newborns: The clinical and molecular characteristics and outcomes. Microorganisms 2021, 9, 2094. [Google Scholar] [CrossRef]
- Kadambari, S.; Trotter, C.L.; Heath, P.T.; Goldacre, M.J.; Pollard, A.J.; Goldacre, R. Group B Streptococcal disease in England (1998-2017): A population based observational study. Clin. Infect Dis. 2021, 72, e791–e798. [Google Scholar] [CrossRef] [PubMed]
- Al Luhidan, L.; Madani, A.; Albanyan, E.A.; Saif, S.A.; Nasef, M.; AlJohani, S.; Madkhali, A.; al Shaalan, M.; Alalola, S. Neonatal group B Streptococcus infection in a tertiary care hospital in Saudi Arabia: A 13-year experience. Pediatr. Infect. Dis. J. 2019, 38, 731–734. [Google Scholar] [CrossRef]
- Medugu, N.; Iregbu, K.; Parker, R.; Plemmons, J.; Singh, P.; Audu, L.; Efetie, E.; Davies, H.; Manning, S. Group B streptococcal colonization and transmission dynamics in pregnant women and their newborns in Nigeria: Implications for prevention strategies. Clin. Microbiol. Infect. 2017, 23, 673.e9–673.e16. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.F.; Tsai, M.H.; Lin, L.C.; Chu, S.M.; Lai, M.Y.; Huang, H.R.; Chiang, M.C.; Yang, P.H.; Lu, J.J. Genomic characterization of serotype III/ST-17 Group B Streptococcus strains with antimicrobial resistance using whole genome sequencing. Biomedicines 2021, 9, 1477. [Google Scholar] [CrossRef]
- Li, S.; Huang, J.; Chen, Z.; Guo, D.; Yao, Z.; Ye, X. Antibiotic prevention for maternal group B Streptococcal colonization on neonatal GBS-related adverse outcomes: A meta-analysis. Front. Microbiol. 2017, 8, 374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, S.; Dukhovny, D.; Mao, W.; Eichenwald, E.C.; Puopolo, K.M. 2010 perinatal GBS prevention guideline and resource utilization. Pediatrics 2014, 133, 196–203. [Google Scholar] [CrossRef] [Green Version]
- Creti, R.; Imperi, M.; Berardi, A.; Pataracchia, M.; Recchia, S.; Alfarone, G.; Baldassarri, L. Neonatal group B streptococcus infections: Prevention strategies, clinical and microbiologic characteristics in 7 years of surveillance. Pediatr. Infect. Dis. J. 2017, 36, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Alhhazmi, A.; Hurteau, D.; Tyrrell, G.J. Epidemiology of invasive group B streptococcal disease in Alberta, Canada, from 2003 to 2013. J. Clin. Microbiol. 2016, 54, 1774–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Wen, G.; Cao, X.; Guo, D.; Yao, Z.; Wu, C.; Ye, X. Molecular characteristics of Streptococcus agalactiae in a mother-baby prospective cohort study: Implication for vaccine development and insights into vertical transmission. Vaccine 2018, 36, 1941–1948. [Google Scholar] [CrossRef]
- Guan, X.; Mu, X.; Ji, W.; Yuan, C.; He, P.; Zhang, L.; Huang, Y.; Li, J.; Chen, J.; Zhong, H.; et al. Epidemiology of invasive group B streptococcal disease in infants from urban area of South China, 2011–2014. BMC Infect. Dis. 2018, 18, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morozumi, M.; Wajima, T.; Takata, M.; Iwata, S.; Ubukata, K. Molecular characteristics of Group B Streptococci isolated from adults with invasive infections in Japan. J. Clin. Microbiol. 2016, 54, 2695–2700. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Sapugahawatte, D.; Yang, Y.; Wong, K.; Lo, N.; Ip, M. Multidrug-Resistant Streptococcus agalactiae Strains Found in Human and Fish with High Penicillin and Cefotaxime Non-Susceptibilities. Microorganisms 2020, 8, 1055. [Google Scholar] [CrossRef]
- Hazafa, A.; Mumtaz, M.; Farooq, M.F.; Bilal, S.; Chaudhry, S.N.; Firdous, M.; Naeem, H.; Ullah, M.O.; Yameen, M.; Mukhtiar, M.S.; et al. CRISPR/Cas9: A powerful genome editing technique for the treatment of cancer cells with present challenges and future directions. Life Sci. 2020, 263, 118525. [Google Scholar] [CrossRef]
- Zhang, F.; Wen, Y.; Guo, X. CRISPR/Cas9 for genome editing: Progress, implications and challenges. Hum. Mol. Genet. 2014, 23, R40–R46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Zhang, L.; Huang, X. Genome modification by CRISPR/Cas9. FEBS J. 2014, 281, 5186–5193. [Google Scholar] [CrossRef]
- Zhang, C.; Quan, R.; Wang, J. Development and application of CRISPR/Cas9 technologies in genomic editing. Hum. Mol. Genet. 2018, 27, R79–R88. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Shin, J.; Cho, B.-K. Applications of CRISPR/Cas System to Bacterial Metabolic Engineering. Int. J. Mol. Sci. 2018, 19, 1089. [Google Scholar] [CrossRef] [Green Version]
- Spencer, B.L.; Deng, L.; Patras, K.A.; Burcham, Z.M.; Sanches, G.F.; Nagao, P.E.; Doran, K.S. Cas9 contributes to group B Streptococcal colonization and diseases. Front. Microbiol. 2019, 10, 1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauruelle, C.; Pastuszka, A.; Mereghetti, L.; Lanotte, P. Group B Streptococcus vaginal carriage in pregnant women as deciphered by clustered regularly interspaced short palindromic repeats analysis. J. Clin. Microbiol. 2018, 56, e01949-17. [Google Scholar] [CrossRef] [Green Version]
- Beauruelle, C.; Pastuszka, A.; Horvath, P.; Perrotin, F.; Mereghetti, L.; Lanotte, P. CRISPR: A Useful Genetic Feature to Follow Vaginal Carriage of Group B Streptococcus. Front. Microbiol. 2017, 8, 1981. [Google Scholar] [CrossRef]
- Lopez-Sanchez, M.J.; Sauvage, E.; Da Cunha, V.; Clermont, D.; Ratsima Hariniaina, E.; Gonzalez-Zorn, B.; Poyart, C.; Rosinski-Chupin, I.; Glaser, P. The highly dynamic crispr1 system of streptococcus agalactiae controls the diversity of its mobilome. Mol. Microbiol. 2012, 85, 1057–1071. [Google Scholar] [CrossRef]
- Lier, C.; Baticle, E.; Horvath, P.; Haguenoer, E.; Valentin, A.-S.; Glaser, P.; Mereghetti, L.; Lanotte, P. Analysis of the type II-A CRISPR-Cas system of Streptococcus agalactiae reveals distinctive features according to genetic lineages. Front. Genet. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGinn, J.; Marraffini, L.A. Molecular mechanisms of CRISPR–Cas spacer acquisition. Nat. Rev. Genet. 2018, 17, 7–12. [Google Scholar] [CrossRef]
- Renard, A.; Barbera, L.; Courtier-Martinez, L.; Dos Santos, S.; Valentin, A.-S.; Mereghetti, L.; Quentin, R.; Van Der Mee-Marquet, N.L. phiD12-Like Livestock-Associated Prophages Are Associated with Novel Subpopulations of Streptococcus agalactiae Infecting Neonates. Front. Cell. Infect. Microbiol. 2019, 9, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi-Jassir, F.; Paul, P.; To, K.-N.; Carreras-Abad, C.; Seale, A.C.; Jauneikaite, E.; Madhi, S.A.; Russell, N.J.; Hall, J.; Madrid, L.; et al. Systematic review of Group B Streptococcal capsular types, sequence types and surface proteins as potential vaccine candidates. Vaccine 2020, 38, 6682–6694. [Google Scholar] [CrossRef] [PubMed]
- Vuillemin, X.; Hays, C.; Plainvert, C.; Dmytruk, N.; Louis, M.; Touak, G.; Saint-Pierre, B.; Adoux, L.; Letourneur, F.; Frigo, A.; et al. Invasive group B Streptococcus infections in non-pregnant adults: A retrospective study, France, 2007–2019. Clin. Microbiol. Infect. 2020, 27, 129.e1–129.e4. [Google Scholar] [CrossRef]
- Hargreaves, K.R.; Flores, C.O.; Lawley, T.D.; Clokie, M.R.J. Abundant and Diverse Clustered Regularly Interspaced Short Palindromic Repeat Spacers in Clostridium difficile Strains and Prophages Target Multiple Phage Types within This Pathogen. mBio 2014, 5, e01045-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.L. Genomic Insights Into the Distribution and Evolution of Group B Streptococcus. Front. Microbiol. 2019, 10, 1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, E.; Pedroso-Roussado, C.; Cristino, J.M.; Ramirez, M.; The Portuguese Group for the Study of Streptococcal Infections; Oliveira, H.; Vaz, T.; Gião, M.; Ferreira, R.; Silva, A.C.; et al. Streptococcus agalactiae Causing Neonatal Infections in Portugal (2005–2015): Diversification and Emergence of a CC17/PI-2b Multidrug Resistant Sublineage. Front. Microbiol. 2017, 8, 499. [Google Scholar] [CrossRef]
- Gajic, I.; Plainvert, C.; Kekic, D.; Dmytruk, N.; Mijac, V.; Tazi, A.; Glaser, P.; Ranin, L.; Poyart, C.; Opavski, N. Molecular epidemiology of invasive and non-invasive group B Streptococcus circulating in Serbia. Int. J. Med Microbiol. 2018, 309, 19–25. [Google Scholar] [CrossRef]
- Pastuszka, A.; Beauruelle, C.; Camiade, E.; Rousseau, G.M.; Moineau, S.; Mereghetti, L.; Horvath, P.; Lanotte, P. Functional Study of the Type II-A CRISPR-Cas System of Streptococcus agalactiae Hypervirulent Strains. CRISPR J. 2021, 4, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Mosterd, C.; Rousseau, G.M.; Moineau, S. A short overview of the CRISPR-Cas adaptation stage. Can. J. Microbiol. 2021, 67, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gori, A.; Harrison, O.B.; Mlia, E.; Nishihara, Y.; Chan, J.M.; Msefula, J.; Mallewa, M.; Dube, Q.; Swarthout, T.D.; Nobbs, A.H.; et al. Pan-GWAS of Streptococcus agalactiae highlights lineage-specific genes associated with virulence and Niche adaptation. mBio 2020, 11, e00728-20. [Google Scholar] [CrossRef] [PubMed]
- Campisi, E.; Rosini, R.; Ji, W.; Guidotti, S.; Rojas-López, M.; Geng, G.; Deng, Q.; Zhong, H.; Wang, W.; Liu, H.; et al. Genomic analysis reveals multi-drug resistance clusters in group B Streptococcus CC17 hypervirulent isolates causing neonatal invasive disease in southern mainland China. Front. Microbiol. 2016, 7, 1265. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.-H.; Hsu, J.-F.; Lai, M.-Y.; Lin, L.-C.; Chu, S.-M.; Huang, H.-R.; Chiang, M.-C.; Fu, R.-H.; Lu, J.-J. Molecular Characteristics and Antimicrobial Resistance of Group B Streptococcus Strains Causing Invasive Disease in Neonates and Adults. Front. Microbiol. 2019, 10, 264. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.; Tsai, M.H.; Lai, M.Y.; Chu, S.M.; Huang, H.R.; Chiang, M.C.; Fu, R.H.; Lu, J.J.; Hsu, J.F. Emerging serotype III sequence type 17 group B streptococcus invasive infection in infants: The clinical characteristics and impacts on outcomes. BMC Infect. Dis. 2019, 19, 538. [Google Scholar] [CrossRef]
- Björnsdóttir, E.; Martins, E.; Erlendsdóttir, H.; Haraldsson, G.; Cristino, J.M.; Kristinsson, K.; Ramirez, M. Changing epidemiology of group B streptococcal infections among adults in Iceland: 1975–2014. Clin. Microbiol. Infect. 2015, 22, 379–e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing, Twenty-Fourth Informational Supplement, M100-S24; SLCI: Wayne, MI, UAS, 2014. [Google Scholar]
- Metcalf, B.J.; Chochua, S.; Gertz, R.E., Jr.; Hawkins, P.A.; Ricaldi, J.; Li, Z.; Walker, H.; Tran, T.; Rivers, J.; Mathis, S.; et al. Short-read whole genome sequencing for determination of antimicrobial resistance mechanisms and capsular serotypes of current invasive Streptococcus agalactiae recovered in the USA. Clin. Microbiol. Infect. 2017, 23, 574.e7–574.e14. [Google Scholar] [CrossRef] [Green Version]
- Shelburne, S.A.; Sahasrabhojane, P.; Saldaña, M.; Yao, H.; Su, X.; Horstmann, N.; Thompson, E.; Flores, A.R. Streptococcus mitisStrains Causing Severe Clinical Disease in Cancer Patients. Emerg. Infect. Dis. 2014, 20, 762–771. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Length of CRISPR-RFLP (bp) | No. of Isolates (%) | Fragment Length (bp) | No. of Isolates (%) | CRISPR1 Spacers | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ~1660 | 1 (1) | ~1250 | ~270 | ~140 | 1 | (1) | 476 | 449 | S2 | S3 | 398 | S4 | 119 | 120 | 82 | 82 | 83 | 84 | 85 | 86 | 87 | 7 |
| ~1360 | 2 (1.9) | ~950 | ~270 | ~140 | 2 | (1.9) | S5 | S11 | S12 | S13 | S14 | S15 | 101 | 102 | 49 | 53 | 54 | 55 | 56 | |||
| ~1310 | 2 (1.9) | ~900 | ~270 | ~140 | 2 | (1.9) | S11 | S12 | S13 | S14 | S15 | 101 | 102 | 49 | 53 | 54 | 55 | 56 | ||||
| ~1290 | 2 (1.9) | ~880 | ~270 | ~140 | 1 | (1) | 277 | S6 | 984 | 254 | 101 | 102 | 49 | 53 | 54 | 55 | 56 | |||||
| 1 | (1) | S7 | 724 | S8 | 243 | 101 | 102 | 49 | 53 | 54 | 55 | 56 | ||||||||||
| ~1230 | 23 (22.3) | ~820 | ~270 | ~140 | 22 | (21.4) | 171 | 984 | 254 | 101 | 102 | 49 | 53 | 54 | 55 | 56 | ||||||
| 1 | (1) | S6 | 984 | 254 | 101 | 102 | 49 | 53 | 54 | 55 | 56 | |||||||||||
| ~1160 | 27 (26.2) | ~750 | ~270 | ~140 | 14 | (13.6) | 984 | 254 | 101 | 102 | 49 | 53 | 54 | 55 | 56 | |||||||
| 3 | (2.9) | 171 | 984 | 254 | 101 | 102 | 53 | 54 | 55 | 56 | ||||||||||||
| 3 | (2.9) | S18 | 243 | 101 | 102 | 49 | 53 | 54 | 55 | 56 | ||||||||||||
| 2 | (1.9) | S9 | 243 | 101 | 102 | 49 | 53 | 54 | 55 | 56 | ||||||||||||
| 2 | (1.9) | S17 | 243 | 101 | 102 | 49 | 53 | 54 | 55 | 56 | ||||||||||||
| 1 | (1) | 171 | 984 | 254 | 102 | 49 | 53 | 54 | 55 | 56 | ||||||||||||
| 1 | (1) | S19 | 243 | 101 | 102 | 49 | 53 | 54 | 55 | 56 | ||||||||||||
| 1 | (1) | S11 | S12 | 101 | 102 | 49 | 53 | 54 | 55 | 56 | ||||||||||||
| ~1090 | 21 (20.4) | ~680 | ~270 | ~140 | 20 | (19.4) | 243 | 101 | 102 | 49 | 53 | 54 | 55 | 56 | ||||||||
| 1 | (1) | S10 | S1 | S12 | S13 | S14 | S15 | 101 | 56 | |||||||||||||
| ~1030 | 4 (3.9) | ~620 | ~270 | ~140 | 1 | (1) | 243 | 101 | 102 | 49 | 53 | 54 | 55 | |||||||||
| 1 | (1) | 101 | 102 | 49 | 53 | 54 | 55 | 56 | ||||||||||||||
| 1 | (1) | 243 | 102 | 49 | 53 | 54 | 55 | 56 | ||||||||||||||
| 1 | (1) | 243 | 101 | 49 | 53 | 54 | 55 | 56 | ||||||||||||||
| ~990 | 5 (4.9) | ~580 | ~270 | ~140 | 2 | (1.9) | 724 | 49 | 53 | 54 | 55 | 56 | ||||||||||
| 1 | (1) | 243 | 101 | 102 | 49 | 55 | 56 | |||||||||||||||
| 1 | (1) | 171 | 49 | 53 | 54 | 55 | 56 | |||||||||||||||
| 1 | (1) | 243 | 101 | 102 | 54 | 55 | 56 | |||||||||||||||
| ~890 | 12 (11.7) | ~480 | ~270 | ~140 | 12 | (11.7) | 49 | 53 | 54 | 55 | 56 | |||||||||||
| ~840 | 4 (3.9) | ~430 | ~270 | ~140 | 4 | (3.9) | 171 | 984 | 254 | 56 | ||||||||||||
| Group | A | B | C |
|---|---|---|---|
| Case number, n (% of total) | 22 (21.4) | 34 (33.0) | 47 (45.6) |
| RFLP patterns | |||
| <1000 bp | 15 (68.2) | 2 (5.9) | 4 (8.5) |
| 1000–1300 bp | 3 (13.6) | 32 (94.1) | 42 (89.4) |
| >1300 bp | 4 (18.2) | 0 (0) | 1 (2.1) |
| Birth body weight (g) | 2914.8 ± 430.2 | 2880.0 ± 644.5 | 3002.6 ± 683.0 |
| Gestational age (weeks) | 38.5 ± 1.8 | 37.6 ± 3.1 | 38.0 ± 3.1 |
| Prematurity | 3 (13.6) | 8 (23.5) | 8 (17.0) |
| Sex (male/female) | 7 (31.8)/15 (68.2) | 24 (70.6)/10 (29.4) | 18 (38.3)/29 (61.7) |
| Any chronic comorbidity | 1 (4.5) | 3 (8.8) | 5 (10.6) |
| Antibiotic susceptibility | |||
| Erythromycin (R) | 17 (77.3) | 2 (82.4) | 47 (100.0) |
| Clindamycin (R) | 15 (68.2) | 2 (70.6) | 47 (100.0) |
| Clinical presentations | |||
| Severe sepsis * | 14 (63.6) ** | 7 (20.6) | 18 (38.3) |
| Meningitis | 11 (50.0) ** | 6 (17.6) | 12 (25.5) |
| Neurological sequelae | 6 (27.3) | 4 (11.8) | 6 (12.8) |
| Final mortality | 1 (4.5) | 1 (0) | 2 (4.3) |
| Spacer | Sequence | Homology Analysis | Homology Percentage | Function |
|---|---|---|---|---|
| Viral DNA (phages), 65.1% (n = 28) | ||||
| 49 | TGCTAAAAGGTAAATTTAACATTCCAGGTA | Streptococcus phage LF2 | 100% | major tail protein |
| 53 | TATTTGATAGCGGTAACGGGTCATATACAA | Streptococcus phage Javan284 | 76% | putative C5 methylase |
| 54 | TGGTGGTATTTATAATGTACGAGCAAATCG | Streptococcus phage Javan52 | 100% | tail fibers protein |
| 55 | GATAAAAAGTGGGAGCTGAATTAAAAGGCA | Streptococcus phage Javan52 | 100% | hypothetical protein |
| 56 | ATTTGAACGATTTTTATATTCCTGATATGT | Streptococcus phage Javan516 | 100% | lysin, N-acetylmuramoyl-l-alanine amidase |
| 82 | CGTACCATCTATCAATTTACCGCAAGCTGT | Streptococcus phage LF2 | 100% | hypothetical protein |
| 83 | CCGATTATTTCCTACATAATACGCACGTTT | Streptococcus phage LF2 | 100% | hypothetical protein |
| 84 | AACTGTGTGATACTTTTCGTTTTTTTCTTT | Streptococcus phage Javan7 | 100% | hypothetical protein |
| 85 | TTTTATAAGTGATAGAGTGTGCAACACCGT | Streptococcus phage JX01 | 100% | putative minor structural protein |
| 87 | TTCTAAATGCTGGTGACTGCTTTGCATAAA | Streptococcus phage LF2 | 100% | hypothetical protein |
| 119 | GCGATGATGGTAAGTCATCATGGACAGCGT | Streptococcus phage Javan48 | 100% | tail fibers protein |
| 120 | TTTTACACACGATGTCAGATATAATGTCAA | Streptococcus phage Javan10 | 100% | membrane protein |
| 243 | TTGACCGCTCGTCCATTTTTTTAATGTAAA | Streptococcus phage Javan48 | 100% | tail fibers protein |
| 254 | ACCTTGCTCCGATGACACCATCGCGAACCT | Streptococcus phage Javan52 | 100% | tail fibers protein |
| 277 | AATTGATTGCCGTTAAAACCGATAGAGGA | Streptococcus phage LF2 | 100% | structural protein |
| 449 | TAAAATCCTGAAACAGAATGGGATTGATAT | Streptococcus phage Javan90 | 82% | antirepressor protein |
| S1 | AATTGATACATTGCAACGTCTAGCAGGAGC | Streptococcus phage LF2 | 100% | tail length tape-measure protein |
| S2 | GTGTGTTCTTCATTTTTATCAAACCAAAA | Streptococcus phage Javan471 | 100% | hypothetical protein |
| S3 | AAGAAATTCGGTAGAGACCCCAGACTCAT | Streptococcus phage Javan46 | 100% | tail length tape-measure protein |
| S6 | ATTAAATCTTCTTTTGAAGTTACTGTACGT | Staphylococcus phage pSco-10 | 70% | hypothetical protein |
| S10 | TTTATATTGTTCAGAAGAATGCCGCAAAAA | Streptococcus phage Javan44 | 100% | Phage-associated protein |
| S11 | TGTGTACGTTGCCTTTCCGTCAGCACCAGC | Streptococcus phage Javan52 | 100% | tail fibers protein |
| S12 | CCATAAACTTGCCAGTAGATGTGTCACGCT | Streptococcus phage Javan648 | 100% | hypothetical protein |
| S13 | ACCATTCGAAGTAGCTAGTTTGATTTCGTA | Streptococcus phage LF2 | 100% | tail fibers protein |
| S14 | TGTCGATGGTGTTCAAATACAAATGTTTTC | Streptococcus phage Javan52 | 100% | hypothetical protein |
| S15 | CTTTACCATTATTGATTTGTTCTTGCTTTT | Streptococcus phage Javan478 | 100% | hypothetical protein |
| S17 | TCGCAATAATTACTATATGCTTAAGCGGAG | Streptococcus phage Javan7 | 96% | Phage protein |
| S18 | AATCCAACAAAAACAACTTGCTTTAAATAA | Streptococcus phage Javan48 | 100% | membrane protein |
| Plasmid, 2.3% (n = 1) | ||||
| 102 | CTGTTCATAAAGAGCAACTAGTGGCAACAT | Bacillus megaterium strain YC4-R4 plasmid unnamed2 | 70% | hypothetical protein |
| Chromosomal sequences, 18.6% (n = 8) | ||||
| 7, 86, 101, 171, 476, 984, S4, S7 | GBS chromosome | x | x | |
| Unmatched, 14.0% (n = 6) | ||||
| 398, 724, S5, S8, S9, S19 | Unmatch | x | x | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, J.-F.; Lu, J.-J.; Lin, C.; Chu, S.-M.; Lin, L.-C.; Lai, M.-Y.; Huang, H.-R.; Chiang, M.-C.; Tsai, M.-H. Clustered Regularly Interspaced Short Palindromic Repeat Analysis of Clonal Complex 17 Serotype III Group B Streptococcus Strains Causing Neonatal Invasive Diseases. Int. J. Mol. Sci. 2021, 22, 11626. https://doi.org/10.3390/ijms222111626
Hsu J-F, Lu J-J, Lin C, Chu S-M, Lin L-C, Lai M-Y, Huang H-R, Chiang M-C, Tsai M-H. Clustered Regularly Interspaced Short Palindromic Repeat Analysis of Clonal Complex 17 Serotype III Group B Streptococcus Strains Causing Neonatal Invasive Diseases. International Journal of Molecular Sciences. 2021; 22(21):11626. https://doi.org/10.3390/ijms222111626
Chicago/Turabian StyleHsu, Jen-Fu, Jang-Jih Lu, Chih Lin, Shih-Ming Chu, Lee-Chung Lin, Mei-Yin Lai, Hsuan-Rong Huang, Ming-Chou Chiang, and Ming-Horng Tsai. 2021. "Clustered Regularly Interspaced Short Palindromic Repeat Analysis of Clonal Complex 17 Serotype III Group B Streptococcus Strains Causing Neonatal Invasive Diseases" International Journal of Molecular Sciences 22, no. 21: 11626. https://doi.org/10.3390/ijms222111626
APA StyleHsu, J. -F., Lu, J. -J., Lin, C., Chu, S. -M., Lin, L. -C., Lai, M. -Y., Huang, H. -R., Chiang, M. -C., & Tsai, M. -H. (2021). Clustered Regularly Interspaced Short Palindromic Repeat Analysis of Clonal Complex 17 Serotype III Group B Streptococcus Strains Causing Neonatal Invasive Diseases. International Journal of Molecular Sciences, 22(21), 11626. https://doi.org/10.3390/ijms222111626